

Abstract
Background
In this study, we describe the generation of a fully human monoclonal antibody (named ‘7NP2’) targeting human fibroblast activation protein (FAP), an antigen expressed in the microenvironment of different types of solid neoplasms.
Methods
7NP2 was isolated from a synthetic antibody phage display library and was improved by one round of mutagenesis-based affinity maturation. The tumor recognition properties of the antibody were validated by immunofluorescence procedures performed on cancer biopsies from human patients. A fusion protein consisting of the 7NP2 antibody linked to interleukin (IL)-12 was generated and the anticancer activity of the murine surrogate product (named mIL12-7NP2) was evaluated in mouse models. Furthermore, the safety of the fully human product (named IL12-7NP2) was evaluated in Cynomolgus monkeys.
Results
Biodistribution analysis in tumor-bearing mice confirmed the ability of the product to selectively localize to solid tumors while sparing healthy organs. Encouraged by these results, therapy studies were conducted in vivo, showing a potent antitumor activity in immunocompetent and immunodeficient mouse models of cancer, both as single agent and in combination with immune checkpoint inhibitors. The fully human product was tolerated when administered to non-human primates.
Conclusions
The results obtained in this work provided a rationale for future clinical translation activities using IL12-7NP2.
Keywords: antibodies, neoplasm; antigens; cytokines; immunotherapy
WHAT IS ALREADY KNOWN ON THIS TOPIC
Fibroblast activation protein (FAP) has been described as the ‘next billion-dollar nuclear theranostics target’, as >28 different tumor types have successfully been imaged in patients with FAP ligands.
Therefore, FAP-based delivery of cytokine payloads may represent an elegant strategy to increase the therapeutic efficacy.
WHAT THIS STUDY ADDS
Here, we describe an antibody-cytokine fusion protein based on interleukin (IL)-12 and an antibody fragment targeting FAP.
The prototype was capable of inducing tumor regression in an immunocompetent mouse model of cancer.
Moreover, a toxicology study in Cynomolgus monkeys revealed that the fully human product had a linear dose-dependence in pharmacokinetic profiles, was safe and potently active.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The data presented in this study provide a rationale for clinical trials using IL12-7NP2 for the treatment of various types of FAP-positive malignancies.
Background
Over the last decades, the relevance of the tumor microenvironment (TME) in cancer progression has been extensively studied.1 Cancer is a complex disease, in which malignant cells interact with structures of various nature with the goal of generating an environment that facilitates their proliferation.2 On one hand, the tumor stroma holds essential functions fostering tumor growth. On the other hand, that same environment typically produces unique antigens that can be exploited for antibody-based pharmacodelivery applications.3
One of the major components of the TME (which represents approximately 20%–40% of the total tumor mass) are ‘activated’ fibroblasts or cancer-associated fibroblasts (CAFs). The latter usually develop a modified phenotype that resembles the fibroblasts involved in wound healing.4 5 CAFs play a key role in cancer progression such as the proliferation of cancer cells, angiogenesis, and promoting the production of immunosuppressive cytokines.6 7 Fibroblast activation protein (FAP) is a target expressed on CAFs, the antigen was discovered in 1986 using a murine monoclonal antibody called F19.8
From a structural point of view, FAP is a 170 kDa homodimeric type II transmembrane glycoprotein, belonging to the family of postproline dipeptidyl aminopeptidase.9 10 FAP can be found in the majority of malignant solid tumors, while being absent in most healthy tissues.11 Thus, it is an attractive target for both imaging and therapeutic applications.10
The antigen has been described as the ‘next billion-dollar nuclear theranostics target’, since >28 different tumor types have successfully been imaged in patients with radiolabeled FAP ligands (eg, 68Ga-FAPI-04, 68Ga-FAPI-46, 68Ga-OncoFAP).12–14 We have previously described the development of OncoFAP, a highly potent small organic FAP ligand with a dissociation constant of 680 pM to the cognate target.14 The tumor-homing performance of OncoFAP has been validated by PET imaging studies with the 68Ga-OncoFAP derivative. The tracer was administered at a dose of 20 µg per patient and was able to selectively localize in primary tumors as well as in metastatic lesions without evidence of trapping in healthy organs and in blood.14
Several recombinant immunoglobulins targeting FAP have been developed over the last decades. F19 was the first antibody able to target human FAP (hFAP). Immunohistochemistry studies revealed that the F19 antibody could strongly stain pancreatic, breast, ovarian, and colorectal cancers.11 15 A radiolabeled preparation of the humanized version of F19, sibrotuzumab, was used to image patients with metastatic cancer of different origins.16 However, the slow clearance of the intact immunoglobulin from the bloodstream impaired the development of the product as an imaging agent. In a phase II clinical trial, focused on the treatment of patients with metastatic colorectal cancer, the unconjugated antibody was found to be well tolerated but showed no therapeutic activity.16 Fisher et al described in 2012 the generation of two novel FAP-specific antibodies, termed ESC11 and ESC14. In preclinical experiments, radiolabeled preparation of the ESC11 antibody (ie, 177Lu-ESC11) showed preferential accumulation in human melanoma xenografts and induced tumor growth retardation.17
A number of therapeutic proteins directed against FAP, with promising preclinical results, have recently been generated and moved to clinical trials.18 19 A bispecific antibody which simultaneously recognizes FAP on tumor cells and 4-1BB ligand (4-1BBL) on lymphocytes was described by Claus et al and induced long-term cancer remissions in mouse tumor models.18 The product is currently being investigated in a phase I clinical trial. MP0310 (NCT04049903) and MP0317 are two early stage clinical products, based on the DARPin platform, which combine a FAP ligand with 4-1BBL and CD40 agonistic ligands, respectively.19
Monoclonal antibodies targeting TME antigens have been considered for the delivery of bioactive payloads, such as proinflammatory cytokines.20 21 Antibody-cytokine fusions (also called immunocytokines) may exploit the tumor-homing properties of the antibody moiety, in order to concentrate the cytokine payload at the site of disease and enhance the therapeutic index.20 21 Proinflammatory cytokines, such as interleukin-2 (IL-2), tumor necrosis factor (TNF), and interleukin-12 (IL-12), have been extensively studied in oncology.22–24 IL-12 strongly promotes natural killer (NK) cells, CD4+, and CD8+ T cells to produce interferon-gamma (IFN-γ), one of the most relevant mediators of anticancer immunity.25 26
Potent anticancer activity has been achieved by several IL-12-delivery systems, such as immunocytokine fusions.27 Targeted IL-12 therapeutics are proving to be more effective and less toxic when delivered to the tumor site thus generating a systemic anticancer immunity from a locally initiated immune response.28 29 The anti-EDB fusion protein huBC1-IL12 has been evaluated for safety during a phase I trial in patients with renal cell carcinoma. Results showed that the maximum tolerated dose (MTD) of weekly infusions was found to be 15 µg/kg, higher compared with the MTD of recombinant human IL-12 (rhIL12) (0.5 µg/kg).30 Another necrosis-targeting IL-12, NHS-IL12, is currently in phase I clinical studies as monotherapy (NCT01417546) or in combination with avelumab (NCT02994953). NHS-IL12 was well tolerated with an MTD of 16.8 µg/kg with 9% of patients experiencing stable disease.31 Both cited fusion proteins (huBC1-IL12 and NHS-IL12) are based on intact immunoglobulin structures.30 31 By contrast, our group has extensively worked on the generation of antibody fragments for the selective delivery of cytokines in order to achieve a more rapid clearance from circulation and better tumor:organ ratios. L19-IL12, an EDB-targeted immunocytokine, has been evaluated in preclinical studies showing tumor growth inhibition and complete remissions in several immunocompetent mouse models of cancer.32 33 The product entered a phase I dose escalation trial in patients with advanced solid carcinomas (NCT04471987).
In this work, we describe the generation of a novel anti-FAP antibody, called 7NP2. The molecule successfully targeted FAP-positive tumors in vivo and served as a building block for the development of an IL-12-antibody fusion protein. mIL12-7NP2 was produced by fusing murine IL-12 to the N-terminus of 7NP2 in tandem diabody format.32 The immunocytokine was capable of inducing durable complete responses in tumor-bearing mice both as single agent and in combination with checkpoint inhibitors. To prepare for future clinical trials, a fusion protein consisting of human IL-12 linked to the 7NP2 antibody was further investigated in a toxicology study in Cynomolgus monkeys.
Methods
Cell lines
All cell lines were obtained between 2018 and 2021, expanded, and stored as cryopreserved aliquots in liquid nitrogen. The human renal cell carcinoma SKRC52 cell line was kindly provided by Professor E Oosterwijk (Radbound University Nijmegen Medical Center, Nijmegen, The Netherlands) and maintained in RPMI medium (Invitrogen) supplemented with fetal calf serum (10%, FCS; Invitrogen) and antibiotic-antimycotic (1%, AA; Invitrogen). Chinese hamster ovary (CHO) cells, human fibrosarcoma HT1080, and murine colon carcinoma CT26 were purchased from the American Type Culture Collection. Cells were cultured according to the supplier’s protocol (for HT1080: Dulbecco’s Modified Eagle Medium+10% fetal bovine serum (FBS)+1% AA for CT26: RPMI+10% FBS+1% AA) and kept for no longer than 10 passages. Authentication of the cell lines was performed by the cell bank before shipment. SKRC52-hFAP cells and HT1080-hFAP were prepared as previously reported.14 NK-92 cells were purchased from DSMZ (code ACC48) and cultured within MEM complete medium (75% α-MEM medium, 12.5% FCS, 12.5% horse serum, 2 mM L-glutamine, and IX antibiotic mix).
In vitro protein characterization
The fusion proteins described in this work were produced by transient gene expression (TGE) in CHO-S cells and purified from the cell culture supernatant by protein A Sepharose (Sino Biological) affinity chromatography, dialyzed against phosphate-buffered saline (PBS), and stored in PBS at −80°C.34 Purified proteins were analyzed by size-exclusion chromatography using a Superdex 200 increase 10/300 GL column on an ÄKTA FPLC (Cytiva, Marlborough, Massachusetts, USA). Sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed using 10% gels under reducing and non-reducing condition. Affinity measurements were performed by SPR using a BIAcore X100 instrument (Cytiva,) on hFAP-coated SA chip.
Cloning, expression, and purification of mIL12-7NP2 and IL12-7NP2
The fusion protein IL12-7NP2 contains the 7NP2 antibody in homodimeric tandem scFv format linked to murine or human IL-12 at N-terminus. The gene encoding for 7NP2 in diabody format and the gene encoding for the murine IL-12 or human IL-12 were PCR amplified, PCR assembled, and cloned into pcDNA 3.1 (+) between NheI/HindIII restriction sites. The two fusion proteins were expressed using TGE in CHO-S cells, purified, and characterized in vitro as described before.
In vitro biological activities
The biological activity of mIL12-7NP2 was evaluated by an IFN-γ release assay on splenocytes as described by Puca et al.32 The biological activity of IL12-7NP2 was evaluated on NK-92 cells and on human peripheral blood mononuclear cells (hPBMCs) isolated from healthy donors (hPBMCs) as described by Corbellari et al.35 For the stimulation of NK-92 cells, cultured cells were treated with different concentrations of IL12-7NP2. After the defined stimulation time (24 hours), a human IFN-γ ELISA assay (Invitrogen, code EHIFNG2, EHIFNG5), was performed to measure the IFN-γ produced in the culture supernatants according to the manufacturer’s instructions and compared with the supplied IL-12 standard.
Immunofluorescence studies
FAP expression was assessed on ice-cold acetone-fixed 10 µm cryostat sections of SKRC52-hFAP tumors, slides from a human tissue microarray (AmbsBio, B712100), and patient-derived colon cancer samples, patient-derived pancreatic cancer samples, and patient-derived brain metastasis biopsies stained with 7NP2 and KSF IgG1-FITC (the KSF antibody is specific to hen egg lysozyme, an irrelevant antigen). Proteins were FITC labeled according to the manufacturer protocol (Sigma final concentration 10 µg/mL); and revealed with rabbit anti-FITC (Bio-Rad, 4510-7804) and antirabbit AlexaFluor488 (Invitrogen; A11008). For the immunofluorescence-based biodistribution study, tumor-bearing mice were injected with 100 µg/mouse of C5 IgG1-FITC, KSF IgG1-FITC, 7NP2 IgG1-FITC or 200 µg/mouse of mIL12-7NP2 and mIL12-KSF when tumor size reached 100–250 mm3 and sacrificed 24 hours after injection. Tumors were excised and embedded in cryo-embedding medium (NEG-50, Thermo Fisher) and cryostat sections (10 µm) were stained and detected with rabbit anti-FITC and antirabbit AlexaFluor488 (for FITC-IgG1) or with ProteinA-AlexaFluor488 (for mIL12-7NP2). Blood vessels were stained with anti-CD31 (red, AlexaFluor 594). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen; D1306). Slides were mounted with fluorescent mounting medium (Dako) and analyzed with Leica DMI6000B (Leica Microsystems).
Experimental animals
A total of 25 female BALB/c nude mice and 35 BALB/c mice, aged 8 weeks with an average weight of 20 g, were used in this work. Mice were purchased from Janvier (Route du Genest, 53940 Le Genest-Saint-Isle, France) and raised in a pathogen-free environment with a relative humidity of 40%–60%, at a temperature between 18°C and 26°C and with daily cycles of 12 hours light/darkness according to guidelines (GV-SOLAS; FELASA). Blinding of the experimental groups was not performed, animals were randomized in experimental groups according to their tumor volume.
Biodistribution experiments
SKRC52-hFAP or HT1080-hFAP (5×106) tumor cells were injected subcutaneously in the right flank of BALB/c nude mice, while SKRC52 or HT1080 wild-type (wt) tumor cells were injected subcutaneously in the left flank. When tumors reached a volume of 100–200 mm3, fusion proteins (100 μg) were radioiodinated with 125I and chloramine T hydrate, purified on a PD10 column and injected into the lateral tail vein as described before.36 Mice were euthanized 24 hours after injection. Organs, blood, and tumors were weighed, and radioactivity was quantified using a Packard Cobra gamma counter. The immunocytokine uptake was calculated and expressed as the percentage of the injected dose per gram of tissue (%ID/g±SEM). Data were corrected for tumor growth.36
Therapy studies
SKRC52-hFAP (6×106) cells were implanted subcutaneously in the flank of BALB/c nude mice. Mice were monitored daily and tumor volume was measured with a caliper and volume was calculated using the formula: tumor size=(length (mm)×width2(mm))/2. When tumors reached a suitable volume (approximately 100 mm3), mice were injected three times into the lateral tail vein with the fusion proteins. mIL12-7NP2 and mIL12-KSF were diluted in PBS (pH 7.4) and administered at a dose of 8 µg/mouse every 48 hours for three times.
CT26-hFAP (5×106) cells were implanted subcutaneously in the flank of BALB/c mice. When tumors reached a suitable volume (approximately 100 mm3), mice were injected into the lateral tail vein with the pharmacological agents. mIL12-7NP2 was diluted in PBS (pH 7.4) and administered at a dose of 10 µg/mouse every 48 hours for three times; anti-programmed cell death protein 1 (anti-PD1) antibody (BioXcell, clone: 29F.1A12, cat: BE0273) was administered at a dose of 200 µg/mouse every 48 hours for three times, 1 day after the mIL12-7NP2 administration.
Non-human primate study
The purpose of the study was to determine the potential toxicity and systemic exposure of IL12-7NP2 when administered intravenously, once weekly, for four consecutive weeks to the Cynomolgus monkeys. Two animals/sex/group were treated at doses of 0.04, 0.2. and 1 mg/kg/week in a volume of administration of 3 mL/kg. Animals were monitored daily for mortality, clinical signs, and food consumption (qualitative). Body weight was measured once weekly. Clinical pathology investigations (hematology, serum chemistry, coagulation, and urinalysis) were performed predose, on days 7/8 (unscheduled) and day 26. Hematology evaluation was also conducted on day 2, after the first treatment. Electrocardiography examination was performed pretest and at the end of the treatment period (day 26). Flow cytometry analysis was performed on days 1 and 22 at predose, 24, and 96 hours after dosing. In addition, samples for systemic exposure (predose, 5, 15, 60, 120, 240, and 360 min after dosing), cytokines (predose, 6, 12, 24, and 96 hours after dosing), neopterin (predose, 24, and 96 hours after dosing), and antidrug antibodies (ADA) evaluation (predose) were collected on days 1 and 22.
Statistical analysis
Data were analyzed using Prism V.9.0 (GraphPad Software). Statistical significances between multiple groups were evaluated with the one-way analysis of variance (ANOVA) followed by Tukey’s post-test. Differences in tumor volume between therapeutic groups were compared using the two-way ANOVA or mixed effects analysis followed by Tukey’s post-test. Kaplan-Meier survival analysis was performed to assess survival differences among the treatment groups and p values were calculated with Gehan-Breslow-Wilcoxon test. P<0.05 was considered statistically significant.
Results
Isolation and characterization of antibodies specific to human FAP
A first monoclonal antibody (named C5) specific to the extracellular domain of hFAP was isolated from a synthetic human scFv library by phage display technology37 with a KD of 130 nM (kon 1.83×105 M-1s-1 and koff 2.38×10–2 s-1) and its binding affinity was further improved by combinatorial mutagenesis of complementarity-determining region 2 (CDR2) loops using a previously described methodology.38 The best candidate (termed 7NP2) in terms of affinity with a KD of 9.8 nM (kon 2.88×105 M-1s-1 and koff 2.83×10–3 s-1) and specificity was chosen for further characterization (online supplemental figure S1).
jitc-2022-005282supp001.pdf (17.9MB, pdf)
Immunofluorescence analysis on specimens of patient with cancer
7NP2 in IgG1 format (online supplemental figure S2) was studied by immunofluorescence analysis on tissue sections. The protein was labeled with fluorescein isothiocyanate (FITC) and used to stain healthy and malignant tissues from patients (online supplemental figure S3). 7NP2 exhibited an intense staining in colon carcinoma samples, but not in healthy colon tissues, confirming the capability of the antibody to recognize FAP in vitro (figure 1A). In addition, pancreas tumor specimens were stained, confirming FAP expression (figure 1B). In this work, we also report the first FAP expression analysis in secondary brain metastasis samples. The 7NP2 antibody was able to recognize FAP in cancer specimens of different origin (figure 1C). Confocal microscopy analysis on SKRC52-hFAP cells showed no internalization of 7NP2 in IgG1 (online supplemental figure S4).
Figure 1.

Microscopic fluorescence analysis of human fibroblast activated protein (hFAP) expression on tissue human microarray and colon healthy and malignant samples from donors with IgG1(7NP2)-fluorescein isothiocyanate (FITC). Microscopic fluorescence analysis of hFAP expression on healthy and malignant colon samples (A), pancreas specimens (B), and brain metastasis (C) from donors detected with IgG1(7NP2)-FITC and IgG1(KSF)-FITC (negative control). Cryosections were stained with anti-FITC (green, AlexaFluor 488); blood vessels were stained with anti-CD31 (red, AlexaFluor 594); cell nuclei were stained with DAPI (blue). 20× magnification, scale bars=100 µm.
Tumor-homing properties and pharmacokinetic profiles of 7NP2 IgG1
The in vivo tumor-targeting performance of the C5 and 7NP2 antibodies in IgG1 format was studied by injecting FITC-labeled preparations of these immunoglobulins in tumor-bearing mice, followed by an immunofluorescence detection procedure. Tissue sections of SKRC52-hFAP tumors and normal organs, obtained 24 hours after intravenous administration, revealed a selective accumulation of both C5 and 7NP2 into the tumor mass (online supplemental figures S5 and S6). Targeting selectivity was also confirmed in mice bearing two tumors, one expressing hFAP (SKRC52-hFAP) and the other one devoid of the target antigen (wt SKRC52) (online supplemental figure S5). When intravenously administered to a Cynomolgus monkey at a dose of 0.1 mg/kg, 7NP2 IgG1 did not show any in vivo trapping events at early timepoints (online supplemental figure S7).
Generation and in vivo validation of mIL12-7NP2
Encouraged by the preliminary results obtained with the 7NP2 antibody, we generated an antibody-cytokine fusion protein using IL-12 as payload. Figure 2A represents a schematic representation of the mIL12-7NP2 prototype based on a tandem diabody format (online supplemental figure S8).32 39 40 The product was well behaved as evidenced by SDS-PAGE and Size Exclusion Chromatography (SEC) profiles (figure 2B, C) and the mIL12 moiety retained its biological activity to induce IFN-γ release in murine splenocytes (figure 2D). The tumor-homing properties of radio-iodinated mIL12-7NP2 were evaluated by quantitative biodistribution analysis in immunocompromised mice bearing SKRC52-FAP and SKRC52 wt tumors (figure 2E) and in mice bearing HT1080-FAP and HT1080 wt (figure 2F).14 A selective accumulation of mIL12-7NP2 was observed in FAP-positive tumors, but not in FAP-negative tumors and in healthy organs. To test the activity of mIL12-7NP2 in an immunocompetent mouse model, we generated a CT26 cell line stably expressing hFAP (online supplemental figure S9). Also in this setting, mIL12-7NP2 showed a preferential accumulation in FAP-positive neoplastic lesions 24 hours after intravenous administration as evidenced by immunofluorescence-based biodistribution analysis. No detectable signal could be observed in normal organs. The negative control protein mIL12-KSF showed no uptake in tumors and organs (figure 2G).
Figure 2.

Preclinical characterization of mIL12-7NP2. (A) Schematic representation of mIL12-7NP2; (B) sodium dodecyl-sulfate polyacrylamide gel electrophoresis, 10% gel in non-reducing (NR) and reducing (R) conditions of purified mIL12-7NP2; (C) size exclusion chromatogram of mIL12-7NP2; (D) interferon-gamma (IFN-γ) induction assay by mIL12-7NP2 in freshly isolated BALB/c splenocytes. Cells were resuspended at 5×106 cells/mL and incubated for 5 days at 37°C and 5% CO2 with or without 0.1 pM of the interleukin-12 (IL-12) derivatives. IFN-γ was measured in cultured supernatants by sandwich ELISA; (E–F) quantitative biodistribution analysis of radioiodinated mIL12-7NP2 in BALB/c nude mice bearing SKRC52 renal cell carcinoma (E) or HT1080 (F) fibrosarcoma. Results are expressed as percentage of injected dose per gram of tissue (%ID/g±SEM; n=4–5); (G) microscopic fluorescence analysis of tumor-targeting performance of mIL12-7NP2 in CT26-hFAP tumor and organs from BALB/c mice. Cryosections were stained with ProteinA-AlexaFluor 488; cell nuclei were stained with DAPI (blue). 20× magnification, scale bars=100 µm; (H) therapeutic performance of mIL12-7NP2 in BALB/c nude mice bearing SKRC52-hFAP human renal cell carcinoma. Data represent mean tumor volume±SEM, n=6 mice per group; (I) therapeutic performance of mIL12-7NP2 in BALB/c mice bearing CT26-hFAP colon carcinoma. Data represent mean tumor volume±SEM, n=3-5 mice per group. CR, complete response; hFAP, human fibroblast activated protein; IP-10; IFN-inducible protein 10; i.v., intravenous; TNF, tumor necrosis factor. *P<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
mIL12-7NP2 induced tumor remission in three out of six immunocompromised mice, bearing SKRC52-hFAP tumors (figure 2H), without apparent evidence of toxicity (online supplemental figure S10). By contrast, no tumor remission was observed in mice treated with the negative control protein mIL12-KSF (survival plots and statistical analysis in online supplemental figure S11). The anticancer activity of mIL12-7NP2 was further evaluated in immunocompetent BALB/c mice bearing CT26 tumors, stably transfected with hFAP. mIL12-7NP2 was able to induce two complete responses out of five mice, while treatment with mIL12-KSF or anti-PD1 was ineffective in this tumor model. Moreover, the combination of mIL12-7NP2 with an anti-PD1 antibody was capable of inducing three complete responses out of three mice (figure 2I, left panel) and was well tolerated (online supplemental figure S12) (survival plots and statistical analysis in online supplemental figure S13). On three injections of 10 µg mIL12-7NP2 every 48 hours in Balb/c mice, an elevation in serum levels of IFN-γ, TNF, IL-6, and IP-10 was observed, while no changes in IL-10 levels were detected compared with saline treatment (figure 2I, right panel). A histopathological analysis conducted 24 hours after the last injection revealed a mild splenomegaly in mice treated with mIL12-7NP2 compared with saline treatment. No further abnormalities were observed in the other organs (online supplemental figure S14). Treatment with mIL12-7NP2 promoted an influx of NK cells and CD8+ T cells in the tumor mass (online supplemental figure S15).
A fully human IL12-7NP2 fusion protein for clinical applications
To prepare for clinical applications, a fully human IL12-7NP2 fusion protein was generated (figure 3A and online supplemental figure S16). The product showed excellent biochemical properties in SDS-PAGE and SEC profiles (figure 3B, C). The biological activity of human IL-12 to induce IFN-γ release was evaluated both in NK-92 cells and on freshly isolated hPBMCs (figure 3D, E). IL12-7NP2 exhibited a high affinity to recombinant FAP in SPR analysis (figure 3F). We compared the pharmacokinetic profiles of IL12-7NP2 at three dosages (1 mg/kg, 0.2 mg/kg, and 0.04 mg/kg) in Cynomolgus monkeys after a single intravenous administration. The fusion protein exhibited a linear clearance profile (figure 3G), with a half-life of ~3 hours, similar to the one obtained with other antibody-cytokine fusion proteins in diabody format.21 The 7NP2 antibody was able to bind in ELISA to recombinant cynoFAP in ELISA with comparable signal to hFAP (online supplemental figure S17).
Figure 3.

In vitro characterization of IL12-7NP2 (A) Schematic representation of IL12-7NP2; (B) sodium dodecyl-sulfate polyacrylamide gel electrophoresis, 10% gel in non-reducing (NR) and reducing (R) conditions of purified IL12-7NP2; (C) size exclusion chromatogram of IL12-7NP2; (D–E) biological activity assay on human peripheral blood mononuclear cells (D) and on NK-92 cells (E) incubated with IL12-7NP2 and interleukin-12 (IL-12) reference standard; (F) surface plasmon resonance of IL12-7NP2 (250 nM) on streptavidin chip coated with human FAP (hFAP) (correction for change in the refraction index was applied); (G) pharmacokinetic analysis conducted in Cynomolgus monkeys injected once at the dose of 1 mg/kg, 0.2 mg/kg, or 0.04 mg/kg of IL12-7NP2. Blood samples were collected on day 1 and day 22 of the safety toxicology study, before dosing, at 5, 15, 60, 120, 240, and 360 min after end of dosing.
Toxicology studies in Cynomolgus monkeys: hematology, coagulation, and serum chemistry analysis
A toxicology study was performed in Cynolmogus monkeys using IL12-7NP2 at three different doses (1 mg/kg, 0.2 mg/kg, and 0.04 mg/kg) once weekly for 4 weeks (day 1, 8, 15, and 22). After the first treatment, one female animal given 1 mg/kg showed marked hypoactivity. This regimen was discontinued (i.e., animals did not receive subsequent injections of IL12-7NP2) and analyses on day 22 were not conducted.
Hematological assessments revealed a slight to moderate, poor dose-related decrease in lymphocyte absolute count (30%–67%) on day 2 in both sexes at 0.2 and 1 mg/kg/week followed by a complete recovery starting from day 8 and up to day 26 (figure 4A). Marked decrease in monocyte absolute count (about 85%) was observed in one male (no. 4128) and in one female (no. 4134) at 1 mg/kg/week on days 7/8 and, to a lesser extent (about 30%–50%) also in the other male and female at 1 mg/kg/week and in male no. 4131 and female no. 4136 dosed at 0.2 mg/kg/week. The count of large unstained cells increased in almost all treated animals on day 8 (figure 4A). A slight to severe, poor dose-related decrease in neutrophil absolute count (25%–89%) was recorded on days 7/8 in both sexes at 0.2 and 1 mg/kg/week and in male no. 4133 at 0.04 mg/kg/week.
Figure 4.

Hematology, coagulation, and serum chemistry analysis of Cynolmoulgus monkeys administered with IL12-7NP2 at different doses. Hematology parameters evaluated at predose, day 2, day 8, and day 26 of (A) white blood cells (WBC) count, lymphocytes (LYAB) count, neutrophils (LUAB) count, and monocytes (MAB) count of Cynomolgus monkeys administered with IL12-7NP2 (1 mg/kg, 0.2 mg/kg, and 0.04 mg/kg); (B) red blood cell (RBC) count, reticulocytes (RAB) count, hemoglobin (HGB), and hematocrit (HCT) of Cynomolgus monkeys administered with IL12-7NP2 (1 mg/kg, 0.2 mg/kg, and 0.04 mg/kg); (C) platelets count (PLT), prothrombin time (PT), partial prothrombin time (PPT), and fibrinogen (FIB) of Cynomolgus monkeys administered with IL12-7NP2 (1 mg/kg, 0.2 mg/kg, and 0.04 mg/kg); (D) serum chemistry parameters evaluated at predose, day 8, and day 26: aspartate aminotrasnferase (AST), alanine aminotransferase (ALT), gamma-glutamyl transferase (GGT), and alkaline phosphatase (AP) of Cynomolgus monkeys administered with IL12-7NP2 (1 mg/kg, 0.2 mg/kg, and 0.04 mg/kg).
A minimal to moderate, dose-related decrease in red blood cells, hemoglobin, and hematocrit (15%–48%) was seen in both sexes at all doses starting from day 2 and up to day 8. A minimal to severe increase in reticulocytes absolute count was observed on day 8 in both sexes at 0.04 mg/kg/week (3-fold to 28-fold), 0.2 mg/kg/week (13-fold to 25-fold), and at 1 mg/kg/week (1.7-fold to 9.9-fold) (figure 4B). A minimal to slight, poor dose-related decrease in platelet count (about 20%–40%) was recorded on day 2 in both males and in one female (no. 4140) at 0.04 mg/kg/week and in both sexes at 0.2 and 1 mg/kg/week followed by an increase starting from day 8 (figure 4C).
A minimal to slight prolongation in activated partial thromboplastin time (1.2-fold to 1.5-fold) was seen in both males and females treated at 1 mg/kg/week sampled unscheduled on days 7 and/or 8. In addition, both females and one male (no. 4130) at 1 mg/kg/week showed a slight to moderate increase in plasma fibrinogen levels (1.4-fold to 2.8-fold) (figure 4C).
Among serum chemistry, slight to severe increases in aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were noted in male animal no. 4128 (about 2.5-fold) and female no. 4134 (11-fold) at 1 mg/kg/week on days 7/8 and, to a lesser extent, also in male no. 4131 at 0.2 mg/kg/week. AST minimally increased also in male animal no. 4132 and in female no. 4136 at 0.2 mg/kg/week. Minimal to slight decreases in alkaline phosphatase and gamma glutamyl transferase were recorded in almost all animals at 0.2 and 1 mg/kg/week on day 8 and at 0.2 mg/kg/week only on day 26 (figure 4D). No changes were recorded at 0.04 and 0.2 mg/kg/week at the end of the study period.
Toxicology studies in Cynomolgus monkeys: flow cytometry analysis
Compared with the predose values, no major changes were recorded in the percentage of T and B lymphocytes along the sampling times. A substantial but transient decrease in the percentage of NK cells (with a 60%–80% reduction compared with basal values) was observed at 24 hours after the first administration in all treatment groups. A moderate decrease (ranging from 40% to 60%) was again recorded 24 hours after the last treatment (day 22), followed by recovery at 96 hours postdose (figure 5).
Figure 5.

Flow cytometry analysis of Cynomolgus monkeys peripheral blood cells. Flow cytometry analysis of Cynomolgus monkeys blood cells; monkeys were administered with 1 mg/kg (A), 0.2 mg/kg (B), or 0.04 mg/kg (C). Samples were collected at predose, day 2, day 5, day 22 (predose, 24, and 96 hours after dosing). Per cent (%) of CD20+, CD16+, CD8+, and CD4+ cells were analyzed. NK, natural killer.
Toxicology studies in Cynomolgus monkeys: cytokine levels in plasma and antidrug antibodies
In order to characterize the cytokine response after IL12-7NP2 administration, IL-6, IP-10, IFN-γ, and neopterin determinations were performed (figure 6A–D). Results have revealed a dose-dependent increase of IL-6, IP-10, IFN-γ, and neopterin in the serum of the treated animals following the administration of IL12-7NP2. Monkey no. 4134 showed elevated ADA levels already on day 1. Most of animals showed positive ADA levels on day 22 likely due to the fully human nature of IL12-7NP2 (online supplemental figure S18).
Figure 6.
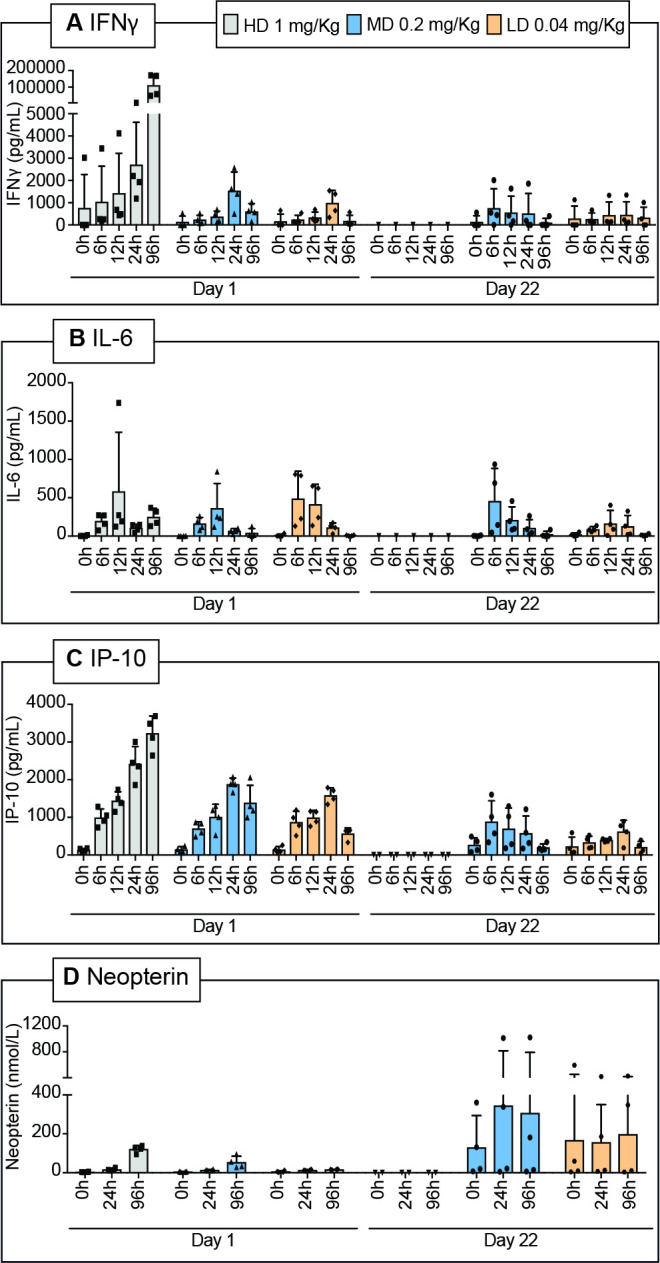
Quantification of interleukin-6 (IL-6), IFN-inducible protein 10 (IP-10), interferon-gamma (IFN-γ), and neopterin in Cynolmogus monkey serum. Blood collection timepoints for cytokine plasma levels (IFN-γ (A), IL-6 (B), IP-10 (C), and neopterin (D)) were at day 1 (predose, 6, 12, 24, and 96 hours after dosing) and at day 22 (predose, 6, 12, 24, and 96 hours after dosing).
Discussion
In this work, we described the in vitro and in vivo properties of a novel FAP-specific immunocytokine called IL12-7NP2. The 7NP2 antibody was isolated by phage display technology from an affinity maturation library generated by combinatorial mutagenesis of the parental antibody’s CDR2 loops.37 The affinity of the 7NP2 antibody (KD=9.8 nM) was improved by >10-fold compared with one of the parental antibody C5 (KD=130 nM). 7NP2 was able to stain FAP in human cancer specimens, such as colon carcinoma, pancreatic cancer, as well as metastatic brain cancer. Encouraged by these results, we generated a fusion protein with IL-12. IL12-7NP2 selectively localized to tumors after intravenous administration and induced an anticancer response in mice bearing FAP-positive neoplasms. Moreover, the fusion protein was found to be well tolerated in Cynomolgus monkeys up to 0.2 mg/kg.
Recombinant hIL12 has been administered to patients with cancer at the MTD dose of 0.5 µg/kg/day and induced objective responses in cutaneous T cell lymphoma, AIDS-related Kaposi’s sarcoma, non-Hodgkin’s lymphoma, renal cell carcinoma, and breast cancer.41–45 We anticipate that a tumor-targeted version may exhibit an improved therapeutic index. In this work, we showed that the IL-12 moiety within FAP-IL12 retained the same in vitro biological activity of rhIL12.
The antibody-based targeted delivery of immunostimulatory cytokines such as IL-12 is particularly attractive when the cognate antigen is accessible, abundant, and genetically stable within the tumor mass, while being undetectable in serum and in normal adult tissues. FAP has been extensively validated by nuclear medicine, without apparent evidence of trapping.
When considering immunocytokine products, it is essential to make a clear distinction between products featuring antibodies in IgG format and products based on antibody fragments. For the first category, a longer circulatory half-life would be expected as a consequence of FcRn interaction, while the in vivo half-life of fusion proteins based on antibody fragments is typically short and similar to the one of the naked cytokine payload.20
For example, the half-life of L19-TNF (based on the L19 antibody in scFv format) was approximately 30 min in patients.46 From a tolerability perspective, recombinant IL-2 received marketing authorization for use at 18 mio IU. L19-IL2 (based on the L19 antibody in diabody format) was safely used at 22.5 mio IU.47 Similarly, recombinant human TNF can be safely given intravenously at ~250 µg/patient.48 In full analogy, L19-TNF can be safely given intravenously at ~780 µg/patient (which corresponds to ~250 µg of recombinant human TNF).46
Collectively, these and other data clearly indicate that immunocytokines based on antibody fragments do not worsen the tolerability profile of the cytokine moiety. We typically prefer to focus on antibody fragments, which are short-lived to minimize the systemic exposure, but still achieving a good tumor uptake.
The safety profile emerging from the toxicology study performed in non-human primates suggests that IL12-7NP2 should be well tolerated and active in patients, at doses higher than the ones previously reported for recombinant IL-12 and IgG-based fusions. Since single agent activities have been reported for non-targeted versions of the cytokine, the new fusion protein will be initially evaluated in monotherapy for its ability to induce durable anticancer responses. Antibody-cytokine fusions may also be considered for combination treatments. We have previously observed that antibody fusions based on IL-2, IL-12, and TNF were able to potently synergize with chemotherapy, radiotherapy, immune-checkpoint inhibitors, or small molecule drug conjugates.49 50 Clinical trials in patients with FAP-positive tumors are planned.
Acknowledgments
The authors thank ScopeM (ETH Zürich) for the use of their electron microscopy facilities. We would like to thank Dr Lorenzo Ghezzi, Dr Jacopo Millul, and Virginie De Woelmont for their help with experimental procedures.
Footnotes
Contributors: Conception and design: LN, EPuc, DN, RD. Development of methodology: LN, EPuc, DN, RD. Acquisition of data: LN, FP, AE, TW, TL, MW, GP, CCarb, GT, MM, NF, RC, CDN, CH, EPuc, CL, SG, CCaro, EPro, DN, RD. Analysis and interpretation of data: LN, EPuc, DN, RD. Study supervision: DN, RD. Guarantors: DN, RD.
Competing interests: DN is a co-founder and shareholder of Philogen (www.philogen.com), a Swiss- Italian Biotech company that operates in the field of ligand-based pharmacodelivery. LN, FP, AE, NF, EPuc, CDN, EPro, RC, MM, and RD are employees of Philochem AG, daughter company of Philogen acting as discovery unit of the group.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
Mouse experiments were performed under a project license (license number 04/2018) granted by the Veterinäramt des Kantons Zürich, Switzerland, in compliance with the Swiss Animal Protection Act (TSchG) and the Swiss Animal Protection Ordinance (TSchV). Procedures on Cynomolgus monkeys (including housing, health monitoring, restrain, dosing, etc) and ethical revision were performed according to the current Italian legislation (Legislative Decree 4 March 2014, n. 26) enforcing the 2010/63/EU Directive on the protection of animals used for biomedical research.
References
- 1.Jin M-Z, Jin W-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther 2020;5:166. 10.1038/s41392-020-00280-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 2020;20:174–86. 10.1038/s41568-019-0238-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brocks B, Garin-Chesa P, Behrle E, et al. Species-crossreactive scFv against the tumor stroma marker "fibroblast activation protein" selected by phage display from an immunized FAP-/- knock-out mouse. Mol Med 2001;7:461–9. 10.1007/BF03401851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Micke P, Ostman A. Tumour-Stroma interaction: cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004;45 Suppl 2:163–75. 10.1016/j.lungcan.2004.07.977 [DOI] [PubMed] [Google Scholar]
- 5.Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol 2014;25:61–8. 10.1016/j.semcancer.2014.02.006 [DOI] [PubMed] [Google Scholar]
- 6.Olumi AF, Grossfeld GD, Hayward SW, et al. Carcinoma-Associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 1999;59:5002–11. 10.1186/bcr138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhome R, Goh RW, Bullock MD, et al. Exosomal microRNAs derived from colorectal cancer-associated fibroblasts: role in driving cancer progression. Aging 2017;9:2666–94. 10.18632/aging.101355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rettig WJ, Chesa PG, Beresford HR, et al. Differential expression of cell surface antigens and glial fibrillary acidic protein in human astrocytoma subsets. Cancer Res 1986;46:6406–12. [PubMed] [Google Scholar]
- 9.Lindner T, Loktev A, Giesel F, et al. Targeting of activated fibroblasts for imaging and therapy. EJNMMI Radiopharm Chem 2019;4:16. 10.1186/s41181-019-0069-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu R, Li H, Liu L, et al. Fibroblast activation protein. Cancer Biol Ther 2012;13:123–9. 10.4161/cbt.13.3.18696 [DOI] [PubMed] [Google Scholar]
- 11.Puré E, Blomberg R. Pro-Tumorigenic roles of fibroblast activation protein in cancer: back to the basics. Oncogene 2018;37:4343–57. 10.1038/s41388-018-0275-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calais J. Fap: the next billion dollar nuclear theranostics target? J Nucl Med 2020;61:163–5. 10.2967/jnumed.119.241232 [DOI] [PubMed] [Google Scholar]
- 13.Kratochwil C, Flechsig P, Lindner T, et al. 68Ga-FAPI PET/CT: Tracer Uptake in 28 Different Kinds of Cancer. J Nucl Med 2019;60:801–5. 10.2967/jnumed.119.227967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Millul J, Bassi G, Mock J, et al. An ultra-high-affinity small organic ligand of fibroblast activation protein for tumor-targeting applications. Proc Natl Acad Sci U S A 2021;118:e2101852118. 10.1073/pnas.2101852118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Busek P, Vanickova Z, Hrabal P, et al. Increased tissue and circulating levels of dipeptidyl peptidase-IV enzymatic activity in patients with pancreatic ductal adenocarcinoma. Pancreatology 2016;16:829–38. 10.1016/j.pan.2016.06.001 [DOI] [PubMed] [Google Scholar]
- 16.Scott AM, Wiseman G, Welt S, et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res 2003;9:1639–47. [PubMed] [Google Scholar]
- 17.Fischer E, Chaitanya K, Wüest T, et al. Radioimmunotherapy of fibroblast activation protein positive tumors by rapidly internalizing antibodies. Clin Cancer Res 2012;18:6208–18. 10.1158/1078-0432.CCR-12-0644 [DOI] [PubMed] [Google Scholar]
- 18.Claus C, Ferrara C, Xu W, et al. Tumor-Targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med 2019;11:eaav5989. 10.1126/scitranslmed.aav5989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Link A, Hepp J, Reichen C, et al. Abstract 3752: preclinical pharmacology of MP0310: a 4-1BB/FAP bispecific DARPin drug candidate promoting tumor-restricted T-cell costimulation. Cancer Res 2018;78:3752. 10.1158/1538-7445.AM2018-3752 [DOI] [Google Scholar]
- 20.Neri D. Antibody-Cytokine fusions: versatile products for the modulation of anticancer immunity. Cancer Immunol Res 2019;7:348–54. 10.1158/2326-6066.CIR-18-0622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.List T, Neri D. Immunocytokines: a review of molecules in clinical development for cancer therapy. Clin Pharmacol 2013;5:29–45. 10.2147/CPAA.S49231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenberg SA. Il-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8. 10.4049/jimmunol.1490019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark J, Vagenas P, Panesar M, et al. What does tumour necrosis factor excess do to the immune system long term? Ann Rheum Dis 2005;64 Suppl 4:iv70–6. 10.1136/ard.2005.042523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol 1995;13:251–76. 10.1146/annurev.iy.13.040195.001343 [DOI] [PubMed] [Google Scholar]
- 25.Gerosa F, Baldani-Guerra B, Lyakh LA, et al. Differential regulation of interleukin 12 and interleukin 23 production in human dendritic cells. J Exp Med 2008;205:1447–61. 10.1084/jem.20071450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity 2003;19:641–4. 10.1016/s1074-7613(03)00296-6 [DOI] [PubMed] [Google Scholar]
- 27.Nguyen KG, Vrabel MR, Mantooth SM, et al. Localized interleukin-12 for cancer immunotherapy. Front Immunol 2020;11:575597. 10.3389/fimmu.2020.575597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyerly HK, Osada T, Hartman ZC. Right time and place for IL12: targeted delivery stimulates immune therapy. Clin Cancer Res 2019;25:9–11. 10.1158/1078-0432.CCR-18-2819 [DOI] [PubMed] [Google Scholar]
- 29.Bramson JL, Hitt M, Addison CL, et al. Direct intratumoral injection of an adenovirus expressing interleukin-12 induces regression and long-lasting immunity that is associated with highly localized expression of interleukin-12. Hum Gene Ther 1996;7:1995–2002. 10.1089/hum.1996.7.16-1995 [DOI] [PubMed] [Google Scholar]
- 30.Lo K-M, Lan Y, Lauder S, et al. huBC1-IL12, an immunocytokine which targets EDB-containing oncofetal fibronectin in tumors and tumor vasculature, shows potent anti-tumor activity in human tumor models. Cancer Immunol Immunother 2007;56:447–57. 10.1007/s00262-006-0203-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fallon J, Tighe R, Kradjian G, et al. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget 2014;5:1869–84. 10.18632/oncotarget.1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puca E, Probst P, Stringhini M, et al. The antibody-based delivery of interleukin-12 to solid tumors boosts NK and CD8+ T cell activity and synergizes with immune checkpoint inhibitors. Int J Cancer 2020;146:2518–30. 10.1002/ijc.32603 [DOI] [PubMed] [Google Scholar]
- 33.Weiss T, Puca E, Silginer M, et al. Immunocytokines are a promising immunotherapeutic approach against glioblastoma. Sci Transl Med 2020;12:eabb2311. 10.1126/scitranslmed.abb2311 [DOI] [PubMed] [Google Scholar]
- 34.Pasche N, Woytschak J, Wulhfard S, et al. Cloning and characterization of novel tumor-targeting immunocytokines based on murine IL7. J Biotechnol 2011;154:84–92. 10.1016/j.jbiotec.2011.04.003 [DOI] [PubMed] [Google Scholar]
- 35.Corbellari R, Stringhini M, Mock J, et al. A novel Antibody-IL15 fusion protein selectively localizes to tumors, synergizes with TNF-based immunocytokine, and inhibits metastasis. Mol Cancer Ther 2021;20:859–71. 10.1158/1535-7163.MCT-20-0853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Luca R, Soltermann A, Pretto F, et al. Potency-matched dual Cytokine-Antibody fusion proteins for cancer therapy. Mol Cancer Ther 2017;16:2442–51. 10.1158/1535-7163.MCT-17-0211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villa A, Lovato V, Bujak E, et al. A novel synthetic naïve human antibody library allows the isolation of antibodies against a new epitope of oncofetal fibronectin. MAbs 2011;3:264–72. 10.4161/mabs.3.3.15616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pini A, Viti F, Santucci A, et al. Design and use of a phage display library. human antibodies with subnanomolar affinity against a marker of angiogenesis eluted from a two-dimensional gel. J Biol Chem 1998;273:21769–76. 10.1074/jbc.273.34.21769 [DOI] [PubMed] [Google Scholar]
- 39.Pasche N, Wulhfard S, Pretto F, et al. The antibody-based delivery of interleukin-12 to the tumor neovasculature eradicates murine models of cancer in combination with paclitaxel. Clin Cancer Res 2012;18:4092–103. 10.1158/1078-0432.CCR-12-0282 [DOI] [PubMed] [Google Scholar]
- 40.Nadal L, Corbellari R, Villa A, et al. Novel human monoclonal antibodies specific to the alternatively spliced domain D of Tenascin C efficiently target tumors in vivo. MAbs 2020;12:1836713. 10.1080/19420862.2020.1836713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guida M, Casamassima A, Monticelli G, et al. Basal cytokines profile in metastatic renal cell carcinoma patients treated with subcutaneous IL-2-based therapy compared with that of healthy donors. J Transl Med 2007;5:51. 10.1186/1479-5876-5-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nunez A, Arroyo N, Godoy L. The role of interleukin-12 in breast cancer (CCR6P.220). J Immunol 2015;194:187.7.25416810 [Google Scholar]
- 43.Rook AH, Wood GS, Yoo EK, et al. Interleukin-12 therapy of cutaneous T-cell lymphoma induces lesion regression and cytotoxic T-cell responses. Blood 1999;94:902–8. [PubMed] [Google Scholar]
- 44.Little RF, Pluda JM, Wyvill KM, et al. Activity of subcutaneous interleukin-12 in AIDS-related Kaposi sarcoma. Blood 2006;107:4650–7. 10.1182/blood-2005-11-4455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ansell SM, Witzig TE, Kurtin PJ, et al. Phase 1 study of interleukin-12 in combination with rituximab in patients with B-cell non-Hodgkin lymphoma. Blood 2002;99:67–74. 10.1182/blood.v99.1.67 [DOI] [PubMed] [Google Scholar]
- 46.Spitaleri G, Berardi R, Pierantoni C, et al. Phase I/II study of the tumour-targeting human monoclonal antibody-cytokine fusion protein L19-TNF in patients with advanced solid tumours. J Cancer Res Clin Oncol 2013;139:447–55. 10.1007/s00432-012-1327-7 [DOI] [PubMed] [Google Scholar]
- 47.Johannsen M, Spitaleri G, Curigliano G, et al. The tumour-targeting human L19-IL2 immunocytokine: preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur J Cancer 2010;46:2926-35. 10.1016/j.ejca.2010.07.033 [DOI] [PubMed] [Google Scholar]
- 48.Roberts NJ, Zhou S, Diaz LA, et al. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget 2011;2:739–51. 10.18632/oncotarget.344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zegers CML, Rekers NH, Quaden DHF, et al. Radiotherapy combined with the immunocytokine L19-IL2 provides long-lasting antitumor effects. Clin Cancer Res 2015;21:1151–60. 10.1158/1078-0432.CCR-14-2676 [DOI] [PubMed] [Google Scholar]
- 50.Schliemann C, Hemmerle T, Berdel AF, et al. Dose escalation and expansion phase I studies with the tumour-targeting antibody-tumour necrosis factor fusion protein L19TNF plus doxorubicin in patients with advanced tumours, including sarcomas. Eur J Cancer 2021;150:143–54. 10.1016/j.ejca.2021.03.038 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2022-005282supp001.pdf (17.9MB, pdf)
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information.