

Abstract

A series of new CN-bridged coordination networks of different dimensionality and topology was obtained through the modification of reaction conditions between [Ni(cyclam)]2+ (cyclam = 1,4,8,11-tetraazacyclotetradecane) and [W(CN)8]4–. The factors determining the reaction pathway are temperature and addition of the LiCl electrolyte. The products include three negatively charged frameworks incorporating Li+ guests: the 1D Li2[Ni(cyclam)][W(CN)8]·6H2O (1) straight chain, the 1D Li2[Ni(cyclam)][W(CN)8]·2H2O (2) zigzag chain, and the 2D Li2[Ni(cyclam)]3[W(CN)8]2·24H2O (3) honeycomb-like network, as well as the 3D two-fold interpenetrating [Ni(cyclam)]5[Ni(CN)4][W(CN)8]2·11H2O (4) network and the 1D [Ni(cyclam)][Ni(CN)4]·2H2O (5) chain, which result from partial decomposition of the starting complexes. Together with the previously characterized 3D [Ni(cyclam)]2[W(CN)8]·16H2O (6) network, they constitute the largest family of CN-bridged coordination polymers obtained from the same pair of building blocks. All compounds exhibit paramagnetic behavior because of the separation of paramagnetic nickel(II) centers through the diamagnetic polycyanidometallates. However, the presence of the photomagnetically active octacyanidotungstate(IV) ions allowed observation of the magnetic superexchange after the violet light excitation (405 nm) for compound 3, which constitutes the first example of the photomagnetic effect in a NiII–[WIV(CN)8] system. The photomagnetic investigations for fully hydrated and dehydrated sample of 3, as well as for the isostructural octacyanidomolybdate(IV)-based network are discussed.
Short abstract
Six coordination networks of different dimensionality and topology can be obtained from the same pair of building blocks: [Ni(cyclam)]2+ and [W(CN)8]4− depending on reaction conditions. The negatively charged 2D Li2[Ni(cyclam)]3[W(CN)8]2·nH2O microporous network is the first example of the photomagnetic effect in a NiII−[WIV(CN)8] system.
Introduction
Multifunctionality at the molecular level is an appealing pathway to miniaturization. Molecule-based systems comprising organic and inorganic fragments organized into extended structures by chemical bonds and intermolecular interactions may combine a wide range of desired properties including magnetic order,1 photosensitivity,2 conductivity,3 luminescence,4 or optical activity.5 Cyanido-bridged coordination polymers are an important and versatile group of functional molecule-based materials.6 Cyanide ligands allow easy design of hybrid organic–inorganic polynuclear assemblies via the building block approach. Organic ligands may introduce additional properties and can be used to manipulate topology and dimensionality, enabling formation of discrete polynuclear structures, chains, and layers, as well as intricate 3D architectures.7 At the same time, cyanide ligands mediate relatively strong interactions between paramagnetic metal centers giving rise to interesting magnetic properties.8
Switching of magnetization by visible light is one of the desirable functionalities that can be achieved in molecule-based magnetics. In contrast to classical magnets, molecular systems show great versatility and chemical susceptibility and can be obtained as optically transparent single crystals. The photomagnetic effect is realized by photoexcitation to metastable spin states, which is often connected with modification of existing or appearance of new magnetic exchange pathways. Photomagnetic behavior of octacyanidomolybdate(IV)-based systems was first discovered in a bimetallic MnII–MoIV chain and interpreted in terms of irreversible MoIV photo-oxidation.9 In a series of CuII–MoIV assemblies, the mechanism of photo-induced charge transfer was proposed.10,11 An alternative concept of MoIV-centered spin transition from singlet to triplet was proposed to explain the photomagnetic behavior of the ZnII2MoIV trinuclear molecule.12 The excited triplet state trapping by reversible photodissociation of CN was proved to occur in [MoIV(CN)8]4– salts.13 The octacyanidotungstate(IV) ion was found to show similar photo-induced spin transition to its MoIV congener in the hexanuclear MnII–MIV and FeII–MIV (M = Mo, W) clusters.14,15 In the latter compounds, site-selective photoswitching on both FeII and MIV metal centers could be achieved by using irradiation with different wavelengths.15 Photo-induced ferrimagnetic order with Tc of 93 K was found in the 3D {[MnII(Him)]2[WIV(CN)8]} framework.16 Interestingly, the photomagnetic effect in this network can be deactivated by the absorption of moisture. However, the [WIV(CN)8]4–-based systems are still sparsely investigated in terms of the photomagnetic effect in comparison to those of [MoIV(CN)8]4–.1 Moreover, NiII–[WIV(CN)8] photomagnetic systems were not reported previously.
The construction of molecule-based materials is usually carried out using the building block self-assembly method. By taking into account the building block characteristics, in particular the geometry, charge, coordination environment of metal centers, and potential ability to create new coordination connections, certain predictions regarding the product dimensionality and topology can be made.17,18 In addition to strong coordination bonds, the presence of weaker intermolecular interactions including hydrogen bonds and/or pi-stacking, greatly affects the resulting product structure.19−22 Selection of an appropriate solvent as well as time and temperature of the reaction is not less important.23−25 Also, the presence and concentration of an additional electrolyte may strongly influence the course of the crystallization process, by increasing the ionic strength of the solution. It often results in the increase of the building blocks’ solubility and the slower crystallization process, which can lead to the formation of larger and better quality product crystals. Moreover, the presence of guest molecules and ions in the solution often results in their incorporation into the product structure, particularly in the case of microporous systems, which can result in the change of network dimensionality or topology.
In our previous studies, we have presented the versatility of the [Ni(cyclam)]2+/3+ (cyclam = 1,4,8,11-tetraazacyclotetradecane) cationic building block using it as a linear linker in combination with various polycyanidometallates. It afforded numerous coordination networks differing in dimensionality and topology with a wide range of desirable properties, including long-range magnetic order, microporosity, sorption ability, solvatomagnetic effect,26−28 ionic conductivity,29,30 and presence of multi-switchable electron-transfer phase transitions with a memory effect.31 We have also shown the possibilities of manipulation of topology by incorporation of guest ions or changing reaction conditions.32,33 In this work, we present an unprecedented success of crystal engineering: six different networks of varied topologies obtained from [Ni(cyclam)]2+ and [W(CN)8]4– building blocks with addition of LiCl by varying reactants’ concentrations and reaction temperature. Moreover, we present the first investigation of the photomagnetic effect for a NiII–WIV network, as well as its NiII–MoIV analogue.
Experimental Section
Materials
The K4[W(CN)8]·2H2O precursor complex was synthesized according to the literature procedure.34 [Ni(cyclam)(NO3)2] was prepared according to the modified published method.35 Ni(NO3)2·6H2O (535.0 mg, 1.84 mmol) was dissolved in boiling absolute ethanol (10 mL). To the above solution, solid cyclam (368.6 mg, 1.84 mmol) was added which resulted in the immediately precipitating violet product. The suspension was stirred for 10 min and left in the freezer for 1 h (−18 °C). The product was filtered and washed with a small amount of cold acetone (4 °C). Yield: 701.0 mg (99.4%). All other reagents and solvents were commercially available and used as supplied.
Synthesis of Compounds
Li2[Ni(cyclam)][W(CN)8]·6H2O (1)
A water solution (5 mL) of [Ni(cyclam)(NO3)2] (19.2 mg, 0.05 mmol) and LiCl (2.5 g, 58.97 mmol), heated to 46 °C, was added to a water solution (5 mL) containing K4[W(CN)8]·2H2O (29.2 mg, 0.05 mmol) and LiCl (2.5 g, 58.97 mmol) heated to 46 °C. The yellow solution was left for crystallization at room temperature. After 24 h, the yellow plate-shaped crystals of 1 were formed. In order to perform the elemental analysis and IR spectra, the product was filtered and washed with small amount of LiCl water solution (5 mL, 2.5 g) because the compound is unstable in pure water. EA: found: C, 26.23; N, 20.04; H, 4.65, calculated for: C18H38ClLi3N12Ni1O7W1 (Li2[Ni(cyclam)][W(CN)8]·6H2O + LiCl·H2O): C, 25.94; N, 20.17; H, 4.60. IR ν(CN): 2114.7, 2128.2 cm–1. The purity of 1 was additionally confirmed by powder X-ray diffraction measurement for the crystalline sample in LiCl water solution (5 mL, 2.5 g).
Li2[Ni(cyclam)][W(CN)8]·2H2O (2)
A water solution (5 mL) of [Ni(cyclam)(NO3)2] (19.2 mg, 0.05 mmol) and LiCl (2.5 g, 58.97 mmol), heated to 105 °C, was added to a water solution (5 mL) of K4[W(CN)8]·2H2O (29.2 mg, 0.05 mmol) and LiCl (2.5 g, 58.97 mmol) heated to 105 °C. The resulting yellow solution was slowly cooled to 40 °C and left for crystallization in a water bath (40 °C). After 24 h, the yellow needle-shaped crystals of 2 were formed. In order to perform the elemental analysis and IR spectra, the product was filtered and washed with small amount of LiCl water solution (5 mL, 2.5 g), due to decomposition that takes place upon even short contact with pure water. EA: found: C, 24.56; N, 19.03; H, 4.82, calculated for: C18H38Cl2Li4N12Ni1O7W1 (Li2[Ni(cyclam)][W(CN)8]·2H2O + 2LiCl·5H2O)): C, 24.69; N, 19.19; H, 4.37. IR ν(CN): 2114.7, 2127.2, 2136.8 cm–1. The purity of 2 was additionally confirmed by powder X-ray diffraction measurement for the crystalline sample in LiCl water solution (5 mL, 2.5 g).
Li2[Ni(cyclam)]3[W(CN)8]2·24H2O (3)
A water solution (10 mL) of [Ni(cyclam)(NO3)2] (9.6 mg, 0.025 mmol) and LiCl (1 g, 23.59 mmol) was added at room temperature to a water solution (10 mL) of K4[W(CN)8]·2H2O (9.9 mg, 0.017 mmol) and LiCl (1 g, 23.59 mmol). The resulting suspension was left at room temperature and after 3 days yellow block-shaped crystals of 3 were formed. In order to perform the elemental analysis and IR spectra, the product was filtered, washed with small amount of cold water, and dried in air, which resulted in partial dehydration. EA: found: C, 27.78; N, 19.60; H, 5.82, calculated for: C46H118Li2N28Ni3O23W2 (Li2[Ni(cyclam)]3[W(CN)8]2·23H2O): C, 27.77; N, 19.72; H, 5.98. IR ν(CN): 2103.5, 2123.7 cm–1. The purity of 3 was additionally confirmed by powder X-ray diffraction measurement for the crystalline sample suspended in the LiCl solution of the same concentration as the mother liquor.
[Ni(cyclam)]5[Ni(CN)4][W(CN)8]2·11H2O (4)
A water solution (10 mL) of [Ni(cyclam)(NO3)2] (40.0 mg, 0.11 mmol) and LiCl (1 g, 23.59 mmol), heated to 84 °C, was added to a water solution (10 mL) of K4[W(CN)8]·2H2O (40.0 mg, 0.07 mmol) and LiCl (1 g, 23.59 mmol) heated to 84 °C. The resulting yellow solution was slowly cooled to 62 °C and left for crystallization in a water bath. After 2 days, the yellow block-shaped crystals of 4 were formed. In order to perform the elemental analysis and IR spectra, the product was filtered, washed with water, and dried in air, which resulted in partial dehydration. EA: found: C, 34.89; N, 23.16; H, 5.77, calculated for: C70H138N40Ni6O9W2 ([Ni(cyclam)]5[Ni(CN)4][W(CN)8]2·9H2O): C, 34.97; N, 23.31; H, 5.79. IR ν(CN): 2091.5, 2105.0, 2119.5, 2138.8 cm–1. The purity of 4 was additionally confirmed by powder X-ray diffraction measurement for the crystalline sample in water.
[Ni(cyclam)][Ni(CN)4]·2H2O (5)
The light pink block-shaped crystals of 5 suitable for single crystal X-ray diffraction were obtained from the filtrate of compound 4 after about 2 weeks at room temperature. Compound 5 was also obtained in an alternative way: a water solution (35 mL) of [Ni(cyclam)(NO3)2] (40.0 mg, 0.11 mmol) and KNO3 (5 g, 49.45 mmol), heated to 65 °C, was added to a water solution (35 mL) of K2[Ni(CN)4] (26.51 mg, 0.11 mmol) and KNO3 (5 g, 49.45 mmol) heated to 65 °C. The yellow solution was slowly cooled to 50 °C and after about 2 h the crystals of 5 were formed. The reaction solution was cooled to the room temperature. In order to perform the elemental analysis and IR spectra, the product was filtered, washed with water, and dried in air, which resulted in partial dehydration. EA: found: C, 39.64; N, 26.06; H, 5.71, calculated for: C14H25N8Ni2O0.5 ([Ni(cyclam)][Ni(CN)4]·0.5H2O): C, 39.03; N, 26.01; H, 5.85. IR ν(CN): 2116.6, 2128.2, 2140.7, 2159.0 cm–1. The purity of 5 was additionally confirmed by powder X-ray diffraction measurement for crystalline sample in water.
[Ni(cyclam)]2[W(CN)8]·16H2O (6)
[Ni(cyclam)]2[W(CN)8]·16H2O (6) was synthesized according to the published method.27
Li2[Ni(cyclam)]3[Mo(CN)8]2·24H2O (7)
Li2[Ni(cyclam)]3[Mo(CN)8]2·24H2O (7) was obtained according to the published method.33
Structure Determination
The single-crystal X-ray diffraction measurements for samples 1–5 were performed on a Bruker D8 Quest Eco diffractometer equipped with a Mo Kα radiation source (λ = 0.71073 Å). The structures were solved by direct methods with SHELXT and the refinement was performed using SHELXL.36 All non-hydrogen atoms were refined anisotropically. The C–H and N–H hydrogen atoms were placed in idealized positions and refined isotropically using the riding model with Uiso(H) equal to 1.2Ueq for the C- or N-atoms. H-atoms for heavily disordered water molecules in the structures of 3 and 4 were not considered but included in the empirical formula. In the structures of 1, 2, and 5, the H-atoms of water molecules were found from the electron density map and refined isotropically without restraints. The Li+ cations in the structures of 1, 2, and 3 were found from the electron density map. The presence of tetrahedral coordination environment was considered in the assignment of Li. Due to high absorption, which effect is not compensated by empirical multi-scan correction, some residual electron density peaks appear around W in the structure of 3. Graphical representations of the structures were prepared with Mercury CSD 4.3.1 software.37 The analysis of coordination polyhedra was performed using SHAPE 2.1.38 The crystallographic data are presented in Table 1.
Table 1. Selected Crystallographic Data for Compounds 1–5.
| compound | 1 | 2 | 3 | 4 | 5 |
| empirical formula | C18H36Li2N12NiO6W | C18H28Li2N12NiO2W | C46H120Li2N28Ni3O24W2 | C70H142N40Ni6O11W2 | C14H28N8Ni2O2 |
| FW | 773.03 | 700.96 | 2007.40 | 2440.19 | 457.86 |
| T (K) | 120 | 120 | 100 | 100 | 100 |
| crystal system | monoclinic | monoclinic | monoclinic | monoclinic | triclinic |
| space group | P2/n | C2/c | C2/m | P21/n | P1̅ |
| a (Å) | 10.3259(2) | 12.2145(10) | 26.2103(14) | 14.3208(6) | 7.7147(7) |
| b (Å) | 9.5362(2) | 14.1512(11) | 15.3800(8) | 14.8322(6) | 8.8748(8) |
| c (Å) | 15.5361(3) | 14.5322(12) | 10.1406(6) | 23.1077(9) | 15.7304(14) |
| α (deg) | 90 | 90 | 90 | 90 | 88.656(5) |
| β (deg) | 104.698(1) | 94.454(3) | 95.268(2) | 93.578(2) | 85.592(5) |
| γ (deg) | 90 | 90 | 90 | 90 | 74.727(5) |
| V (Å3) | 1479.78(5) | 2504.3(4) | 4070.6(4) | 4898.7(3) | 1035.89(16) |
| Z | 2 | 4 | 2 | 2 | 2 |
| ρcalc (mg/m3) | 1.735 | 1.859 | 1.638 | 1.654 | 1.468 |
| μ (mm–1) | 4.573 | 5.383 | 3.579 | 3.538 | 1.842 |
| F(000) | 768 | 1376 | 2044 | 2492 | 480 |
| crystal size (mm) | 0.30 × 0.11 × 0.07 | 0.50 × 0.05 × 0.05 | 0.20 × 0.12 × 0.06 | 0.26 × 0.21 × 0.04 | 0.14 × 0.13 × 0.09 |
| θ range (deg) | 3.45–30.6 | 3.35–28.7 | 2.43–25.1 | 2.59–30.5 | 2.38–27.9 |
| reflections collected | 23852 | 25358 | 16913 | 63270 | 20863 |
| independent | 4293 | 3229 | 3729 | 14257 | 4939 |
| observed | 3978 | 3096 | 3422 | 12089 | 3216 |
| parameters | 183 | 171 | 259 | 589 | 253 |
| Rint | 0.037 | 0.038 | 0.055 | 0.041 | 0.053 |
| GOF on F2 | 1.077 | 1.065 | 1.059 | 1.057 | 1.148 |
| R1 [I > 2σ(I)] | 0.0254 | 0.0162 | 0.0481 | 0.0386 | 0.0446 |
| wR2 (all data) | 0.0506 | 0.0384 | 0.1284 | 0.0821 | 0.0829 |
Physical Measurements
Powder X-ray diffraction data were collected on a PANalytical X’Pert PRO MPD diffractometer (samples 1–3 immersed in LiCl water solution of the same concentration as used in syntheses) and on a Bruker D8 Advance Eco diffractometer (samples immersed in pure water, stable in air, and dehydrated), both equipped with a Cu Kα radiation source (λ = 1.541874 Å), in the Debye–Scherrer geometry. The microcrystalline samples were sealed in glass capillaries and measured in the 5–50° 2θ range at room temperature. The reference powder patterns from SC-XRD measurements were generated using Mercury CSD 4.3.1 software.37 Elemental analyses of CHN were performed on an ELEMENTAR Vario Micro Cube CHNS analyzer. IR spectra in the range of 4000–500 cm–1 were collected on a Thermo Scientific Nicolet iN10 MX FTIR microscope operating in the transmission mode. Thermogravimetric analysis (TGA) was performed on a Mettler Toledo TGA/SDTA 851e analyzer under an argon atmosphere in a temperature range of 35–300 °C with a heating rate of 2 °C·min–1. The water sorption/desorption processes were characterized by the dynamic vapor sorption method using the SMS DVS Resolution apparatus. The isotherms for 3 and 4 were measured in a 0–94% relative humidity range at a temperature of 25 °C. Every measurement step was performed until a stable mass was achieved (dm/dt = 0.002 mg·min–1). Magnetic susceptibility measurements were performed on a Quantum Design MPMS-3 Evercool magnetometer in magnetic fields up to 70 kOe. The experimental data were corrected for diamagnetism of the sample and the sample holder. The fully hydrated microcrystalline samples were sealed in double polyethylene bags under pure water (4, 5) or lithium chloride water solution of the same concentration as used in syntheses (1, 2, 3, 7) to prevent dehydration. The dehydrated under dry nitrogen flow samples of 3deh and 7deh were protected with paraffin oil and placed in polycarbonate capsules under an argon atmosphere in a glovebox. Samples for photomagnetic measurements were ground to a fine powder and spread onto a colorless adhesive tape in form of a thin layer protected with a Scotch tape. The hydrated phases were additionally protected with small amount of lithium chloride solution and dehydrated forms were treated similarly to bulk samples. In order to prevent 3 and 7 from dehydration in the magnetometer sample chamber, the samples were inserted at 250 K and then vacuum-pumped. The irradiation was performed using 405 and 450 nm laser diodes (both wavelengths fall within the d–d absorption bands of [M(CN)8]4– (M = Mo, W) complexes).
Results and Discussion
Crystal Engineering
The reaction between [Ni(cyclam)]2+ and [W(CN)8]4– in water–methanol solution under slow diffusion conditions was found to lead to a 3D diamond-like [Ni(cyclam)]2[W(CN)8]·16H2O network.27 The fact that a quick precipitation reaction from the same solution led to a product with a different PXRD pattern was a clear indication of the existence of other possible reaction pathways. In order to investigate that possibility, we performed the reaction between the same building blocks in water solution with the addition of LiCl at various temperatures. All syntheses were preliminarily carried out at the 3:2 ratio between [Ni(cyclam)]2+ and [W(CN)8]4–. The addition of the LiCl electrolyte and elevated reaction temperature were expected to prevent instantaneous precipitation of the product and facilitate crystallization of the thermodynamically most stable network under given reaction conditions. As a result of applied modifications, five new compounds differing in network dimensionality and topology were obtained: a 1D Li2[Ni(cyclam)][W(CN)8]·6H2O (1) straight chain and a Li2[Ni(cyclam)][W(CN)8]·2H2O (2) zigzag chain, a 2D Li2[Ni(cyclam)]3[W(CN)8]2·24H2O (3) honeycomb-like network, a 3D two-fold interpenetrating [Ni(cyclam)]5[Ni(CN)4][W(CN)8]2·11H2O (4) network, and a 1D [Ni(cyclam)][Ni(CN)4]·2H2O (5) chain. In order to obtain pure products with good reproducibility, the synthetic conditions were optimized in terms of lithium chloride concentration, reaction temperature, and the building blocks’ ratio. The different reaction pathways are summarized in Figure 1.
Figure 1.

Scheme of reaction pathways between [Ni(cyclam)]2+ and [W(CN)8]4– building blocks leading to different topology networks; Ni: yellow balls, W: dark blue balls, bridging CN: gray and light blue balls, terminal ligands: sticks. Guest molecules and ions omitted for clarity.
In the case of compounds 1–3, the formation of a negatively charged coordination skeleton and incorporation of the Li+ cations into the structure is observed. The reaction carried out in highly concentrated LiCl solution (close to saturation at 20 °C) leads to the formation of 1D alternating bimetallic NiII-WIV chains. Depending on the reaction temperature straight (1) or zigzag shape (2) chain structures are formed. The optimized building block ratio for the synthesis of 1 and 2 is 1:1. The compounds are stable in the mother liquor or concentrated LiCl solution. However, they recrystallize immediately in contact with water, to form a neutral [Ni(cyclam)]2[W(CN)8]·nH2O network,27 with some unidentified additional phase in the case of 2, as shown by PXRD (Figures S1 and S2). Such high sensitivity to water is caused by the presence of the guest Li+ cations in the structure, which are easily removed by water, causing reorganization of the coordination network. We have observed similar behavior in related compounds containing guest cations.29,32,331 and 2 remain stable in air when filtered off, which was checked by repeated SC-XRD unit cell determination on dry crystals. Because they cannot be washed with water, dry compounds 1 and 2 contain small amount of hydrated LiCl on the surface. The elemental analysis results show contamination with lithium chloride at ca. 7% for 1 and 20% for 2. The difference may be caused by the smaller size of crystals of 2.
The 2D Li2[Ni(cyclam)]3[W(CN)8]2·24H2O (3) honeycomb-like network was obtained at room temperature with lower concentration of LiCl than in the case of 1D compounds 1 and 2. The stoichiometric 3:2 ratio between the building blocks was found optimal. The reaction initially resulted in fine yellow suspension, which after a few days recrystallized to yellow block-shaped crystals of 3. Crystals of 3 are stable in the mother liquor or LiCl solution of the same concentration (Figure S3), but disintegrate when placed in water. However, in contrast to compounds 1 and 2, sample of 3 remains crystalline when filtered off and washed with small amount of water. The elemental analysis has shown that drying in air resulted in the loss of one crystallization water molecule per formula unit. Further dehydration under dry nitrogen flow results in structural transformation, typical for related 2D networks,26,28 which is confirmed by changes in the PXRD pattern. The stability of 3 is similar to that of the analogous compound based on the [Mo(CN)8]4– building block,33 as well as the family of its isotypic Mx[Ni(cyclam)]3[Nb(CN)8]2·nH2O (M = NH4+, Li+, Na+, Mg2+, Ca2+, Sr2+, Ba2+, x = 1 or 2) congeners.29,32
The 3D two-fold interpenetrating [Ni(cyclam)]5[Ni(CN)4][W(CN)8]2·11H2O (4) network was obtained at the same LiCl concentration as compound 3, but the solutions were mixed at 84 °C and no initial precipitate was observed. The optimal building block ratio for the synthesis of 4 was found to be 1:1. Formation of 4 is the result of partial decomposition of the building blocks and the formation of the [Ni(CN)4]2– complex. In contrast to compounds 1–3, the lithium cations are not embedded in the structure of 4 and therefore its crystals are stable in water as shown by PXRD measurements (Figure S4). The crystals are stable when dried in air, but in the flow of dry nitrogen they disintegrate and small shifts of the PXRD peaks are visible, indicating that some structural changes take place upon dehydration.
If the crystals of 4 are filtered off and the filtrate is left at room temperature, an additional product of the formula [Ni(cyclam)][Ni(CN)4]·2H2O (5) is obtained after about 2 weeks in the form of light pink crystals. The same compound was obtained by a different method and structurally characterized earlier.39 We also obtained 5 in an alternative synthesis by mixing equimolar amounts of [Ni(cyclam)]2+ and [Ni(CN)4]2– dissolved in 1.4 mol·dm–3 potassium nitrate water solution at 65 °C. The purity of 5 and its stability in water was checked by PXRD measurements (Figure S5). Upon drying in air, compound 5 losses most of the crystallization water and undergoes structural transformation, which was confirmed by PXRD and EA.
Structure Description
1D Straight Chain of Li2[Ni(cyclam)][W(CN)8]·6H2O (1)
Li2[Ni(cyclam)][W(CN)8]·6H2O (1) crystallizes in the monoclinic system, the space group P2/n (Table 1). The asymmetric unit contains 1/2 of the [W(CN)8]4– ion located at the two-fold rotation axis, 1/2 of the [Ni(cyclam)]2+ ion at the center of inversion, as well as one Li+ cation, and three crystallization water molecules in general positions (Figure S6). The WIV ions are coordinated by eight cyanide ligands, two of which are connected to nickel centers and four to lithium cations. The geometry of the [W(CN)8]4– building block is close to an ideal square antiprism (D4d, CShM = 0.166, Table S1). The NiII ions, equatorially coordinated by four N-atoms of the cyclam ligand and axially by two N-atoms of CN bridges, show slightly distorted octahedral geometries (Oh, CShM = 0.172, Table S2), due to the elongation of the axial bonds and angular distortions caused by coordination of cyclam (Table S4). The lithium cations are surrounded by two water molecules and two cyanido ligands of neighboring octacyanidotungstate(IV) anions, in the geometry close to an ideal tetrahedron (Td, CShM = 0.780, Table S3). The structure of 1 consists of one-dimensional chains of alternating cyanido-bridged [Ni(cyclam)]2+ and [W(CN)8]4– moieties that run along the [−101] direction (Figure 2).
Figure 2.
Structure of 1. View along the [010] direction (a) and along the coordination chains in the [−101] crystallographic direction (b).
The coordination chains are slightly undulating with the Ni1–W1–Ni1 angle equal to 153.17°. The CN bridges are bent (160.0°) resulting in a distance between the Ni and W metal centers of 5.3258 Å (Table S4). The negatively charged chains are linked through lithium ions into layers located on the (020) crystallographic plane. Within the layer, the neighboring chains are separated by 7.49 Å and their relative shift is 7.11 Å along the [−101] direction, whereas the distance between supramolecular layers is equal to 9.54 Å. The presence of semi-coordination bonds between lithium cations and N-atoms of [W(CN)8]4– ions of neighboring chains has an impact on the distorted tetrahedral geometry of lithium centers. The distortion is also caused by the presence of hydrogen bonds between the water molecules connected to lithium centers of different layers that results in an O1–Li1–O2 angle equal to 99.39°, which is almost 10° less than for the ideal tetrahedron.
1D Zigzag Chain of Li2[Ni(cyclam)][W(CN)8]·2H2O (2)
Li2[Ni(cyclam)][W(CN)8]·2H2O (2) crystallizes in the monoclinic system, the space group C2/c. The asymmetric unit shown in Figure S7 is similar to compound 1. It is also composed of one-half of the [Ni(cyclam)]2+ complex in a special position at the center of inversion, one-half of the [W(CN)8]4– ion located at the two-fold axis, and one lithium cation in a general position. However, in contrast to 1 the asymmetric unit of 2 contains not three but only one crystallization water molecule in a general position. As in the structure of 1, the tungsten center is coordinated by eight cyanide ligands, two of which are connected to nickel ions, but instead of four, only two of the remaining ones are connected to lithium ions. The Ni1 and Li1 coordination environment is the same as in compound 1. CShM analysis (Tables S2 and S3) shows that for Ni1 octahedral geometry distortion is slightly higher than for 1 (Oh, CShM = 0.195). The geometry of the tungsten complex is different than in 1, instead of square antiprism (D4d, CShM = 1.820), the complex is closer to the triangular dodecahedron (D2d, CShM = 0.319). The structure of 2 also consists of one-dimensional chains that are formed of alternating [Ni(cyclam)]2+ and [W(CN)8]4– building blocks, but with the distinct zigzag shape (Figure 3) with the Ni1–W1–Ni1 angle of 87.52°. The CN bridges connected to nickel centers are also more bent (155.47°), which result in closer than in 1 distance between neighboring nickel and tungsten metal centers (5.2527 Å). The coordination chains run along the crystallographic c direction. Two lithium cations are connected by two water molecules forming [Li2(H2O)2]2+ units, which link the chains by semi-coordination bonds to terminal cyanido ligands, resulting in a three-dimensional supramolecular network.
Figure 3.

Structure of 2. View along the [100] direction showing the zigzag shape of the coordination chains (a) and view along the chains in the [001] direction (b).
2D Honeycomb-like Network of Li2[Ni(cyclam)]3[W(CN)8]2·24H2O (3)
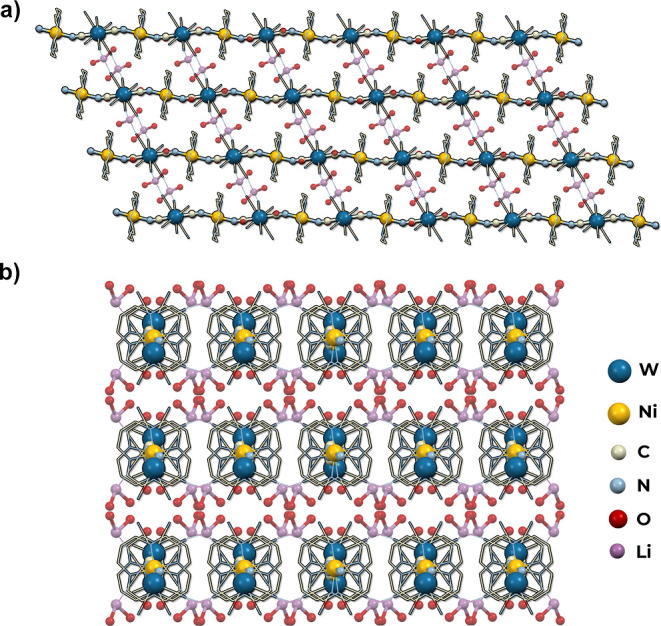
Li2[Ni(cyclam)]3[W(CN)8]2·24H2O (3) crystallizes in the monoclinic system, the space group C2/m. The asymmetric unit (Figure S8) is composed of one-half of the [W(CN)8]4– ion located in a special position on the mirror plane, one-half of the [Ni(cyclam)]2+ complex (Ni2) located at an inversion center, and one-quarter of the nickel complex (Ni1) located at the intersection of mirror plane and two-fold axis. It also contains the lithium ion, disordered over two positions, surrounded by four water molecules, one of which is located on a mirror plane. There are four other water molecules, one located in a general position (O5), two on a mirror plane (O7, O8), and one on a two-fold axis (O6). Each anionic [W(CN)8]4– building block is connected by CN bridges to three [Ni(cyclam)]2+ cations, acting as a three-connected network node. The coordination geometry of tungsten centers is approximately square antiprismatic (D4d, CShM = 0.396, Table S1). The nickel cations are equatorially coordinated by N-atoms of cyclam and connected to two tungsten centers through CN bridges, acting as cationic linear linkers of the geometry of slightly disordered octahedron (Oh, CShM = 0.124 and 0.159, Table S2). The cyanido bridges are bent with the angle of Ni–N–C below 160° (Table S4). The structure of 3 is composed of two-dimensional layers of honeycomb-like topology with Schläfli symbol 63 (Figure 4). The distance between parallel slightly corrugated layers located on the (201) planes is 7.65 Å. They are intersected by one-dimensional channels filled with crystallization water molecules and lithium cations that compensate the negative charge of the coordination skeleton. The channels take up 29.8% of the unit cell volume and run along in the c crystallographic direction, crossing the coordination layers at an angle of 49°, which is related to the relative relocation of the neighboring layers by 6.65 Å in the [10–2] crystallographic direction. Each lithium cation is surrounded by four water molecules in geometry close to an ideal tetrahedron (Td, CShM = 0.286, Table S3). The distortion is probably caused by one water molecule (O2) which is shared between two lithium cations resulting in elongation of the Li1–O2 distance. Moreover, there is a net of hydrogen bonds between water molecules, terminal cyanido ligands, and NH-groups of cyclam that additionally stabilize the network. The compound 3 is part of the isotypic family of honeycomb-like networks based on the [M(CN)8]4– (M = Nb, Mo) and [Ni(cyclam)]2+ complex containing guest ions (NH4+, Li+, Na+, Mg2+, Ca2+, Sr2+, Ba2+).29,32,33
Figure 4.

Structure of 3. View along the channels in the [001] direction (a) and view along the [010] direction showing parallel layers (b).
3D Two-fold Interpenetrating Network of [Ni(cyclam)]5[Ni(CN)4][W(CN)8]2·11H2O (4)
[Ni(cyclam)]5[Ni(CN)4][W(CN)8]2·11H2O (4) crystallizes in the monoclinic system, the space group P21/n. The coordination network is composed of three building units: cationic [Ni(cyclam)]2+, anionic [W(CN)8]4–, and [Ni(CN)4]2–. The asymmetric unit presented in Figure S9 consists of the [Ni(cyclam)]2+ cation (Ni2) and [Ni(CN)4]2– anion (Ni4) both of the occupancy of one-half in special positions on the inversion centers, as well as one [W(CN)8]4– anion, two [Ni(cyclam)]2+ cations (Ni1, Ni3), and five and a half crystallization water molecules in general positions. Each [W(CN)8]4– anion forms four cyanide bridges to the [Ni(cyclam)]2+ complexes, acting as a four-connected network node of the geometry close to an ideal triangular dodecahedron (D2d, CShM = 0.372, Table S1). The cationic [Ni(cyclam)]2+ complexes, axially bound by two CN bridges, show slightly disordered octahedral geometry (Table S2). The coordination of [Ni(CN)4]2– is close to an ideal square planar geometry (D4h, CShM = 0.185, Table S3). Both cationic and anionic complexes of nickel ions act as linear linkers. The structure of 4 consists of two separate 3D sub-networks of diamond-like topology with Schläfli symbol 66 (Figure 5). Each sub-network is composed of 2D honeycomb-like layers build of [Ni(cyclam)]2+ and [W(CN)8]4– building blocks that form hexagonal rings, much more distorted than in the case of compound 3, which is reflected in the low Ni–N–C angle values (Table S4). Thus, the whole two-fold structure could be described as the set of alternating layers that lay on the ⟨−101⟩ crystallographic planes. The layers of one sub-network are connected through long trimetallic linkers composed of {[Ni(cyclam)]2+–[Ni(CN)4]2––[Ni(cyclam)]2+} units. Those bridging units pass through the centers of hexagonal openings of the other sub-network. The distance between the honeycomb-like layers of one sub-network is equal to 25.01 Å. There are isolated cavities in the structure filled with crystallization water molecules that take up 10.1% of the unit cell volume. The water molecules create a net of hydrogen bonds connected with terminal CN ligands of polycyanidometallates. The compound 4 is isostructural with an analogous network based on the [Mo(CN)8]4– building block.33
Figure 5.

Structure of 4. View along the [010] direction (a) showing two interpenetrating sub-networks (one marked by yellow highlighting) and view of a single honeycomb-like layer on the (−101) plane with linkers of the second sub-network passing through the hexagonal ring (b). Crystallization water omitted for clarity.
1D Straight Chain of [Ni(cyclam)][Ni(CN)4]·2H2O (5)
[Ni(cyclam)][Ni(CN)4]·2H2O (5) crystallizes in the triclinic system, the space group P1̅. The structure is composed of cationic [Ni(cyclam)]2+ and anionic [Ni(CN)4]2– building blocks arranged in alternating chains. All metal ions occupy special positions at the centers of inversion. There are two independent positions of the [Ni(cyclam)]2+ complex (Ni2, Ni4) and two independent positions of the [Ni(CN)4]2– complex (Ni1, Ni3) (Figure S10). The structure also contains two water molecules in general positions. The cationic [Ni(cyclam)]2+ complexes, axially bound by N-atoms of cyanido ligands show the smallest distortion of octahedral geometry among structures 1–5 (Oh, CShM = 0.114 (Ni2) and 0.109 (Ni4), Table S2). The [Ni(CN)4]2– ions are characterized by almost an ideal square planar geometry (D4h, CShM = 0.003 (Ni1) and 0.002 (Ni3), Table S3). The structure of 5 consists of one-dimensional chains of alternating cyanido-bridged [Ni(cyclam)]2+ and [Ni(CN)4]2– complex ions that runs along the [−110] direction (Figure 6). The coordination chains are slightly undulating with the angle of Ni–N–C of 165.6°. The distance between chains in the c crystallographic direction is equal to 7.84 Å. The crystallization water molecules form hydrogen bonds only with N-atoms of terminal cyanido ligands and NH-groups of cyclam, resulting in the formation of a three-dimensional supramolecular network. In contrast to 5, the earlier characterized related chains [Ag(cyclam)][Pt(CN)4] and [Cu(cyclam)][M(CN)4] (M = Ni, Pd, Pt) do not contain crystallization water and their coordination skeleton has a zigzag shape.40,41
Figure 6.

Structure of 5. View along the [100] direction (a) and along the coordination chains in the [−110] direction (b).
Thermal Stability and Sorption Properties
As described above, the crystalline samples of 1 and 2 collapse and decompose upon even short contact with pure water. Therefore, after the crystals are filtered from the mother liquor, their surface is contaminated with lithium chloride, which is extremely hygroscopic and creates various hydrates depending on the relative humidity.42 TGA for both compounds (Figure S11) shows sharp mass loss beginning under dry argon flow at ambient temperature. This initial stage is composed of two unresolved steps with Tmax at 44 and 79 °C (1) or 47 and 63 °C (2). From 95 °C (1) or 65 °C (2) to about 150 °C, a stability phase is observed. Then, a further well-defined mass loss step with Tmax of 160 °C is observed, which is very small for 1 and much larger for 2. The mass loss below 200 °C can be attributed to dehydration of both the coordination polymer and LiCl. Because of the fact that there is no stability period at the beginning of the experiment and the presence of LiCl impurity, the quantitative interpretation of the TG results is not possible. Larger overall mass loss upon dehydration observed for 1 is consistent with higher amount of crystallization water in this compound. The mass loss step at 160 °C can be attributed mainly to the dehydration of LiCl.42 After a long plateau, decomposition of both compounds begins at 250 °C.
Similar to 1 and 2, the TG curve of 3 (Figure S12) shows sharp two-step initial decrease with Tmax at 39 and 78 °C, indicating that the dehydration begins under the dry argon flow at ambient temperature. After a plateau between 85 and 120 °C, an additional prolonged dehydration step with Tmax of 155 °C occurs, which we believe to be connected with the strong bonding of H2O coordinated to the Li+ cations. Then, after a short stability period, decomposition begins around 225 °C. For compound 4, the beginning of dehydration is also not visible in TGA (Figure S12). It is completed in two unresolved steps with Tmax at 60 and 84 °C. From 95 to 250 °C, a long stability period of an anhydrous phase is present, followed by decomposition. The fact that there is no additional dehydration step around 150 °C is consistent with lack of Li+ guest cations. TGA of the crystalline sample of 5 shows a flat line in the temperature range of 35–300 °C, indicating that the dehydration process in the dry argon flow is very fast. It is consistent with the EA results, which show that partial dehydration of 5 takes place upon drying in air.
The water sorption properties of samples 3 and 4 were studied using the dynamic vapor sorption gravimetric method at a constant temperature of 25 °C and variable relative humidity (RH). The isotherms showing hydration and dehydration processes are presented in Figure 7. Each measurement step in the relative humidity range of 0–94% was performed until the stable sample mass was achieved. The amount of crystallization water molecules in fully hydrated phases designated on the basis of measured isotherms corresponds to the structural models of 3 and 4. The desorption isotherm of 3 shows three distinct steps, indicating the existence of metastable phases containing 18 H2O at 12–16% RH and 12 H2O at 6–10% RH. In the dehydrated under dry nitrogen flow phase, there are still two water molecules per formula unit, which are probably strongly bound to lithium cations and cannot be removed at ambient temperature. Taking into account that in the original structure of 3, there are seven water molecules bound to two Li+ per formula unit, we assume that upon dehydration structural transformation with the rearrangement of the Li+ coordination sphere takes place, possibly including formation of semi-coordination bonds to terminal cyanide ligands of [W(CN)8]4–. The sorption isotherm of 3 shows immediate high water intake at 2% RH, which is characteristic for microporous materials. In contrast to the three-step desorption isotherm, it is composed of just two steps, with the metastable phase, containing 12 H2O, between 8 and 14% RH. Due to the difference between the sorption and desorption processes, a small hysteresis loop appears in the 10–18% RH range. In the higher humidity region, the sorption and desorption isotherms almost overlap. The desorption isotherm of compound 4 shows that all 11 crystallization water molecules are released under dry nitrogen flow, which corresponds to about 9% of mass change, calculated on the basis of anhydrous polymer mass. The mass decrease upon water desorption is at first very slow and down 12% RH only three water molecules are released. Then, the loss of the remaining eight water molecules takes place in two sharp steps below 10% (∼5H2O) and 2% RH (∼3H2O). The sorption isotherm of 4 is characterized by a relatively small water intake (∼1H2O) up to 4% RH, which reflects the presence of only small isolated cavities in the structure, as opposed to the microporous channels of 3. Between 4 and 12% RH, a sharp sorption step up to 8H2O is observed, followed by a slow and almost linear mass increase over the higher humidity range with a small step at 22% RH. The differences between sorption and desorption processes result in an irregular small hysteresis. The presence of sharp steps and hysteresis in the isotherms of both compounds indicates that the structures are not rigid, but undergo humidity-driven structural transformations adapting to the changes in the number of guest water molecules. Interestingly, the water sorption isotherms of 3 and 4, although similar, are not identical to those of their Mo-based isotypic congeners.33
Figure 7.

Water sorption isotherms at the temperature of 25 °C for compound 3 (top) and 4 (bottom). Scale on the right indicates the approximate number of crystallization water molecules per formula unit.
Magnetic Properties
The magnetic properties of compounds 1–5 were characterized by DC susceptibility measurements at 1 kOe in the temperature range of 1.8–300 K and magnetization measurements at 1.8 K in magnetic fields up to 70 kOe. All microcrystalline samples were carefully protected from dehydration by immersion in pure water or lithium chloride solution of the same concentration as used in their syntheses. Every compound is composed of paramagnetic [Ni(cyclam)]2+ complex ions (S = 1, g = 2.2) that are separated by diamagnetic [W(CN)8]4– or [Ni(CN)4]2– (in the case of 4 and 5). Thus, the χMT products show a constant value from about 20 K up to 250 K, which is typical for paramagnetic behavior (Figure 8).
Figure 8.

Temperature dependence of χMT product for samples 1–5 at a constant magnetic field of 1 kOe.
The experimental values of high-temperature χMT products are in good agreement with the theoretical ones related to the respective chemical formulas and an integer number of NiII ions with S = 1 and g = 2.2. The decrease of χMT at low temperature is connected with zero field splitting effects and the presence of weak antiferromagnetic interactions between nickel(II) centers through diamagnetic polycyanidometallates. The magnetization curves also show typical paramagnetic run and do not saturate in the field of 70 kOe (Figures S13–S16). Moreover, the magnetic properties of 3 and 4 well corresponds to the characteristics of their octacyanidomolybdate(IV) congeners.33
Photomagnetic Investigations
The light-induced magnetic experiments were performed for a fully hydrated sample of Li2[Ni(cyclam)]3[W(CN)8]2·24H2O (3) and for its octacyanidomolybdate(IV) congener Li2[Ni(cyclam)]3[Mo(CN)8]2·24H2O (7), as well as for the dehydrated phases containing two crystallization water molecules per formula unit (3deh and 7deh). All samples were carefully protected from changing the hydration level.
The sample of 3 shows a negligible change of magnetization even after 26 h of irradiation (Figure S17a), manifested by a negligible lowering of magnetization at high magnetic fields in the M(H) plot (Figure S17b). The χMT(T) curves (Figure S17c) before and after irradiation are nearly identical in the 1.8 to 10 K range, after which a negligible artificial decrease for irradiated 3 is observed. Hence, the light-induced effect is not present or negligible in the fully hydrated phase of compound 3.
A significant photomagnetic effect can be observed for the dehydrated phase 3deh. Figure S18a presents the increase of the magnetic moment during irradiation indicating the conversion of the low-spin diamagnetic [WIV(CN)8] moiety to the high-spin S = 1 species. Such switching was observed before for MnII- and FeII-based systems.15,16,43 The M(H) dependence registered at 1.8 K shows a pronounced lowering of magnetization (ca. 4% at 70 kOe) after 42 h of irradiation (Figure S18b). This suggests that the magnetic interactions between the photo-induced high-spin tungsten(IV) species connected to NiII ions via CN bridges is most probably antiferromagnetic JWNi* < 0. The divergence of the M(H) curves takes place at about 10 kOe. The clear increase of the χMT product in the 5–150 K range confirms the persistence of the photo-induced state (Figure 9). The effect caused by the irradiation can be reversed thermally. After heating the sample to 250 K, the initial χMT(T) and M(H) curves are fully restored (Figures 9 and S18).
Figure 9.
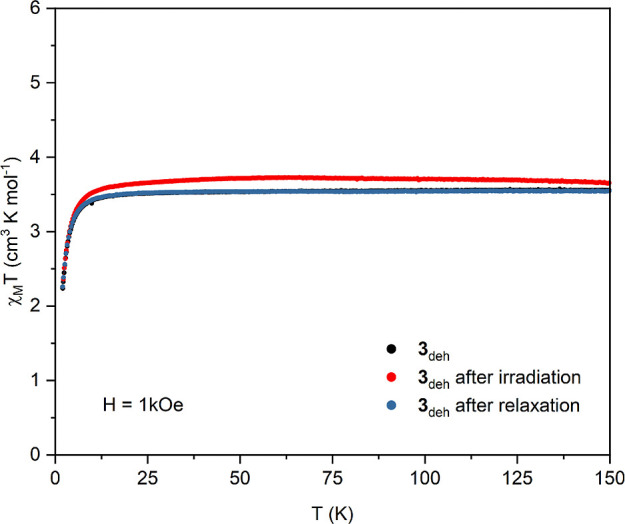
Temperature dependence of χMT product in a magnetic field of 1 kOe for dehydrated phase 3deh (black points), after 42 h of blue (450 nm) light irradiation (red points), and after thermal relaxation at 250 K (blue points).
The fully hydrated Li2[Ni(cyclam)]3[Mo(CN)8]2·24H2O (7) shows small magnetization changes in response to 28 h of irradiation (Figure S19a), with slightly higher χMT values in the 1.8–50 K range as compared to the “dark” state (Figure S19b). Slightly higher magnetization is also observed in the M(H) plot recorded after irradiation (Figure S19c). As in the case of 3deh, the observed photomagnetic effect is most probably caused by the change of the spin state of the MoIV centers accompanied by the cyanido ligand dissociation.13 After relaxation at 250 K, the M(H) and χMT(T) plots overlap perfectly with those recorded before irradiation, hence photo-induced changes are reversible. Additional analysis is hindered by a very small change of the magnetization caused by light irradiation.
The strongest photomagnetic effect was observed after dehydration of compound 7 resulting in the Li2[Ni(cyclam)]3[Mo(CN)8]2·2H2O (7deh) phase. The magnetic moment of 7deh increases in response to blue light excitation leading to much higher M(H) values in the whole investigated magnetic field range. The magnetization at 70 kOe attains the value of 6.95 Nβ which is about 15% larger than that before the irradiation (Figures 10 and S20). The χMT(T) curve for the irradiated sample shows significant increase in comparison to the “dark” one. This can be ascribed to the behavior similar to the light-induced spin state trapping effect in FeII complexes.44 The diamagnetic S = 0 MoIV center undergoes light-induced spin state switching from the low-spin state to the S = 1 high-spin state. Such behavior was observed for a Zn2Mo complex.12 Analysis of both dependences M(H) and χMT(T) suggests that the magnetic interactions between the S = 1 NiII and the photo-induced S = 1 MoIV are ferromagnetic JMoNi* > 0. This is clearly visible in the photo-induced χMT(T) curve (red points in Figure 11) as a flat maximum at around 15 K. Heating the sample after the photomagnetic experiment to 250 K does not restore the pristine state completely, suggesting a slight decomposition of the compound which was also observed for some octacyanidomolybdate(IV)–manganese(II) photomagnetic systems, where the photodissociation of the CN ligand was behind the photomagnetic switching mechanism (Figure 11).45
Figure 10.

Magnetic field dependence of magnetization at 1.8 K for dehydrated phase 7deh (black points), after 16 h of blue (450 nm) light irradiation (red points), and after thermal relaxation at 250 K (blue points).
Figure 11.

Temperature dependence of χMT product in a magnetic field of 1 kOe for dehydrated phase 7deh (black points), after 16 h of blue (450 nm) light irradiation (red points), and after thermal relaxation at 250 K (blue points).
Conclusions
Our work is an unprecedented example of crystal engineering of CN-bridged assemblies. We present a record number of six coordination networks of different dimensionality and topology obtained from the same pair of building blocks: [Ni(cyclam)]2+ and [W(CN)8]4–. The products include coordination polymers with negatively charged frameworks compensated by the presence of the Li+ cations: 1D Ni–W chains of straight (1) and zigzag (2) shape and a 2D honeycomb-like network (3). Neutral frameworks formed as a result of partial decomposition of the building blocks: a remarkable 3D two-fold interpenetrating network (4) and a 1D Ni–Ni chain (5) as well as a 3D Ni–W diamond-like network (6) characterized before.27 The control of topology and product purity in this complicated multi-pathway reaction system is realized by addition of LiCl electrolyte and temperature adjustment. Moreover, for the first time we present the photomagnetic switching of octacyanidotungstate(IV) and octacyanidomolybdate(IV) photomagnetic chromophores embedded into the nickel(II)-based coordination polymers (3, 7). All previous examples involve other combinations of metal ions: CuII–MIV(CN)8,46,47 ZnII–MIV(CN)8,12,48 MnII–MIV(CN)8,14,49 FeII–MIV(CN)8,15 CdII–MIV(CN)8,50 or PtIV–MIV(CN)820 (M = Mo, W). Similar to MnII–MIV(CN)8 coordination polymers,49 the photomagnetic response of MIV(CN)8 moieties in the reported NiII–MIV(CN)8 frameworks is significantly stronger for the dehydrated phases as compared to the pristine ones. This supports the hypothesis that the light-induced spin state switching in MIV(CN)8 photomagnetic chromophores might be associated with the cyanido ligand photodissociation and the change of the coordination number of the metal center. Additionally, the magnetic interaction of NiII with the photoexcited MIV through the CN bridges can be either ferromagnetic or antiferromagnetic depending on the bridge geometry. Similar observations were made for NiII–MV(CN)8 (M = Mo or W)26,51−55 and NiII–NbIV(CN)8 magnetic coordination frameworks,27,29,32,56 where the second metal center MV or NbIV is an S = 1/2 ion.
Acknowledgments
The authors acknowledge the financial support of the Polish Ministry of Science and Higher Education under the “Diamond Grant” program (0193/DIA/2017/46).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c01629.
Detailed crystallographic data, additional structure diagrams, PXRD data, thermogravimetric characteristics, and magnetic data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Chorazy S.; Zakrzewski J. J.; Magott M.; Korzeniak T.; Nowicka B.; Pinkowicz D.; Podgajny R.; Sieklucka B. Octacyanidometallates for multifunctional molecule-based materials. Chem. Soc. Rev. 2020, 49, 5945–6001. 10.1039/d0cs00067a. [DOI] [PubMed] [Google Scholar]
- Pardo R.; Zayat M.; Levy D. Photochromic organic-inorganic hybrid materials. Chem. Soc. Rev. 2011, 40, 672–687. 10.1039/c0cs00065e. [DOI] [PubMed] [Google Scholar]
- Meng X.; Wang H.-N.; Song S.-Y.; Zhang H.-J. Proton-conducting crystalline porous materials. Chem. Soc. Rev. 2017, 46, 464–480. 10.1039/c6cs00528d. [DOI] [PubMed] [Google Scholar]
- Farinola G. M.; Ragni R. Electroluminescent materials for white organic light emitting diodes. Chem. Soc. Rev. 2011, 40, 3467–3482. 10.1039/c0cs00204f. [DOI] [PubMed] [Google Scholar]
- Lu H.; Kobayashi N. Optically Active Porphyrin and Phthalocyanine Systems. Chem. Rev. 2016, 116, 6184–6261. 10.1021/acs.chemrev.5b00588. [DOI] [PubMed] [Google Scholar]
- Molecular Magnetic Materials; Sieklucka B., Pinkowicz D., Eds.; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Nowicka B.; Korzeniak T.; Stefańczyk O.; Pinkowicz D.; Chorąży S.; Podgajny R.; Sieklucka B. The impact of ligands upon topology and functionality of octacyanidometallate-based assemblies. Coord. Chem. Rev. 2012, 256, 1946–1971. 10.1016/j.ccr.2012.04.008. [DOI] [Google Scholar]
- Ferrando-Soria J.; Vallejo J.; Castellano M.; Martínez-Lillo J.; Pardo E.; Cano J.; Castro I.; Lloret F.; Ruiz-García R.; Julve M. Molecular magnetism, quo vadis? A historical perspective from a coordination chemist viewpoint☆. Coord. Chem. Rev. 2017, 339, 17–103. 10.1016/j.ccr.2017.03.004. [DOI] [Google Scholar]
- Rombaut G.; Golhen S.; Ouahab L.; Mathonière C.; Kahn O. Structural and photomagnetic studies of a 1-D bimetallic chain [MnII2(L)2(H2O)][MoIV(CN)8]·5H2O (L = macrocycle): analogy with the photo-oxidation of K4[MoIV(CN)8]·2H2O. J. Chem. Soc., Dalton Trans. 2000, 20, 3609–3614. 10.1039/b003983g. [DOI] [Google Scholar]
- Rombaut G.; Verelst M.; Golhen S.; Ouahab L.; Mathonière C.; Kahn O. Structural and Photomagnetic Studies of Two Compounds in the System Cu2+/Mo(CN)84-: From Trinuclear Molecule to Infinite Network. Inorg. Chem. 2001, 40, 1151–1159. 10.1021/ic0008722. [DOI] [PubMed] [Google Scholar]
- Herrera J. M.; Marvaud V.; Verdaguer M.; Marrot J.; Kalisz M.; Mathonière C. Reversible Photoinduced Magnetic Properties in the Heptanuclear Complex [MoIV(CN)2(CN-CuL)6]8+: A Photomagnetic High-Spin Molecule. Angew. Chem., Int. Ed. 2004, 43, 5468–5471. 10.1002/anie.200460387. [DOI] [PubMed] [Google Scholar]
- Bridonneau N.; Long J.; Cantin J.-L.; von Bardeleben J.; Pillet S.; Bendeif E.-E.; Aravena D.; Ruiz E.; Marvaud V. First evidence of light-induced spin transition in molybdenum(IV). Chem. Commun. 2015, 51, 8229–8232. 10.1039/c5cc01714a. [DOI] [PubMed] [Google Scholar]
- Qi X.; Pillet S.; Graaf C.; Magott M.; Bendeif E.-E.; Guionneau P.; Rouzières M.; Marvaud V.; Stefańczyk O.; Pinkowicz D.; Mathonière C. Photoinduced Mo-CN Bond Breakage in Octacyanomolybdate Leading to Spin Triplet Trapping. Angew. Chem., Int. Ed. 2020, 59, 3117–3121. 10.1002/anie.201914527. [DOI] [PubMed] [Google Scholar]
- Mathonière C.; Podgajny R.; Guionneau P.; Labrugere C.; Sieklucka B. Photomagnetism in Cyano-Bridged Hexanuclear Clusters [MnII(bpy)2]4[MIV(CN)8]2,xH2O (M = Mo, x = 14, and M = W, x = 9). Chem. Mater. 2005, 17, 442–449. 10.1021/cm048785l. [DOI] [Google Scholar]
- Arczyński M.; Stanek J.; Sieklucka B.; Dunbar K. R.; Pinkowicz D. Site-Selective Photoswitching of Two Distinct Magnetic Chromophores in a Propeller-Like Molecule To Achieve Four Different Magnetic States. J. Am. Chem. Soc. 2019, 141, 19067–19077. 10.1021/jacs.9b09576. [DOI] [PubMed] [Google Scholar]
- Magott M.; Reczyński M.; Gaweł B.; Sieklucka B.; Pinkowicz D. A Photomagnetic Sponge: High-Temperature Light-Induced Ferrimagnet Controlled by Water Sorption. J. Am. Chem. Soc. 2018, 140, 15876–15882. 10.1021/jacs.8b09322. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Zaworotko M. J. Crystal Engineering of Hybrid Coordination Networks: From Form to Function. Trends Chem. 2020, 2, 506–518. 10.1016/j.trechm.2020.02.013. [DOI] [Google Scholar]
- Braga D. Crystal Engineering, Where from? Where To?. Chem. Commun. 2003, 2751–2754. 10.1039/b306269b. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. 10.1021/ja403264c. [DOI] [PubMed] [Google Scholar]
- Magott M.; Sarewicz M.; Buda S.; Pinkowicz D. Heterotrimetallic Cyanide-Bridged 3d-4d-5d Frameworks Based on a Photomagnetic Secondary Building Unit. Inorg. Chem. 2020, 59, 8925–8934. 10.1021/acs.inorgchem.0c00737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z.-F.; Wang J.-Y.; Pei J. Control of π–π Stacking via Crystal Engineering in Organic Conjugated Small Molecule Crystals. Cryst. Growth Des. 2018, 18, 7–15. 10.1021/acs.cgd.7b01385. [DOI] [Google Scholar]
- Aakeröy C. B.; Champness N. R.; Janiak C. Recent advances in crystal engineering. CrystEngComm 2010, 12, 22–43. 10.1039/B919819A. [DOI] [Google Scholar]
- Seoane B.; Castellanos S.; Dikhtiarenko A.; Kapteijn F.; Gascon J. Multi-scale crystal engineering of metal organic frameworks. Coord. Chem. Rev. 2016, 307, 147–187. 10.1016/j.ccr.2015.06.008. [DOI] [Google Scholar]
- Zhong D.-C.; Wen Y.-Q.; Deng J.-H.; Luo X.-Z.; Gong Y.-N.; Lu T.-B. Uncovering the Role of Metal Catalysis in Tetrazole Formation by an In Situ Cycloaddition Reaction: An Experimental Approach. Angew. Chem., Int. Ed. 2015, 54, 11795–11799. 10.1002/anie.201505118. [DOI] [PubMed] [Google Scholar]
- Ou G.-C.; Yuan X.-Y.; Li Z.-Z.; Li W.-Y.; Zeng F.; Deng J.-H.; Zhong D.-C. Vanadium Polyoxoanions within Coordination Polymers Based on a Macrocyclic Nickel Complex: Structural Diversities and Single-Crystal to Single-Crystal Transformation. Eur. J. Inorg. Chem. 2016, 3500–3505. 10.1002/ejic.201600284. [DOI] [Google Scholar]
- Nowicka B.; Reczyński M.; Rams M.; Nitek W.; Kozieł M.; Sieklucka B. Larger pores and higher Tc: {[Ni(cyclam)]3[W(CN)8]2·solv}n - a new member of the largest family of pseudo-polymorphic isomers among octacyanometallate-based assemblies. CrystEngComm 2015, 17, 3526–3532. 10.1039/c5ce00287g. [DOI] [Google Scholar]
- Nowicka B.; Bałanda M.; Reczyński M.; Majcher A. M.; Kozieł M.; Nitek W.; Łasocha W.; Sieklucka B. A water sensitive ferromagnetic [Ni(cyclam)]2[Nb(CN)8] network. Dalton Trans. 2013, 42, 2616–2621. 10.1039/c2dt32299d. [DOI] [PubMed] [Google Scholar]
- Nowicka B.; Reczyński M.; Bałanda M.; Fitta M.; Gaweł B.; Sieklucka B. The Rule Rather than the Exception: Structural Flexibility of [Ni(cyclam)]2+-Based Cyano-Bridged Magnetic Networks. Cryst. Growth Des. 2016, 16, 4736–4743. 10.1021/acs.cgd.6b00800. [DOI] [Google Scholar]
- Reczyński M.; Heczko M.; Kozieł M.; Ohkoshi S.; Sieklucka B.; Nowicka B. Proton-Conducting Humidity-Sensitive NiII-NbIV Magnetic Coordination Network. Inorg. Chem. 2019, 58, 15812–15823. 10.1021/acs.inorgchem.9b02141. [DOI] [PubMed] [Google Scholar]
- Reczyński M.; Nowicka B.; Näther C.; Kozieł M.; Nakabayashi K.; Ohkoshi S.; Sieklucka B. Dehydration-Triggered Charge Transfer and High Proton Conductivity in (H3O)[NiIII(cyclam)][MII(CN)6] (M = Ru, Os) Cyanide-Bridged Chains. Inorg. Chem. 2018, 57, 13415–13422. 10.1021/acs.inorgchem.8b01992. [DOI] [PubMed] [Google Scholar]
- Reczyński M.; Pinkowicz D.; Nakabayashi K.; Näther C.; Stanek J.; Kozieł M.; Kalinowska-Tłuścik J.; Sieklucka B.; Ohkoshi S.; Nowicka B. Room-Temperature Bistability in a Ni–Fe Chain: Electron Transfer Controlled by Temperature, Pressure, Light, and Humidity. Angew. Chem., Int. Ed. 2021, 60, 2330–2338. 10.1002/anie.202012876. [DOI] [PubMed] [Google Scholar]
- Heczko M.; Reczyński M.; Näther C.; Nowicka B. Tuning of magnetic properties of the 2D CN-bridged NiII-NbIV framework by incorporation of guest cations of alkali and alkaline earth metals. Dalton Trans. 2021, 50, 7537–7544. 10.1039/d1dt00367d. [DOI] [PubMed] [Google Scholar]
- Heczko M.; Sumińska E.; Sieklucka B.; Nowicka B. A two-fold 3D interpenetrating cyanido-bridged network based on the octa-coordinated [Mo(CN)8]4- building block. CrystEngComm 2019, 21, 5067–5075. 10.1039/c9ce00851a. [DOI] [Google Scholar]
- Matoga D.; Szklarzewicz J.; Mikuriya M. [PPh4]3[W(CN)7(O2)]·4H2O as the Representative of the [M(L)7(LL)] Class for Nine-Coordinate Complexes. Inorg. Chem. 2006, 45, 7100–7104. 10.1021/ic060523t. [DOI] [PubMed] [Google Scholar]
- Berry D. E.; Girard G.; McAuley A. The Synthesis and Reactions of Nickel(III) Stabilized by a Nitrogen-Donor Macrocycle. J. Chem. Educ. 1996, 73, 551–554. 10.1021/ed073p551. [DOI] [Google Scholar]
- Sheldrick G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. 10.1107/s2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae C. F.; Bruno I. J.; Chisholm J. A.; Edgington P. R.; McCabe P.; Pidcock E.; Rodriguez-Monge L.; Taylor R.; van de Streek J.; Wood P. A. Mercury CSD 2.0 - new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. 10.1107/s0021889807067908. [DOI] [Google Scholar]
- Llunell M.; Cirera J.; Alemany P.; Alvarez S.. SHAPE v. 2.1; University of Barcelona: Spain, 2013.
- Tsymbal L. V.; Andriichuk I. L.; Shova S.; Lampeka Y. D. Crystal structure of [{[Ni(C10H24N4)][Ni(CN)4]}·2H2O]n, a one-dimensional coordination polymer formed from the [Ni(cyclam)]2+ cation and the [Ni(CN)4]2- anion. Acta Crystallogr. 2021, 77, 1140–1143. 10.1107/s2056989021010902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Černák J.; Kuchár J.; Stolárová M.; Kajňaková M.; Vavra M.; Potočňák I.; Falvello L. R.; Tomás M. Preparation, spectroscopic and magnetic characterization of Cu(cyclam)M(CN)4 complexes exhibiting one-dimensional crystal structures (cyclam = 1,4,8,11-tetraazacyclotetradecane, M = Ni, Pd, Pt). Transit. Met. Chem. 2010, 35, 737–744. 10.1007/s11243-010-9387-5. [DOI] [Google Scholar]
- Munakata M.; Zhong J. C.; Ino I.; Kuroda-Sowa T.; Maekawa M.; Suenaga Y.; Oiji N. 1-D cyano-bridged heterometallic complexes consisting of 1,4,8,11-tetraazacyclotetradecanesilver(II) and tetracyanopalladium(II) or tetracyanoplatinum(II). Inorg. Chim. Acta 2001, 317, 268–275. 10.1016/s0020-1693(01)00366-8. [DOI] [Google Scholar]
- Bhattacharya A. K.; Hartridge A.; Mallick K. K. The role of lattice water in determining the reactivity of various crystalline aluminas. J. Mater. Sci. 1997, 32, 1113–1116. 10.1023/a:1018507228858. [DOI] [Google Scholar]
- Magott M.; Pinkowicz D. Chiral porous CN-bridged coordination polymer mimicking MOF-74 and showing magnetization photoswitching. Chem. Commun. 2021, 57, 9926–9929. 10.1039/d1cc03870b. [DOI] [PubMed] [Google Scholar]
- Chorazy S.; Charytanowicz T.; Pinkowicz D.; Wang J.; Nakabayashi K.; Klimke S.; Renz F.; Ohkoshi S.; Sieklucka B. Octacyanidorhenate(V) Ion as an Efficient Linker for Hysteretic Two-Step Iron(II) Spin Crossover Switchable by Temperature, Light, and Pressure. Angew. Chem., Int. Ed. 2020, 59, 15741–15749. 10.1002/anie.202007327. [DOI] [PubMed] [Google Scholar]
- Magott M.; Stefańczyk O.; Sieklucka B.; Pinkowicz D. Octacyanidotungstate(IV) Coordination Chains Demonstrate a Light-Induced Excited Spin State Trapping Behavior and Magnetic Exchange Photoswitching. Angew. Chem., Int. Ed. 2017, 56, 13283–13287. 10.1002/anie.201703934. [DOI] [PubMed] [Google Scholar]
- Pai T.; Stefanczyk O.; Kumar K.; Mathonière C.; Sieklucka B.; Ohkoshi S. Experimental and theoretical insights into the photomagnetic effects in trinuclear and ionic Cu(II)–Mo(IV) systems. Inorg. Chem. Front. 2022, 9, 771–783. 10.1039/d1qi01469b. [DOI] [Google Scholar]
- Korzeniak T.; Sasmal S.; Pinkowicz D.; Nitek W.; Pełka R.; Czernia D.; Stefańczyk O.; Sieklucka B. Chiral Photomagnets Based on Copper(II) Complexes of 1,2-Diaminocyclohexane and Octacyanidomolybdate(IV) Ions. Inorg. Chem. 2020, 59, 5872–5882. 10.1021/acs.inorgchem.9b03511. [DOI] [PubMed] [Google Scholar]
- Qi X.; Guionneau P.; Lafon E.; Perot S.; Kauffmann B.; Mathonière C. New Photomagnetic Ionic Salts Based on [MoIV(CN)8]4– and [WIV(CN)8]4– Anions. Magnetochemistry 2021, 7, 97. 10.3390/magnetochemistry7070097. [DOI] [Google Scholar]
- Magott M.; Gaweł B.; Sarewicz M.; Reczyński M.; Ogorzały K.; Makowski W.; Pinkowicz D. Large breathing effect induced by water sorption in a remarkably stable nonporous cyanide-bridged coordination polymer. Chem. Sci. 2021, 12, 9176–9188. 10.1039/d1sc02060a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniak T.; Jankowski R.; Kozieł M.; Pinkowicz D.; Sieklucka B. Reversible Single-Crystal-to-Single-Crystal Transformation in Photomagnetic Cyanido-Bridged Cd4M2 Octahedral Molecules. Inorg. Chem. 2017, 56, 12914–12919. 10.1021/acs.inorgchem.7b01708. [DOI] [PubMed] [Google Scholar]
- Hilfiger M. G.; Zhao H.; Prosvirin A.; Wernsdorfer W.; Dunbar K. R. Molecules based on M(V) (M = Mo, W) and Ni(II) ions: a new class of trigonal bipyramidal cluster and confirmation of SMM behavior for the pentadecanuclear molecule {NiII[NiII(tmphen)(MeOH)]6[Ni(H2O)3]2[μ-CN]30[WV(CN)3]6. Dalton Trans. 2009, 5155–5163. 10.1039/b903691a. [DOI] [PubMed] [Google Scholar]
- Lim J. H.; Yoo H. S.; Yoon J. H.; Koh E. K.; Kim H. C.; Hong C. S. Structure and magnetic properties of cyanide-bridged NiII9MoV6 cluster modified by bidentate capping ligands. Polyhedron 2008, 27, 299–303. 10.1016/j.poly.2007.09.006. [DOI] [Google Scholar]
- Lim J. H.; Yoon J. H.; Kim H. C.; Hong C. S. Surface Modification of a Six-Capped Body-Centered Cube Ni9W6 Cluster: Structure and Single-Molecule Magnetism. Angew. Chem., Int. Ed. 2006, 45, 7424–7426. 10.1002/anie.200601759. [DOI] [PubMed] [Google Scholar]
- Visinescu D.; Desplanches C.; Imaz I.; Bahers V.; Pradhan R.; Villamena F. A.; Guionneau P.; Sutter J.-P. Evidence for increased exchange interactions with 5d compared to 4d metal ions. Experimental and theoretical insights into the ferromagnetic interactions of a series of trinuclear [{M(CN)8}3-/NiII] compounds (M = MoV or WV). J. Am. Chem. Soc. 2006, 128, 10202–10212. 10.1021/ja061911x. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan T. S.; Desplanches C.; Rajamani R.; Guionneau P.; Ducasse L.; Ramasesha S.; Sutter J.-P. Tetranuclear [{Ni(HL3)}{W(CN)8}]2 Square: A Case of Antiferromagnetic {NiIIWV} Interactions. Inorg. Chem. 2008, 47, 4854–4860. 10.1021/ic800114y. [DOI] [PubMed] [Google Scholar]
- Pinkowicz D.; Pełka R.; Drath O.; Nitek W.; Bałanda M.; Majcher A. M.; Poneti G.; Sieklucka B. Nature of Magnetic Interactions in 3D {[MII(pyrazole)4]2[NbIV(CN)8]·4H2O}n (M = Mn, Fe, Co, Ni) Molecular Magnets. Inorg. Chem. 2010, 49, 7565–7576. 10.1021/ic100937h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.