

Abstract
The ATX-LPA-LPAR1 signaling pathway plays a universal role in stimulating diverse cellular responses, including cell proliferation, migration, survival, and invasion in almost every cell type. The ATX-LPAR1 axis is linked to several metabolic and inflammatory diseases including cancer, fibrosis, and rheumatoid arthritis. Numerous selective ATX or LPAR1 inhibitors have been developed and so far, their clinical efficacy has only been evaluated in idiopathic pulmonary fibrosis. None of the ATX and LPAR1 inhibitors have advanced to clinical trials for cancer and rheumatoid arthritis. Nonetheless, several research groups, including ours, have shown considerable benefit of simultaneous ATX and LPAR1 inhibition through combination therapy. Recent research suggests that dual-targeting therapies are superior to combination therapies that use two selective inhibitors. However, limited reports are available on ATX-LPAR1 dual inhibitors, potentially due to co-expression of multiple different LPARs with close structural similarities at the same target. In this review, we discuss rational design and future directions of dual ATX-LPAR1 inhibitors.
Keywords: autotaxin (ATX), lysophosphatidic acid (LPA), lysophosphatidic acid receptor subtype-1 (LPAR1), dual inhibitors, combination therapy, cancer, metastasis, idiopathic pulmonary fibrosis (IPF)
1. Introduction
Ectonucleotide pyrophosphatase 2 (ENPP2) commonly referred to as Autotaxin (ATX) was initially discovered through its lysophospholipase D activity [1]. ATX hydrolyzes extracellular lysophospholipids, primarily lysophosphatidylcholine (LPC) due to its high abundance in biological fluids, to generate the lipid mediator lysophosphatidic acid (LPA) [2,3]. LPA induces several cellular responses, including cell proliferation, migration, survival, invasion, metastasis, and production of cytokines, by modulating six distinct LPA G-protein coupled receptors (LPAR) (Figure 1) [1,2,3,4,5,6]. Activation of the ATX-LPA-LPAR signaling pathway is critical for the maintenance of stemness in pluripotent somatic and cancer stem cells [7,8], the development of neurological systems, the generation of blood vessels, wound healing, and tissue repair [4,5]. Consequently, dysregulation of this signaling pathway is often linked to metabolic and inflammatory disorders, including cancer, tumor immunity [9,10,11,12], fibrosis, neuropathic pain and neurodegeneration [13,14,15], inflammation, autoimmune diseases, and metabolic syndrome [4,5,16]. In this context, genetic studies have demonstrated a decisive role of LPAR1 and ATX in mouse models of lung fibrosis [3,16,17,18,19,20]. A study of idiopathic pulmonary fibrosis (IPF) patients has indicated aberrant levels of LPA in the bronchoalveolar lavage (BAL) fluid [21]. Overexpression of ATX has also been documented in patients with IPF [17]. Apart from IPF, extensive research has shown that blocking the ATX-LPA-LPAR axis can have promising therapeutic benefits on a variety of illnesses, including cancers of the breast, ovary, pancreas and liver, cardiovascular diseases, pregnancy induced hypertension (preeclamsia), inflammation induced systemic bone loss, rheumatoid arthritis, and suppression of antitumor immunity [1,2,3,4,5,16,17,21,22,23,24,25,26]. Therefore, there has been extensive interest in developing new therapeutics targeting the ATX-LPAR signaling pathway [1,2,3,22,27]. In concert with these efforts, Katsifa et al. have demonstrated that long-term pharmacologic suppression and widespread genetic deletion of ATX in adult mice are well tolerated, allaying possible toxicity concerns of ATX therapeutic targeting [28]. In addition, Lin et al. have demonstrated that genetic deletion of ATX in mice suppresses experimental colitis [29]. This review discusses recent advances in ATX inhibitors and the advantages of simultaneous inhibition of ATX and LPAR1.
Figure 1.

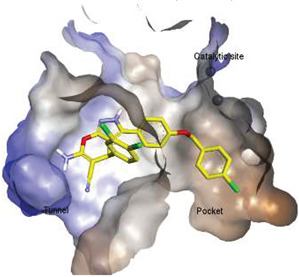
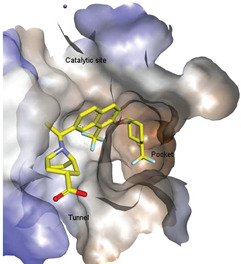
ATX-LPA-LPAR signaling axis and ATX binding sites. (A). ATX catalyzes the formation of LPA from LPC and subsequently activates LPARs. (B). Depiction of ATX surface and domains (PDB: 3NKN). Abbreviations used: SMB, somatomedin-like domain; NUC, nucleotidase-like domain. (C). ATX tripartite binding site with GLPG 1690 (red) and LPC (yellow).
2. Structural Aspects of ATX Inhibitors
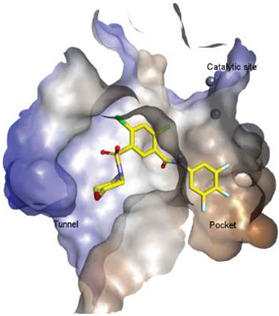
Lipid-like and non-lipid ATX inhibitors have been reported over the last decade, with co-crystal structures revealed for inhibitors LPA (14:1, PDB: 3NKN, Figure 1) and HA-155 (PDB: 2XRG, Figure 2) [30,31]. ATX inhibitors can be divided into five different classes based on their binding mode, namely: class I, orthosteric site; class II, hydrophobic pocket; class III, allosteric tunnel; class IV, tunnel-pocket hybrids; and class V, tunnel-active site hybrids, which exhibit a distinct binding mode in the tripartite interacting site in the T-shaped binding pocket of ATX (Figure 1) [4,32]. A significant number of these inhibitors, including HA-155 and PF-8380 (PDB: 5OLB, Figure 2), belong to the category of orthosteric-site inhibitors as their mode of action is to completely block the substrate by binding to the active site and the hydrophobic pocket [32,33]. Hydrophobic pocket ATX inhibitors have been designed to compete with substrate binding without requiring the headgroup aimed for the active site, such as PAT-494 (PDB: 4ZGA, Figure 2) [4,34]. Our group has reported several potent small molecule non-lipid hydrophobic pocket ATX inhibitors, such as 3b and 3f (Figure 2), that exhibit efficacy in vivo [1,27,33]. ATX tunnel binders such as UDCA (Figure 2) demonstrate moderate efficacy due to their non-competitive binding to the hydrophobic tunnel [32]. A recent expansion approach had led to the development of potent tunnel-pocket hybrid inhibitors such as GLPG1690 (PDB: 5MHP, Figure 2) [2,4,35]. The latest efforts have provided access to the first tunnel-active site hybrid by combining important structural motifs of ATX tunnel binders with that of ATX orthosteric inhibitors (PDB: 7Z0N, Figure 2) [32].
Figure 2.

Classification of recently developed non-lipid small molecule ATX inhibitors.  .
.
3. ATX-LPAR1 GPCR Axis in Idiopathic Pulmonary Fibrosis and Cancer
Despite significant progress in the development of these different classes of ATX inhibitors, only five drug candidates that target the ATX-LPAR signaling pathway have been evaluated in clinical trials [4,5]. The pocket- and tunnel-binding hybrid GLPG1690 reached a Phase-III clinical trial for the treatment of IPF before the trial was halted in the developmental stage [4]. According to reports, patients taking GLPG1690 for at least six to nine months exhibit a higher mortality risk than patients taking a placebo [35]. Hence, the trial was discontinued in the late stage as the risk-benefit profile was no longer favorable. Currently, the orally active non-competitive ATX inhibitor cudetaxestat (BLD-0409, compound 10, Table 1), which binds in the ATX tunnel, is in a phase II clinical trial for the treatment of IPF (NCT05373914) [36,37]. BLD-0409 shows significant reduction in lung fibrosis, fibrotic markers, and levels of profibrotic LPA in pulmonary fibrotic mouse models [38]. BLD-0409 blocks ATX in a dose-dependent manner, unlike the linear effects of GLPG1690 [4,21]. It is noteworthy that the pocket-tunnel binding compound GLPG1690 competes with the ATX substrate LPC for the hydrophobic pocket region. As LPC occupies the pocket and active site (Figure 1), it is believed that the efficacy of GLPG1690 may be limited when LPC is present at higher concentrations [37,38]. Unlike GLPG1690, BLD-0490 blocks ATX by binding to the allosteric site without interacting with the substrate and may retain its potency at elevated levels of LPC. Due to aberrantly high levels of LPA and overexpression of LPAR1 in IPF patients, LPAR1 inhibitor such as BMS-986020 have also been evaluated in clinical trials for the treatment of IPF [39,40]. A 26-week clinical trial demonstrated that treatment of IPF patients with BMS-986020 results in significant improvement in forced vital capacity (NCT01766817) [39,41]. However, hepatobiliary toxicity has also been reported in some patients receiving BMS-986020. Consequently, a second-generation LPAR1 inhibitor, BMS-986278, was developed and results from phase I studies have shown no evidence of hepatobiliary toxicities. BMS-986278 is currently being evaluated in phase II clinical trials (NCT04308681) for the treatment of IPF [19]. Lastly, the selective oral LPAR1 antagonist SAR100842 was evaluated in clinical trials for their safety and efficacy in reducing skin sclerosis in patients with diffuse cutaneous systemic sclerosis (NCT01651143) [42].
Table 1.
Potent ATX inhibitors targeting hydrophobic pocket and tunnel.
| ID | Structure | Biological Efficacy (IC50: nM) | Mode of Binding | Cocrystal Structure (PDB) |
|---|---|---|---|---|
| 1 |
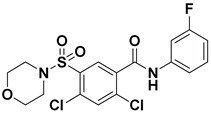
|
LPC Assay: 43.6 (ATX) | Pocket binder (Class II) | N.D. |
| 2 |
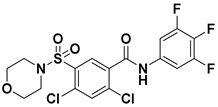
|
FS-3 Assay: 9 (ATX) 14450 (LPAR1) |
Pocket binder (Class II)
|
N.D. |
| 3 |
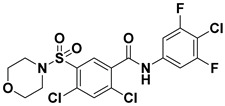
|
FS-3 Assay: 84 (ATX) 6100 (LPAR1) |
Pocket binder (Class II) | N.D. |
| 4 |
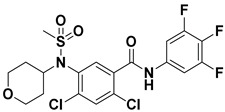
|
FS-3 Assay: 219 (ATX) | Pocket binder (Class II) | N.D. |
| 5 |
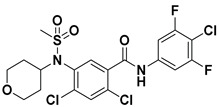
|
FS-3 Assay: 218 (ATX) |
Pocket binder (Class II)
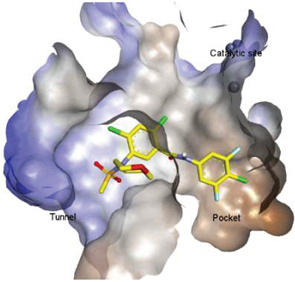
|
N.D. |
| 6 |
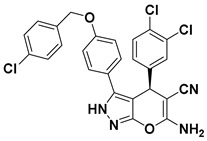
|
LPC Assay: 87 (ATX) |
Pocket binder (class II)
|
N.D. |
| 7 |
|
LPC Assay: 24.2 (ATX) | Pocket Binder (Class II) | N.D. |
| 8 |
|
LPC Assay: 15.3 (ATX) |
Pocket Binder (Class II)
|
N.D. |
| 9 |
|
LPC Assay: >300 | Tunnel Binder (Class III) | 5LQQ |
| 10 |
|
LPC Assay: 81 (ATX) |
Tunnel Binder (Class III)
|
5LQQ |
| 11 |
|
LPC Assay: 1 (ATX) |
Pocket-Tunnel Hybrid (Class IV)
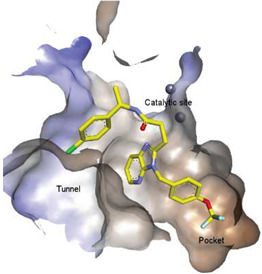
|
5LIA |
| 12 |
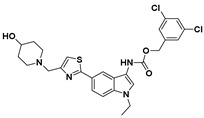
|
LPC Assay: 1.01 (ATX) |
Pocket-Tunnel Hybrid (Class IV)
|
N.D. |
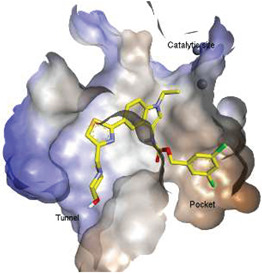
| 13 |
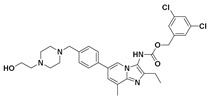
|
FS-3 Assay: 3.4 (ATX) | Pocket-Tunnel Hybrid (Class IV) | N.D. |
| 14 |
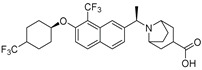
|
Plasma hlPA: 53 (ATX) ATX FRET: 1 (ATX) |
Pocket-Tunnel Hybrid (Class IV)
|
7MFH |
N.D. Not determined.
In the context of cancer, research findings over the last decade suggest that two factors, namely (i) resistance to chemo- and radiation-therapy and (ii) the role of the tumor microenvironment (TME) represent the greatest obstacles to inhibition of cancer progression and therapeutic efficacy [5]. A major barrier to therapeutic interventions lies in the existence of cancer stem cells (CSC), whose slower rates of proliferation, self-renewal capabilities, and upregulation of drug efflux transporters contribute to therapy resistance [5,43]. Growing evidence suggests that LPA activates CSC-associated genes, including stem-cell surface markers, antioxidants, and drug transporters [5]. The ATX-LPAR axis is also upregulated in CSC and the inhibition of ATX and LPAR1 attenuates the CSC-like characteristics of ovarian and breast cancers [1,5,8,44]. These observations are corroborated by reports that established the critical role of LPA in maintaining stemness [45,46,47]. In addition to earlier reports demonstrating a fundamental role in tumorigenesis and epithelial-mesenchymal transition [48], recent findings suggest that the ATX-LPAR axis plays a decisive role in influencing cells in the TME. The inhibitory action of LPA in blocking T cell receptor activation, thereby reducing tumor immunity, has been clearly established [9,10,11,12]. Consequently, there is increasing interest in developing new ATX and LPAR1 inhibitors to suppress cancer progression. Efforts to identify new candidates targeting the ATX-LPAR axis received a significant boost from the solving of the crystal structure of LPAR1 co-crystalized with an inhibitor [49].
4. Progress with Different Classes of ATX Inhibitors
4.1. Orthosteric Inhibitors (Class I)
Lipid-like ATX inhibitors that primarily belong to class I have achieved limited success in preclinical and regulatory development as a result of their high partition coefficient (logP > 5), making them potentially incompatible with clinical development [1,4,27,33]. Discovery of the ATX inhibitor co-crystal structure has provided insights into the active site surfaces and many research groups have used this structure to design small molecule non-lipid class-I ATX inhibitors, such as HA155 and PF-8380 (Figure 1) [50,51,52,53,54]. These inhibitors consist of a core spacer, a hydrophobic tail to occupy the hydrophobic pocket, and a carboxylic acid or its bioisostere to interact with zinc ions located in the active site. However, none of the orthosteric (class I) inhibitors have successfully completed advanced clinical trials, potentially due to off target effects. Hence, recent approaches have focused on developing non-carboxylic acid, non-lipid ATX inhibitors targeting the hydrophobic pocket (class II), the tunnel (class III, allosteric site), or pocket-tunnel hybrid sites (class IV).
4.2. Hydrophobic Pocket Inhibitors (Class II)
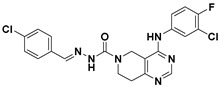
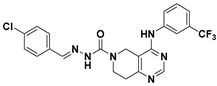
Hydrophobic pocket inhibitors (class II) obstruct binding of the LPC substrate to ATX [1,4,33]. To the best of our knowledge, our group was the first to report a hydrophobic pocket ATX inhibitor (1, Table 1), obtained through virtual and high-throughput screening [33]. The benzamide head group of these compounds reaches deep into the hydrophobic pocket and the morpholine ring sits at the entrance of the tunnel. We have performed structure-activity relationship (SAR) optimization (2–5, Table 1) to improve in vitro and in vivo efficacies of this class of compounds [1,27]. These compounds significantly inhibit melanoma metastasis into the lungs and re-sensitize taxol-resistant breast CSC to paclitaxel [1]. Pantsar et al. (2017) performed a virtual screening and biological evaluation to identify a pyranopyrazole derivative (6, Table 1) of (S) conformation that exhibits high potency inhibition of ATX [55]. Molecular docking experiments suggest that the 3,4-dichlorophenyl head group enters the hydrophobic pocket, the 6-amino-dihydropyranopyrazole-5-carbonitrile moiety acts as the linker, and the 4-chlorobenzyloxy group reaches closer to the tunnel. Zhai and colleagues (2018, 2020) reported dihydropyridopyrimidine carbohydrazide as potent ATX-EGFR dual inhibitors (7 and 8, Table 1) [56,57]. These inhibitors fit primarily deep into the hydrophobic pocket and the other building block extends closer to the tunnel.
4.3. Allosteric Tunnel Inhibitors (Class III)
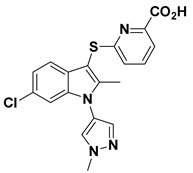
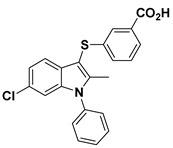
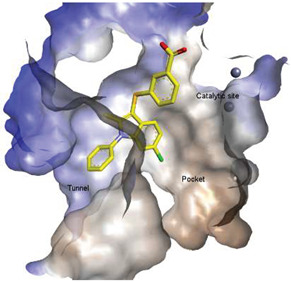
Allosteric tunnel binders deter the release and transport of hydrolyzed LPA [4]. This class of inhibitors represents non-competitive ATX inhibitors that fail to compete with the LPC substrate [58]. Miller et al. (2017) reported that the highly potent N-substituted indole derivatives (9 and 10, Table 1) bind at the allosteric site (tunnel), away from the catalytic site [58]. The optimized compound with N-pyrazole substitution (10, Table 1) exhibits good water solubility and lipophilic ligand efficiency (LLE) [58].
4.4. Pocket-Tunnel Hybrids (Class IV)
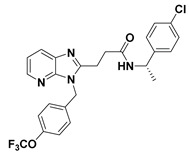
Shah et al. (2016) used HTS to identify imidazopyridine derivatives and performed hit-to-lead synthetic optimization to obtain a potent compound (11, Table 1) [59]. Analysis of X-ray cocrystal structures revealed that the trifluoromethoxy benzene group penetrates deep into the hydrophobic pocket and the (S)-4-chlorophenylethylamine group interacts with the tunnel. The (S)-confirmation is very important, as it enhances potency manifolds. Recently Zhai and colleagues (2020, 2022) reported potent class IV derivatives of GLPG1690 with flexible carbamate and urea moieties that combine important structural motifs of GLPG1690 and PF-8380 (12 and 13, Table 1) [2,3]. These inhibitors demonstrate excellent antitumor efficacy against Hep3B and RAW264.7 cell lines that are known to overexpress ATX mRNA. Recently, Ma et al. (2021) reported the BIO-32546 compound (14, Table 1), which exhibits ATX inhibitory efficacy in the low nanomolar range [22]. X-crystallographic analysis suggests that these classes of compounds belong to pocket-tunnel hybrid (class IV), since they occupy the pocket with a 4-trifluoromethylcyclohexyloxy moiety and the tunnel with an 8-azabicyclo [3.2.1] octane-3-carboxylic acid moiety.
4.5. Tunnel-Pocket Hybrid Inhibitors (Class V)
Clark et al. (2022) recently introduced tunnel-pocket hybrid inhibitors (class V) that combine important structural motifs of non-competitive tunnel binders and competitive orthosteric inhibitors (PDB: 7Z0N) [32]. These inhibitors occupy the tunnel and catalytic site and behave in a competitive manner with the LPC substrate. This series of partially orthosteric-allosteric inhibitors did not demonstrate interactions with the catalytic site. The indirect inhibition of LPAR1 internalization was accomplished by these inhibitors through modulation of LPA-mediated ATX allostery.
It has now been accepted by a large part of the scientific community that inhibitors in Classes II, III, and IV more effectively block the effects of ATX than do those in Class I. These binding modes also allow design and development of more selective ATX inhibitors due to their distance from the zinc ion-containing active site.
5. Biological Prospective of ATX-LPAR1 Dual Inhibition
LPA exerts growth factor-like effects in the majority of cell types. Different LPA species can activate six LPARs to regulate a range of biological responses, including cell proliferation, survival, migration, and invasion. LPA is generated either by hydrolysis of LPC by ATX or via the de novo pathway of glycerol 3-phosphate esterification by glycerol-3-phosphtae acyltransfarase-1 (GPAT-1). ATX, LPAR1 and LPAR2 are overexpressed in different tumor types, including ovarian cancer, osteocarcinoma, metastatic melanoma, neuroblastoma, breast cancer, pancreatic cancer, prostate cancer, and hepatocellular carcinoma [43,60]. Expression of ATX and/or LPAR can be further increased upon chemo- or radiotherapy, as shown for breast cancer [44,61].
Therapy to inhibit ATX, LPAR1 or LPAR2 significantly reduced the incidence of metastasis and resistance to chemotherapy [5,8]. We showed that a combination therapy with an LPAR1 inhibitor (Ki16425) and an ATX inhibitor (BMP22) is more effective in inhibiting melanoma metastasis than single therapy [62]. Although the expression of ATX and LPAR by cancer cells can dictate tumor progression, we and others found that host/stromal ATX and LPAR in the TME play equally important roles in influencing cancer progression, either directly or through the regulation of tumor immunity [9,10,11,12]. We showed that a knockout (KO) mouse that lacked LPAR1 demonstrated strong resistance to melanoma metastasis, suggesting that stromal LPAR1 modulates B16F10 melanoma metastasis to the lung [62]. Notably, B16F10 melanoma cells express ATX at high levels but do not express LPAR1, indicating that the therapeutic success of combined inhibition of LPAR1 and ATX with Ki16425 and BMP22, respectively, in suppressing melanoma metastasis is due to targeting of the ATX-LPAR axis in both cancer and stromal cells.
These findings are consistent with our previous result in which a lipid-like ATX-LPAR pan-antagonist (BrP-LPA, Figure 3) strongly inhibited B16F10 melanoma metastasis into the lung [63]. Likewise, Peyruchaud and coworkers demonstrated that inhibition of LPAR1 or ATX substantially blocked breast cancer bone metastasis [64,65]. In this context, breast cancer cells that overexpress LPAR1 do not express ATX. Hence, these findings suggest that stromal/host ATX and LPAR1 could be targeted simultaneously to further inhibit tumor progression and metastasis [66]. Iwaki et al. (2020) reported that ATX inhibitors enhance the antitumor efficacy of paclitaxel in a breast cancer model [67]. Similarly, we found that simultaneous inhibition of ATX and LPAR1 with a dual inhibitor increased the sensitivity of breast CSC to paclitaxel [1]. Indeed, blocking the ATX-LPAR1 axis reduces the expression of genes associated with multi-drug resistance transporter and antioxidant [68]. Moreover, studies by Erstad et al. (2017) showed in a rat model of hepatic fibrosis and hepatocellular carcinoma (HCC) that targeting the ATX-LPAR1 axis by inhibiting either ATX or LPAR1 decreased fibrosis and HCC development [69].
Apart from cancer, ATX-LPAR1 combination therapy or dual inhibitors of ATX and LPAR1 could also be used to treat IPF, where this signaling pathway has major implications in driving fibrosis. Ninou et al. (2018) showed for the first time that combined ATX-LPAR1 antagonism reduced bleomycin-induced pulmonary fibrosis [17]. Although the effect is minor in comparison to single therapy, it certainly suggests that simultaneous inhibition of ATX and LPAR1 could be a promising therapeutic approach that warrants further investigation. In the case of rheumatoid arthritis (RA), Nikitopoulou et al. observed reduced synovial inflammation and hyperplasia in ATX KO mice [70]. Likewise, Miyabe et al. found that LPAR1 KO mice were protected from RA with significant reduction in synovial inflammation, cartilage damage and bone erosion [71]. Subsequently, two independent groups demonstrated that inhibition of LPAR1 with Ki16425 or inhibition of the ATX-LPAR axis with the ATX-LPAR pan-antagonist Br-LPA reduced the clinical severities of collagen-induced arthritis or RA [72,73]. Altogether, these results encourage implication of ATX-LPAR1 combination therapy or dual inhibitors of ATX-LPAR1 as treatment for various cancers, IPF and rheumatoid arthritis.
6. Strategies for Designing Dual Targeting Inhibitors
Solving of the LPAR1-inhibitor cocrystal structure (PDB: 4Z35) spurred the development of new LPAR1 based inhibitors and novel ATX inhibitors with LPAR1 inhibitory activity. Our group reported first a lipid-like ATX-LPAR pan-antagonist that significantly reduced tumor volume and blood vessel density in breast cancer patients, relative to paclitaxel [63]. We recently reported a small molecule dual inhibitor of ATX-LPAR1 (3f, Figure 3) that is a weak inhibitor of LPAR1. A substantial number of scientific communities believe that dual inhibitors have significant advantages over combination therapy with single inhibitors for a number of reasons, including (i) lower risk of drug-drug interactions [74]; (ii) predictable PK profiles [75]; (iii) lower chance of target-based resistance of dual acting inhibitors than single inhibitors [76]; (iv) dual action inhibitors may overcome compensatory mechanisms [77]; and (v) development of dual acting inhibitors may require fewer clinical trials [78,79]. Based on these concepts, the development of dual inhibitors has drawn significant attention in both academia and industry over the past decade. In an effort to address the problems with single-target drugs or combination therapy, numerous research groups have worked to develop multitarget therapeutic agents, particularly dual inhibitors [74,76], which are designed to preserve the benefits of combination therapy while avoiding its drawbacks [76]. Due to the greater challenge associated with designing dual inhibitors, increased efforts with multidirectional strategies have been pursued [76]. Primarily three strategies have been extensively employed to develop dual agents, namely (i) drug repurposing, (ii) active pharmacophore amalgamation, and (iii) in silico modeling-based approaches [79]. Drug repurposing seeks to identify new therapeutic indications for known drugs [80]. This process is noted for its cost effectiveness and efficient developmental process with respect to the conventional drug discovery process. A number of dual inhibitors discovered during the drug repurposing process share structural similarities of their binding pockets [81]. Pharmacophore amalgamation has been primarily accomplished using two approaches: pharmacophore linking or pharmacophore merging. The pharmacophore linking approach has gained popularity, as it allows the connection of pivotal pharmacophores from multiple selective parent inhibitors into a single candidate [82]. This is a straightforward strategy to bring together crucial functional groups of two different molecules by introducing a suitable linker. The pharmacophore linking strategy is especially useful for the design of potent dual inhibitors from two selective inhibitors that lack a common pharmacophore. However, this approach often leads to higher molecular weight candidates with potentially adverse bioavailability and physiochemical properties [83] and the linkers can interfere with the interactions between the binding sites and the pharmacophores [76]. In contrast, the pharmacophore merging approach allows us to construct a common feature pharmacophore by extracting overlapping features from different selective inhibitors, thereby resulting in hybrid candidates [80]. The pharmacophore merge strategy requires bioactive molecules that possess similar pharmacophore features, often leading to lower molecular weight candidates with favorable physiochemical properties for further hit-to-lead optimization [82]. However, some structural modifications to the parent active molecule may result in diminished efficacy, necessitating precise identification of pharmacophore characteristics. Recently, computational based approaches have been widely employed to expedite the discovery of dual inhibitors [84,85], Computational approaches, including structure-based or ligand-based drug discovery, are greatly advantageous in the absence of either multiple active inhibitors or active site information of a new target. These computational approaches allow us to perform structure/ligand-based pharmacophores, molecular docking, and molecular dynamics (MD) simulation-based virtual screening to find a candidate with high binding affinity and binding free energy for multiple targets.
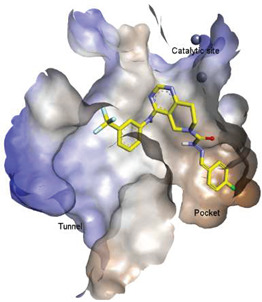
Figure 3.

ATX inhibitors with LPAR inhibitory efficacy. (A). ATX-LPAR pan antagonist BrP-LPA. (B). ATX/LPAR1 dual inhibitor 3f. (C). Mode of binding of 3f in LPAR1 binding site (PDB: 4Z35). (D). Binding mode of 3f in ATX hydrophobic pocket (PDB: 5MHP).
7. The Ins and Outs of ATX Activity Measurement
In the field, methods for the measurement of ATX lysophospholipase D activity have evolved over time [86]. Initially, thin layer chromatography was used to assess generation of radiolabeled LPA by ATX, using 14C-palmitoyl-LPC as a substrate [87]. This technique was later modified to use fluorescently labeled LPC to avoid the use of radiolabeled products [88]. Further, there are several methods that allow for the overall quantification of LPA from biological fluids ranging from immunoassays [89] to mass spectrometric analyses [90,91] as well as MALDI-TOF [92]. These techniques, though effective and able to quantify specific LPA species, generally, lack the capacity for moderate- to high-throughput outputs amenable to screening potential ATX inhibitory compounds. Additionally, global LPA quantification in biological samples is not a direct measure of ATX activity, as other mechanisms for LPA production exist in vivo. As such, LPA quantification is better implemented for downstream characterization rather than identification of ATX inhibitors. Among all of these methods, two standard techniques have emerged by which the lysophospholipase D activity of ATX can be measured via moderate throughput, 96-well platforms. Each assay presents unique advantages and disadvantages.
ATX LPLD activity can be measured indirectly by assessing the production of choline in the cleavage of LPC to LPA in an Amplex Red assay. Utilizing an enzymatic cascade, choline oxidase catalyzes metabolism of the choline byproduct to generate hydrogen peroxide, which subsequently reacts with the Amplex Red reagent in the presence of horseradish peroxidase to generate the fluorescent target resorufin, which that is ultimately quantified [93]. The Amplex Red assay has the advantage of utilizing endogenous LPC substrates of varying chain lengths to assess ATX activity in a more biologically relevant way. However, due to the indirect nature of the assay system, it is impossible to use the Amplex Red assay to determine the mechanism of action of an inhibitor. Additionally, due to the enzymatic cascade involved, it is more difficult to discern whether the potential inhibitory compound acts on ATX itself or one (or more) of the other enzymes involved in the cascade in the absence of additional testing.
ATX LPLD activity can also be measured directly using the fluorescence resonance energy transfer (FRET)-based synthetic LPC analog FS-3 (Echelon Biosciences). In this technique, the fluorophore moiety of the FS-3 substrate is quenched or “silenced” in the native molecule but cleavage by the LPLD activity of ATX separates the fluorophore from the quencher, resulting in detectable fluorescence [94]. By providing a direct readout of ATX LPLD activity, the FS-3 assay is amenable to both moderate-throughput screening of numerous inhibitory compounds and determining inhibitory mechanism of action. However, because FS-3 is a synthetic substrate, the biological implications of ATX LPLD activity determined in this way may be less clear.
Ultimately, when searching for ATX inhibitors, utilization of both assay systems is preferred. FS-3 may be used as an initial screening mechanism, followed by confirmation of activity versus endogenous LPC in the Amplex Red assay. Once validated in the Amplex Red platform, compounds may be further characterized for potency and mechanism of action determination in the FS-3 assay. This two-pronged approach provides the most informative measures of inhibitory capacity while maintaining moderate-throughput workflow.
8. Conclusions and Future Directions
The ATX-LPAR1 axis plays a unique role in the pathogenesis and progression of cancer, IPF, and RA. Although there are multiple reports of potent selective inhibitors of ATX or LPAR1, their application in the clinical setting is still at its infancy. The tunnel-pocket hybrid ATX inhibitor GLPG1690 was the only inhibitor in its class to advance to a phase III clinical trial for IPF before being withdrawn, as it was believed to be ineffective at the terminal stage of the disease, which is characterized by higher concentrations of the ATX substrate, LPC. Currently, a non-competitive tunnel binding ATX inhibitor, BLD-0409, is the subject of a phase II clinical trial for the treatment of IPF and its success remains to be determined. A number of LPAR1 inhibitors, including BMS-986278, are in clinical trials for IPF, but further studies are imperative to determine their prolonged efficacy. Notably, ATX stimulates lung epithelial cell migration via both LPA-dependent and -independent pathways [95].
Hence, growing evidence suggests that simultaneous inhibition of ATX and LPAR1 will be significantly advantageous in preventing disease pathogenesis. Several studies have shown that dual inhibitors have substantial benefit over combination therapy with two selective inhibitors. Most importantly, dual inhibitors may circumvent drug-drug interactions and issues with unpredictable PK profiles of two different drugs. Although recent advancement in computer-aided drug development led to the development of diverse dual inhibitors with unique targets, there are limited reports on the use of ATX-LPAR1 dual inhibitors. One of the issues in designing ATX-LPAR1 specific dual inhibitors can be attributed to co-expression of multiple different LPARs with close structural similarities in the same target or stromal cells. Although our group has reported the ATX-LPAR pan-antagonist Br-LPA, specific inhibition of ATX-LPAR1 is preferred due to a comprehensive range of LPA actions. In this regard, we recently developed two small molecule dual inhibitors of ATX-LPAR1 with weak inhibitory activity against LPAR1. Solution of the LPAR1-inhibitor cocrystal structure has stimulated a computational approach that involves virtual screening by structure based-pharmacophore, molecular docking, and MD simulation followed by biological evaluations to find dual inhibitor hits for further synthetic optimizations of therapeutic utilities.
Acknowledgments
S.B. would like to thank funding from generous startup package from the Middle Tennessee State University and the MTSU Molecular Biosciences Ph.D. program. S.C.L. would like to thank the William and Ella Owens Medical Research Foundation. G.T. is grateful to the Harriet Van Vleet Endowment in Basic Oncology research and NCI grant CA092670. The authors thank Kyle Johnson-Moore in the UTHSC Office of Scientific Writing for editing the manuscript.
Author Contributions
S.B. conceptualization, methodology, writing—original draft preparation, review and editing, funding acquisition. S.L. writing—original draft preparation, review and editing, funding acquisition. D.D.N. methodology, writing—original draft preparation, review and editing. G.J.T. writing—original draft preparation, review and editing, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were included or generated in this review.
Conflicts of Interest
Authors declare no conflict of interest.
Funding Statement
This study was funded by Middle Tennessee State University and the MTSU Molecular Biosciences Ph.D. program (S.B.), the William and Ella Owens Medical Research Foundation (S.L.), the Harriet Van Vleet Endowment in Basic Oncology research and NCI grant CA092670 (G.T.).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Banerjee S., Norman D.D., Lee S.C., Parrill A.L., Pham T.C.T., Baker D.L., Tigyi G.J., Miller D.D. Highly Potent Non-Carboxylic Acid Autotaxin Inhibitors Reduce Melanoma Metastasis and Chemotherapeutic Resistance of Breast Cancer Stem Cells. J. Med. Chem. 2017;60:1309–1324. doi: 10.1021/acs.jmedchem.6b01270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lei H., Wang X., Zhao G., Li T., Cui Y., Wu H., Yang J., Jiang N., Zhai X. Design, synthesis and promising anti-tumor efficacy of novel imidazo[1,2-a]pyridine derivatives as potent autotaxin allosteric inhibitors. Eur. J. Med. Chem. 2022;236:114307. doi: 10.1016/j.ejmech.2022.114307. [DOI] [PubMed] [Google Scholar]
- 3.Lei H., Guo M., Li X., Jia F., Li C., Yang Y., Cao M., Jiang N., Ma E., Zhai X. Discovery of Novel Indole-Based Allosteric Highly Potent ATX Inhibitors with Great In Vivo Efficacy in a Mouse Lung Fibrosis Model. J. Med. Chem. 2020;63:7326–7346. doi: 10.1021/acs.jmedchem.0c00506. [DOI] [PubMed] [Google Scholar]
- 4.Jia Y., Li Y., Xu X.-D., Tian Y., Shang H. Design and Development of Autotaxin Inhibitors. Pharmaceuticals. 2021;14:1203. doi: 10.3390/ph14111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tigyi G., Dacheux M.A., Lin K.-H., Yue J., Norman D., Benyó Z., Lee S.C. Anti-cancer strategies targeting the autotaxin-lysophosphatidic acid receptor axis: Is there a path forward? Cancer Metastasis Rev. 2021;40:3–5. doi: 10.1007/s10555-021-09955-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valdés-Rives S.A., González-Arenas A. Autotaxin-Lysophosphatidic Acid: From Inflammation to Cancer Development. Mediat. Inflamm. 2017;2017:9173090. doi: 10.1155/2017/9173090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tigyi G., Lin K.H., Jang I.H., Lee S.C. Revisiting the role of lysophosphatidic acid in stem cell biology. Exp. Biol. Med. 2021;246:1802–1809. doi: 10.1177/15353702211019283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seo E.J., Kwon Y.W., Jang I.H., Kim D.K., Lee S.I., Choi E.J., Kim K.H., Suh D.S., Lee J.H., Choi K.U., et al. Autotaxin Regulates Maintenance of Ovarian Cancer Stem Cells through Lysophosphatidic Acid-Mediated Autocrine Mechanism. Stem Cells. 2016;34:551–564. doi: 10.1002/stem.2279. [DOI] [PubMed] [Google Scholar]
- 9.Hu J., Oda S.K., Shotts K., Donovan E.E., Strauch P., Pujanauski L.M., Victorino F., Al-Shami A., Fujiwara Y., Tigyi G., et al. Lysophosphatidic acid receptor 5 inhibits B cell antigen receptor signaling and antibody response. J. Immunol. 2014;193:85–95. doi: 10.4049/jimmunol.1300429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee S.C., Dacheux M.A., Norman D.D., Balazs L., Torres R.M., Augelli-Szafran C.E., Tigyi G.J. Regulation of Tumor Immunity by Lysophosphatidic Acid. Cancers. 2020;12:1202. doi: 10.3390/cancers12051202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathew D., Kremer K.N., Strauch P., Tigyi G., Pelanda R., Torres R.M. LPA5 Is an Inhibitory Receptor That Suppresses CD8 T-Cell Cytotoxic Function via Disruption of Early TCR Signaling. Front. Immunol. 2019;10:1159. doi: 10.3389/fimmu.2019.01159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oda S.K., Strauch P., Fujiwara Y., Al-Shami A., Oravecz T., Tigyi G., Pelanda R., Torres R.M. Lysophosphatidic Acid Inhibits CD8 T-cell Activation and Control of Tumor Progression. Cancer Immunol. Res. 2013;1:245–255. doi: 10.1158/2326-6066.CIR-13-0043-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yung Y.C., Stoddard N.C., Mirendil H., Chun J. Lysophosphatidic Acid signaling in the nervous system. Neuron. 2015;85:669–682. doi: 10.1016/j.neuron.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rivera R.R., Lin M.E., Bornhop E.C., Chun J. Conditional Lpar1 gene targeting identifies cell types mediating neuropathic pain. Faseb J. 2020;34:8833–8842. doi: 10.1096/fj.202000317R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ueda H., Matsunaga H., Olaposi O.I., Nagai J. Lysophosphatidic acid: Chemical signature of neuropathic pain. Biochim. Biophys. Acta. 2013;1831:61–73. doi: 10.1016/j.bbalip.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Ninou I., Magkrioti C., Aidinis V. Autotaxin in Pathophysiology and Pulmonary Fibrosis. Front. Med. 2018;5:180. doi: 10.3389/fmed.2018.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ninou I., Kaffe E., Müller S., Budd D.C., Stevenson C.S., Ullmer C., Aidinis V. Pharmacologic targeting of the ATX/LPA axis attenuates bleomycin-induced pulmonary fibrosis. Pulm. Pharmacol. Ther. 2018;52:32–40. doi: 10.1016/j.pupt.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Murphy B., Sum C.-S., Wang T., Heiry R., Kalinowski S., Hung C.-P., Chu C.-H., Azzara A., Milinda Z., Burns L. LPA1 antagonist BMS-986278 for idiopathic pulmonary fibrosis: Preclinical pharmacological in vitro and in vivo evaluation. Eur. Respir. J. 2019;54((Suppl. 63)):PA5383. [Google Scholar]
- 19.Corte T.J., Lancaster L., Swigris J.J., Maher T.M., Goldin J.G., Palmer S.M., Suda T., Ogura T., Minnich A., Zhan X. Phase 2 trial design of BMS-986278, a lysophosphatidic acid receptor 1 (LPA1) antagonist, in patients with idiopathic pulmonary fibrosis (IPF) or progressive fibrotic interstitial lung disease (PF-ILD) BMJ Open Respir. Res. 2021;8:e001026. doi: 10.1136/bmjresp-2021-001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swaney J., Chapman C., Correa L., Stebbins K., Bundey R., Prodanovich P., Fagan P., Baccei C., Santini A., Hutchinson J. A novel, orally active LPA1 receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010;160:1699–1713. doi: 10.1111/j.1476-5381.2010.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montesi S.B., Mathai S.K., Brenner L.N., Gorshkova I.A., Berdyshev E.V., Tager A.M., Shea B.S. Docosatetraenoyl LPA is elevated in exhaled breath condensate in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2014;14:5. doi: 10.1186/1471-2466-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma B., Zhang L., Sun L., Xin Z., Kumaravel G., Marcotte D., Chodaparambil J.V., Wang Q., Wehr A., Jing J., et al. Discovery of Potent Selective Nonzinc Binding Autotaxin Inhibitor BIO-32546. ACS Med. Chem. Lett. 2021;12:1124–1129. doi: 10.1021/acsmedchemlett.1c00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattarai S., Sharma S., Ara H., Subedi U., Sun G., Li C., Bhuiyan M.S., Kevil C., Armstrong W.P., Minvielle M.T., et al. Disrupted Blood-Brain Barrier and Mitochondrial Impairment by Autotaxin-Lysophosphatidic Acid Axis in Postischemic Stroke. J. Am. Heart Assoc. 2021;10:e021511. doi: 10.1161/JAHA.121.021511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flammier S., Peyruchaud O., Bourguillault F., Duboeuf F., Davignon J.-L., Norman D.D., Isaac S., Marotte H., Tigyi G., Machuca-Gayet I., et al. Osteoclast-Derived Autotaxin, a Distinguishing Factor for Inflammatory Bone Loss. Arthritis Rheumatol. 2019;71:1801–1811. doi: 10.1002/art.41005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erenel H., Yilmaz N., Cift T., Bulut B., Sozen I., Aslan Cetin B., Gezer A., Ekmekci H., Kaya B., Tuten A. Maternal serum autotaxin levels in early- and late-onset preeclampsia. Hypertens. Pregnancy. 2017;36:310–314. doi: 10.1080/10641955.2017.1388389. [DOI] [PubMed] [Google Scholar]
- 26.Matas-Rico E., Frijlink E., van der Haar Àvila I., Menegakis A., van Zon M., Morris A.J., Koster J., Salgado-Polo F., de Kivit S., Lança T., et al. Autotaxin impedes anti-tumor immunity by suppressing chemotaxis and tumor infiltration of CD8(+) T cells. Cell Rep. 2021;37:110013. doi: 10.1016/j.celrep.2021.110013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banerjee S., Norman D.D., Deng S., Fakayode S.O., Lee S.C., Parrill A.L., Li W., Miller D.D., Tigyi G.J. Molecular modelling guided design, synthesis and QSAR analysis of new small molecule non-lipid autotaxin inhibitors. Bioorg. Chem. 2020;103:104188. doi: 10.1016/j.bioorg.2020.104188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katsifa A., Kaffe E., Nikolaidou-Katsaridou N., Economides A.N., Newbigging S., McKerlie C., Aidinis V. The Bulk of Autotaxin Activity Is Dispensable for Adult Mouse Life. PLoS ONE. 2015;10:e0143083. doi: 10.1371/journal.pone.0143083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin S., Haque A., Raeman R., Guo L., He P., Denning T.L., El-Rayes B., Moolenaar W.H., Yun C.C. Autotaxin determines colitis severity in mice and is secreted by B cells in the colon. Faseb J. 2019;33:3623–3635. doi: 10.1096/fj.201801415RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hausmann J., Kamtekar S., Christodoulou E., Day J.E., Wu T., Fulkerson Z., Albers H.M., van Meeteren L.A., Houben A.J., van Zeijl L., et al. Moolenaar, W.H.; Perrakis, A. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011;18:198–204. doi: 10.1038/nsmb.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishimasu H., Okudaira S., Hama K., Mihara E., Dohmae N., Inoue A., Ishitani R., Takagi J., Aoki J., Nureki O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011;18:205–212. doi: 10.1038/nsmb.1998. [DOI] [PubMed] [Google Scholar]
- 32.Clark J.M., Salgado-Polo F., Macdonald S.J.F., Barrett T.N., Perrakis A., Jamieson C. Structure-Based Design of a Novel Class of Autotaxin Inhibitors Based on Endogenous Allosteric Modulators. J. Med. Chem. 2022;65:6338–6351. doi: 10.1021/acs.jmedchem.2c00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fells J.I., Lee S.C., Norman D.D., Tsukahara R., Kirby J.R., Nelson S., Seibel W., Papoian R., Patil R., Miller D.D., et al. Targeting the hydrophobic pocket of autotaxin with virtual screening of inhibitors identifies a common aromatic sulfonamide structural motif. FEBS J. 2014;281:1017–1028. doi: 10.1111/febs.12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuozzo J.W., Clark M.A., Keefe A.D., Kohlmann A., Mulvihill M., Ni H., Renzetti L.M., Resnicow D.I., Ruebsam F., Sigel E.A., et al. Novel Autotaxin Inhibitor for the Treatment of Idiopathic Pulmonary Fibrosis: A Clinical Candidate Discovered Using DNA-Encoded Chemistry. J. Med. Chem. 2020;63:7840–7856. doi: 10.1021/acs.jmedchem.0c00688. [DOI] [PubMed] [Google Scholar]
- 35.Drugmaker Galapagos Keeps ‘Appetite’ for Risk after Clinical Setbacks. Reuters. Reported by SarahMorland in Gdansk and Edited by Larry King. [(accessed on 20 July 2022)]. Available online: https://www.reuters.com/article/us-galapagos-outlook-idUKKBN2AJ218.
- 36.Bano S., Al-Rashida M., Alharthy R.D., Khan I.A., Iqbal J. Nucleotide pyrophosphatase/phosphodiesterases (NPPs) including NPP1 and NPP2/ ATX as important drug targets: A patent review (2015–2020) Exp. Opin. Ther. Pat. 2022;32:743–751. doi: 10.1080/13543776.2022.2058874. [DOI] [PubMed] [Google Scholar]
- 37.Figueiredo M. With Positive Early Findings, Phase 2 Cudetaxestat Trial Expected Soon. Pulmonary Fibrosis News. May 24, 2022.
- 38.Blade Therapeutics Announces FDA Activation of IND Application to Investigate Cudetaxestat, a Non-Competitive Autotaxin Inhibitor, in Idiopathic Pulmonary Fibrosis (IPF) [(accessed on 20 July 2022)]. Available online: https://www.businesswire.com/news/home/20210825005020/en/Blade-Therapeutics-Announces-FDA-Activation-of-IND-Application-to-Investigate-Cudetaxestat-a-Non-Competitive-Autotaxin-Inhibitor-in-Idiopathic-Pulmonary-Fibrosis-IPF.
- 39.Abdel-Magid A.F. Lysophosphatidic Acid Receptor 1 Antagonists for the Treatment of Fibrosis. ACS Med. Chem. Lett. 2019;10:1378–1379. doi: 10.1021/acsmedchemlett.9b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palmer S.M., Snyder L., Todd J.L., Soule B., Christian R., Anstrom K., Luo Y., Gagnon R., Rosen G. Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial of BMS-986020, a Lysophosphatidic Acid Receptor Antagonist for the Treatment of Idiopathic Pulmonary Fibrosis. CHEST. 2018;154:1061–1069. doi: 10.1016/j.chest.2018.08.1058. [DOI] [PubMed] [Google Scholar]
- 41.Decato B.E., Leeming D.J., Sand J.M.B., Fischer A., Du S., Palmer S.M., Karsdal M., Luo Y., Minnich A. LPA1 antagonist BMS-986020 changes collagen dynamics and exerts antifibrotic effects in vitro and in patients with idiopathic pulmonary fibrosis. Respir. Res. 2022;23:61. doi: 10.1186/s12931-022-01980-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allanore Y., Distler O., Jagerschmidt A., Illiano S., Ledein L., Boitier E., Agueusop I., Denton C.P., Khanna D. Lysophosphatidic Acid Receptor 1 Antagonist SAR100842 for Patients With Diffuse Cutaneous Systemic Sclerosis: A Double-Blind, Randomized, Eight-Week Placebo-Controlled Study Followed by a Sixteen-Week Open-Label Extension Study. Arthritis Rheumatol. 2018;70:1634–1643. doi: 10.1002/art.40547. [DOI] [PubMed] [Google Scholar]
- 43.She S., Zhang Q., Shi J., Yang F., Dai K. Roles of Autotaxin/Autotaxin-Lysophosphatidic Acid Axis in the Initiation and Progression of Liver Cancer. Front. Oncol. 2022;12:922945. doi: 10.3389/fonc.2022.922945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tigyi G.J., Yue J., Norman D.D., Szabo E., Balogh A., Balazs L., Zhao G., Lee S.C. Regulation of tumor cell—Microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis. Adv. Biol. Regul. 2019;71:183–193. doi: 10.1016/j.jbior.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Gonzalo F.R., Izpisua Belmonte J.C. Albumin-associated lipids regulate human embryonic stem cell self-renewal. PLoS ONE. 2008;3:e1384. doi: 10.1371/journal.pone.0001384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stavish D., Boiers C., Price C., Frith T.J.R., Halliwell J., Saldana-Guerrero I., Wray J., Brown J., Carr J., James C., et al. Generation and trapping of a mesoderm biased state of human pluripotency. Nat. Commun. 2020;11:4989. doi: 10.1038/s41467-020-18727-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee D., Suh D.-S., Lee S.C., Tigyi G.J., Kim J.H. Role of autotaxin in cancer stem cells. Cancer Metastasis Rev. 2018;37:509–518. doi: 10.1007/s10555-018-9745-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu S., Umezu-Goto M., Murph M., Lu Y., Liu W., Zhang F., Yu S., Stephens L.C., Cui X., Murrow G., et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell. 2009;15:539–550. doi: 10.1016/j.ccr.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chrencik J.E., Roth C.B., Terakado M., Kurata H., Omi R., Kihara Y., Warshaviak D., Nakade S., Asmar-Rovira G., Mileni M., et al. Crystal Structure of Antagonist Bound Human Lysophosphatidic Acid Receptor 1. Cell. 2015;161:1633–1643. doi: 10.1016/j.cell.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parrill A.L., Echols U., Nguyen T., Pham T.C., Hoeglund A., Baker D.L. Virtual screening approaches for the identification of non-lipid autotaxin inhibitors. Bioorg. Med. Chem. 2008;16:1784–1795. doi: 10.1016/j.bmc.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 51.Hoeglund A.B., Bostic H.E., Howard A.L., Wanjala I.W., Best M.D., Baker D.L., Parrill A.L. Optimization of a Pipemidic Acid Autotaxin Inhibitor. J. Med. Chem. 2010;53:1056–1066. doi: 10.1021/jm9012328. [DOI] [PubMed] [Google Scholar]
- 52.Albers H.M.H.G., van Meeteren L.A., Egan D.A., van Tilburg E.W., Moolenaar W.H., Ovaa H. Discovery and Optimization of Boronic Acid Based Inhibitors of Autotaxin. J. Med. Chem. 2010;53:4958–4967. doi: 10.1021/jm1005012. [DOI] [PubMed] [Google Scholar]
- 53.Albers H.M.H.G., Hendrickx L.J.D., van Tol R.J.P., Hausmann J., Perrakis A., Ovaa H. Structure-Based Design of Novel Boronic Acid-Based Inhibitors of Autotaxin. J. Med. Chem. 2011;54:4619–4626. doi: 10.1021/jm200310q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawaguchi M., Okabe T., Okudaira S., Nishimasu H., Ishitani R., Kojima H., Nureki O., Aoki J., Nagano T. Screening and X-ray Crystal Structure-based Optimization of Autotaxin (ENPP2) Inhibitors, Using a Newly Developed Fluorescence Probe. ACS Chem. Biol. 2013;8:1713–1721. doi: 10.1021/cb400150c. [DOI] [PubMed] [Google Scholar]
- 55.Pantsar T., Singha P., Nevalainen T.J., Koshevoy I., Leppänen J., Poso A., Niskanen J.M.A., Pasonen-Seppänen S., Savinainen J.R., Laitinen T., et al. Design, synthesis, and biological evaluation of 2,4-dihydropyrano[2,3-c]pyrazole derivatives as autotaxin inhibitors. Eur. J. Pharm. Sci. 2017;107:97–111. doi: 10.1016/j.ejps.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 56.Jing T., Miao X., Jiang F., Guo M., Xing L., Zhang J., Zuo D., Lei H., Zhai X. Discovery and optimization of tetrahydropyrido[4,3-d]pyrimidine derivatives as novel ATX and EGFR dual inhibitors. Bioorg. Med. Chem. 2018;26:1784–1796. doi: 10.1016/j.bmc.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 57.Jiang N., Zhou Y., Zhu M., Zhang J., Cao M., Lei H., Guo M., Gong P., Su G., Zhai X. Optimization and evaluation of novel tetrahydropyrido[4,3-d]pyrimidine derivatives as ATX inhibitors for cardiac and hepatic fibrosis. Eur. J. Med. Chem. 2020;187:111904. doi: 10.1016/j.ejmech.2019.111904. [DOI] [PubMed] [Google Scholar]
- 58.Miller L.M., Keune W.-J., Castagna D., Young L.C., Duffy E.L., Potjewyd F., Salgado-Polo F., Engel García P., Semaan D., Pritchard J.M., et al. Structure–Activity Relationships of Small Molecule Autotaxin Inhibitors with a Discrete Binding Mode. J. Med. Chem. 2017;60:722–748. doi: 10.1021/acs.jmedchem.6b01597. [DOI] [PubMed] [Google Scholar]
- 59.Shah P., Cheasty A., Foxton C., Raynham T., Farooq M., Gutierrez I.F., Lejeune A., Pritchard M., Turnbull A., Pang L., et al. Discovery of potent inhibitors of the lysophospholipase autotaxin. Bioorg. Med. Chem. Lett. 2016;26:5403–5410. doi: 10.1016/j.bmcl.2016.10.036. [DOI] [PubMed] [Google Scholar]
- 60.Balijepalli P., Sitton C.C., Meier K.E. Lysophosphatidic Acid Signaling in Cancer Cells: What Makes LPA So Special? Cells. 2021;10:2059. doi: 10.3390/cells10082059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gupta P.B., Onder T.T., Jiang G., Tao K., Kuperwasser C., Weinberg R.A., Lander E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee S.-C., Fujiwara Y., Liu J., Yue J., Shimizu Y., Norman D.D., Wang Y., Tsukahara R., Szabo E., Patil R., et al. Autotaxin and LPA1 and LPA5 Receptors Exert Disparate Functions in Tumor Cells versus the Host Tissue Microenvironment in Melanoma Invasion and Metastasis. Mol. Cancer Res. 2015;13:174–185. doi: 10.1158/1541-7786.MCR-14-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang H., Xu X., Gajewiak J., Tsukahara R., Fujiwara Y., Liu J., Fells J.I., Perygin D., Parrill A.L., Tigyi G., et al. Dual Activity Lysophosphatidic Acid Receptor Pan-Antagonist/Autotaxin Inhibitor Reduces Breast Cancer Cell Migration In vitro and Causes Tumor Regression In vivo. Cancer Res. 2009;69:5441–5449. doi: 10.1158/0008-5472.CAN-09-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leblanc R., Lee S.C., David M., Bordet J.C., Norman D.D., Patil R., Miller D., Sahay D., Ribeiro J., Clezardin P., et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood. 2014;124:3141–3150. doi: 10.1182/blood-2014-04-568683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leblanc R., Peyruchaud O. The role of platelets and megakaryocytes in bone metastasis. J. Bone Oncol. 2016;5:109–111. doi: 10.1016/j.jbo.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sahay D., Leblanc R., Grunewald T.G., Ambatipudi S., Ribeiro J., Clézardin P., Peyruchaud O. The LPA1/ZEB1/miR-21-activation pathway regulates metastasis in basal breast cancer. Oncotarget. 2015;6:20604–20620. doi: 10.18632/oncotarget.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iwaki Y., Ohhata A., Nakatani S., Hisaichi K., Okabe Y., Hiramatsu A., Watanabe T., Yamamoto S., Nishiyama T., Kobayashi J., et al. ONO-8430506: A Novel Autotaxin Inhibitor That Enhances the Antitumor Effect of Paclitaxel in a Breast Cancer Model. ACS Med. Chem. Lett. 2020;11:1335–1341. doi: 10.1021/acsmedchemlett.0c00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venkatraman G., Benesch M.G., Tang X., Dewald J., McMullen T.P., Brindley D.N. Lysophosphatidate signaling stabilizes Nrf2 and increases the expression of genes involved in drug resistance and oxidative stress responses: Implications for cancer treatment. Faseb J. 2015;29:772–785. doi: 10.1096/fj.14-262659. [DOI] [PubMed] [Google Scholar]
- 69.Erstad D.J., Tager A.M., Hoshida Y., Fuchs B.C. The autotaxin-lysophosphatidic acid pathway emerges as a therapeutic target to prevent liver cancer. Mol. Cell Oncol. 2017;4:e1311827. doi: 10.1080/23723556.2017.1311827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nikitopoulou I., Oikonomou N., Karouzakis E., Sevastou I., Nikolaidou-Katsaridou N., Zhao Z., Mersinias V., Armaka M., Xu Y., Masu M., et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 2012;209:925–933. doi: 10.1084/jem.20112012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miyabe Y., Miyabe C., Iwai Y., Takayasu A., Fukuda S., Yokoyama W., Nagai J., Jona M., Tokuhara Y., Ohkawa R., et al. Necessity of Lysophosphatidic Acid Receptor 1 for Development of Arthritis. Arthritis Rheum. 2013;65:2037–2047. doi: 10.1002/art.37991. [DOI] [PubMed] [Google Scholar]
- 72.Orosa B., García S., Martínez P., González A., Gómez-Reino J.J., Conde C. Lysophosphatidic acid receptor inhibition as a new multipronged treatment for rheumatoid arthritis. Ann. Rheum. Dis. 2014;73:298–305. doi: 10.1136/annrheumdis-2012-202832. [DOI] [PubMed] [Google Scholar]
- 73.Nikitopoulou I., Kaffe E., Sevastou I., Sirioti I., Samiotaki M., Madan D., Prestwich G.D., Aidinis V. A metabolically-stabilized phosphonate analog of lysophosphatidic acid attenuates collagen-induced arthritis. PLoS ONE. 2013;8:e70941. doi: 10.1371/journal.pone.0070941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Proschak E., Stark H., Merk D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019;62:420–444. doi: 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]
- 75.Benek O., Korabecny J., Soukup O. A Perspective on Multi-target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020;41:434–445. doi: 10.1016/j.tips.2020.04.008. [DOI] [PubMed] [Google Scholar]
- 76.Shuai W., Wang G., Zhang Y., Bu F., Zhang S., Miller D.D., Li W., Ouyang L., Wang Y. Recent Progress on Tubulin Inhibitors with Dual Targeting Capabilities for Cancer Therapy. J. Med. Chem. 2021;64:7963–7990. doi: 10.1021/acs.jmedchem.1c00100. [DOI] [PubMed] [Google Scholar]
- 77.Wang G., Zhao Y., Liu Y., Sun D., Zhen Y., Liu J., Fu L., Zhang L., Ouyang L. Discovery of a Novel Dual-Target Inhibitor of ERK1 and ERK5 That Induces Regulated Cell Death to Overcome Compensatory Mechanism in Specific Tumor Types. J. Med. Chem. 2020;63:3976–3995. doi: 10.1021/acs.jmedchem.9b01896. [DOI] [PubMed] [Google Scholar]
- 78.Smith A.D., Roda D., Yap T.A. Strategies for modern biomarker and drug development in oncology. J. Hematol. Oncol. 2014;7:70. doi: 10.1186/s13045-014-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun D., Zhao Y., Zhang S., Zhang L., Liu B., Ouyang L. Dual-target kinase drug design: Current strategies and future directions in cancer therapy. Eur. J. Med. Chem. 2020;188:112025. doi: 10.1016/j.ejmech.2019.112025. [DOI] [PubMed] [Google Scholar]
- 80.Banerjee S., Yadav S., Banerjee S., Fakayode S.O., Parvathareddy J., Reichard W., Surendranathan S., Mahmud F., Whatcott R., Thammathong J., et al. Drug Repurposing to Identify Nilotinib as a Potential SARS-CoV-2 Main Protease Inhibitor: Insights from a Computational and In Vitro Study. J. Chem. Inform. Model. 2021;61:5469–5483. doi: 10.1021/acs.jcim.1c00524. [DOI] [PubMed] [Google Scholar]
- 81.Arnst K.E., Banerjee S., Chen H., Deng S., Hwang D.-J., Li W., Miller D.D. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med. Res. Rev. 2019;39:1398–1426. doi: 10.1002/med.21568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morphy R., Rankovic Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 83.Cavalli A., Bolognesi M.L., Minarini A., Rosini M., Tumiatti V., Recanatini M., Melchiorre C. Multi-target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008;51:347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 84.Zhang W., Pei J., Lai L. Computational Multitarget Drug Design. J. Chem. Inf. Model. 2017;57:403–412. doi: 10.1021/acs.jcim.6b00491. [DOI] [PubMed] [Google Scholar]
- 85.Sonawane V., Mohd Siddique M.U., Jadav S.S., Sinha B.N., Jayaprakash V., Chaudhuri B. Cink4T, a quinazolinone-based dual inhibitor of Cdk4 and tubulin polymerization, identified via ligand-based virtual screening, for efficient anticancer therapy. Eur. J. Med. Chem. 2019;165:115–132. doi: 10.1016/j.ejmech.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 86.Jesionowska A., Cecerska E., Dolegowska B. Methods for quantifying lysophosphatidic acid in body fluids: A review. Anal. Biochem. 2014;453:38–43. doi: 10.1016/j.ab.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 87.Ferry G., Tellier E., Try A., Grés S., Naime I., Simon M.F., Rodriguez M., Boucher J., Tack I., Gesta S., et al. Autotaxin is released from adipocytes, catalyzes lysophosphatidic acid synthesis, and activates preadipocyte proliferation. Up-regulated expression with adipocyte differentiation and obesity. J. Biol. Chem. 2003;278:18162–18169. doi: 10.1074/jbc.M301158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Baker D.L., Fujiwara Y., Pigg K.R., Tsukahara R., Kobayashi S., Murofushi H., Uchiyama A., Murakami-Murofushi K., Koh E., Bandle R.W., et al. Carba analogs of cyclic phosphatidic acid are selective inhibitors of autotaxin and cancer cell invasion and metastasis. J. Biol. Chem. 2006;281:22786–22793. doi: 10.1074/jbc.M512486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen J.H., Zou F., Wang N.D., Xie S.W., Zhang X. Production and application of LPA polyclonal antibody. Bioorg. Med. Chem. Lett. 2000;10:1691–1693. doi: 10.1016/S0960-894X(00)00325-5. [DOI] [PubMed] [Google Scholar]
- 90.Baker D.L., Umstot E.S., Desiderio D.M., Tigyi G.J. Quantitative analysis of lysophosphatidic acid in human blood fractions. Ann. N. Y. Acad. Sci. 2000;905:267–269. doi: 10.1111/j.1749-6632.2000.tb06557.x. [DOI] [PubMed] [Google Scholar]
- 91.Shan L., Jaffe K., Li S., Davis L. Quantitative determination of lysophosphatidic acid by LC/ESI/MS/MS employing a reversed phase HPLC column. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008;864:22–28. doi: 10.1016/j.jchromb.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 92.Bollinger J.G., Ii H., Sadilek M., Gelb M.H. Improved method for the quantification of lysophospholipids including enol ether species by liquid chromatography-tandem mass spectrometry. J. Lipid Res. 2010;51:440–447. doi: 10.1194/jlr.D000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Umezu-Goto M., Kishi Y., Taira A., Hama K., Dohmae N., Takio K., Yamori T., Mills G.B., Inoue K., Aoki J., et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ferguson C.G., Bigman C.S., Richardson R.D., van Meeteren L.A., Moolenaar W.H., Prestwich G.D. Fluorogenic Phospholipid Substrate to Detect Lysophospholipase D/Autotaxin Activity. Org. Lett. 2006;8:2023–2026. doi: 10.1021/ol060414i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao J., He D., Berdyshev E., Zhong M., Salgia R., Morris A.J., Smyth S.S., Natarajan V., Zhao Y. Autotaxin induces lung epithelial cell migration through lysoPLD activity-dependent and -independent pathways. Biochem. J. 2011;439:45–55. doi: 10.1042/BJ20110274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were included or generated in this review.