

Abstract
Hyperglycemia has been shown to counterbalance the beneficial effects of tissue plasminogen activator (tPA) and increase the risk of intracerebral hemorrhage in ischemic stroke. Thioredoxin interacting protein (TXNIP) mediates hyperglycemia-induced oxidative damage and inflammation in the brain and reduces cerebral glucose uptake/utilization. We have recently reported that TXNIP-induced NLRP3 (NOD-like receptor pyrin domain-containing-3) inflammasome activation contributes to neuronal damage after ischemic stroke. Here, we tested the hypothesis that tPA induces TXNIP-NLRP3 inflammasome activation after ischemic stroke, in hyperglycemic mice. Acute hyperglycemia was induced in mice by intraperitoneal (IP) administration of a 20% glucose solution. This was followed by transient middle cerebral artery occlusion (t-MCAO), with or without intravenous (IV) tPA administered at reperfusion. The IV-tPA exacerbated hyperglycemia-induced neurological deficits, ipsilateral edema and hemorrhagic transformation, and accentuated peroxisome proliferator activated receptor-γ (PPAR-γ) upregulation and TXNIP/NLRP3 inflammasome activation after ischemic stroke. Higher expression of TXNIP in hyperglycemic t-MCAO animals augmented glucose transporter 1 (GLUT-1) downregulation and increased vascular endothelial growth factor-A (VEGF-A) expression/matrix metallopeptidase 9 (MMP-9) signaling, all of which result in blood brain barrier (BBB) disruption and increased permeability to endogenous immunoglobulin G (IgG). It was also associated with a discernible buildup of nitrotyrosine and accumulation of dysfunctional tight junction proteins: zonula occludens-1 (ZO-1), occludin and claudin-5. Moreover, tPA administration triggered activation of high mobility group box protein 1 (HMGB-1), nuclear factor kappa B (NF-κB), and tumor necrosis factor-α (TNF-α) expression in the ischemic penumbra of hyperglycemic animals. All of these observations suggest a powerful role for TXNIP-NLRP3 inflammasome activation in the tPA-induced toxicity seen with hyperglycemic stroke.
Keywords: Hyperglycemic stroke, Thioredoxin interacting protein, Tissue plasminogen activator, BBB disruption
Introduction
Admission hyperglycemia is now considered an independent predictor of worse outcomes in patients diagnosed with ischemic stroke [1, 2]. Based on the available clinical evidence, thrombolytic therapy is known to be associated with a higher rate of hemorrhagic transformation in hyperglycemic stroke patients, as confirmed in a recent propensity analysis [1]. However, safety outcome trials do not support intensive glycemic control by insulin administration [3, 4], raising more concerns in clinical management. Given the high prevalence of stress hyperglycemia in both diabetic (80–100%) and non-diabetic (30–50%) stroke patients [5, 6], there is a desperate need for adjunctive therapies to minimize the side effects of thrombolytic therapy in hyperglycemic stroke.
Proper blood brain barrier (BBB) function depends on the integrity of its components, mainly the microvascular endothelial cells involved in its formation. These highly specialized endothelial cells are connected by complex junctional structures, namely tight junctions (TJs) and adherent junctions (AJs), and surrounded by pericytes and perivascular astrocytes [7]. Restoration of oxygen and glucose flow to ischemic tissue, during reperfusion, drive reactive oxygen species (ROS) generation, which in turn modify the structure and distribution of TJ proteins leading to increased BBB permeability [8, 9]. Recombinant tissue plasminogen activator (tPA), as the most commonly used drug for clot lysis, is vasoactive, neurotoxic, and pro-oxidant when contacts the lining endothelial cells and extracellular matrix following effective recanalization [10]. This is further aggravated after ischemia in hyperglycemic conditions. Hyperglycemic reperfusion overloads the glucose flow, as a fuel for ROS, after ischemic brain injury and escalates BBB leakage [11]. Thioredoxin (TRX), an essential antioxidant, is inhibited by thoiredoxin interacting protein (TXNIP), an inducible α-arrestin family protein that directly responds to the intracellular concentrations of glucose [12]. Upon stimulation, by oxidative stress or high glucose, TXNIP restrains thioredoxin’s antioxidant activity, exacerbating ROS propagation, to accelerate reperfusion injury. In fact, TXNIP upregulation is known to mediate hyperglycemia-induced oxidative stress [13] and enhance the endocytosis and degradation of glucose trasporter-1 (GLUT-1), to suppress glucose uptake by brain cells [14]. Beyond its role in glucose metabolism, TXNIP has divergent effectors, to drive ROS propagation and inflammation, making it an important therapeutic target in case of endothelial damage induced by metabolic stress [15, 16]. Earlier findings have shown that TXNIP expression was increased in animals subjected to focal ischemia, which was further augmented in hyperglycemic stroke [12, 17]. Nevertheless, the role of TXNIP in tissue plasminogen activator (tPA) induced BBB disruption in hyperglycemic stroke is yet to be determined.
In the context of acute oxidative stress, like that associated with tPA, TRX-TXNIP dissociation could also allow TXNIP to interact with the NOD-like receptor protein (NLRP3) inflammasome, leading to its oligomerization with apoptosis-associated speck-like (ASC), subsequent caspase-1 maturation, and IL-1β secretion. Consequently, it has been shown that NLRP3 inflammasome mediates the hemorrhagic transformation (HT) in ischemic reperfusion (I/R) injury with [18] or without thrombolytic therapy [19].
Based on these findings, we hypothesized that TXNIP is involved in tPA-induced hemorrhagic conversion in hyperglycemic conditions. Therefore, the aim of this preclinical study was to examine the effect of tPA on TXNIP/NLRP3 inflammasome activation in hyperglycemic stroke animals. To determine whether our findings are linked to tPA therapy or to the process of reperfusion per se, we utilized an intraluminal ischemia/reperfusion mouse model, with and without tPA, administered at reperfusion [20]. Our primary data in hyperglycemic stroke animals indicated that tPA promotes TXNIP/NLRP3 inflammasome activation and hemorrhagic transformation, along with poor neurological outcomes. TXNIP has also been suggested to possess other downstream effectors that promote endothelial damage, independent of NLRP3 inflammasome activation [21]. TXNIP appears to affect vascular endothelial-growth factor (VEGF) signaling, particularly under hyperglycemic conditions [22, 23]. This is known to induce matrix metalloproteinases (MMPs) transcription [24], which has an established role in tPA-induced BBB disruption [25] and emphasized our hypothesis that hyperglycemic-TXNIP contributes to tPA-induced BBB toxicity. Therefore, we further examined the plausible association between TXNIP and specific markers of VEGF release/signaling, BBB disruption, and inflammation in tPA therapy.
Experimental Procedures
Animals and Experimental Groups
Wild-type C57Bl/6 mice (Jackson Laboratory, Bar Harbor, ME, USA) were used in the study. All experiments were conducted according to procedures approved by the Institutional Animal Care and Use Committee (IACUC) at UTHSC, Memphis TN. The studies are reported in accordance with the ARRIVE (Animal Research: Reporting in Vivo Experiments) guidelines [26]. Adult male (8–10 weeks) C57Bl/6 mice were housed in standard humidity (45–50%) and temperature (21–25 °C) and 12-h light/dark cycle with food and water ad libitum. These animals were subjected to MCAO or sham surgery and assigned to one of three different experimental groups: MCAO only, MCAO + HG, or MCAO+ HG + tPA. Each group was comprised of 6 animals (n = 6/group). Acute HG (blood glucose 300–400 mg/dl) was achieved through intraperitoneal (IP) injection of a 0.2 ml, 20% glucose solution 15 min before MCAO. Blood glucose was measured from the tail tip using a glucometer (Contour Blood Glucose Monitoring System). The tPA (Activase® (alteplase), Genentech, Inc., San Francisco, CA, USA) was dissolved in sterile water and administered intravenously at a dose of 10 mg/kg with a 10% bolus and 90% continuous infusion over 20 min, started at 1 h after MCAO, using a Harvard 11 Plus Syringe Pump (Harvard apparatus, USA). After administration of tPA, or vehicle, mice were returned to their cages. Body temperature was maintained at 37 ± 0.5 C with heating pad for the duration of surgery. After 24 h of MCAO, animals were deeply anesthetized with ketamine/xylazine mixture (85% and 15%, respectively) and transcardially perfused with ice cold PBS. Animals were then decapitated and their brains were collected. Serial coronal sections were obtained and analyzed with TTC staining, western blotting, and histology. Furthermore, a total of 22 mice were used in the present study; one animal died from each in the t-MCAO and HG t-MCAO group, respectively, and 2 animals from the HG t-MCAO + tPA group. Animals that died were excluded from further evaluation.
Induction of Transient Focal Cerebral Ischemia
Adult animals (22–25 g) were subjected to transient middle cerebral artery occlusion (t-MCAO) using the intraluminal suture model, as described previously [19]. Briefly, animals were anesthetized using 2–5% isoflurane inhalation. A midline incision was made on the ventral surface of the neck, and the right common carotid arteries were isolated and ligated with 6.0 silk suture. The internal carotid artery and the pterygopalatine artery were temporarily occluded with a microvascular clip. Middle cerebral artery occlusion (MCAO) was achieved using a 6–0 silicon-coated nylon suture (Doccol, Sharon, MA), advanced through the internal carotid artery to block the origin of the middle cerebral artery. For all the groups, animals with a reduction in cerebral blood flow of less than 40% from baseline were excluded. After 1 h, animals were re-anesthetized to remove the sutures and allow reperfusion to ischemic brain areas. Anesthesia duration was similar in all groups. Animals were kept at 37 °C for their comfort and recovery after surgery.
Assessment of Functional Neurological Deficit Score
Neurological deficits were evaluated in a blinded manner after 24 h I/R, just before animals were euthanized for ex-vivo evaluations. Using the modified neurological deficit score [17], animals with no apparent deficits obtained 0; signs of forelimb flexion, 1; reduced resistance to push, 2, and with circling, 3. For consistent MCAO completion, only animals with a score of ~ 3 at reperfusion were included in further analysis.
Assessment of Infarct Size and Edema
Infarct size and edema were measured by a blinded investigator. Seven 1-mm thick coronal sections from each brain were stained with 2% TTC solution (2,3,5-triphenyltetrazolium chloride-Sigma-Aldrich, St. Louis, MO) for 2 min and scanned. Infarction and total hemispheric areas were blindly measured using Image J software and corrected for edema, based on recent optimizations using the formula: [(infarct area/ipsilateral area) × contralateral area] [27]. Hemispheric edema was calculated as the difference in area between the ischemic hemisphere and contralateral hemisphere.
Hemoglobin Excess and Hemorrhagic Transformation
Bleeding was quantified using two different methods. 1) Cerebrovascular disruption and the subsequent blood cell infiltration to the brain tissue was quantified using a colorimetric hemoglobin detection assay (QuantiChrom Hemoglobin Assay Kit, BioAssay Systems; Haywood, CA) to address hemorrhagic transformation after stroke. Ipsilateral TTC-stained brain samples were separated and homogenized in a 10% glycerol-Tris buffer saline solution containing 0.5% Tween-20. Samples were analyzed for uniformly colored hemoglobin and read at 562 nm using a standard microplate reader (Synergy HT, BioTek instruments). Hemoglobin concentrations were calculated according to the manufacturer’s instructions and recorded in μg/dL, based on the standard, and was represented as hemoglobin (Hb) excess in the ischemic hemisphere in comparison with contralateral hemisphere. 2) Hemorrhagic occurrence rate (presence of macroscopic bleeding) was performed by visual inspection at the time of sacrifice. This was determined and compared between the treatment groups.
Immunoglobulin Extravasation
Extravasation of endogenous IgG was performed, to accurately address BBB permeability following stroke. Penumbral proteins (30 μg/well) were size-fractionated on SDS-Page gels and electro blotted on a PVDF membrane. Blots were then incubated with horseradish peroxidate (HRP)-conjugated anti-mouse IgG antibody (1 : 10,000; A9044; Sigma USA) for 1 h at room temperature and processed for visualizing the immunoreactive signal. The protein bands were quantified using ImageJ software.
Western Blotting
Peri-infarct (penumbral) and infarcted cortical tissues were used for western blot analysis. Using a brain matrix, the brains were rapidly dissected into 4.0 mm coronal sections (approximately 0.5 mm and – 3.5 mm from Bregma). Brain tissue was homogenized and processed for western blotting as previously described [19]. Thirty-micrograms of protein were loaded in each lane and separated followed by transfer to nitrocellulose membranes. The membranes were blocked for non-specific binding and probed with primary antibodies against NLRP3, Caspase-1, ASC (1:1000; AG-20B-0014; AG-20B-0042; AG-25B-0006 Adipogen Life Sciences), TXNIP (1:1000; NBP1-54578SS; Novus Biologicals), ZO-1, Occludin (1:1000; 40–2200; 33–1500; Thermoscientific), PPARα, HMGB1 (1:1000; SC7273; SC56698; Santa Cruz Biotechnology), Claudin5, VEGF (1:1000; 35–2500; AB-1876-1; Invitrogen), cleaved IL-1β, TNFα, Phospho-NFκBp65 (ser536), NFκBp65, TRX, and GLUT1(1:1000; 12,242; 11,948; 3033; 3034; 2429; 12,939, Cell Signaling Technology), at 4 °C for overnight. Following TBS-T washes, the membranes were incubated with horseradish peroxidate (HRP)-conjugated anti-mouse IgG antibody and anti-rabbit antibody (1 : 10,000; A9044; A9169; SigmaUSA) for 1 h at room temperature. The bands were then visualized by means of an enhanced chemiluminescent substrate system (Thermo fisher scientific). Protein levels were analyzed densitometrically using Image J software, normalized to loading controls, and expressed as fold change.
Immunofluorescence Staining
At 24 h after MCAO, mice were anesthetized with ketamine/xylazine and transcardially perfused with 30 ml of PBS followed by 50 ml of 10% buffered formalin (Fischer Scientific). Brains were removed and post-fixed in the same fixative overnight at 4 °C and then with 30% sucrose in PBS for 72 h. The brains were sectioned in the coronal plane, at a thickness of 10 μm, and were blocked with Serum-Free Protein Block (X0909, DAKO) followed by incubation with primary antibodies against TXNIP (1:100; NBP1-54578SS; Novus biologicals) and cleaved IL-1β (1:100; CST-12242, Cell signaling technology, USA) overnight at 4 °C in a humid chamber. Sections were washed, incubated with fluorescent anti mouse secondary antibodies (1:200; 072–04–18-03; Dylight-549, KPL) for 1 h at room temperature and mounted with ProLong™ Diamond Antifade Mountant with DAPI (Invirogen), and viewed using a Zeiss 710 confocal laser scanning microscope. Negative controls were prepared by omitting the primary antibodies.
Slot Blot for Nitrotyrosine
As a widely used indicator of superoxide-dependent peroxynitrite formation, nitrotyrosine (NO2-Tyr) immunoreactivity was measured by slot blot analysis. Peri-infarct (penumbral) sample homogenates (20 μg) were immobilized onto a nitrocellulose membrane, using a slot blot microfiltration unit. The membranes were then blocked with 5% nonfat milk, incubated with an anti-nitrotyrosine antibody (1:1000; 05–233; Millipore) and visualized with Pierce Super Signal Kit. The optical density of various samples was quantified using densitometry Image J software [17].
Statistical Analysis
The results were expressed as mean ± SEM. Differences among experimental groups were evaluated by student’s t test or ANOVA followed by Tukey’s post-hoc test. Kruskal-Wallis test was used in non-parametric comparisons. Significance was defined by a p < 0.05.
Results
tPA Induces Poor Neurological Outcome and Accentuated BBB Breakdown in Hyperglycemic t-MCAO
All animals showed clear hyperglycemia (blood glucose 300–400 mg/dl), at the time of middle cerebral artery occlusion and reperfusion. Blood glucose concentrations were not affected by tPA infusion (Fig. 1a). Based on our 3-factor neurological assessment score, acute hyperglycemia aggravates neurological deficits, 24 h after ischemic stroke. These deficits were further intensified in those receiving intravenous tPA at reperfusion (Fig. 1b). This is in line with previous findings indicating that intravenous tPA is associated with BBB toxicity that is unrelated to its thrombolytic effects [20]. The escalating neurological deficits seen in hyperglycemic t-MACO and tPA + t-MCAO animals were associated with an incremental increase in ipsilateral brain edema, with no difference in infarct size (Fig. 1c–e), suggesting that poor outcomes are more likely linked to cerebrovascular injury [28]. Consistently, we found more ipsilateral hemorrhages in the coronal sections obtained from brains of hyperglycemic animals treated with tPA (Fig. 2a). Based on further examinations, however, the differences in parenchymal hemoglobin content were limited to an incremental trend, suggesting that BBB disruption is probably more disperse in tPA treated mice (Fig. 2b). For a more precise evaluation of BBB damage, extravasation of endogenous IgG heavy and light chains were evaluated by immunoblotting and confirmed that tPA worsens the BBB disruption induced by hyperglycemic reperfusion (Fig. 2c).
Fig. 1.
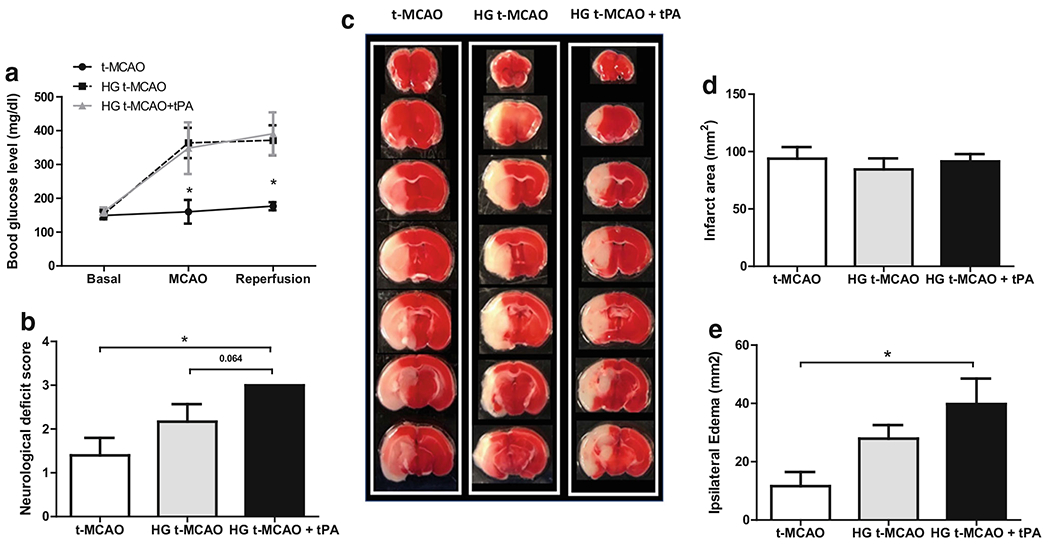
Intravenous tPA induces ipsilateral edema and poor neurological outcomes in hyperglycemic stroke mice. Adult C57Bl/6 mice underwent t-MCAO, 15 min after a single 2 ml IP injection of normal saline or 20% glucose solution. Blood glucose check confirmed the HG state at both occlusion and reperfusion, during which some HG t-MCAO animals received IV tPA (a). IV tPA worsened the neurological function of HG t-MCAO animals (p = 0.064) (b), while there were no remarkable changes detected in TTC stained sections (c) and infarct size (d). The significant escalation in ipsilateral edema suggests occurrence of cerebrovascular damage by tPA in HG t-MCAO mouse brains (e). All values are presented as mean ± SEM. *p < 0.05. IP, intraperitoneal; IV, intravenous; tPA, tissue plasminogen activator; HG, hyperglycemic; NG, normoglycemic; t-MCAO, transient middle cerebral artery occlusion
Fig. 2.
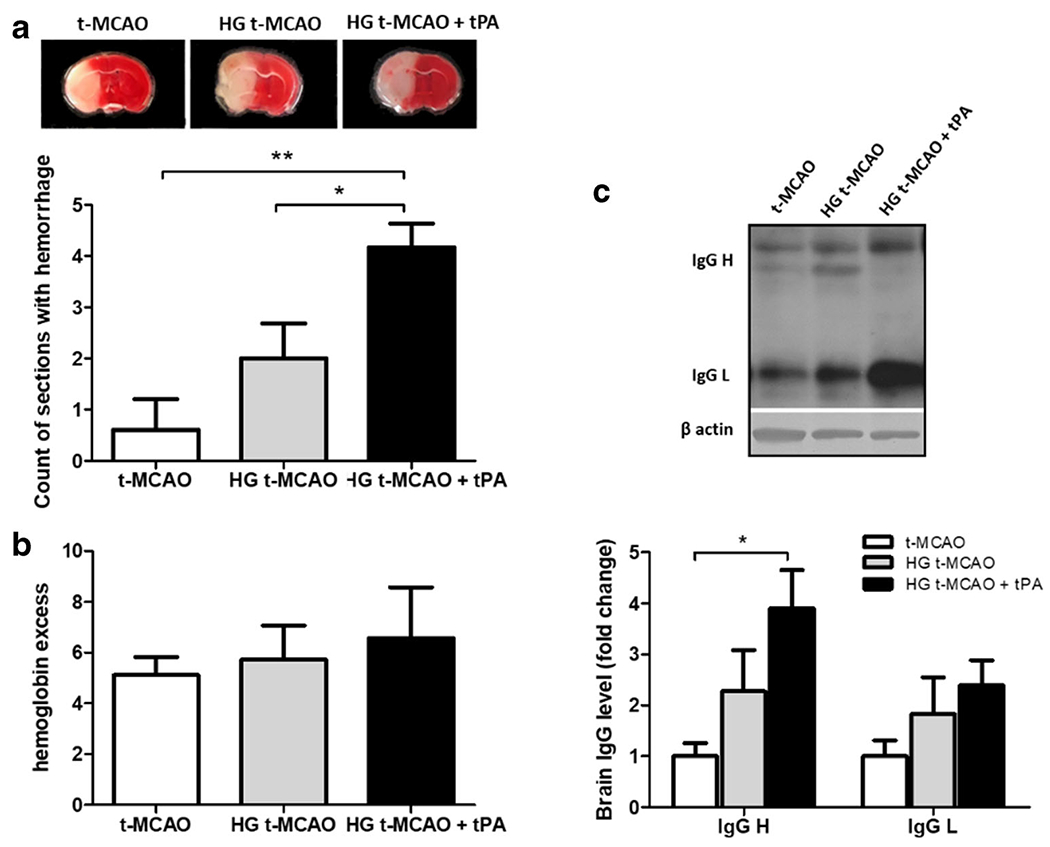
tPA augments BBB breakdown in hyperglycemic stroke brains. Coronal brain sections from HG animals subjected to t-MCAO displayed a much greater number of dispersed hemorrhagic lesions compared with their NG counterparts. This was even more pronounced in those treated with tPA (a). However parenchymal hemoglobin excess did not show significant differences between experimental groups (b), enhanced extravasation of endogenous IgG heavy chain confirmed increased BBB permeability by tPA (c). All values are presented as mean ± SEM. *p < 0.05, **p < 0.01. BBB, Blood brain barrier; tPA, tissue plasminogen activator; HG, hyperglycemic; NG, normoglycemic; t-MCAO, transient middle cerebral artery occlusion
Hyperglycemia Moderately Promotes BBB Leakage, Following t-MCAO, in Parallel with TXNIP/NLRP3 Inflammasome Stimulation
We have previously shown that TXNIP is higher expressed, following experimental stroke in normoglycemic animals [17]. Our immunoblotting analysis of ipsilateral t-MCAO samples indicates that acute hyperglycemia may further elevate TXNIP expression (Fig. 3a). High glucose levels have been suggested to induce TXNIP, partly through peroxisome proliferator-activated receptor gamma (PPAR-γ) transcriptional activity, which in turn regulates PPAR-γ signaling [29, 30]. Consistently, we detected a subtle rise in PPAR-γ protein expression (Fig. 3b). Although, increased PPAR-γ activity is known to be neuroprotective, enhanced protein expression does not necessarily conclude more DNA binding activity [31]. In case of hyperglycemic reperfusion tPA toxicity, increased PPAR-γ expression did not retain enough DNA-binding to inhibit the higher TXNIP expression, as we observed in brain samples. However, given the parallel poor outcomes, we postulate that the potentially neuroprotective transcripts are not strong enough to overweigh the parallel deteriorating signals like TXNIP, inflammasome, and tPA-induced BBB injury as well. Interestingly, the moderate TXNIP upregulation in hyperglycemic stroke was associated with significant NRLP3 inflammasome activation and cleavage of caspase-1/IL-1β precursors (Fig. 3c–g). The rise in NLRP3/caspase-1/IL-1β appears to be a result of NLRP3 inflammasome stimulation rather than priming, as we observed no change in ASC and non-cleaved caspase-1 (Fig. 3d and e). The enhanced cytosolic TXNIP expression was also found to co-localize with more cytokine release in the peri-infarct (penumbra) region of hyperglycemic stroke brains (Fig. 3h), in parallel with increased MMP-9 expression (Fig. 5) and ipsilateral edema (see Fig. 1e).
Fig. 3.
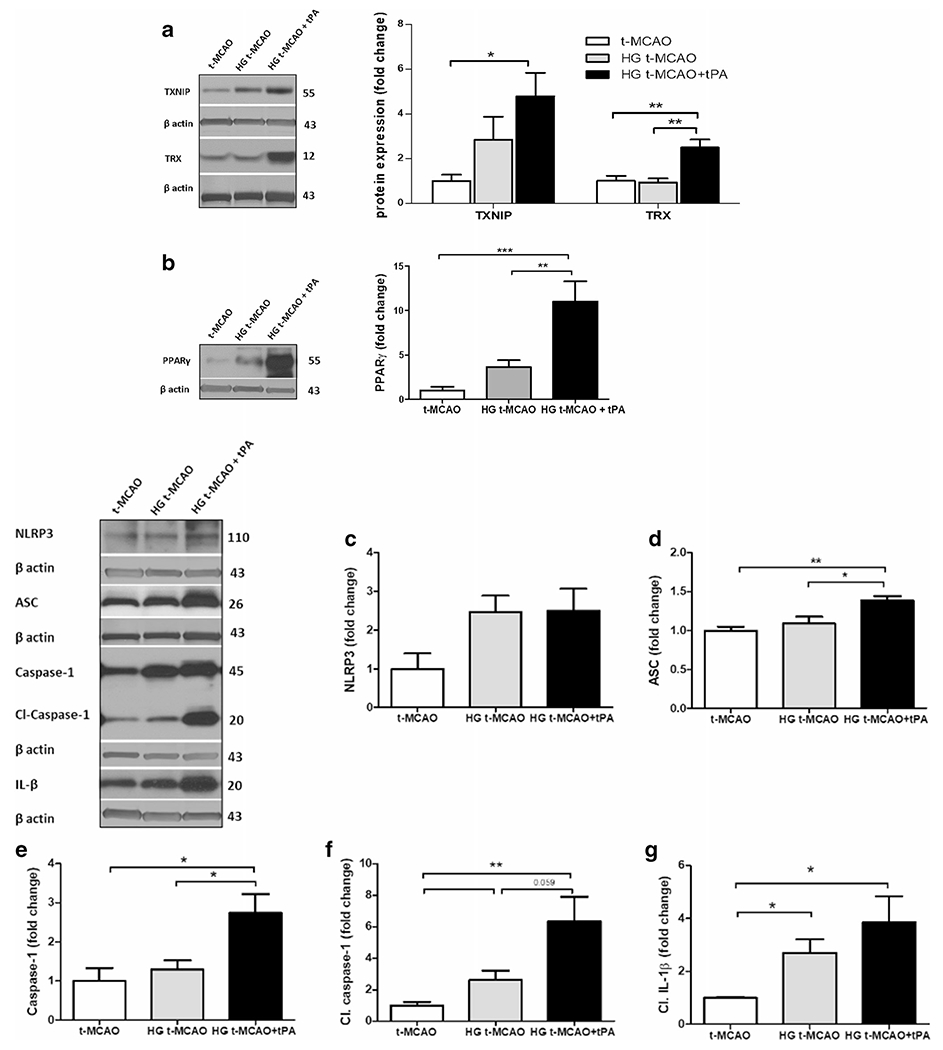
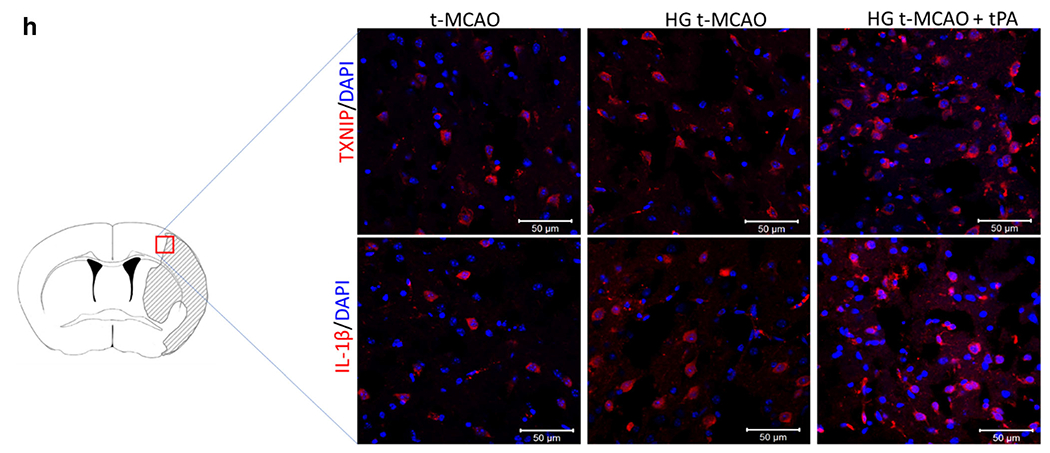
TXNIP/NLRP3 inflammasome activity is upregulated in hyperglycemic stroke and further exacerbated with tPA. A marginal increase in TXNIP was detected in HG t-MCAO compared with NG brains. IV tPA-induced a remarkable increase in both TXNIP and TRX levels (a). This was associated with substantial penumbral PPAR-γ upregulation (b). Further examinations of NLRP3 inflammasome assembly, indicated that only brains of HG t-MCAO animals that received tPA showed a significant upregulation of NLRP3 (c), ASC (d), and caspase-1 (e), implying NLRP3 inflammasome priming. Hyperglycemia could marginally stimulate NLRP3 inflammasome activity in stroke brains, as indicated by its cleavage products cleaved-caspase 1 (f) and IL-1β (g), both of which built-up in those treated with tPA at reperfusion. Immunostaining of t-MCAO penumbral region further confirmed discernible TXNIP/IL-1β upregulation in HG animals, particularly those treated with tPA at reperfusion (H). All values are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. tPA, tissue plasminogen activator; HG, hyperglycemic; IV Intravenous; NG, normoglycemic; t-MCAO, transient middle cerebral artery occlusion; TXNIP, thioredoxin interacting protein; TRX, thioredoxin; PPAR-γ, peroxisome proliferator activated receptor-γ; NLRP3, NOD-like receptor pyrin domain-containing-3; ASC, apoptosis-associated speck-like protein; IL-1β, interleukin 1-β
Fig. 5.
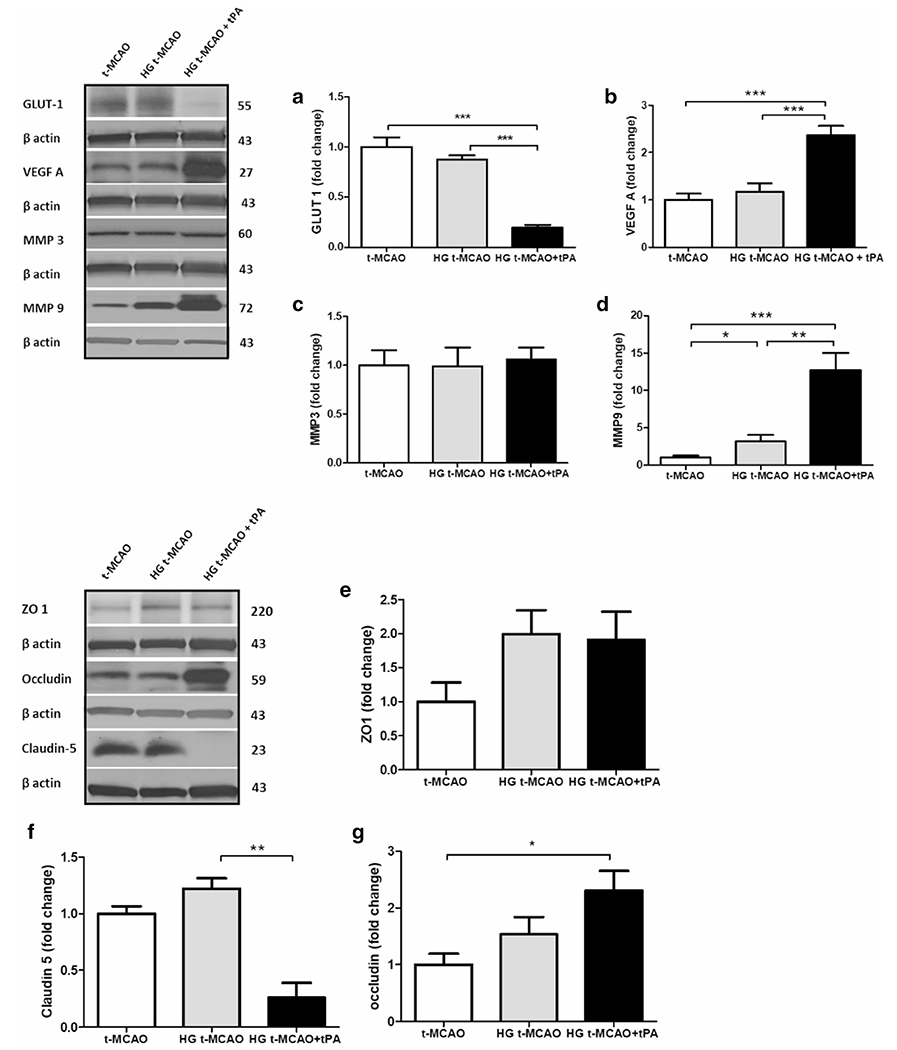
Intravenous tPA differentially impairs GLUT-1 expression and stimulates VEGF-A/MMP-9 signaling in hyperglycemic stroke. While hyperglycemic stroke, on its own, did not induce any changes in GLUT-1, IV tPA profoundly decreased penumbral GLUT-1 levels (a) in parallel with a remarkable VEGF-A induction (b). The VEGF-A upregulation was not associated with a higher MMP-3 expression in HG animals (c) but paralleled significant MMP-9 expression (d). However, this did not affect the mildly induced ZO-1 expression seen with hyperglycemic stroke but (e) may partly explain the tPA-induced claudin-5 degradation (f) and accumulation of non-functional tight junction protein occludin (g) with tPA in the context of hyperglycemia. All values are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. IV, intravenous; tPA, tissue plasminogen activator; HG, hyperglycemic; t-MCAO, transient middle cerebral artery occlusion; GLUT-1, glucose transporter 1; VEGF-A, vascular endothelial growth factor-A; MMP-9; ZO-1, zonula occludens-1
tPA Accentuates TXNIP/NLRP3 Inflammasome Assembly, Following Hyperglycemic Reperfusion, along with HMGB-1 and Nitrotyrosine Buildup
tPA led to a drastic increase in BBB permeability, when infused early, at the time of hyperglycemic reperfusion (see Fig. 2c). This coincided with a rise in PPAR-γ/ TXNIP expression and NLRP3/caspase-1/IL-1β cascade stimulation (see Fig. 3), which we recently showed to be partly responsible for the hemorrhagic transformation associated with ischemic stroke [19]. Enhanced levels of NLRP3, ASC, and un-cleaved caspase-1 are implicative of enhanced NLRP3 inflammasome priming, probably arising from enhanced inflammatory cytokines and immune cell recruitment. Interestingly, even though HMGB-1 was suggested to play a role in hyperglycemia induced endothelial injury, components of the HMGB-1/NF-κB/TNF-α pathway were not upregulated in hyperglycemic stroke brains, except in those that received tPA at reperfusion (Fig. 4a–c). Nitrotyrosine levels, determined by slot blot analysis as an index of the pro-oxidant/ antioxidant status [32], were markedly elevated in brains of tPA treated animals (Fig. 4d). This provided direct evidence of oxidative damage, in parallel with TXNIP hyperactivity.
Fig. 4.
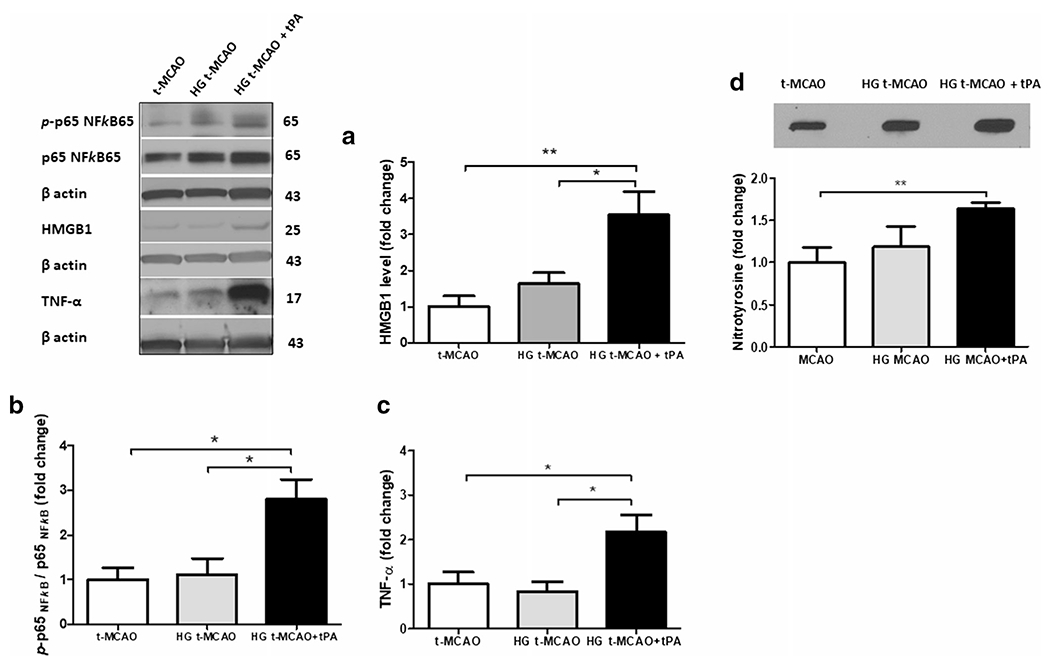
Intravenous tPA induces HMGB-1 upregulation in hyperglycemic stroke and aggravates NF-κB activation and oxidative stress. IV tPA resulted in a significant upregulation of HMGB-1 protein in HG t-MCAO animals (a) leading to consequent NF-κB activation, as determined by p-p65 NF-kB / p65 NF-kB ratio (b) as further confirmed by substantial TNF-α release (c). HMGB-1/NF-κB/TNF-α activation by tPA concurred with discernible oxidative stress as indicated by nitrothyrosine slot-blotting (d). All values are presented as mean ± SEM. *p < 0.05, **p < 0.01. IV, intravenous; tPA, tissue plasminogen activator; HG, hyperglycemic; t-MCAO, transient middle cerebral artery occlusion; HMGB-1, high mobility group box protein 1; NF-κB, nuclear factor kappa B; TNF-α, tumor necrosis factor-α
The Distinct Influence of tPA on Hyperglycemic Reperfusion Injury Is Associated with GLUT-1 Degradation, VEGF-A Release, and TXNIP Upregulation
Despite significant rise in antioxidant capacity of TRX in tPA treated animals (Fig. 3a), the nitrotyrosine levels were still high in brain samples, indicating that tPA administration in hyperglycemic animals post-stroke was associated with profound oxidative stress and inflammation. It is also important to note that while hyperglycemia could impair BBB integrity following stroke, tPA-infusion nearly doubled that BBB breakdown in hyperglycemic reperfusion (Fig. 2c). This was associated with a ten-fold and five-fold increase in PPAR-γ and TXNIP levels respectively (see Fig. 3a and b). Infact, TXNIP upregulation did not affect GLUT-1 expression in hyperglycemic stroke brains, except in those that received tPA (Fig. 5a), suggesting a required TXNIP threshold to induce tangible GLUT-1 degradation. Importantly, hyperglycemic stroke did not induce significant changes in VEGF-A expression unless the animals were treated with tPA, at reperfusion (Fig. 5b). These animals showed significant elevations in VEGF-A, which did not affect MMP-3 expression (Fig. 5c), despite its ability to induce a ten-fold increase in MMP-9 expression (Fig. 5d). Existing evidence, particularly in-vitro, indicates that MMP-9 increases BBB breakdown by degradation of tight junction (TJ) proteins [33]. However, our observations showed that MMP-9 upregulation, particularly in tPA treated animals, was associated with an elevation in TJ proteins (Fig. 5e–g), especially claudin-5 which better reflected tPA-induced TJ degradation (Fig. 5f). On the other hand, zonula occludens-1 (ZO-I) did not show considerable differences between hyperglycemic stroke brains with and without tPA (Fig. 5a). The moderate increase we observed in occludin (Fig. 5g) might be explained with the severity of I/R injury and subsequent angiogenic signals. This may lead to accumulation of premature TJ proteins, particularly in the core region which may be picked in penumbral sampling. The escalated IgG extravasation (Fig. 2c) may have been a consequence of the dysfunctional protein accumulation following endothelial inflammation [34].
Discussion
The BBB disruption that ensues from thrombolytic therapy with tPA [35] is a complex consequence of ischemic insult and reperfusion injury. A high blood glucose level is detrimental, as it contributes to tPA-induced cerebrovascular damage [36]. This experimental study concludes that tPA profoundly aggravates TXNIP upregulation and BBB breakdown, in response to experimental hyperglycemic stroke. Our results illustrate a sharp increase in neurovascular inflammation. This entails stimulation of NLRP3 inflammasome assembly with activation of HMGB-1/NF-κB and VEGF/MMP-9 pathways, each of which may mediate the detrimental effects of TXNIP in tPA-associated BBB disruption after hyperglycemic stroke (Fig. 6).
Fig. 6.
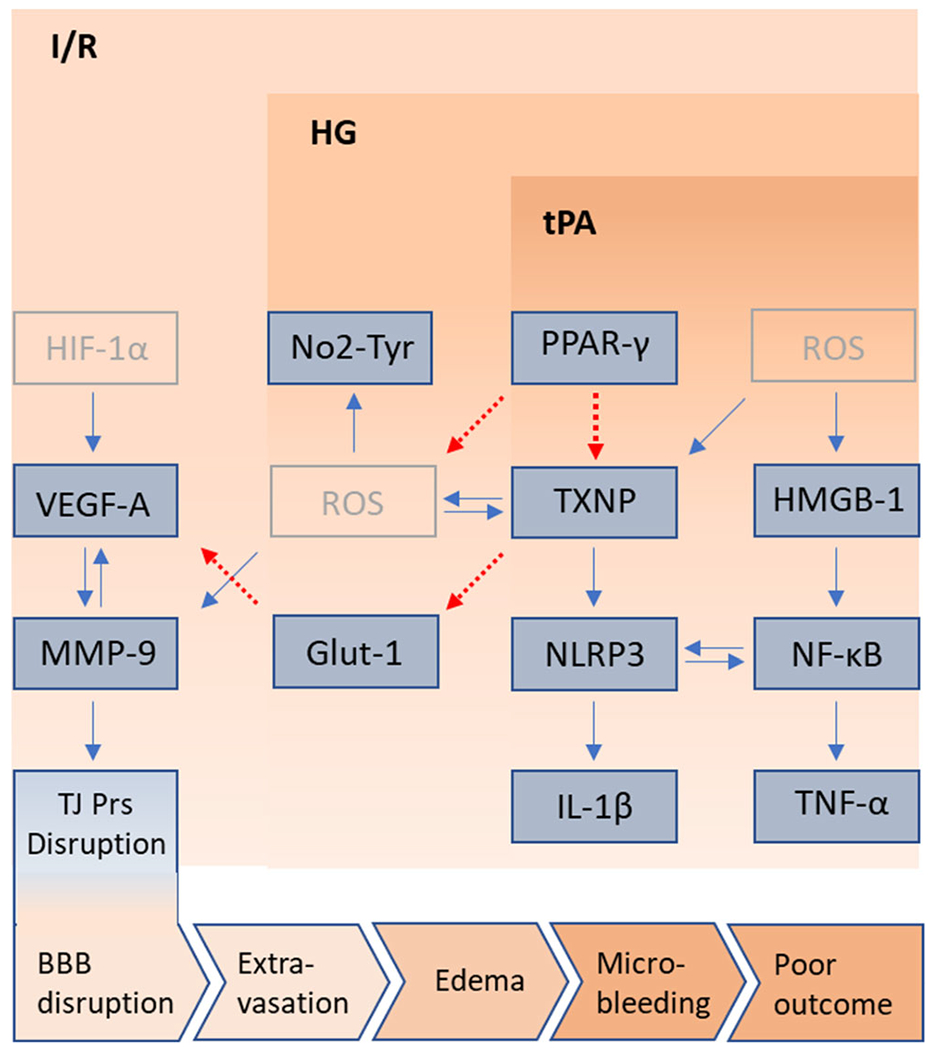
Schematic representation of potential pathways involved in tPA toxicity in hyperglycemic stroke. The upper and lower pink blocks represent the worsening outcomes of ischemia/reperfusion (I/R) by hyperglycemia (HG) and early administration of tPA. Besides few pale colored putative pathways, blue boxes illustrate the culprit molecules presumably interconnected with stimulatory (blue arrows) or inhibitory (dotted red arrows) pathways. Molecules presented in dark blue were differentially affected in the brains of HG stroke animals, treated with tPA. To represent the cumulative effect of detrimental pathways, induced by HG and tPA at reperfusion, the pathways involved are depicted, to hypothetically explain the poor outcomes in stroke. Based on the present work, HG enhances PPAR-γ levels and TXNIP/NLRP3/IL-1β activation. TXNIP is a key amplifier of ROS build up, leading to higher MMP-9 activitity which also works downstream of I/R induced HIF-1-α/VEGF-A signaling, to disturb TJ proteins’ integrity. Another pathway involves TXNIP indirectly activating the release of VEGF-A by repressing GLUT-1 transcription. Probably through more ROS generation, tPA differentially stimulates TXNIP and HMGB-1 in hyperglycemic I/R. HMGB-1/NF-κB/TNF-α is evidently liked to NLRP3 inflammasome priming and activation. With a synergistic effect with HMGB-1 on NLRP3 inflammasome, TXNIP upregulation may also contribute to ROS propagation in tPA treated brains, as determined by enhanced NO2-Tyr. Furthermore, strong tPA-induced TXNIP upregulation may drastically impair glucose transport and strengthen aberrant VEGF-A signaling, to drive more BBB disruption and edema, explaining the hemorrhagic transformation that occurs in brains of tPA treated hyperglycemic stroke animals. GLUT-1, glucose transporter 1; HG: hyperglycemic; HMGB-1, high mobility group box protein 1; IL-1β, interleukin 1-β; I/R, ischemic reperfusion; MMP, matrix metalloprotease; NF-κB, nuclear factor kappa B; NLRP3, NOD-like receptor pyrin domain-containing-3; PPAR-γ, peroxisome proliferator activated receptor-γ; TJ proteins, tight junction proteins; t-MCAO, transient middle cerebral artery occlusion; TNF-α, tumor necrosis factor-α; tPA, tissue plasminogen activator; TXNIP, thioredoxin interacting protein; VEGF-A, vascular endothelial growth factor-A
Chronic hyperglycemia has long been known to drive the endothelial complications of diabetes and metabolic syndrome through divergent pathways, including HMGB-1/NF-κB [37, 38] and TXNIP/NLRP3 signaling cascades [39, 40]. The well-documented contribution of hyperglycemia to I/R damage [11] has been mostly attributed to NADPH oxidase-dependent ROS production and abnormal protein glycosylation, consequent to glucose overflow during reperfusion [41]. This leads to MMP-3/9 activation, tight junction disruption, and ultimately BBB dysfunction [11, 42]. Our findings demonstrate that hyperglycemic I/R also promotes TXNIP signaling following stroke. This is further aggravated by tPA which moderately accentuates BBB disruption, with no effect on infarct size. TXNIP, a pivotal regulator of several biological processes, is inducible by either ROS or high glucose levels, both of which are essential components in hyperglycemic reperfusion injury. Although not sufficient to repress GLUT-1 expression, TXNIP upregulation in our hyperglycemic t-MCAO animals was enough to stimulate NLRP3 inflammasome cleavage activity to build up caspase-1 and raise IL-1β secretion. Through regulating IL-1β and other chemokines, NLRP3 greatly contributes to neurovascular neutrophil recruitment following I/R injury. Consistently, the moderate TXNIP/NLRP3 inflammasome assembly seen in our hyperglycemic t-MCAO animals was associated with an increased BBB permeability, as determined by IgG immunoreactivity in brain parenchyma.
The existing knowledge of mechanisms underlying tPA-associated BBB disruption has been limited to normoglycemic observations [43]. Accordingly, tPA toxicity mainly arises from its protease activity, which induces profound peroxynitrite generation tailed by MMP activation, mediated by several intermediate pathways including HMGB-1/TLRs and a number of protein kinases (Src, ROCK, and GSK-3b) [44, 45]. Importantly, in a relevant study about tPA toxicity in the context of hyperglycemia, experimental findings suggest that BBB disruption following ischemic reperfusion injury directly correlate with the severity of hyperglycemia. Accordingly, it has been shown to exacerbate tPA-induced hemorrhage through an increase in NADPH oxidase-mediated superoxide production. Accordingly hyperglycemic reperfusion, 10 min following tPA infusion, is linked to massive ROS generation, which may reasonably trigger TXNIP activity [46]. Based on our experimental data, tPA-induced BBB toxicity, in hyperglycemic animals, was associated with a dramatic rise in TXNIP/NLRP3/IL-1β activation and tripled HMGB-1/NF-κB/TNF-α levels. However, our experiments alone may not conclude that tPA-induced BBB disruption depends on TXNIP/NLRP3 inflammasome hyperactivity. In fact, involvement of NLRP3 inflammasome has already been supported by recent experiments conducted in normoglycemic animals, where the specific NLRP3 inflammasome inhibitor MCC950 has been shown to mitigate the hemorrhagic transformation induced by delayed tPA-administration [18]. This is well corroborated with reports indicating that IMM-H004, a derivative of coumarin, ameliorates tPA-induced HT [47], while repressing NLRP3/Caspase-1/IL-1β signaling [48]. Consistently, recent reports [19, 42] support the vasculoprotective effects of MCC950 following intraluminal stroke. Here, we found that TXNIP upregulation was strong enough to downregulate GLUT-1 in tPA treated hyperglycemic animals. Although increased PPAR-γ expression is neuroprotective, increased protein expression does not confirm increased DNA binding activity. This was demonstrated that increased expression of PPAR-γ was inversely associated with PPAR-γ binding activity in the ischemic hemisphere following t-MCAO [49]. This concurred with remarkably high BBB permeability, as identified by increased IgG immunoreactivity. It is of note, tPA-induced TXNIP upregulation was not only associated with higher NLRP3 inflammasome activity (cleaved caspase-1 and IL-1β) but it also enhanced priming of NLRP3 inflammasome components (ASC and caspase-1). This might be a consequence of the noticeable upregulation of HMGB-1 in tPA treated animals, which, along with NLRP3/IL-1β, may amplify toll like receptor (TLR)/NF-κB signaling by a variety of damage signals. HMGB1 is a nuclear non-histone DNA-binding protein that might be either actively secreted by immune cells or passively released from necrotic tissue, as a damage-associated molecular pattern (DAMP), to elicit microglial activation and inflammatory response. HMGB-1 has been demonstrated to closely associate with NLRP3 inflammasome activation [50, 51] and play a detrimental role in ischemia reperfusion injury [52, 53] and BBB disruption [54]. In fact, an antagonizing HMGB1-binding peptide has been documented to mitigate tPA-induced neurovascular complications in normoglycemic rats [55].
MMP-3 and MMP-9 are established effectors of tPA. These endopeptidases, with respective broad and narrow specificity for components of the neurovascular unit, cause degradation of the basal lamina and TJ proteins leading to BBB damage. MMP-9 has been extensively studied and shown to mediate HT after stroke in both clinical [56, 57] and preclinical studies [58, 59]. Importantly, experimental findings have demonstrated a pivotal role for MMP-3 activation in the reperfusion injury associated with hyperglycemic stroke and further aggravated with tPA [60]. This was recently shown to be mediated by nitrative stress [61]. Here, we showed that tPA-induced BBB breakdown enhanced MMP-9 expression in our hyperglycemic stroke animals, without changing MMP-3 levels. Intravenous tPA may enhance MMP-9 through reperfusion-linked ROS generation [62], as was illustrated by elevated levels of brain nitrotyrosine in animal. Moreover, in specific contexts, tPA may also act as a ligand for microglial low density lipoprotein receptor–related protein (LPR) [58] or protease activated receptor 1 (PAR1) [62] to enhance MMP-9 release. Beyond these, ischemia-associated VEGF release may intimately contribute to transcription of MMPs [63, 64], which accounts for part of the endothelial damage in ischemic stroke [65]. Cerebral VEGF secretion is fine-tuned through diverse pathways [66]. Regarding the trivial changes in VEGF seen in our hyperglycemic stroke animals, glucose alone may not be sufficient to explain the profound VEGF release associated with tPA therapy [67]. Higher TXNIP expression was documented to reduce VEGF-A [68] while is essential in VEGF-A/VEGF-R2 signaling [23, 69].
Our findings, illustrated a drastic GLUT-1 repression that may be meaningful enough to explain tPA-induced toxicity in hyperglycemic stroke. Reduced GLUT-1 expression is a major characteristic of diabetes that drastically compromises glucose uptake by endothelial cells. While complete deletion of GLUT-1 is lethal [70], specific pharmacological GLUT-1 blocker worsen BBB disruption [71]. Intriguingly, reduction of BBB specific GLUT-1 expression reduces brain glucose uptake and has been shown to evoke a compensatory rise in VEGF-A concentrations [72]. In support of this, GLUT-1 repression has been shown to inversely correlate with systemic VEGF release, in clinical reports [73]. In our hyperglycemic stroke animals, either the severe intracellular shortage of glucose or the drastic rise in VEGF/MMP-9 and inflammation were critically significant elements in tPA-induced BBB breakdown.
Few limitations are inherent to this work. The induced hyperglycemia (between 300 and 400 mg/dL) and the administered tPA (10 mg/kg) dosage are relatively high compared to those seen clinically. The steep blood glucose level, which was nearly double that seen in hyperglycemic stroke patients may have instigated severe endothelial damage and BBB breakdown that was further aggravated by tPA. Therefore, our findings address a kind of “accelerated” glucose toxicity in the context of thrombolytic therapy. However for the much less fibrinolytic activity in rodents [74], tPA is commonly used in same dosage of 10 mg/kg in murine studies. Furthermore, to address BBB disruption, it was ideal to use a non-craniotomy stroke model as MCAO in which severity of cortical damage may obscure some differences between the groups. The other important thing to note is the potentially great difference between tPA toxicity in a clot-occluded vessel and perfused artery as we used in intraluminal transient stroke model. Given the main question to elucidate tPA toxicity in hyperglycemic reperfusion, we intentionally used a successfully reperfused artery model. Thus, our conclusions, although closely replicating rapid reperfusion with tPA treatment, might be optimally relevant to spontaneous reperfusion before treatment. Ultimately without using an appropriate specific TXNIP inhibitor [75], the question remains as to whether TXNIP upregulation is responsible for tPA associated cerebrovascular damage in stroke, with hyperglycemia. Further experimental studies are also planned to evaluate the potential benefit of pharmacological inhibition of TXNIP in a clinically relevant embolic model of MCAO and recanalization with tPA.
Tissue plasminogen activator (tPA)-induced hemorrhagic events, resulting in cerebrovascular injury, are common in hyperglycemic stroke and pose a serious concern in this patient population. Our data primarily confirm that tPA worsens hyperglycemic reperfusion damage to BBB, even when administered early, in its therapeutic time window. Our preclinical findings show that tPA further augments hyperglycemic stroke-associated TXNIP upregulation and NLRP3 inflammasome assembly. Coupled with our earlier findings implicating TXNIP’s role in ischemic stroke [17], the present data indicate that TXNIP/NLRP3/IL-1β greatly reflects the increasing DAMPs in I/R injury. With their drastic repressive effect on GLUT-1 and VEGF-A levels, we postulate that the tPA/TXNIP interaction accelerates endogenous responses to metabolic stress and disrupts essential neurovascular unit components, required for maintenance of a healthy BBB. With several mechanisms proposed, the magnitude of each has to be weighed to postulate optimal therapeutic targets.
Funding information
This work was supported by National Institute of Health: R01-NS097800 (TI); startup funds: Department of Anatomy and Neurobiology, UTHSC Memphis TN (TI).
Abbreviations
- ASC
Apoptosis-associated speck-like protein
- BBB
Blood brain barrier
- DAMPs
Damage-associated molecular patterns
- GLUT-1
Glucose transporter-1
- HG
Hyperglycemic
- HMGB-1
High mobility group box protein-1
- HT
Hemorrhagic transformation
- IL-1β
Interleukin 1-β
- I/R
Ischemic reperfusion
- LPR
Lipoprotein receptor–related protein
- MMP
Matrix metalloprotease
- NF-κB
Nuclear factor kappa B
- NLRP3
NOD-like receptor pyrin domain-containing-3
- PAR1
Protease activated receptor 1
- PPAR-γ
Peroxisome proliferator activated receptor-γ
- TJ proteins
Tight junction proteins
- TLR
Tol like receptor
- tMCAO
Transient middle cerebral artery occlusion
- TNF-α
Tumor necrosis factor-α
- tPA
Tissue plasminogen activator
- TRX
Thioredoxin
- TXNIP
Thioredoxin interacting protein
- VEGF-A
Vascular endothelial growth factor-A
- ZO-1
Zonula occludens-1
References
- 1.Tsivgoulis G, Katsanos AH, Mavridis D, Lambadiari V, Roffe C, Macleod MJ, Sevcik P, Cappellari M et al. (2019) Association of baseline hyperglycemia with outcomes of patients with and without diabetes with acute ischemic stroke treated with intravenous thrombolysis: a propensity score–matched analysis from the SITS-ISTR registry. Diabetes 68(9):1861–1869 [DOI] [PubMed] [Google Scholar]
- 2.Yong M, Kaste M (2008) Dynamic of hyperglycemia as a predictor of stroke outcome in the ECASS-II trial. Stroke 39(10):2749–2755 [DOI] [PubMed] [Google Scholar]
- 3.Johnston KC, Bruno A, Pauls Q, Hall CE, Barrett KM, Barsan W, Fansler A, Van de Bruinhorst K et al. (2019) Intensive vs standard treatment of hyperglycemia and functional outcome in patients with acute ischemic stroke: the SHINE randomized clinical trial. JAMA 322(4):326–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruno A, Durkalski VL, Hall CE, Juneja R, Barsan WG, Janis S, Meurer WJ, Fansler A et al. (2014) The stroke hyperglycemia insulin network effort (SHINE) trial protocol: a randomized, blinded, efficacy trial of standard vs. intensive hyperglycemia management in acute stroke. Int J Stroke 9(2):246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allport L, Baird T, Butcher K, MacGregor L, Prosser J, Colman P, Davis S (2006) Frequency and temporal profile of poststroke hyperglycemia using continuous glucose monitoring. Diabetes Care 29(8):1839–1844 [DOI] [PubMed] [Google Scholar]
- 6.Furie KL, Ay H (2018) Initial evaluation and management of transient ischemic attacks and minor ischemic stroke. UpToDate; Waltham, MA [Google Scholar]
- 7.Chow BW, Gu C (2015) The molecular constituents of the blood–brain barrier Trends Neurosci 38(10):598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kago T, Takagi N, Date I, Takenaga Y, Takagi K, Takeo S (2006) Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun 339(4):1197–1203 [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA (2007) Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab 27(4):697–709 [DOI] [PubMed] [Google Scholar]
- 10.Kaur J, Zhao Z, Klein GM, Lo EH, Buchan AM (2004) The neurotoxicity of tissue plasminogen activator? J Cereb Blood Flow Metab 24(9):945–963 [DOI] [PubMed] [Google Scholar]
- 11.Zhang S, An Q, Wang T, Gao S, Zhou G (2018) Autophagy-and MMP-2/9-mediated reduction and redistribution of ZO-1 contribute to hyperglycemia-increased blood–brain barrier permeability during early reperfusion in stroke. Neuroscience 377:126–137 [DOI] [PubMed] [Google Scholar]
- 12.Kim GS, Jung JE, Narasimhan P, Sakata H, Chan PH (2012) Induction of thioredoxin-interacting protein is mediated by oxidative stress, calcium, and glucose after brain injury in mice. Neurobiol Dis 46(2):440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT (2004) Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem 279(29):30369–30374 [DOI] [PubMed] [Google Scholar]
- 14.Waldhart AN, Dykstra H, Peck AS, Boguslawski EA, Madaj ZB, Wen J, Veldkamp K, Hollowell M et al. (2017) Phosphorylation of TXNIP by AKT mediates acute influx of glucose in response to insulin. Cell Rep 19(10):2005–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bedarida T, Domingues A, Baron S, Ferreira C, Vibert F, Cottart C-H, Paul J-L, Escriou V et al. (2018) Reduced endothelial thioredoxin-interacting protein protects arteries from damage induced by metabolic stress in vivo. FASEB J 32(6):3108–3118 [DOI] [PubMed] [Google Scholar]
- 16.Ren X, Wang N-n, Qi H, Y-y Q, C-h Z, Brown E, Kong H, Kong L (2018) Up-regulation thioredoxin inhibits advanced glycation end products-induced neurodegeneration. Cell Physiol Biochem 50(5):1673–1686 [DOI] [PubMed] [Google Scholar]
- 17.Ishrat T, Mohamed IN, Pillai B, Soliman S, Fouda AY, Ergul A, El-Remessy AB, Fagan SC (2015) Thioredoxin-interacting protein: a novel target for neuroprotection in experimental thromboembolic stroke in mice. Mol Neurobiol 51(2):766–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Z, Yu S, Chen X, Zheng P, Hu T, Duan Z, Liu X, Liu Q et al. (2018) Suppression of NLRP3 attenuates hemorrhagic transformation after delayed rtPA treatment in thromboembolic stroke rats: involvement of neutrophil recruitment. Brain Res Bull 137:229–240 [DOI] [PubMed] [Google Scholar]
- 19.Ismael S, Zhao L, Nasoohi S, Ishrat T (2018) Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci Rep 8(1):5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hafez S, Hoda MN, Guo X, Johnson MH, Fagan SC, Ergul A (2015) Comparative analysis of different methods of ischemia/reperfusion in hyperglycemic stroke outcomes: interaction with tPA. Transl Stroke Res 6(3):171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nasoohi S, Ismael S, Ishrat T (2018) Thioredoxin-interacting protein (TXNIP) in cerebrovascular and neurodegenerative diseases: regulation and implication. Mol Neurobiol 55(10):7900–7920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abdelsaid MA, Matragoon S, El-Remessy AB (2013) Thioredoxin-interacting protein expression is required for VEGF-mediated angiogenic signal in endothelial cells. Antioxid Redox Signal 19(18):2199–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duan J, Du C, Shi Y, Liu D, Ma J (2018) Thioredoxin-interacting protein deficiency ameliorates diabetic retinal angiogenesis. Int J Biochem Cell Biol 94:61–70 [DOI] [PubMed] [Google Scholar]
- 24.Zhang HT, Zhang P, Gao Y, Li CL, Wang HJ, Chen LC, Feng Y, Li RY et al. (2017) Early VEGF inhibition attenuates blood-brain barrier disruption in ischemic rat brains by regulating the expression of MMPs. Mol Med Rep 15(1):57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanazawa M, Takahashi T, Nishizawa M, Shimohata T (2017) Therapeutic strategies to attenuate hemorrhagic transformation after tissue plasminogen activator treatment for acute ischemic stroke. J Atheroscler Thromb 24(3):240–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8(6):e1000412. 10.1371/journal.pbio.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McBride DW, Klebe D, Tang J, Zhang JH (2015) Correcting for brain swelling’s effects on infarct volume calculation after middle cerebral artery occlusion in rats. Transl Stroke Res 6(4):323–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elgebaly MM, Ogbi S, Li W, Mezzetti EM, Prakash R, Johnson MH, Bruno A, Fagan SC et al. (2011) Neurovascular injury in acute hyperglycemia and diabetes: a comparative analysis in experimental stroke. Transl Stroke Res 2(3):391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.S-i O, Masutani H, Liu W, Horita H, Wang D, Kizaka-Kondoh S, Yodoi J (2006) Thioredoxin-binding protein-2-like inducible membrane protein is a novel vitamin D3 and peroxisome proliferator-activated receptor (PPAR) γ ligand target protein that regulates PPARy signaling. Endocrinology 147(2):733–743 [DOI] [PubMed] [Google Scholar]
- 30.Qi W, Chen X, Holian J, Tan CY, Kelly DJ, Pollock CA (2009) Transcription factors Krüppel-like factor 6 and peroxisome proliferator-activated receptor-γ mediate high glucose-induced thioredoxin-interacting protein. Am J Pathol 175(5):1858–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Victor N, Wanderi E, Gamboa J, Zhao X, Aronowski J, Deininger K, Lust W, Landreth G et al. (2006) Altered PPARγ expression and activation after transient focal ischemia in rats. Eur J Neurosci 24(6):1653–1663 [DOI] [PubMed] [Google Scholar]
- 32.Takizawa S, Fukuyama N, Hirabayashi H, Nakazawa H, Shinohara Y (1999) Dynamics of nitrotyrosine formation and decay in rat brain during focal ischemia-reperfusion. J Cereb Blood Flow Metab 19(6):667–672 [DOI] [PubMed] [Google Scholar]
- 33.Li C, Wang X, Cheng F, Du X, Yan J, Zhai C, Mu J, Wang Q (2019) Geniposide protects against hypoxia/reperfusion-induced blood-brain barrier impairment by increasing tight junction protein expression and decreasing inflammation, oxidative stress, and apoptosis in an in vitro system. Eur J Pharmacol 854:224–231 [DOI] [PubMed] [Google Scholar]
- 34.Bauer AT, Bürgers HF, Rabie T, Marti HH (2010) Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab 30(4):837–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fredriksson L, Lawrence DA, Medcalf RL (2017) tPA modulation of the blood-brain barrier: a unifying explanation for the pleiotropic effects of tPA in the CNS. Semin Thromb Hemost 43(2):154–168. 10.1055/s-0036-1586229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hafez S, Coucha M, Bruno A, Fagan SC, Ergul A (2014) Hyperglycemia, acute ischemic stroke, and thrombolytic therapy. Transl Stroke Res 5(4):442–453. 10.1007/s12975-014-0336-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu R, Luo Q, You W, Jin M (2018) MicroRNA-106 attenuates hyperglycemia-induced vascular endothelial cell dysfunction by targeting HMGB1. Gene 677:142–148 [DOI] [PubMed] [Google Scholar]
- 38.Zhang W, Wang Y, Kong Y (2019) Exosomes derived from mesenchymal stem cells modulate miR-126 to ameliorate hyperglycemia-induced retinal inflammation via targeting HMGB1. Invest Ophthalmol Vis Sci 60(1):294–303 [DOI] [PubMed] [Google Scholar]
- 39.Lu L, Lu Q, Chen W, Li J, Li C, Zheng Z (2018) Vitamin D3 protects against diabetic retinopathy by inhibiting high-glucose-induced activation of the ROS/TXNIP/NLRP3 Inflammasome pathway J Diabetes Res. 10.1155/2018/8193523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen W, Zhao M, Zhao S, Lu Q, Ni L, Zou C, Lu L, Xu X et al. (2017) Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflamm Res 66(2):157–166 [DOI] [PubMed] [Google Scholar]
- 41.Robbins NM, Swanson RA (2014) Opposing effects of glucose on stroke and reperfusion injury: acidosis, oxidative stress, and energy metabolism. Stroke 45(6):1881–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desilles JP, Syvannarath V, Ollivier V, Journe C, Delbosc S, Ducroux C, Boisseau W, Louedec L et al. (2017) Exacerbation of thromboinflammation by hyperglycemia precipitates cerebral infarct growth and hemorrhagic transformation. Stroke 48(7):1932–1940. 10.1161/STROKEAHA.117.017080 [DOI] [PubMed] [Google Scholar]
- 43.Suzuki Y, Nagai N, Umemura K (2016) A review of the mechanisms of blood-brain barrier permeability by tissue-type plasminogen activator treatment for cerebral ischemia. Front Cell Neurosci 10:2. 10.3389/fncel.2016.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen H, Chen X, Luo Y, Shen J (2018) Potential molecular targets of peroxynitrite in mediating blood–brain barrier damage and haemorrhagic transformation in acute ischaemic stroke with delayed tissue plasminogen activator treatment. Free Radic Res 52(11–12):1220–1239 [DOI] [PubMed] [Google Scholar]
- 45.Wang X, Tsuji K, Lee S-R, Ning M, Furie KL, Buchan AM, Lo EH (2004) Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke 35(11_suppl_1):2726–2730 [DOI] [PubMed] [Google Scholar]
- 46.Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA (2011) Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by increasing superoxide production. Ann Neurol 70(4):583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuo W, Chen J, Zhang S, Tang J, Liu H, Zhang D, Chen N (2014) IMM-H004 prevents toxicity induced by delayed treatment of t PA in a rat model of focal cerebral ischemia involving PKA-and PI 3 K-dependent A kt activation. Eur J Neurosci 39(12):2107–2118 [DOI] [PubMed] [Google Scholar]
- 48.Ai Q-D, Chen C, Chu S-F, Zhang Z, Luo Y, Guan F-F, Lin M-Y, Liu D et al. (2019) IMM-H004 therapy for permanent focal ischemic cerebral injury via CKLF1/CCR4-mediated NLRP3 inflammasome activation. Transl Res 212:36–53 [DOI] [PubMed] [Google Scholar]
- 49.Victor NA, Wanderi EW, Gamboa J, Zhao X, Aronowski J, Deininger K, Lust WD, Landreth GE et al. (2006) Altered PPARgamma expression and activation after transient focal ischemia in rats. Eur J Neurosci 24(6):1653–1663. 10.1111/j.1460-9568.2006.05037.x [DOI] [PubMed] [Google Scholar]
- 50.Chi W, Chen H, Li F, Zhu Y, Yin W, Zhuo Y (2015) HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-κB pathway in acute glaucoma. J Neuroinflammation 12(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT-H, Taxman DJ, Duncan JA, Ting JP-Y (2009) NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and-independent pathways. J Immunol 183(3):2008–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T, Sato Y, Hiraga N et al. (2007) Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J 21(14):3904–3916 [DOI] [PubMed] [Google Scholar]
- 53.Song Y, Jun J-H, Shin E-J, Kwak Y-L, Shin J-S, Shim J-K (2017) Effect of pregabalin administration upon reperfusion in a rat model of hyperglycemic stroke: mechanistic insights associated with high-mobility group box 1. PLoS One 12(2):e0171147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, Takahashi HK, Liu K, Wake H, Liu R, Maruo T, Date I, Yoshino T et al. (2011) Anti-high mobility group box-1 monoclonal antibody protects the blood–brain barrier from ischemia-induced disruption in rats. Stroke 42(5):1420–1428 [DOI] [PubMed] [Google Scholar]
- 55.Li M, Chen S, Shi X, Lyu C, Zhang Y, Tan M, Wang C, Zang N et al. (2018) Cell permeable HMGB1-binding heptamer peptide ameliorates neurovascular complications associated with thrombolytic therapy in rats with transient ischemic stroke. J Neuroinflammation 15(1):237–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yi X, Sui G, Zhou Q, Wang C, Lin J, Chai Z, Zhou J (2019) Variants in matrix metalloproteinase-9 gene are associated with hemorrhagic transformation in acute ischemic stroke patients with atherothrombosis, small artery disease, and cardioembolic stroke. Brain Behav 9(6):e01294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shimada Y, Shimura H, Tanaka R, Yamashiro K, Koike M, Uchiyama Y, Urabe T, Hattori N (2018) Phosphorylated recombinant HSP27 protects the brain and attenuates blood-brain barrier disruption following stroke in mice receiving intravenous tissue-plasminogen activator. PLoS One 13(5):e0198039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang C, An J, Haile WB, Echeverry R, Strickland DK, Yepes M (2009) Microglial low-density lipoprotein receptor-related protein 1 mediates the effect of tissue-type plasminogen activator on matrix metalloproteinase-9 activity in the ischemic brain. J Cereb Blood Flow Metab 29(12):1946–1954 [DOI] [PubMed] [Google Scholar]
- 59.Chen S, Chen Z, Cui J, McCrary ML, Song H, Mobashery S, Chang M, Gu Z (2018) Early abrogation of gelatinase activity extends the time window for tPA thrombolysis after embolic focal cerebral ischemia in mice. eNeuro 5(3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hafez S, Abdelsaid M, El-Shafey S, Johnson MH, Fagan SC, Ergul A (2016) Matrix metalloprotease 3 exacerbates hemorrhagic transformation and worsens functional outcomes in hyperglycemic stroke. Stroke 47(3):843–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hafez S, Abdelsaid M, Fagan SC, Ergul A (2018) Peroxynitrite-induced tyrosine nitration contributes to matrix metalloprotease-3 activation: relevance to hyperglycemic ischemic brain injury and tissue plasminogen activator. Neurochem Res 43(2):259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gerzanich V, Kwon MS, Woo SK, Ivanov A, Simard JM (2018) SUR1-TRPM4 channel activation and phasic secretion of MMP-9 induced by tPA in brain endothelial cells. PLoS One 13(4):e0195526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hollborn M, Stathopoulos C, Steffen A, Wiedemann P, Kohen L, Bringmann A (2007) Positive feedback regulation between MMP-9 and VEGF in human RPE cells. Invest Ophthalmol Vis Sci 48(9):4360–4367 [DOI] [PubMed] [Google Scholar]
- 64.Rodrigues M, Xin X, Jee K, Babapoor-Farrokhran S, Kashiwabuchi F, Ma T, Bhutto I, Hassan SJ et al. (2013) VEGF secreted by hypoxic Muller cells induces MMP-2 expression and activity in endothelial cells to promote retinal neovascularization in proliferative diabetic retinopathy Diabetes 62(11):3863–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Won S, Lee JH, Wali B, Stein DG, Sayeed I (2014) Progesterone attenuates hemorrhagic transformation after delayed tPA treatment in an experimental model of stroke in rats: involvement of the VEGF–MMP pathway. J Cereb Blood Flow Metab 34(1):72–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Simons M, Gordon E, Claesson-Welsh L (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17(10):611–625 [DOI] [PubMed] [Google Scholar]
- 67.Caprnda M, Kubatka P, Saxena S, Valaskova J, Stefanickova J, Kobyliak N, Zulli A, Kruzliak P (2017) The impact of hyperglycemia on VEGF secretion in retinal endothelial cells. Folia Med 59(2):183–189 [DOI] [PubMed] [Google Scholar]
- 68.Farrell MR, Rogers LK, Liu Y, Welty SE, Tipple TE (2010) Thioredoxin-interacting protein inhibits hypoxia-inducible factor transcriptional activity. Free Radic Biol Med 49(9):1361–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dunn LL, Simpson PJ, Prosser HC, Lecce L, Yuen GS, Buckle A, Sieveking DP, Vanags LZ et al. (2014) A critical role for thioredoxin-interacting protein in diabetes-related impairment of angiogenesis. Diabetes 63(2):675–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, Sengillo JD, Hillman S et al. (2015) GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 18(4):521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muneer PA, Alikunju S, Szlachetka AM, Murrin LC, Haorah J (2011) Impairment of brain endothelial glucose transporter by methamphetamine causes blood-brain barrier dysfunction. Mol Neurodegener 6(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S, Mauer J, Steculorum SM et al. (2016) Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell 165(4):882–895 [DOI] [PubMed] [Google Scholar]
- 73.Schüler R, Seebeck N, Osterhoff MA, Witte V, Flöel A, Busjahn A, Jais A, Brüning JC et al. (2018) VEGF and GLUT1 are highly heritable, inversely correlated and affected by dietary fat intake: consequences for cognitive function in humans. Mol Metab 11:129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Korninger C, Collen D (1981) Studies on the specific fibrinolytic effect of human extrinsic (tissue-type) plasminogen activator in human blood and in various animal species in vitro. Thromb Haemost 46(02):561–565 [PubMed] [Google Scholar]
- 75.Thielen L, Chen J, Xu G, Jing G, Grayson T, Jo S, Shalev A (2018) Novel small molecule TXNIP inhibitor protects against diabetes. Diabetes 67(Supplement 1). 10.2337/db18-87-OR [DOI] [Google Scholar]