

Abstract
The single-cell revolution in the field of genomics is in full bloom, with clever new molecular biology tricks appearing regularly that allow researchers to explore new modalities or scale up their projects to millions of cells and beyond. Techniques abound to measure RNA expression, DNA alterations, protein abundance, chromatin accessibility, and more, all with single cell resolution and often in combination. Despite such a rapidly changing technology landscape, there are several fundamental principles that are applicable to the majority of experimental workflows to help users avoid pitfalls and exploit the advantages of the chosen platform. In this overview article, we describe a variety of popular single-cell genomics technologies and address some common questions pertaining to study design, sample preparation, quality control, and sequencing strategy. Since the majority of relevant publications currently revolve around single-cell RNA-seq, we will prioritize this genomics modality in our discussion.
Keywords: Single-Cell, Genomics, Transcriptomics, Sequencing, scRNA-seq, scATAC-seq
1. Introduction
The single-cell era of genomics debatably began in 2009 with the sequencing of the transcriptome from a single mouse blastomere (Tang et al., 2009). Since then, a plethora of new laboratory devices and clever barcoding techniques have emerged that enable ‘omics’-scale experiments in increasingly larger numbers of individually resolved cells. “Single-cell” varieties of nearly every conceivable type of genomics assay have now been developed, some of which have been commercialized with huge success and brought into widespread use. A variety of these techniques have been deployed to build colossal organism-wide cell atlases, starting with tractable model organisms like worms (Cao et al., 2017) and flies (H. Li et al., 2021), with ongoing efforts to build increasingly comprehensive references for more developmentally complex species such as mice (Han et al., 2018; Tabula Muris Consortium, 2020; Tabula Muris Consortium et al., 2018) and humans (Domcke et al., 2020; Regev et al., 2017; K. Zhang et al., 2021). As more studies employ single cell technology in specialized research areas, these reference atlases serve as essential touchstones to guide our understanding of the normal range of cellular states and perturbations that happen during development and disease.
The study of tumor evolution via DNA copy number alterations (CNA) was one of the earliest applications of single-cell genomics technologies (Baslan et al., 2012; Navin et al., 2011). Since then, genomic DNA-based single-cell assays for CNAs, point mutations, and methylation have seen several cycles of innovation, but the challenges inherent to exploring gigabase-scale genomes in thousands of cells have limited their adoption. In contrast, single-cell RNA sequencing (scRNA-seq) has proven to be scalable, simple to perform and expand upon, and able to address a broad range of biological questions (Aldridge & Teichmann, 2020; Lim, Lin, & Navin, 2020; Tanay & Regev, 2017). Recently, single-cell assays of chromatin accessibility (scATAC-seq) have seen increasing usage, especially in combination with scRNA-seq in ‘multi-modal’ or ‘multi-omic’ workflows that capture both readouts from the same cells. Recent commercialization of a combined snRNA-seq/scATAC-seq protocol by 10X Genomics has lowered the barrier to entry for this type of assay, and is already seeing widespread adoption across immunology and neurobiology (Allaway et al., 2021; Granja et al., 2019; Trevino et al., 2021; You et al., 2021). Likewise, clever tricks to measure protein abundance have been developed and commercialized that can be combined with nearly any other genomic assay (Mimitou et al., 2021; Peterson et al., 2017; Stoeckius et al., 2017). The trend toward combining additional layers of single-cell data is always increasing, as sequencing costs drop and innovations emerge.
Unsurprisingly, experimental methods capable of making thousands of measurements within thousands or even millions of individual cells are, by their nature, complex and challenging to undertake. Single-cell studies also tend to be much more expensive and time consuming than their bulk-tissue counterparts, providing few opportunities to revise and retry should the data turn out to be unsatisfactory. Careful consideration must thus be put toward every step of a project: study design philosophy, sample collection logistics, tissue dissociation, sample cleanup, cell-type enrichment, sequencing depth and format, and other steps that are universal regardless of the specific molecular biology of a given method.
The intent of this Overview is to provide guidance to experimental design, execution, and analysis of single cell genomics experiments as a general class whenever possible, but with an intentional bias towards discussing the particulars of scRNA-seq, due to its dominance in the current literature. Most single-cell genomics assays will share certain similarities with regards to sample preparation and study design considerations, and we will attempt to provide a general perspective on these topics. We will not attempt to walk through the core steps of any given single-cell workflow, as this level of technical detail is beyond the scope of this article. Instead, we will focus on the “interstitial” protocol considerations, that is, on general best practices and guidelines that are common across a variety of experiment types, which are often glossed over in step-by-step protocols. Cumulatively, we hope these basic principles will help build the foundation for a successful experiment. At the time of writing this overview, commercial droplet microfluidics instruments, particularly the 10X Genomics Chromium platform, comprised the majority of newly published single-cell experiments; as such, this guide will prioritize discussing issues relevant to these platforms while also touching upon competing or alternative technologies.
2. Technology Platforms
When planning a single-cell experiment, the choice of which technology to use should be influenced by the goals of the analysis: what modalities are being measured, and can this platform be adapted for the required readout? How much sensitivity is required to detect the molecules of interest? How many cells are needed? How difficult is it to get started using the protocol? Is integrating your results with published datasets on the same platform a priority? Some general characteristics of different technology platforms are summarized in Table 1 and are discussed in this section.
Table 1. Comparing technology platforms.
Each broad class of single-cell technology offers advantages and disadvantages in terms of throughput and cost, flexibility to be adapted to different modalities (RNA, ATAC, DNA methylation, etc.), sensitivity, protocol simplicity (often owing to robust commercialized kits), and widespread adoption rate, which expands the existing knowledgebase about the platform and allows for easy comparison with other similar datasets.
| Throughput (cost/labor per cell) | Flexibility | Sensitivity / Max Depth | Protocol Simplicity / Accessibility | Adoption / Available public datasets | |
|---|---|---|---|---|---|
| Droplet | ++ | + | ++ | +++ | +++ |
| Sorted/Plate-based | + | +++ | +++ | ++ | ++ |
| Microwell | ++ | ++ | ++ | + | + |
| Split / Pool | +++ | ++ | ++ | ++ | ++ |
The primary factor separating different technology platforms is the means by which single cells are partitioned and barcoded (Figure 1 and Table 1). These platforms range in complexity and cost: some require no specialized equipment but large amounts of skilled hands-on time and optimization, while others require expensive commercial devices that greatly simplify and speed up the workflow. The partitioning method, in turn, influences the practicality and scalability of different enzymatic reactions for generating libraries targeting mRNA, open chromatin, transcription factors, or others. Some platforms may be fundamentally incapable of certain genomics readouts, while other more flexible ones may never achieve the cell numbers possible with high-throughput methods.
Figure 1. Summary of common single-cell technology platforms.
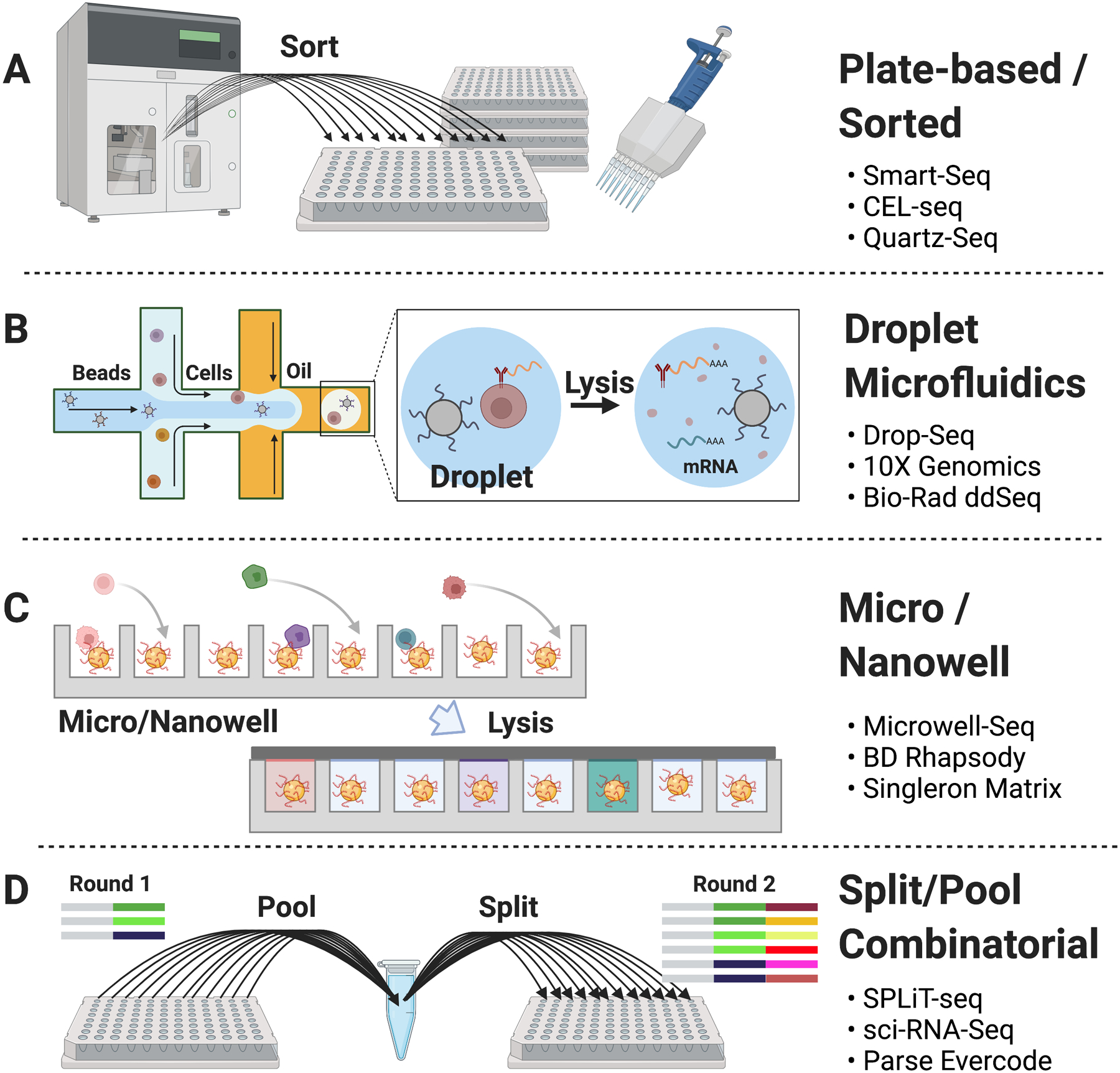
(A) Plate-based methods involve sorting or manually depositing cells into wells of a standard microplate, which are then processed as individual libraries by hand or with automation. (B) Droplet microfluidic devices such as the 10X Genomics Chromium Controller partition cells into emulsion droplets along with gel beads containing barcoded primers. Enzymatic barcoding by reverse transcription or ligation occurs in the emulsion, and subsequent library steps can be performed as a single pool. (C) Micro- and nanowell approaches allow a dilute cell suspension to settle into picoliter-sized wells along with oligo-conjugated beads under conditions that favor one bead and cell per well. (D) Combinatorial split/pool methods generally start with fixed and permeabilized cells that are distributed across a starting plate. A well-specific DNA barcode is appended, and all cells are then pooled to allow uniform mixing before re-distributing to a new plate, where a second barcode is added serially.
2.1. Plate-based/Sorted Cells
The earliest examples of single-cell genomics used the straightforward strategy of carefully depositing cells individually into separate reaction chambers, and this approach still remains popular because of its simplicity and flexibility. Cells can be sorted into 96- or 384-well plates with conventional flow sorters, or even by mouth pipetting (Dong et al., 2018; Eberwine et al., 1992; Fan et al., 2020; Spaethling et al., 2017). Enzymatic steps are processed separately in each well, which allows for nearly any conceivable genomics workflow, but this comes at high reagent cost, due to volume limitations of pipets. Robotic automation, however, can increase throughput where available, and ultra-low volume acoustic liquid handlers can be used to minimize reagent costs (Minussi et al., 2021). Unlike other approaches, plate-based methods are easily adapted to sequencing full-length mRNA transcripts rather than short sequence tags, but at high cost per cell. Plate-based methods also tend to provide richer data per captured cell, and are frequently used to complement larger —but sparser— atlas-scale datasets produced by other technology platforms.
2.2. Droplet Microfluidics
In this strategy, cells are partitioned by a microfluidic device into picoliter-sized droplets within an oil emulsion. DNA-barcoded beads are co-encapsulated along with cells, and barcodes are enzymatically coupled to target molecules, usually by reverse transcription of polyadenylated RNA or by ligation to fragmented DNA. The approach was pioneered in academic labs under the names ‘Drop-seq’ (Macosko et al., 2015) and ‘inDrop’, and was later commercialized by 10X Genomics, Bio-Rad, and others. Cell yields are limited by the statistics of random co-encapsulation of barcoded beads and cells (see section 4.2 for more detail), and by the overall diversity of barcodes available in the bead pool. Since the synthesis of a highly diverse library of barcoded beads is non-trivial, this is generally left to commercial vendors. Several methods have been described for generating them in the laboratory (De Rop et al., 2022; Delley & Abate, 2021), albeit with significant start-up, labor, and quality control costs. This makes custom workflows with user-designed primer/adapter sequences a challenge, but the availability of robust and highly standardized commercial solutions greatly facilitates cross-study integrated analyses. Droplet-based experiments account for the majority of single-cell datasets published in recent years, including the Tabula Muris (Tabula Muris Consortium et al., 2018) and Tabula Sapiens (Consortium & Quake, 2021) projects, and much of the Human Cell Atlas (Regev et al., 2017).
2.3. Micro/nanowell
Here, nanoliter-sized reaction wells are patterned onto a fabricated chip and cells are typically randomly seeded into wells as a dilute suspension obeying Poisson-distributed statistics. Barcoded beads are deposited into the same wells, cells are lysed, and barcodes are then coupled enzymatically to their target molecules. Custom microwell devices can be synthesized from PDMS or other polymers, but, as in droplet methods, barcoded bead synthesis is typically outsourced to commercial vendors. Costs per cell and total cell numbers are similar to droplet methods. Commercialized platforms include the Rhapsody (BD Biosciences) and Matrix (Singleron Biosciences). Of note, the >500K Mouse Cell Atlas utilized a custom Microwell-Seq platform (H. Chen et al., 2021; Han et al., 2018).
2.4. Split/Pool
In this strategy, a barcode is built directly onto RNA/DNA in fixed cells through either serial ligation, reverse transcription (RT), transposition, or a combination of reactions. Barcode diversity is achieved through splitting a cell pool into random batches and appending a unique barcode to each batch, followed by pooling and then randomly splitting and barcoding again during one or more subsequent rounds, such that no two cells receive the same combination of barcode segments. In this way, RNA or DNA molecules from a given cell all share a unique string of barcodes that distinguishes it from all other cells in the starting pool. Many split/pool barcoding strategies have been implemented to sequence mRNA, open chromatin regions, and methylated DNA, and for multimodal applications. In addition, they require no specialized equipment for partitioning, but barcode sets and enzymes can add up to a significant up-front investment. The combinatorial nature of this barcoding strategy can theoretically be used to construct an enormous variety of unique sequences and, thus, can be used to capture several times more cells in a given experiment than other methods. By exploiting economies of scale, they can achieve extremely low per-cell costs, but can have the downside of requiring longer and more expensive sequencing run lengths to fully cover the barcode region. Examples of this strategy include sci-RNA-seq (Cao et al., 2017), sci-ATAC-seq (Cusanovich et al., 2015), SPLiT-seq (Rosenberg et al., 2018), Quantum Barcoding (QBC), CoBATCH (Q. Wang et al., 2019), and BAG-seq (S. Li et al., 2020).
3. Study Design Considerations
The availability of robust commercial solutions has triggered a boom in new single-cell studies across a wide range of disciplines. The analytical goals of these studies can, similarly, span a wide range, from descriptive atlas-making efforts to quantitative comparisons across conditions and cell types and beyond. When designing a new single-cell experiment, it is important to consider the capabilities and limitations of available technology platforms together with the goals of the analysis. In this section, we will discuss some basic study design considerations, including differences in resource allocation for coverage- versus counting-based applications, statistical power analysis, biological replicates, batching processing, multiplexing, and accounting for sex variations across samples.
3.1. Resource Allocation.
Single-cell genomics experiments are expensive, and it is important to understand the breakdown of how money and resources are spent across the various steps of a workflow. Different platforms and modalities can vary dramatically in their bottom-line costs per sequenced cell, and proper experimental design will depend heavily on the breakdown of the necessary library construction, sequencing, and analysis costs. For the majority of currently popular scRNA-seq methods, a helpful rule-of-thumb for cost planning is to expect that sample preparation and sequencing will each consume roughly 50% of the available budget. This ratio, of course, can be adjusted relative to the goals of the study: is surveying a broader sample of single cells the priority, or rather the depth of sequencing given to each cell? In contrast, some applications require a much larger proportion to be spent on sequencing. One example is DNA copy number variation analysis, which requires up to 20 times more reads per cell than scRNA-seq for a standard analysis (Minussi et al., 2021).
Microfluidic technologies have made it easy to scale a project to larger cell numbers, with the obvious tradeoff being that it becomes more expensive to sequence each cell deeply. “Depth-first” and “Breadth-first” philosophies each have their respective use cases, so it is important to evaluate the experimental goals and optimize the study design to best match them. For instance, a study focusing on weakly expressed lineage-determining transcription factors may require high reads-per-cell to improve detection of rare transcripts. In contrast, a study exploring global gene expression changes across cell types in response to perturbations may benefit more from larger cell numbers with modest sequencing depth. Several studies have proposed general principles for optimal budget allocation, and should be considered prior to planning an experiment, regardless of the sequencing platform used (Schmid et al., 2021; Svensson, Beltrame, & Pachter, 2019; M. J. Zhang, Ntranos, & Tse, 2020).
3.2. Cost per cell
The total cost per cell is an aggregate function of the capital or start-up investment, the complexity of the cell partitioning step, overall reagent consumption, and the necessary sequencing coverage. Digital counting-based methods (10X Genomics, Drop-Seq, CEL-seq2, Quartz-Seq2, SPLiT-seq) are vastly more cost efficient that gene coverage-based methods (Smart-seq2, NEBNext), due to both the lower sequencing depth requirements and the more efficient reagent utilization given the ability to pool reaction mixtures at an earlier stage. Cost estimates can vary dramatically depending on institutional or service provider price structures, but to a first approximation, droplet-based digital counting methods range from USD $0.40 to $1.50 per sequenced cell, while a plate-sorted coverage-based method might cost $20–$60 per cell. Self-built microfluidic apparatuses such as those designed for DropSeq and InDrop can help save on barcoding costs and provide experiment flexibility, but such custom equipment has begun to fall out of favor because of limitations in standardization, consistency, scalability, challenges in training and maintenance, and trailing performance compared to newer chemistry versions being released by commercial competitors.
For extremely large-scale experiments, combinatorial split-pool approaches to cell barcoding boast excellent economies of scale and can be used upstream of a variety of genomics modalities (Cao et al., 2017; S. Li et al., 2020; Rosenberg et al., 2018). In principle, such methods can be used to build >1M cell datasets at extremely low barcoding costs per cell (as low as $0.01), but they present some trade-offs compared with microfluidic approaches, namely 1) they tend to be labor intensive and require significant initial investment to set up and validate, 2) sequencing costs tend to be higher due to requirements for the longer reads necessary to deconvolute the combinatorial barcodes, and 3) their bioinformatics pipelines tend to require more expertise, especially compared with commercial alternatives. While widespread adoption of split-pool workflows has been slow, commercial vendors such as Parse Biosciences are now beginning to offer optimized kits and analysis tools that promise to streamline the split/pool workflow and lower the barriers to entry for extremely large-scale experiments.
3.3. Coverage- vs Counting-Based Applications
If isoform specificity, allelic expression, or mutation detection are a priority, full-length scRNA-seq methods such as SMART-seq or NEBNext are required. In these workflows, each single-cell library is processed separately in its own tube or plate well, cumulatively requiring larger volumes of expensive enzymes and barcodes. Barcoded libraries are pooled at the end of the workflow and sequenced together, providing full-transcript coverage akin to conventional bulk RNA-seq. In contrast, digital gene expression approaches opt to incorporate barcodes at an early step, allowing samples to be pooled and conveniently processed in a single tube rather than in dozens or hundreds of separate reaction chambers. In digital scRNA-seq, barcodes are appended to mRNAs at the RT step, then fragmented and PCR-amplified such that only the barcoded end is retained. Only a small, strongly biased portion of a given mRNA is covered by sequencing (either 3’- or 5’-biased, depending on the chemistry); hence, isoform utilization is poorly represented, and gene expression data tends to be collapsed to the gene level. Robust quantification of mRNA abundance is achieved by tallying molecular barcodes, commonly referred to as “unique molecular identifiers” (UMIs, see below).
3.4. UMIs
Counting-based applications such as scRNA-seq generally rely on UMIs, also known as molecular barcodes or varietal tags (Levy & Wigler, 2014). Typically, a UMI is a short stretch of 8–12 random synthetic nucleotides embedded within one of the DNA oligos in the sequencing workflow, typically, the reverse-transcription primer. PCR amplification from the same molecule will reproduce an identical UMI; these are tracked bioinformatically, and duplicate UMIs associated with a common cell barcode and parent molecule (e.g., mRNA) are collapsed into a single tally. UMIs significantly improve accuracy of counting-based applications (G. K. Fu, Hu, Wang, & Fodor, 2011; Y. Fu, Wu, Beane, Zamore, & Weng, 2018; Hafemeister & Satija, 2019), but are impractical for full-length cDNA sequencing methods that require fragmentation of the parent cDNA molecule for short-read Illumina sequencing.
3.5. Statistical power analysis
Ideally, one should plan a single-cell study from the outset, with a clear statement of goals and a plan for adequately powering downstream statistical analysis. Employing a power calculation or study simulation tool such as powsimR (Vieth, Ziegenhain, Parekh, Enard, & Hellmann, 2017), scPower (Schmid et al., 2021), scDesign2 (Sun, Song, Li, & Li, 2021), and POWSC (Su, Wu, & Wu, 2020) can be a useful way to predict the feasibility of the stated goals, (e.g. detection of rare cell types, differential expression testing, eQTL analysis) and allocate resources to additional biological replicates, higher cell counts, deeper sequencing, or the like, as needed. Bear in mind that such calculations rely heavily on assumptions about the sources and extent of technical noise, which can vary dramatically across different types of samples. Solid tumors, in particular, tend to have highly variable morphology that makes the cell type composition of a given surgical section difficult to predict, making reliable prediction of statistical power a challenge.
3.6. Biological Replicates
Not unexpectedly, the first wave of single-cell genomics papers was heavily skewed to map-making efforts describing every tractable model organism and tissue. In these “landscape” exploratory studies, biological replicates generally serve a less important role compared with hypothesis-driven experiments. Replicates might be used to add richness and diversity to the superset of observed cell types and states, but are not used directly for statistical tests comparing experimental conditions, and might even be pooled and treated as a single large sample. As single cell technologies approach maturity, more studies are taking on the challenges of hypothesis-driven research that require rigorous application of good statistical practice. Inevitably, one faces the question: how many replicates are necessary?
Unfortunately, there is no straightforward answer to this question. Best practices in single-cell differential expression analysis have been established that argue for the superiority of using biological replicates binned into ‘pseudobulk’ pools (Squair et al., 2021). Thus, scRNA-seq will benefit from additional biological replicates similar to bulk RNA-seq methods using count-based data such as DESeq2 (Love, Huber, & Anders, 2014) and edgeR (Robinson, McCarthy, & Smyth, 2010) for certain hypothesis-driven analysis goals. For instance, in a study where the goal is to ascertain the effect of treatment “X” on cell type “A”, the statistical power is limited both by the number of independent replicates of the treatment conditions as well as the frequency of cell type “A” within the population. If each sample contains few type “A” cells, then the pseudobulk pool of type “A” will contain relatively few mRNA counts, and the statistics of differential expression will suffer. This can be improved by enriching the single-cell preparation for type “A” cells to increase the total pseudobulk mRNA counts from each individual, with the tradeoff of sacrificing some of the cellular heterogeneity in the overall sample.
However, single-cell genomics techniques are exorbitantly expensive and inefficient for formulating a statistical argument. While single-cell data has unparalleled utility as an exploratory tool, if one’s goal is to test a relatively straightforward preconceived hypothesis (for instance, “Treatment with ‘Compound X’ increases the production of inflammatory cytokines in macrophages.”), a single-cell experiment may not only be an overkill, but also underpowered compared with other more sensitive, repeatable, and affordable assays. High technical variance during the sample preparation and barcoding phases can easily overshadow biological variation (Tung et al., 2017), often more severely than in simpler, more traditional types of experiments, such as western blot, qPCR, or bulk RNA-seq. Worse, each additional biological replicate of a single-cell experiment can cost thousands of USD, while alternative methods might cost one or two orders of magnitude less per replicate. Nevertheless, studies comparing tissue composition and gene expression programs across independent variables are increasingly becoming the norm, as techniques become more robust and protocols are standardized.
3.7. Batch Processing
In an experiment involving multiple independent samples, as much care as possible should be taken to balance batches of samples across the experimental variables. As an example of unbalanced design, consider an experiment investigating gene expression in the hippocampus of treated and control mice: sacrificing, dissecting, dissociating, washing, and counting each animal can take significant time, and might need to be spread across days or even into morning and afternoon batches. Idiosyncrasies accumulate within a given batch that can add significant technical variation to the data that might mask subtle biological variation (i.e., batch effects). Balancing each batch with an equal number of treatment and control animals can greatly improve downstream analysis by using statistical methods that can compensate for the batch as a covariate, provided it is not confounded with the treatment variable through poor study design.
3.8. Multiplexing
Several clever multiplexing strategies have been designed to ameliorate a portion of the exorbitant costs of biological replicates (Figure 2). These generally rely on some mechanism to separately track or barcode independent samples through the steps of library preparation. Two popular methods include genetic demultiplexing and cell hashing, which we discuss below.
Figure 2. Multiplexing strategies.
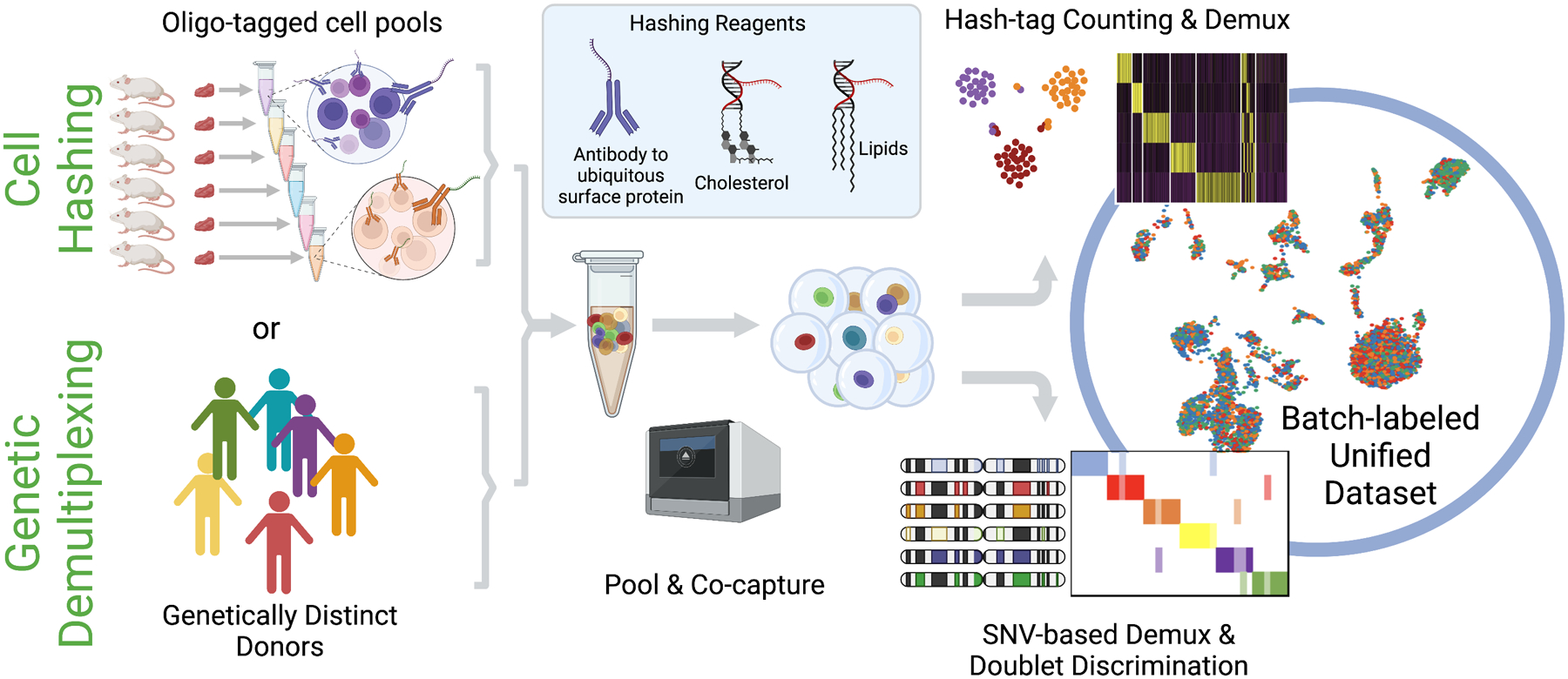
For large-scale studies involving multiple individuals or conditions, samples can be multiplexed using methods such as “Cell Hashing” or “Genetic Demultiplexing” to minimize costs and batch-related artifacts. (Top) In Cell Hashing, batches are labeled by coupling DNA-barcoded hashing reagents to the surface of cells. Examples of hashing reagents include antibodies targeting ubiquitous surface proteins that are chemically conjugated to a DNA oligonucleotide, or lipid- and/or cholesterol-modified oligos that can be embedded within the plasma membrane. Following batch labeling, samples can be pooled and co-captured in the same single cell reaction using, for example, the 10X Genomics platform. Batch barcodes (a.k.a. “hash tags”) are extracted and counted from the resulting sequencing library, and then used to demultiplex the sample downstream. (Bottom) Alternatively, genetically diverse samples such as human patients can be demultiplexed based on their unique single nucleotide variant (SNV) profile. Cells from different donors can be mixed, co-captured, and sequenced as a single sample. Donor-specific SNV profiles, if available, can be compared with the read-level data from each cell, and a probability score is assigned for each of the donors. These probability scores are used to assign cells to different donors, and also to identify and reject “doublet” barcodes that likely contain mRNA from two or more cells. If SNV profiles have not been previously generated, read-level variant calls can still be used to assign cells into different genotype bins, though these bins cannot be matched back to the actual identity of the donors.
Genetic Demultiplexing.
For experiments in humans or other genetically heterogeneous model systems, variant calls gathered from independent DNA-seq or RNA-seq data can be exploited to probabilistically assign each barcode to its likely matching donor. Several open-source tools, including Demuxlet (Kang et al., 2018), Vireo (Huang, McCarthy, & Stegle, 2019), SoupOrCell (Heaton et al., 2020), scSplit (J. Xu et al., 2019), and Demuxalot (Rogozhnikov et al., 2021) can robustly bin cells into distinct donor populations, even with the limited coverage and shallow depth offered by 3’-enriched scRNA-seq and, in some cases, without the requirement of a pre-built variant reference for each donor.
Cell Hashing.
In circumstances where genetic variation cannot reliably be exploited for sample demultiplexing, for instance, in longitudinal studies where multiple samples are drawn from the same individual, or in genetically homogeneous animal models, a clever barcoding scheme called “cell hashing” can be used to exogenously couple a DNA or RNA barcode to cells in a given sample (Stoeckius et al., 2018). Here, antibodies targeting universally expressed cell surface epitopes are conjugated to barcoded DNA oligos and used to decorate the surface of every cell in a given batch. Batches can comprise replicate individuals, treatment conditions, tissue location, time points, or any other conceivable variable to be separately tracked in the single-cell data. The latent DNA-templated DNA polymerase activity of RT enzymes allows these “Hash tag oligos” to be incorporated into the barcoded single-cell library as if it were a standard mRNA, allowing for efficient post facto demultiplexing.
One limitation of antibody-based cell hashing, however, is the challenge of building a truly universal panel of antibodies that will detect every cell in a heterogeneous mixture. Even highly expressed, near-ubiquitous markers, such as the widely used class I MHC complex, can be unpredictably downregulated in certain stem-like lineages, cancer cells, or other special cases. To solve this problem, lipid- or cholesterol-anchored oligos have been employed that can non-specifically incorporate into generic lipid bilayers, including the nuclear envelope (McGinnis et al., 2019). Lipid-targeted “Hash-tags” can, thus, be easily adapted for scRNA-seq, snRNA-seq, scATAC-seq, or even other workflows. A similar chemistry has recently been commercialized by 10X Genomics under the product name “CellPlex”.
The “cell hashing” concept has since been extended to other single cell modalities and chemistries, including ATAC-seq and multi-omic approaches (K. Wang et al., 2021). The proliferation of new batch-barcoding methodologies highlights the pressing need for single-cell technologies to be able to address the concerns of replicability and statistical robustness at manageable cost.
3.9. Sex balance
Much recent attention has been brought to the desirability of minimizing sex bias in biological research (Lee, 2018; Woitowich, Beery, & Woodruff, 2020). Unfortunately, the exorbitant costs of single cell genomics make adding this extra variable (and the associated replicates) challenging. Strong sex-driven differences in gene expression derived both from sex chromosomes and autosomes exist in every human tissue (Lopes-Ramos et al., 2020) and, even in the absence of other confounders, these differences will heavily influence unsupervised clustering and visualization methods. Given unlimited resources, experiments should ideally be carried out with enough replicates to adequately control for sex-specific variation in the data, particularly in studies exploring the effect of an independent variable (e.g. “treated vs untreated”) across individuals, where underlying variation due to sex can confound the signal from the experimental variable. Given limited resources, it maybe be tempting to opportunistically populate a study with a random sampling of sexes and rely on informatic correction methods such as linear regression to minimize variation due to sex-related gene expression. This practice, however, should be avoided, since without large numbers of biological replicates to properly control for sex, attempts to regress away the sex covariate are unlikely to achieve the desired result. Even in a study design incorporating equal numbers of male and female replicates for each experimental condition, spurious differences in cell capture efficiency from sample to sample can lead to strongly imbalanced numbers of cells deriving from each sex. The simplest and most pragmatic solution is to judiciously isolate sex from the independent variable. For example, a pilot animal single-cell study exploring the effects of an experimental compound might begin by first including only sex-matched individuals (that have naturally also been matched for other confounding variables such as age, cage conditions, etc.) and then following up with further experiments to explore sex-specific variation.
3.10. Proteogenomics
Most types of single-cell genomics chemistries have now been adapted to be able to simultaneously measure the abundance of targeted protein panels along with mRNA or chromatin profiles. Generally, antibodies targeting proteins of interest are conjugated to barcoded oligonucleotide adapters that contain priming sites compatible with the specific chemistry of the primary assay, for instance, a short synthetic poly(A) tail for standard oligo-(dT) primed scRNA-seq. Developed concurrently by two groups under the names “CITE-seq” (Cellular Indexing of Transcriptomes and Epitopes, (Stoeckius et al., 2017)) and “REAP-seq” (RNA Expression and Protein Sequencing, (Peterson et al., 2017)), commercial versions of the strategy have now been released by 10X Genomics and Becton-Dickinson, as “Feature Barcoding” and “AbSeq”, respectively.
The key advantage of proteogenomics approaches is the superior dynamic range compared with mRNA measurements. Most mRNAs in scRNA-seq are detected with only a handful of UMIs per cell; in contrast, hundreds of protein molecules may accumulate per mRNA, and the resulting expression signal scales in turn. Proteogenomic labeling can exhibit very similar sensitivity and dynamic range for surface markers as flow cytometry, and the resulting protein counts matrices can closely mimic FACS plots (Stoeckius et al., 2017), making cross-disciplinary comparisons with FACS-driven fields, such as immunology, more sound.
Antibody-oligo tags for proteogenomics can, in principle, be synthesized in any laboratory, —using streptavidin conjugation kits or Click chemistry (Stoeckius et al., 2018), with no specialized equipment. Commercial vendors such as BioLegend are continually expanding their catalog of validated conjugated antibodies compatible with a variety of single-cell genomics applications, including pre-made panels comprising dozens of protein targets.
4. Sample Handling Considerations
While the molecular biology of single-cell genomics protocols can vary dramatically, every method is fundamentally limited by the quality and appropriateness of the methods used to prepare the single cell suspension. The choice of dissociation method, cleanup strategy, and even cell counting approaches can mean the difference between generating a publishable dataset and a sub-standard, artifact-riddled mess. In many cases, these decisions must be made on the day of the capture and barcoding, after sample processing has already begun, in response to differences in the condition of the sample compared to what was expected. Below, we discuss tissue dissociation, cell counting, and enrichment strategies that can be used to meet the needs of different single cell experiments.
4.1. Tissue dissociation
Preparation of a viable single-cell suspension is arguably the most challenging step of any single-cell workflow. Dissociation methods can radically affect the composition and quality of the final sample, and different approaches can be exploited to bias a sample towards a desired cell type. For microfluidics approaches, clearance of non-cellular debris is of critical importance, as large or oblong particles can lead to clogs or “wetting failures”, wherein the disruption of steady laminar flow prevents droplet formation and prevents formation of the emulsion required for barcoding.
4.1.1. Enzymatic dissociation.
Proteolytic extraction of cells from the extracellular matrix is a near-universal first step of any single-cell experiment. Different proteases and dissociation conditions can be optimized for specific tissues to obtain the best possible viability (discussed in more detail in section 4.2), while effectively releasing the desired cell types of interest into a uniform, clump-free single cell suspension. The specificity of different commonly used enzymes makes them amenable to different tissue types. For instance, dispases cleave fibronectin and type IV collagen (Stenn, Link, Moellmann, Madri, & Kuklinska, 1989), collagenases break down collagen in the extracellular matrix (Frantz, Stewart, & Weaver, 2010), trypsin degrades proteins in the cell-cell junctions at the lysine and arginine residues, and papain degrades tight junctions (Reichard & Asosingh, 2019). The right dissociation enzyme or mix of enzymes can be optimized for different tissue types, extracellular matrix compositions, and desired cell types. Gentler protease blends of collagenases and other enzymes release more immune cells, while harsher blends release more stromal cells (including fibroblasts), and the addition of trypsin to dissociations can increase the recovery of certain other cell types, like epithelial cells (Waise et al., 2019). Many comparison studies of various commercial enzyme blends have been done in different tissues, highlighting the variation in viability and cell type composition that can result from these various dissociation protocols. These findings, however, tend to be extremely tissue specific (Fischer et al., 2018; Slyper et al., 2020; Volovitz et al., 2016). Among the more broadly used enzymes are trypsin and commercial trypsin variants such as TrypLE. They are widely used in many single-cell publications across many tissues as the primary enzyme for dissociation, often in combination with EDTA and/or DNase-1. These tissues include esophagus epithelium (Madissoon et al., 2019), pancreatic islet cells (Tatsuoka et al., 2020), human kidney (H. Wu et al., 2018), embryonic mouse tissue (Cheng et al., 2019), mouse cornea (Kaplan et al., 2019), and many others. When performing multimodal experiments, additional consideration must be taken when optimizing a dissociation protocol. Dispase, trypsin, and papain can degrade the antigens on the cell surface, while collagenases and certain commercial trypsin blends like TrypLE can leave cell surface markers intact enough for analysis by flow cytometry (Reichard & Asosingh, 2019).
4.1.2. Chelating Agents.
EDTA and EGTA are frequently included in dissociation and flow cytometry buffers to block divalent cation-dependent cell adhesion molecules such as integrins and cadherins, which mediate high-affinity interactions between cells and anchor them to extracellular matrix proteins (Reichard & Asosingh, 2019; Sheridan & Lefrançois, 2012; Tsuji et al., 2017). EDTA, however, inhibits some enzymes, including collagenase (Swann, Reynolds, & Galloway, 1981), and when used for dissociation before scRNA-seq, it must be washed out of any buffers before the reverse transcription reaction, as high concentrations of EDTA will inhibit it (10x Genomics recommends no more than 0.1mM EDTA to be used in the cell buffer).
4.1.3. DNase treatment.
DNA released by dead cells is a major contributor to clumping and aggregation in dissociated tissue (Reichard & Asosingh, 2019). Degrading this exposed DNA with enzymes such as DNAse-1 can be very effective in rescuing low-viability samples from total sample loss. However, DNases must be removed before the reverse transcription step, as it can degrade the cDNA and, therefore, may not be useful when cell numbers are limited and additional washing steps must be avoided.
4.1.4. Cold-adapted (psychrophilic) proteases (CAPs).
CAPs have been shown to reduce the impact of a dissociation signature on scRNA-seq. While most enzyme dissociation protocols are performed at 37 °C, cold-adapted protease-based protocols use enzymes active at low temperatures, so that the dissociation can be performed at 6 °C. The most commonly used CAP is a serine protease derived from Bacillus licheniformis, a soil bacterium that grows on Himalayan glaciers (Adam, Potter, & Potter, 2017). Psychrophilic proteases from other Bacillus species as well as trypsin from Atlantic cod have also been tested for single cell dissociations (Potter & Steven Potter, 2019). When compared with collagenase digestion at 37 °C, the low temperature protocol with the cold-adapted protease yielded the same cell types but lower overall expression of potential artifacts, such as immediate early response genes, and without inducing a measurable cold-shock response (Adam, Potter, & Potter, 2017; Denisenko et al., 2020; Machado, Relaix, & Mourikis, 2021; O’Flanagan et al., 2019). Ironically, the widespread use of warm dissociation methods can make data comparison with cold-dissociated samples challenging due to the large differential in the stress signature and, thus, some investigators may prefer to adhere to warm protocols. Forward-looking atlas-making efforts would be well advised to consider cold-dissociation methods, as they likely better reflect the true ground state of the transcriptome.
4.1.5. Plants and cell walls.
Most high-throughput scRNA-seq methods rely on detergent-based lysis of the plasma membrane to solubilize mRNA. Plants, algae, and fungi have thick, detergent-resistant polysaccharide cell walls that must first be permeabilized or removed using established protocols for the relevant model organism. The resulting protoplasts and spheroplasts are often quite fragile and, thus, it is recommended to minimize shear stress and centrifugal force during handling. Many successful reports of high-throughput, droplet-based scRNA- and scATAC-seq for Arabidopsis (Dorrity et al., 2021; Jean-Baptiste et al., 2019; Lopez-Anido et al., 2021; Ryu, Huang, Kang, & Schiefelbein, 2019), maize (Marand, Chen, Gallavotti, & Schmitz, 2021; X. Xu et al., 2021), budding yeast (Jariani et al., 2020), and other organisms now exist.
4.2. Cell Counting and Visual Inspection
In most single cell workflows, including microfluidic, microwell, and split-pool approaches, cells and barcodes are brought together by random statistical processes rather than by manual, controlled pipetting. Though random, the distribution of cells in suspension during the partitioning step tends to obey simple Poisson statistics, thus giving the experimenter the ability to control and predict the outcome of the barcoding process merely by controlling the sample concentration. Thus, precise quantification of the concentration of high-quality cells in the preparation is paramount in these protocols. In our experience, poor cell counting is a leading cause of low-quality data, and is a topic worthy of further discussion. In this section, we will discuss the instrumentation, staining reagents, and guiding principles necessary to properly count cells for a single-cell experiment, and accurately distinguish live cells from dead cells and debris.
4.2.1. Counting instruments.
Image-based cell counters, as opposed to flow- or electrical impedance-based counters, are nearly essential for single-cell workflows. Manual hemocytometers and automated image-based cell counters are suitable for both visually inspecting the sample preparation and accurately measuring cell concentration. Typically, tissue dissociation carries through extensive undigested debris that can easily be mistaken for viable cells without expert guidance. Inspection of the preparation for cell integrity, aggregation, and debris, in conjunction with an accurate cell count, is critical for the success of any single-cell experiment. Below we discuss both sorter- and imaging-based counting approaches.
Sorter-based counting.
While it is common to carry out sample enrichment using a flow cytometer as an initial step to many single-cell workflows, the estimated counts produced by most flow sorting strategies are inadequate for precise tuning of the statistically-driven random capture process. Jet-in-air sorters are prone to some amount of cell breakage during the sort step, and imperfect gating strategies and sorter inefficiencies compound to make this a generally unreliable counting method (kb.10xgenomics.com - a, n.d). Moreover, sorters deposit cells along with a non-trivial volume of sheath fluid, which can result in a sample that is too dilute for direct loading into a microfluidics device or other single-cell workflow. Thus, sorted samples often have to be concentrated and re-counted by hemocytometer or image-based counters prior to use. While this provides an opportunity to revise the count and check for cell integrity before running an expensive experiment, some cells are invariably lost to breakage, failure to pellet, or adherence to pipet or tube wells during handling. When possible, it is best to carry out a few “dry-runs” to become familiar with the expected amount of sample loss at this final stage. Sorting a considerable excess of the desired number of cells is the safest way to ensure that enough remain after washing and resuspension to achieve an optimal cell concentration.
Imaging cell counters.
Automated, microscopy-based cell counters are a workhorse instrument of any laboratory carrying out single-cell experiments, and are available from a number of commercial vendors. An ideal cell counter will produce a high-resolution brightfield image of the cell preparation and, ideally, offers multi-color fluorescence imaging, for use of a variety of viability stains. Since single-cell preparations are frequently precious and limited in quantity, a low-volume counting chamber that minimizes the amount of sample spent for counting can be essential. Many comparable devices are available that satisfy these requirements, including the ThermoFisher Countess, Nexcelom Cellometer, DeNovix CellDrop, and Logos Luna product lines. Perhaps more important than the specific choice of instrument, is a tight integration of the counting instrument into the sample preparation workflow, and an empirical understanding of how its cell counting results can be used to predict cell yields and experimental outcomes.
Direct imaging of the cell preparation also affords the chance to identify an inadequate single-cell suspension. Incomplete tissue digestion can produce a high fraction of small aggregates containing two or more cells that would likely be co-captured with a single barcode in droplet, microwell, or any other single-cell workflow. Manual inspection of several microscopic fields to assess the overall aggregation level is recommended before proceeding. Doublet and larger aggregate quantification can, in principle, be carried out with FACS instruments and other counting methods, but are generally a poor substitute for direct observation with an imaging system.
4.2.2. Viability Stains.
Cell viability screening is a critical decision point in any scRNA-seq protocol, where the data is extremely sensitive to the consequences of cell death. The rate at which cells begin to die varies dramatically across tissue types and dissociation conditions. Thus, viability should be monitored as frequently as possible throughout handling: immediately after dissociation, after column-based enrichment steps, after flow cytometry, and after any other significant handling step where cells may have begun to trigger cell death pathways. There is no firm rule for the minimum acceptable sample viability; an experimenter needs to weigh for themself the tradeoff between collecting data with a known amount of dead-cell contamination versus collecting new specimens, optimizing the protocol for better viability, and trying again down the road.
An ideal viability protocol should be rapid, to minimize handling and cell stress and, thus, dye exclusion methods tend to be preferred. For mammalian models, standard Trypan Blue exclusion is fast, accessible, and adequate for single-cell workflows. Alternatively, fluorescent counterstains such an Acridine Orange / Propidium Iodide (AO/PI) mixture provide contrasting colors for live, intact cells vs dye-permeable dead cells. Some evidence suggests that AO/PI stain outperforms Trypan Blue as a viability marker (Hanamsagar et al., 2020; Mascotti, McCullough, & Burger, 2000), so this method is preferred if a suitable cell counter is available. Assays that require enzymatic conversion of fluorogenic molecules such as Calcein AM or caspase substrates can, in principle, provide better sensitivity versus background staining, but with the tradeoff of requiring relatively long incubation times during which the cell state is being artificially perturbed.
Digestion of plant tissues into protoplasts for scRNA-seq can frequently produce cell suspensions with brightly staining or autofluorescent cell wall components and organelles that can obfuscate counting with certain nucleic acid stains like AO/PI. Fluorescein diacetate (FDA) has been proposed as an alternative dye that mitigates some of these issues (Noland & Mohammed, 1997). In general, for plant tissue preps where large debris is unavoidable, manually counting unstained or Trypan Blue-stained cells with a hemocytometer may be preferred as a means of discriminating cells from artifacts. Whatever the counting method, it is important to optimize and perfect this step prior to running a very expensive and delicate scRNA-seq experiment.
4.2.3. Debris.
Amorphous and rigid non-cellular debris is a common byproduct of most tissue dissociation protocols, due to incomplete solubilization of the dense extracellular matrix. While this debris is generally inert from the standpoint of interfering with most assay chemistries, debris can lead to failure for a few reasons:
Large debris or too much debris can cause microfluidics or sorters to clog. Large debris may be removed using filters, while smaller debris can often be partially removed by washing with low-speed centrifugation.
Amorphous debris may be erroneously mistaken for cells, leading to an inaccurate cell count and underloading of the sample.
Debris may contain DNA or RNA, which will contaminate the data. This could be cellular debris that has DNA or RNA associated with it, or it could be red blood cells, which often skew cell counts on automated counters, leading to inappropriate sample loading. Alternatively, it could be contamination of another organism, such as bacteria or yeast.
If excessive debris is a recurring problem, cleanup by density gradient columns or cushions is probably the most reliable and broadly applicable approach (see section 4.4.4 below).
4.3. Problematic cell types
Certain cell types have biological properties that make them a challenge, particularly for scRNA-seq. Neutrophils and other granulocytes contain relatively few mRNA molecules, and high amounts of RNAses and proteolytic enzymes, making them exceedingly difficult to study without proper handling (Deerhake, Reyes, Xu-Vanpala, & Shinohara, 2021; Qi et al., 2021; Xie et al., 2020). For microfluidic applications, extremely large or irregularly shaped cells such as hepatocytes or cardiomyocytes could have trouble fitting through narrow flow channels, causing clogs or shearing the cells apart. This is even more problematic for pancreatic acinar cells, which are large, fragile, full of digestive enzymes, and can burst and contaminate the entire sample, causing widespread cell death and RNA degradation. The intricate architecture of neurons generally causes problems when attempting to dissociate them intact from brain tissue and, as a consequence, single-nucleus RNA-seq has been widely adopted as a harsh but more reproducible work-around (see, “Single Nucleus RNA-seq” section below). It is always important to assess the compatibility of a given workflow with the cell types to be examined, and whether any alternative isolation strategy or protocol alterations can be attempted to mitigate the challenges.
4.4. Cell Type Enrichment and Sample Cleanup
The high cost-per-cell for even the most economical single-cell methods means that it is usually well worth the time and effort to perform some type of enrichment to sequence only the desired cells in the sample. Most commonly, one may wish to enrich for live and intact cells, though it may also be beneficial to enrich for certain cell types of interest to improve the representation of heterogeneous cell states and statistical significance in differential expression calculations. This can be done by using commercial immunomagnetic enrichment or depletion kits, sorting, or specialized enrichment steps like gradient fractionation. Each additional manipulation added to a single-cell isolation presents advantages and disadvantages, which must be considered carefully with respect to the goals of the experiment and the nature of the biological sample. In this section, we will discuss some helpful guidelines that are relevant to most single-cell methodologies.
4.4.1. Affinity Enrichment and Depletion.
Magnetic, antibody-based affinity columns are potentially the quickest and gentlest means of enriching cells of interest. Popular commercial solutions known to be compatible with single-cell workflows include the MACS (Miltenyi Biotec) and EasySep (Stem Cell Technologies) product lines. Pre-designed antibody panels are available to enrich or deplete a variety of common cell types but, in principle, any combination of antibodies can be provided by the end user.
Positive vs. Negative Selection.
In some cases, antibodies used for cell type enrichment can have the undesirable side-effect of triggering signaling cascades via interactions with surface markers. To avoid perturbations in scRNA-seq gene expression signatures caused by antibody labeling, negative selection can be used to selectively deplete all or most of the other unwanted cell types in the sample, leaving the population under study untouched. Negative selection also allows more flexibility when using proteogenomic (e.g. CITE-seq, REAP-seq) approaches, since the surface epitopes were not blocked during the enrichment step.
If positive selection is preferred, it is important to consider whether the enrichment method introduces complications with any delicate downstream steps. For instance, magnetic bead-based protocols tightly bind the targeted cells and beads together, and they remain associated for the duration of the experiment. In most cases, these beads are quite small compared with the diameter of the cell and, generally, do not interfere with downstream chemistry steps. However, it is important to first confirm with the manufacturer that the bead size will not interfere with size-restricted applications, and should generally be smaller than a typical cell diameter.
Dead cell depletion.
Dead and dying cells lose the ability to maintain phospholipid asymmetry on their plasma membranes, and phosphatidylserine (PS) that is normally restricted to the inner leaflet rapidly begins to translocate to the outer leaflet, where it is exposed to the extracellular medium. This can provide a handle for affinity depletion using Annexin V, which tightly and specifically binds PS (Fadok et al., 1992; Koopman et al., 1994). Rapid magnetic dead-cell depletion kits are available that can be applied to most cell types if the viability of the cell suspension is low. In many cases, the resulting column flow-through can be dramatically enriched for live cells. However, these methods can hurt overall cell recovery, and are risky to employ if working with low cell numbers. Moreover, in preparations where cell death is proceeding rapidly during handling, even the short time taken for enrichment may not offset the rate at which cells are dying. One should also be cautious of buffer compatibility: in particular, EDTA is present in some buffers, which chelates divalent cations and must be washed away prior to reverse transcription or other Mg2+ dependent enzymatic steps.
4.4.2. Red Blood Cells (RBCs).
Most animal tissue preparations will contain many RBCs, which harbor a significant number of mRNA and are often seen as a nuisance in scRNA-seq studies. If left alone, RBCs might co-encapsulate with other cell types, or burst during sample handling and release RNAs that contaminate other cells in the batch. Affinity-based RBC depletion kits are available from many manufacturers. Alternatively, RBCs can be selectively lysed in ammonium chloride buffer (Miller, 2016) during preparation of the single-cell suspension. Such RBC lysis steps are generally well tolerated for scRNA-seq provided residual ammonium chloride or other potentially incompatible compounds are removed by washing, and that they are rapid enough that gene expression profiles are unlikely to be perturbed significantly.
4.4.3. Fluorescence-activated cell sorting (FACS).
Employing an upstream flow sorting step in any single-cell workflow can serve three purposes simultaneously: 1) Enrichment of desired cells based on fluorescent markers, 2) Elimination of problematic debris, and 3) Concentration of cells without the need for additional risky centrifugation steps. With a streamlined staining workflow and a validated gating strategy, sorting can often provide the quickest route to a clean, usable sample.
Flow sorting is also more customizable than the magnetic enrichment and depletion kits. Consider a hypothetical experiment where two distinct cell types must be enriched from the same sample, but one of the cell types is comparatively rare, and the sample is precious. Most sorters can simultaneously sort two or more gated populations, wasting nothing as the sample passes through the instrument. The final sorted populations can then be re-combined in any desired ratio, boosting the representation of the rarer cell type. Such sample re-composing would be extraordinarily difficult without FACS. Moreover, cells can be sorted by additional features such as viability, size, granularity, and multiple endogenous and exogenous fluorescent markers.
Instrumentation.
While many high-end sorters such as the BD FACSAria line can spectrally resolve dozens of fluorophores, single-cell experiments may be more amenable to simpler, low-dimensional color panels to preserve heterogeneity for the downstream experiment. Sony Biotechnology offers a lower-cost alternative (the SH800 family) that can use up to six colors and sort into both tube and plate formats, making it flexible for a variety of workflows. More recently, several manufacturers have released specialized microfluidics-based sorters with fewer color options and output formats, but are optimized for ease-of-use and much gentler conditions, which minimize cell death during handling. These include the MACSQuant Tyto (Miltenyi), WOLF product line (Nanocellect), the Sort (On-Chip Bio), and the S3e (Bio-Rad). These sorters are aimed at individual labs and single-cell core facilities rather than specialized flow cytometry facilities, and can help streamline workflow logistics, avoid queuing for sorter time, and minimize the need for trained sorting specialists.
Drawbacks.
Most conventional flow sorters subject cells to relatively high pressures and shear stresses that, if not handled carefully, could lead to premature lysis or trigger an acute stress response that alters the transcriptome. Antibody staining also requires additional incubation times that may further alter gene expression patterns, though generally these steps can be performed on ice, to minimize perturbations. After sorting, it is critical to verify the cell counts prior to single-cell processing, as the count estimates produced by sorters are often unreliable, unless the sorting strategy has been extensively validated.
4.4.4. Optiprep/Gradient Enrichment.
Centrifugation through a density gradient medium can be used to separate cell types based on buoyancy, as well as deplete dead cells or debris from a single cell suspension. Specifically in scRNA-seq applications, stepwise iodixanol (Optiprep) gradients have been used to enrich for live Drosophila hemocytes (Tattikota et al., 2020), enrich for stellate cells during dissociation of pancreatic tissue (Dominguez et al., 2020), and to remove myelin and other debris for snRNA-seq (Del-Aguila et al., 2019). Other density gradient media have also been validated for single-cell applications, including sucrose (Ayhan, Douglas, Lega, & Konopka, 2021), Nycodenz (Dominguez et al., 2020), Percoll (Guldner, Golomb, Wang, Wang, & Zhang, 2021), and Ficoll (Mereu et al., 2020). Gradients are also effective means of separating nuclei and debris, and are generally well tolerated for snRNA-seq, especially in challenging tissue such as postmortem human brain (Maitra et al., 2021). Bear in mind that the high solute concentrations may interfere with downstream enzymatic steps, and may need to be extensively washed or diluted before processing. Serumwerk, a provider of density gradient media, offers a collection of cell type–specific isolation protocols that may serve as a helpful resource when designing a new sample prep workflow: https://diagnostic.serumwerk.com/downloads/.
4.4.5. Low Speed Centrifugation.
Live and dead cells can have differing buoyancies even in standard wash buffers, and this can be exploited as a simple enrichment step. Spinning at or below 200xg can gently pellet large, dense live cells, while dead cells are retained in suspension (Hanamsagar et al., 2020). Centrifugation speed is an important consideration, as it can cause the unintentional enrichment of certain cell types, as certain cell populations will pellet at lower speeds while others will pellet at higher speeds. Moreover, higher speed centrifugation can result in lower cell viability of the sample (Pavel, Sandra, Jaroslav, Mikael, & Radek, 2019), so empirical optimization is required.
5. Cryopreservation and Fixation
Performing scRNA-seq on fresh tissue is not always possible, so cryopreservation or chemical fixation can, in many cases, be used to temporarily preserve cells prior to the single-cell experiment (Figure 3). Preservation has many advantages: it facilitates sample batching to reduce some types of technical artifacts, particularly in longitudinal or time-course studies; it allows flexibility in the case of human tissues collected from surgeries at remote sites or during unusual hours; it aids in logistics across multi-center studies; it enables sample pre-screening and confirmation, e.g. via histology; and greatly simplifies splitting of the tissue across multiple assay types. Recently, this practice has been put to good use in collecting cohorts of frozen COVID-19 infected lung biopsies that were aggregated into a large-scale, multi-patient atlas of lethal disease (Melms et al., 2021).
Figure 3. Preservation strategies.
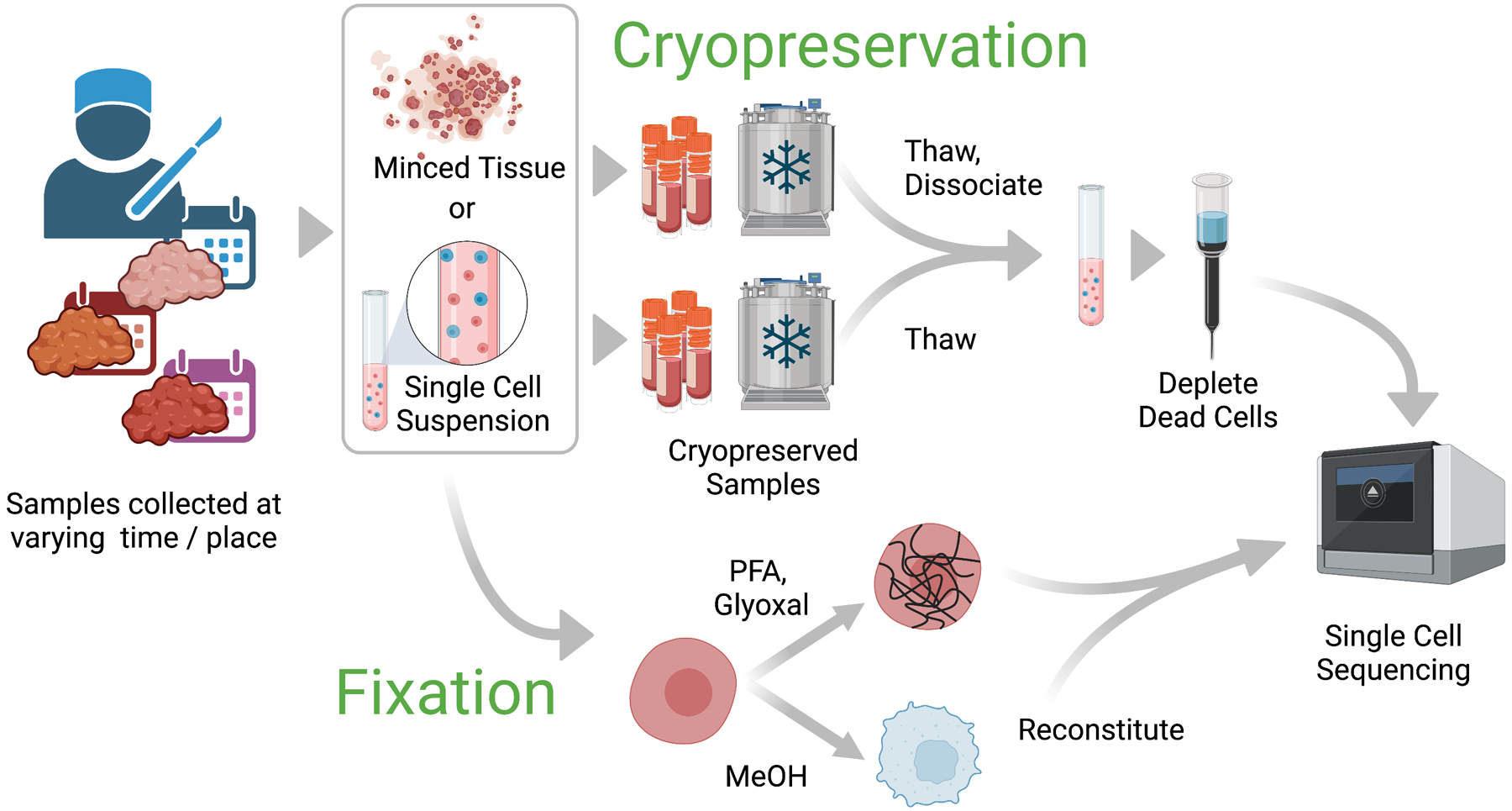
Cells or tissue can often be preserved prior to a single cell experiment to allow for samples to be acquired at different times or locations and then processed synchronously. Either fully dissociated cell suspensions or partially intact, minced tissue can be frozen in liquid nitrogen and stored for weeks or months prior to thawing. If minced tissue pieces were frozen initially, single cell suspensions can be prepared after thawing by standard dissociation protocols used for fresh tissue. Alternatively, cells can be chemically preserved with a variety of fixatives that have been demonstrated to be compatible with many single-cell workflows, such as paraformaldehyde (PFA), glyoxal, or methanol (MeOH). Protocols to reconstitute the fixed single cells prior to sequencing vary depending on the fixative and the chemistry of the single-cell application.
In exchange for this flexibility, however, the overall quality of the data tends to suffer relative to fresh samples, and it risks imparting biases in favor of cells that fare better under the preservation method used. Preservation protocols often need to be optimized for specific tissues and, critically, validated for the subsequent single-cell workflow. In this section, we discuss the methods and trade-offs of storing cells by cryopreservation, hypothermic preservation, and fixation. Using flash frozen tissue to perform snRNA-seq will be discussed in a later section.
5.1. Cryopreservation
Cryopreservation techniques have been well optimized for most single-cell genomics workflows, including scRNA-seq (D. Chen et al., 2021; Denisenko et al., 2020; Guillaumet-Adkins et al., 2017; Morsey et al., 2021; Wohnhaas et al., 2019; S. Z. Wu et al., 2021) and ATAC-seq (Fujiwara, Baek, Varticovski, Kim, & Hager, 2019; Rocks et al., 2021). In general, standard cell-culture cryopreservation practices can be applied to a single-cell suspension for downstream scRNA-seq; i.e., slow freezing in media containing cryoprotectants such as DMSO and high serum content (Kielberg, n.d.). Ideal freezing conditions should be determined empirically for each type of sample, optimizing for cell viability and yield after thawing. Many types of tissue samples can be effectively preserved either as solid, finely separated chunks or as enzymatically dissociated single cell suspensions, with only minor effects on sample quality (S. Z. Wu et al., 2021).
The biological consequences of cryopreservation, while minor, should not be neglected during proper study design. In heterogeneous tissues, cell types may show differing sensitivity to the freeze-thaw process, leading to potential skewing of the sample composition towards the hardier cell types that remain viable after thawing. In scRNA-seq, global gene expression levels are roughly comparable between fresh and frozen samples, though acute changes in stress-related gene expression programs can confound analysis across preservation methods. Induction of immediate-early response genes such as the transcription factors FOS and JUN during handling can obscure biological variability and must be corrected for with computational methods (Morsey et al., 2021; van den Brink et al., 2017). Additionally, the freeze/thaw process can result in cell breakage, which can contaminate the cell suspension with dissolved RNA unless extensively washed (see the “Ambient RNA” section, below), requiring further care in handling. As with any element of good experimental design, it is critical to closely match the preservation conditions and handling across samples in a large, multi-sample study.
5.2. Hypothermic Preservation
As an alternative to cryopreservation, several groups have reported controlled studies demonstrating the feasibility of short-term (< 3d) storage of intact tissue in specialized hypothermic storage media at 4°C (Madissoon et al., 2019; W. Wang, Penland, Gokce, Croote, & Quake, 2018). In these studies, both 10X Genomics and Smart-Seq2 scRNA-seq protocols seem to be highly tolerant to hypothermic storage, evidenced by very minor changes in both gene expression profiles and cell viability. Longer storage duration does correlate with a higher fraction of ambient RNA (see section below) released from lysed cells, but this relatively minor tradeoff makes hypothermic storage a favorable protocol for coordinating human patient samples that often are collected on irregular schedules.
5.3. Fixation
As an alternative to freezing, some tissues and genomics applications may be amenable to preservation by fixation. Both chemical crosslinking and methanol dehydration methods of fixation have been reported in single-cell workflows, with varying levels of success. It is important to note that fixation methods tend to introduce substantial biases to the molecular readout, making data integration with non-fixed samples more challenging. Here, we will discuss advantages and disadvantages of two commonly used fixation techniques, chemical fixation and methanol fixation.
5.3.1. Chemical fixation.
Fixation by covalent crosslinkers is required for many split-pool barcoding methods in scRNA-seq and multiomic methods such as SHARE-seq (Ma et al., 2020), but is not frequently used for droplet based scRNA-seq that rely on first-strand cDNA synthesis from intact RNA. In split-pool methods, the fixation is critical to the barcoding step, and the fixation step may need to be optimized to improve library complexity and reduce background RNA contamination (Ma et al., 2020; Rosenberg et al., 2018). Paraformaldehyde solutions of 1–4% v/v are most commonly used, though alternative fixatives such as glyoxal (Bageritz et al., 2021) and dithio-bis(succinimidyl propionate) (DSP, a.k.a. Lomant’s reagent) (Attar et al., 2018) have been reported. The main challenge with chemical fixations is the reduced accessibility of poly-A RNA substrate to RT enzyme and impaired processivity, which can severely reduce cDNA yield, length, and mappability. This can be circumvented by reversing crosslinks by treating with high temperature and proteases at a point between initial barcoding reactions and final cDNA amplification. Split-pool methods such as SPLiT-seq have a step-wise workflow that provides an easy opportunity to reverse crosslinks. Recently, a derivative of Drop-seq called FD-seq introduced a technique to enable crosslink reversal after droplet encapsulation (Phan et al., 2021), but popular droplet platforms that rely on conventional poly-A-primed library construction such as 10X Genomics do not seem amenable to formaldehyde fixation without significant protocol modifications. In 2022, 10X Genomics announced a new fixed RNA workflow that uses a panel of hybridization probes that are ligated in situ to quantify unique mRNA molecules, rather than using standard cDNA library chemistry.
5.3.2. Methanol fixation.
Somewhat more widespread success has been reported for methanol-based fixation methods, which immobilize proteins and nucleic acids by a mixture of dehydration and precipitation (Alles et al., 2017a; Troiano, Ciovacco, & Kacena, 2009). Generally, a prepared single-cell suspension is brought drop-wise to 80% methanol before cryopreservation or refrigerated storage for an indefinite duration. Cells are later recovered by controlled rehydration, but recovery of intact RNA is strongly dependent on the tissue type and buffer conditions. Buffers comprising PBS + 0.01% w/v bovine serum albumin or saline sodium citrate (SSC) have been compared and contrasted for various cell types, with mixed results (Alles et al., 2017a; Denisenko et al., 2020). Thus, this process should be thoroughly optimized prior to a single-cell experiment.
Methanol-based fixation has also recently been demonstrated to enable a novel intracellular proteogenomic strategy called INS-seq (Katzenelenbogen et al., 2020). Here, cells are fixed with methanol and ammonium sulfate, and permeabilized to allow binding of oligo-conjugated antibodies to intracellular antigens, similar to CITE-seq and REAP-seq. The method is compatible with the 10X Genomics platform, thus enabling joint measurement of transcriptomes and intracellular protein abundance in the same cells.
5.4. Fixation vs. Preservation.
The ideal choice of preservation technique may vary depending on the tissues of interest and other nuances, but most comparison studies have found cryopreservation to better recapitulate the gene expression profiles of fresh tissue (Wohnhaas et al., 2019). Cryopreservation has sometimes been found to deplete certain cell types if the freezing conditions are not optimized for the specific tissue (Denisenko et al., 2020), but returned similar cell type percentages to the fresh tissue in other studies. In contrast, fixation methods have been found in multiple studies to result in more ambient RNA background (see section 7.2.2), and have sometimes been reported to significantly reduce RNA yield compared with fresh tissue (Alles et al., 2017b; Denisenko et al., 2020; Wohnhaas et al., 2019). Fixation can also interfere with other downstream steps, such as cell type enrichment by flow cytometry or screening for viability.
6. Quality Control and Key Decision Points
Careful monitoring of key quality control (QC) metrics throughout a single-cell experiment is critical to avoid wasteful expenditure of resources on poor quality samples. There are three main QC-driven decision points in a typical scRNA-seq workflow that provide an opportunity to cut losses and try again. These decision points naturally fall at points along a workflow where significant amounts of resources (in terms of time, money, or preciousness of the samples) are about to be committed to the subsequent step. Briefly, these QC checkpoints are: 1) sample preparation and single cell suspension quality, 2) cDNA/library quality, and 3) pilot-sequencing quality (Figure 4). Deciding whether to proceed with or to abort a borderline experiment depends chiefly on how quickly a replacement sample can be prepared, but even in the case of one-of-a-kind samples, certain minimum thresholds should be met to avoid wasting resources on utterly useless data. In this section, we will discuss how to make effective use of each of these three checkpoints and avoid the familiar pitfall of “garbage in, garbage out.”
Figure 4. Overview of a generalized single-cell workflow: resource investment and key checkpoints.
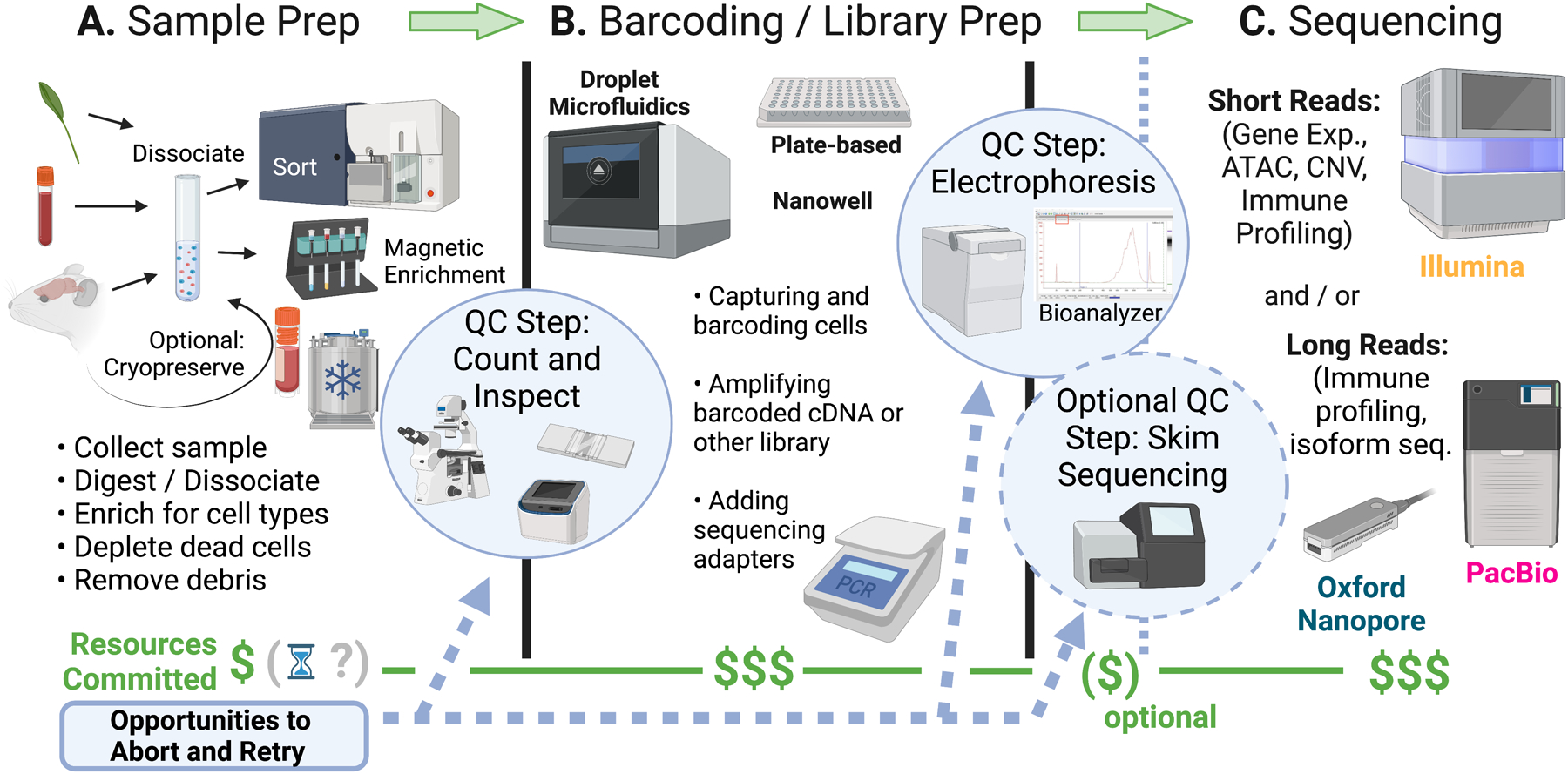
Most single-cell experimental workflows can be subdivided into three distinct phases: sample prep, barcoding and library prep, and sequencing. Each phase entails a significant investment of resources and presents a critical quality control checkpoint that provides opportunities to abort and retry if the samples appear suboptimal. (A) The sample prep stage encompasses all aspects of study design, sample acquisition, storage, and processing upstream of the “genomics” portion of the workflow, and can comprise the majority of the time investment involved in a project. On the day of the experiment, samples are dissociated into single-cell suspensions, and potentially passed through a flow sorter or magnetic column to obtain an enriched population of interest. The cell suspensions should be assessed (e.g., by microscopy) for relevant parameters including purity, viability, cleanliness of the suspension, and clumping. If the suspension looks unsatisfactory or if there are too few intact cells, this is the best time to abort before large amounts of resources are committed in the downstream steps. (B) Barcoding and library prep involves a series of enzymatic reactions that take place in emulsion droplets, PCR plates, nanowells, or other type of isolated compartment. Depending on the library chemistry, this step can consume roughly half of the costs associated with the experiment. Libraries are generally amplified by PCR with the addition of barcoded adapters, and should be assessed at a second QC checkpoint by electrophoresis (e.g., using a Bioanalyzer). (C) Sequencing of the single-cell libraries also consumes a significant percentage of the overall budget. Depending on the application, libraries are sequenced using either short reads (for gene expression, ATAC, CNV, immune profiling, or other applications) on an Illumina instrument, or long reads (e.g., for isoform-resolved RNA-seq, immune profiling) using an Oxford Nanopore or PacBio instrument. For large projects, an optional “skim-sequencing” step can be added for quality control. A few million reads per library is often sufficient to tell whether barcoding proceeded properly, and can provide a crude estimate of captured cell numbers and predicted sample quality.
6.1. Single Cell Suspension Quality
Failure to properly optimize tissue dissociation can manifest in several ways: poor viability, low cell counts, high levels of non-cellular debris, and large cellular aggregates. If any of these parameters are unsatisfactory, recovering from the problem in a ‘live-experiment’ setting is often fruitless and endangers all samples in an experiment through extended exposure to stress conditions.
It is essential to inspect sample preparations under a microscope before committing to any expensive protocol steps. Clumpy or aggregated suspensions result from incomplete enzymatic digestion. Large aggregates are especially problematic for droplet-based systems, as they can clog the microfluidics channels and ruin the emulsion. An excessive number of small, 2–3 cell clumps will also severely confound downstream data analysis, since they will be barcoded together and display a blend of transcriptomes. While it is impossible to remove all such ‘sticky doublets’ from your sample, the observed ratio of clumps to singlets should be evaluated prior to committing to the experiment (Figure 5). Many single-cell protocols suggest filtering out clumps using a standard 40 μm flow cytometry cell strainer or similar filter device. While these can remove larger aggregates, it is essential to visually inspect the results after straining. Often, such strainers do not adequately filter away small cell clusters, in which case further enzymatic dissociation or doublet discrimination via flow cytometry may be called for.
Figure 5. Cell suspension quality control.
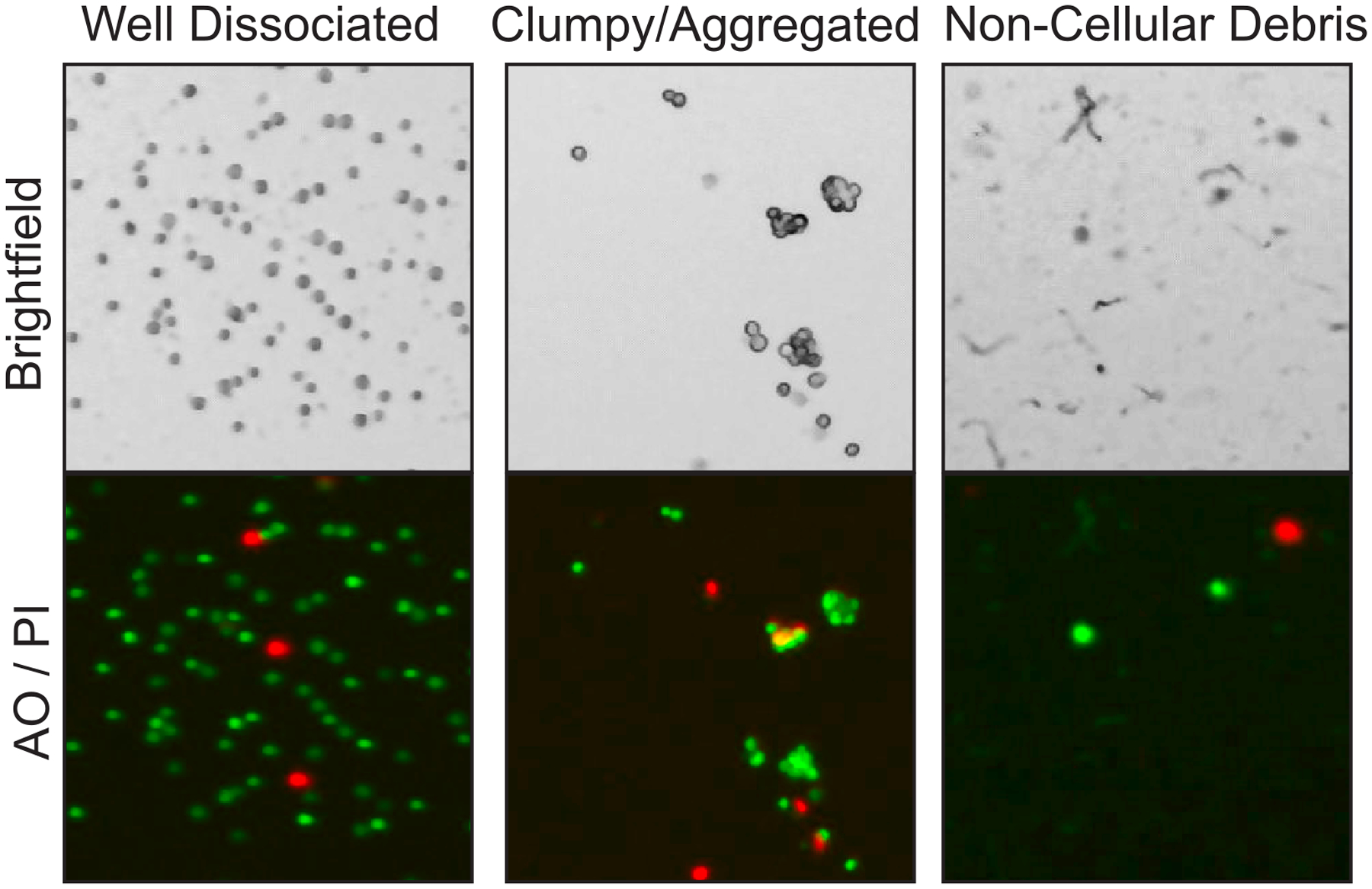
A well-dissociated single cell suspension will be largely free of debris, and cells will have a smooth, round appearance (top left). Cells can be stained with AOPI to visualize live cells as green and dead cells as red (bottom left). A poorly dissociated suspension (middle) will leave many aggregated clumps of several cells, which is not ideal for single-cell methods. Preparations with excessive non-cellular debris (right) should be cleaned by gradient centrifugation, FACS, or some other method to avoid microfluidic clogs or cross-contamination due to material stuck to cell fragments or other non-cellular debris.
6.2. cDNA / library quality
Once the initial barcoding step has been performed, the second checkpoint comes when assessing the quality of the resulting cDNA or ATAC tagmentation pattern, generally using Agilent Bioanalyzer or Tapestation electrophoresis systems (Picelli et al., 2014).
In scRNA-seq workflows, a high-quality full-length cDNA library can be visualized as a broad peak from 1kb-10kb, spanning the natural range of cDNA lengths in the cells of origin (Figure 6A). The mass yield and average molarity of this library can be estimated by the instrument software, and used to normalize and adjust sample input for the downstream steps of library finalization. Low cell viability can be distinguished by broader and lower molecular-weight peaks of several hundred base pairs, indicating RNA degradation. Large spikes at the < 150bp range generally arise from PCR adapter artifacts, and are especially pronounced in low-quality libraries with insufficient input cDNA. A library displaying low-QC characteristics at this stage may still produce a valid, sequenceable library after fragmentation, but will generally contain high levels of PCR duplication, few UMIs, and few unique genes per cell (Figure 6B). Considering the cost, low-quality samples are generally better left unsequenced.
Figure 6. Library quality control.

Libraries should be screened using an electrophoresis instrument such as the Agilent Bioanalyzer prior to sequencing to verify that the DNA fragment size falls within the expected range of the protocol. (A) One common pitfall of scRNA-seq libraries is degraded cDNA from dead or lysed cells, visualized by a shift in the molecular weight towards 1,000 bp and below. (B) Abundant PCR adapter artifacts can also swamp out gene-body reads, resulting in poor quality libraries, as visualized by reduced estimated number of genes detected per cell. (C, right) Likewise, ATAC-seq libraries should be checked for the expected “nucleosome ladder” pattern. Under-tagmented libraries (C, left) can be size-selected to yield an acceptable profile for sequencing.
ATAC-based workflows should also be screened for appropriate quality. Ideally, chromatin tagmentation should produce a characteristic nucleosome “ladder”, with broad peaks of ~250 bp (~150 bp for each nucleosome monomer, plus 50–100 bp for the adapter sequence, depending on the protocol). Over-fragmentation, mainly caused by a molar excess of Tn5 enzyme, will produce a single mononucleosome peak, while under-fragmentation will produce 1 kb+ poly-nucleosomal fragments (Figure 6C, left). Longer library fragments will grossly underperform on Illumina sequencing instruments due to kinetic competition during cluster formation.
In many cases, measures can be taken to deplete problematic artifacts prior to sequencing. Bead-based DNA size selection protocols (e.g. Ampure XP or SpriSelect, Beckman Coulter) can be tailored to deplete low- or high-molecular weight contaminants with good yield on the desired size range (Figure 6C, right). Bead-based depletions, however, have imprecise cutoff thresholds, and if more precision is required to remove an artifact with similar molecules weight to a desired band, agarose or acrylamide gel electrophoresis should be used. Generally, careful intervention at this stage can dramatically improve sequencing performance later on.
6.3. Pilot-Sequencing Quality
For large-scale projects requiring several billions of reads, performing a low-depth first sequencing pass can act as a final quality-control checkpoint. The purpose here is to address some basic questions about library quality, for example: Is the barcode structure correct? What is the fraction of duplicated reads? What is the overall genome mapping rate? How many unique cell barcodes are detected, and how are the reads distributed among them? Many of these concerns can be satisfactorily addressed with only a few million sequencing reads per library, or about 5–10% of the final required depth needed for the project.
A typical single-cell project might involve many separate biological samples, each comprising thousands of single cells. When pooling samples together for a sequencing run, it is difficult to accurately estimate relative molarities of each library based only on average molecular weight and concentration. Additionally, samples may comprise dramatically different numbers of cells, leading to varying amounts of reads per cell across each of the samples in the pool. Pilot sequencing can provide a valuable opportunity to assess sample quality, re-pool according to new molarity estimates, or drop samples altogether.
Even at very low depths, “skim” sequencing can provide a useful estimate for the number of successfully captured cells in each sample. The relative molar ratios of each sub-library in the pooled sample can be used to estimate the yield for larger scale sequencing runs, which can, in turn, be divided among the rough cell number estimate to provide a projection of the final expected number of reads per cell. At this stage, adjusting the pooling ratios of individual under- or over-represented samples can dramatically improve the odds of achieving a well-balanced study after the expensive final sequencing step.
7. Batch Artifacts
In any genomics experiment, the cumulative effects of minor, often intangible, technical differences across sample batches prepared at different times or under different conditions can contribute a non-trivial amount of variation in the resulting data. The exact definition of a sample “batch” is commonly debated, and the term is often used as a catch-all describing a variety of technical covariates, including the date and time of the experiment, the individual(s) handling samples, reagent lots, among others. Batch artifacts can be especially pronounced in single-cell experiments, which usually entail delicate, sensitive workflows that provide ample opportunities for unintended variation. It is important to keep in mind the potential sources of technical, batch-dependent variation, and minimize them wherever possible using a combination of good laboratory practice, robust experimental design, and bioinformatic compensation.
7.1. Common sources of batch artifacts.
Nearly every step of a single-cell workflow presents potential sources of batch variation. During tissue dissociation, slight alterations in digestive enzyme concentration, incubation time, temperature, or agitation method can bias the liberation of certain cell types over others, leading to different single-cell suspension compositions from ostensibly similar tissues (Denisenko et al., 2020). Prolonged dissociation at higher temperatures can also induce stress response pathways, the extent of which can differ substantially across batches (Adam, Potter, & Potter, 2017). The exponential kinetics of PCR steps are particularly sensitive to subtle variations in master mix formulation, temperature control, and cycle numbers, which can influence the uniform representation of unique molecules in the library. Fragmentation steps, such as enzymatic or sonication-based methods used in 3’-digital RNA-seq or Tn5 transposition in ATAC-seq, are highly sensitive to incubation time and temperature. Many library workflows also include one or more cleanup steps that make use of nucleic acid binding beads. Efficient and reproducible binding and elution from such beads requires mindful adherence to the manufacturer’s protocol; for example, allowing them to overdry before elution can dramatically reduce yields and cause precious UMIs to be permanently lost from the pool.
Minimizing variation in these time-sensitive steps can be as simple as setting up a sensible, well laid-out workspace with reliable access to necessary items and using laboratory timers to strictly adhere to reproducible intervals. Temperature variation can be mitigated by pre-setting thermal cyclers or heating blocks, and moving samples directly to and from wet ice. Samples should be processed in parallel whenever possible, potentially even at the time of euthanizing and dissecting in the case of animal studies, where large differences in post-mortem interval could perturb the underlying biology.
Batch effects can technically also be introduced at the sequencing step, though this has become less of an issue in recent years as DNA sequencing technology matures. Modern libraries typically use DNA barcodes to distinguish pooled samples rather than splitting across physical lanes that could suffer from manufacturing defects or pipetting errors. Using barcodes, all samples of a given study can be easily pooled and sequenced together, such that any technical variation is shared across the study. Barcoded libraries can be repeatedly sequenced to achieve greater depth, and the performance of current sequencing instruments is such that flow cell differences do not significantly contribute to batch effects (Tung et al., 2017). The main consideration is to ensure that no samples in the study are grossly under- or over-sequenced in terms of UMIs per cell, reads per cell, or whatever the appropriate metric for the application.
7.2. Ambient RNA
Of all potential sources of batch-related noise in single-cell RNA-seq, contamination of the extracellular medium with dissolved, “ambient” RNA is possibly the most significant. Ambient RNA is released from dying or mechanically lysed cells and becomes uniformly distributed in the sample medium. Following controlled lysis, it is co-barcoded along with the desired intracellular RNA, mixing with the captured cell’s own transcriptome. In an unoptimized workflow, the extent of ambient RNA contamination can vary dramatically across samples, even on the same day and in the hands of a single experimenter. Large, fragile cells with high mRNA content are the most problematic, particularly secretory cell types such as plasma cells, pancreatic acinar cells, and others, depending on the tissue being studied. To avoid ambient RNA contamination, it can be helpful to minimize mechanical stress (e.g. over-pipetting) during sample preparation and to finish with one or more low-speed centrifugal washing steps just prior to loading the single-cell capture device.
With high ambient RNA contamination, cross-sample comparisons become highly batch-confounded, and ambient genes will dominate differential expression tests across all cell types in the study. Informatically minimizing the consequences of ambient RNA is an idiosyncratic challenge that usually requires domain knowledge of the tissues being studied. In droplet-based platforms such as 10X Genomics, the extent of ambient RNA contamination can be estimated by carefully examining the residual gene counts detected in “empty” droplets, but only if this raw data can be accessed. Most publicly available datasets exclude empty droplet information unless raw sequencing data is requested from the original authors. Several statistical and machine learning-based tools have been published that perform subtraction of the estimated ambient RNA fraction (Fleming, Marioni, & Babadi, 2019; Yang et al., 2020; Young & Behjati, 2020), and produce improved differential expression analysis and data integration results. Ambient RNA subtraction methods have proven useful for data integration across large scale tissue atlases assembled under varying conditions in different labs, such as the recently released Fly Cell Atlas (H. Li et al., 2021).
8. Single Nucleus RNA Sequencing (snRNA-seq)
snRNA-seq is an increasingly popular protocol variant that can produce results when preparing a freshly dissociated single cell suspension is non-optimal (Nadelmann et al., 2021; Thibivilliers, Anderson, & Libault, 2020). In cases of fragile cell types, such as neurons, nuclear dissociation is strongly preferred, in order to avoid biases in recovered cell types or fragmentation of cell bodies, since it bypasses standard enzymatic dissociation steps. Perhaps the biggest advantage of working with nuclei, is that it greatly facilitates working with frozen tissues that have not been fixed or cryopreserved as single cells. This allows for easy batching of samples across time or location by flash-freezing small tissue punches at the time of harvest for parallel processing later for snRNA-seq. This advantage has been exploited for a number of large-scale tissue atlas projects (Bakken et al., 2018; Eraslan et al., 2021; Kanton et al., 2019; Lake et al., 2018).
The other key advantage to snRNA-seq is compatibility with additional genomics modalities in high-throughput “multi-omics” applications. After plasma membrane removal, native histone-wrapped genomic DNA is accessible for tagmentation by Tn5 or other enzymatic manipulations. Nuclear isolation also exposes intracellular antigens to barcode-conjugated antibodies, which can be quantified simultaneously with mRNA after light fixation (Katzenelenbogen et al., 2020). All three modalities can even be combined from single nuclei to capture a combined readout of transcriptome, protein expression, and chromatin state (Mimitou et al., 2021; Swanson et al., 2021). It should be noted that some multiomics approaches such as SHARE-seq have demonstrated that nuclear isolation is not a strict requirement, and one can rely on gentle fixation and permeabilization of whole cells to expose chromatin during transposition (Ma et al., 2020).
8.1. General Protocol Considerations for Nuclei
General methods for isolating nuclei prior to single-cell analysis have been extensively described (Krishnaswami et al., 2016; Nadelmann et al., 2021; Nott, Schlachetzki, Fixsen, & Glass, 2021; Santos et al., 2021; Slyper et al., 2020). In general, snRNA-seq requires no substantial change to the chemistry of standard scRNA-seq, aside from adjusting the number of cycles during the cDNA amplification stage to account for the lower mRNA content of nuclei. Even when following published guidelines, it is good practice to thoroughly optimize a nuclear isolation protocol to maximize yield, nuclear integrity (see QC, below), and RNA recovery. Aside from the reduced mRNA yield, another disadvantage is that very few lineage-selective protein markers exist to facilitate sorting or affinity purification of nuclei for cell type enrichment. Thus, enrichment will generally have to be carried out on dissociated, intact cells prior to nuclear isolation and can generally not be achieved with nuclei prepared directly from solid tissue.
8.2. Tissue-Specific Considerations for Nuclei
Like dissociation protocols for standard scRNA-seq, snRNA-seq nuclei isolation should be optimized to address tissue specific challenges like differences in extracellular matrix composition or cell type composition. Here, we will focus on a few common tissue types and the necessary considerations that must be taken to isolate nuclei for snRNA-seq from them.
Brain.
Due to the challenge of dissociating brain tissue into live intact single cells, and because both fresh and frozen brain tissue lends itself well to detergent-based nuclei extraction, many researchers choose snRNA-seq over scRNA-seq. Compared with other organs, brain tissue readily disintegrates in most lysis buffers, and mechanical or enzymatic breakup of the tissue is less important; tissue can generally be disrupted by mild pipetting. The resulting single nuclei suspension will contain debris, which is not compatible with droplet-based snRNA-seq and, therefore, the most critical step is its removal, generally through gradient centrifugation or positive selection of nuclei by FACS (Krishnaswami et al., 2016; Thrupp et al., 2020; Welch et al., 2019). Excessive myelin fragments can also be cleared by commercially available bead-depletion kits (Pennartz, Reiss, Biloune, Hasselmann, & Bosio, 2009; Yamazaki et al., 2021).
Nuclease-rich tissue.
Nuclear RNA-seq of nuclease-rich tissue such as pancreas, present additional challenges due to rapid degradation of exposed RNA not protected by a plasma membrane. In these tissues, FACS upstream of the lysis may be necessary to remove problematic cell types and to clean the preparation of degradative enzymes. Rapid processing under strictly ice-cold conditions in the presence of high concentrations of RNAse inhibitors may also help improve RNA yield.
8.3. Sorting Nuclei
Sorting away the myelin and debris can be an effective one-step strategy to prepare nuclei for snRNA-seq. A wide range of intercalating DNA dyes can clearly mark nuclei versus debris, including DAPI, propidium iodide (PI), 7-AAD, and others. Since nuclei are somewhat hardier to shear stresses than intact cells, smaller sorter nozzles or chip diameters can be exploited to increase sort rates and reduce dead volume in the final sorted sample (Santos et al., 2021). The more concentrated eluate can allow users to skip risky downstream wash steps and sort directly into the sample loading buffer, e.g., for 10X Genomics, without diluting the reaction mixture beyond optimal levels.
Lineage enrichment can also be carried out directly on nuclei, though to a lesser extent than with intact-cell sorting. Some FACS-compatible lineage markers have been widely validated, such as the pan-neuronal marker NeuN (Dammer et al., 2013), while other marker panels to further distinguish microglia and oligodendrocytes have been explored with some success (Nott et al., 2021).
For workflows involving ATAC-seq, it should be noted that some nuclear stains used for sorting, including DAPI and ethidium homodimer, have been reported to interfere with Tn5-mediated transposition (kb.10xgenomics.com - b, n.d.). Careful optimization of both the type of sorting stain and its concentration should be performed prior to a single-cell experiment.
8.4. Disadvantages of snRNA-seq
When choosing to perform snRNA-seq over scRNA-seq there are a few disadvantages to take into consideration. In this section, we will discuss how RNA degradation and the differences between cytoplasmic and nuclear RNA may make snRNA-seq less optimal for certain types of tissues and studies.
Degradation.
Removal of the plasma membrane exposes mRNAs to RNAses that may be present in the tissue homogenate (Slyper et al., 2020), which, in the cases of digestive tissues like the pancreas, can dramatically increase the challenge of recovering intact RNA for sequencing. Nuclei also tend to be more susceptible to aggregation than intact cells, and careful optimization of detergent, salt, and carrier protein concentrations must be performed if working with an unfamiliar tissue type, to prevent total sample loss by catastrophic clumping.
Nuclear vs cytoplasmic RNA.
While a large portion of the cellular mRNA content is contained within the cytoplasm, enough nascent pre-mRNA is present in the nucleus to produce a transcriptome that is largely representative of the full complement of mature mRNAs (Bakken et al., 2018). As expected, nuclear RNA contains a much higher proportion of unspliced pre-mRNA, which due to the degenerate nature of the sequences that often comprise introns, can lead to generally lower mapping rates (Bakken et al., 2018; Broseus & Ritchie, 2020; Thrupp et al., 2020). Importantly, nuclei preparations can be fine-tuned by carefully adjusting detergent conditions to optimize co-purification of mature mRNAs that are stably bound to the outer nuclear membrane or rough ER (Drokhlyansky et al., 2020). Such preparations can increase total mRNA recovery and boost the exon/intron detection ratio, which may better reflect the functional transcriptome of the cells of interest. Despite the advantages some protocol variants may offer, it may be better, in some cases, to match the isolation conditions of a chosen reference study than to improve the results by changing the protocol and introducing systematic challenges for integrated data analysis.
8.5. QC and Counting of Nuclei
High-quality nuclei appear as smooth circles with well-defined borders under high magnification, with no signs of shriveling, fragmentation, or leakage of nuclear contents, the latter as evidenced by a diffuse blur of irregular material around the nuclear periphery. Dyes that stain nucleic acids such as PI and Ethidium Homodimer are effective at staining nuclei for accurate counting, as they do not stain debris. In contrast, Trypan Blue may not provide sufficient contrast between large debris fragments and nuclei for the purposes of automated cell counters. After counting, snRNA-seq is largely the same as scRNA-seq. Some protocols such as DroNc-seq may optimize flow rates and microfluidics specifically for nuclei (Habib et al., 2017). However, commercial microfluidic platforms, including the Chromium scRNA-seq products, are compatible with snRNA-seq without modification and are now used more broadly than those requiring specialized equipment. The main change to the library preparation that may be needed is an increase in the number of cDNA amplification cycles to account for the lower RNA content of nuclei.
9. Sequencing considerations
Once a single-cell library is generated, the question still remains of how to properly sequence it to achieve the desired goals without wasting resources. Since sequencing can account for roughly half of the budget of a single-cell experiment, it is important to economize wherever possible, and use only the minimal sequencing strategy that satisfies the needs of the project. For instance, certain applications will require longer reads to improve mappability or coverage, while others can tolerate shorter reads, freeing up budget for additional depth. Each single-cell methodology also utilizes its own barcode structure, creating a wide variety of required read formats, some more cost-efficient than others. Libraries will also vary in composition, depending on both biological and technical factors, and choosing the appropriate sequencing depth requires knowledge of each. In this section, we will discuss these considerations and provide insight on how to design an effective sequencing strategy.
Single-cell experiments demand a lot of sequencing depth. As studies grow to the million-cell scale and beyond, the only viable sequencing platforms are those capable of delivering tens of billions of sequencing reads at an affordable cost. Illumina instruments are the current standard for the majority of single-cell applications because they yield the highest numbers of high-accuracy reads per unit cost, with the tradeoff that the reads are relatively short compared with competing technologies like Oxford Nanopore (ONT) and Pacific Biosciences (PacBio). The throughput of Illumina sequencers spans a range from 1M to 20B reads per run, and sequencing kits are sold in discrete read length scales ranging from 50 to 600 bp, depending on the instrument. A typical single-cell experiment with thousands of cells will require hundreds of millions to billions of reads to adequately saturate the analysis so, in practice, Illumina’s largest scale instrument families, NextSeq and NovaSeq, are the most common choices. Recently, however, alternative single-cell library construction strategies have begun to emerge to take advantage of the long-read capabilities of ONT and PacBio. Understanding how to properly tailor a sequencing strategy to suit the goals and requirements of the project by selecting the optimal read lengths, sequencing depth, and technology platform is critical to maximizing the return on investment and producing high-quality data.
9.1. How Deeply Should I Sequence?
Planning an appropriate sequencing strategy depends both on the scientific goals of the project, as well as the technical constraints of the single-cell library type. Is the main objective to conduct a survey of cell types across different conditions? Is it to discover new gene expression patterns and cell states within a subtly varying heterogeneous population? To pick out a few rare cells from the tissue milieu? Or to quantify certain lowly-expressed genes? Will this particular library benefit from ultra-deep sequencing, or will it suffer from diminishing returns as money is spent on more reads?
9.1.1. Sequencing saturation.
The first thing to consider when planning a targeted sequencing depth is the expected complexity of the libraries. Even a very low-quality batch of input cDNA can be amplified by PCR to produce enough DNA for sequencing: such libraries, however, are full of duplicate molecules and will typically saturate the detection of novel UMIs very quickly, and over-sequencing would be a waste of resources. Likewise, certain cell types naturally contain fewer mRNA transcripts than others and, thus, can be expected to yield lower total UMI counts and reach saturation more quickly. Library saturation is easily estimated by downsampling using tools such as Picard (Broad Institute, n.d.; Daley & Smith, 2013), and the resulting asymptotic saturation curve can provide a reliable estimate for the recovery rate of new UMIs with additional sequencing (Figure 7). When planning an experiment, it may be beneficial to first examine sequencing saturation curves for similar experiments (both in terms of tissue type and technology platform) using publicly available data. This can give an idea for the expected number of UMIs for a given amount of sequencing, and how to budget accordingly for the appropriate depth. Depending on the single-cell technology used, appropriate sequencing depths can range anywhere from 10,000 to millions of reads per cell.
Figure 7. Sequencing saturation curve.

In any sequencing experiment, the yield of new, non-duplicated molecules follows an asymptotic saturation curve (left). With deeper and deeper sequencing, fewer unique molecules are observed, and the return on the investment drops accordingly (right). Saturation curves can be estimated from an initial round of low-yield sequencing, or by comparing with similar sample and library types, which can be used to guide the choice of final targeted depth.
9.1.2. Sequencing Depth and Analysis Goals.
Frequently, the goal of a single-cell genomics project is to survey the landscape of cell types within a tissue or across conditions. Unsupervised clustering methods are surprisingly robust to data sparsity, and sometimes these goals can be met with relatively modest per-cell sequencing depth. In heterogeneous tissues comprising cells from an assortment of lineages (immune, epithelial, mesenchymal, etc.), adequate cell-type assignment might be achieved with as few as 1,000 UMIs per cell, with diminishing returns in clustering accuracy at higher depths (Figure 8A, B). In contrast, deeper sequencing provides a better return on investment for detecting novel marker genes (Figure 8C). These relationships are strongly dependent upon cell type, both in terms of overall mRNA content and their transcriptional complexity (e.g., secretory cells expressing thousands of copies of only a handful of genes). scATAC-seq and other modalities show similar robustness to downsampling (Fang et al., 2021), suggesting that easier tasks like cell type assignment can be accomplished with relatively sparse sequencing. For scRNA-seq, a convenient optimal depth of one read per protein coding gene in the genome per cell (e.g., approximately 25,000 reads for a typical human cell) has been proposed as a general guideline that covers the most common use cases (M. J. Zhang et al., 2020). In practice, scATAC-seq libraries are generally sequenced to a similar depth.
Figure 8. Depth requirements.
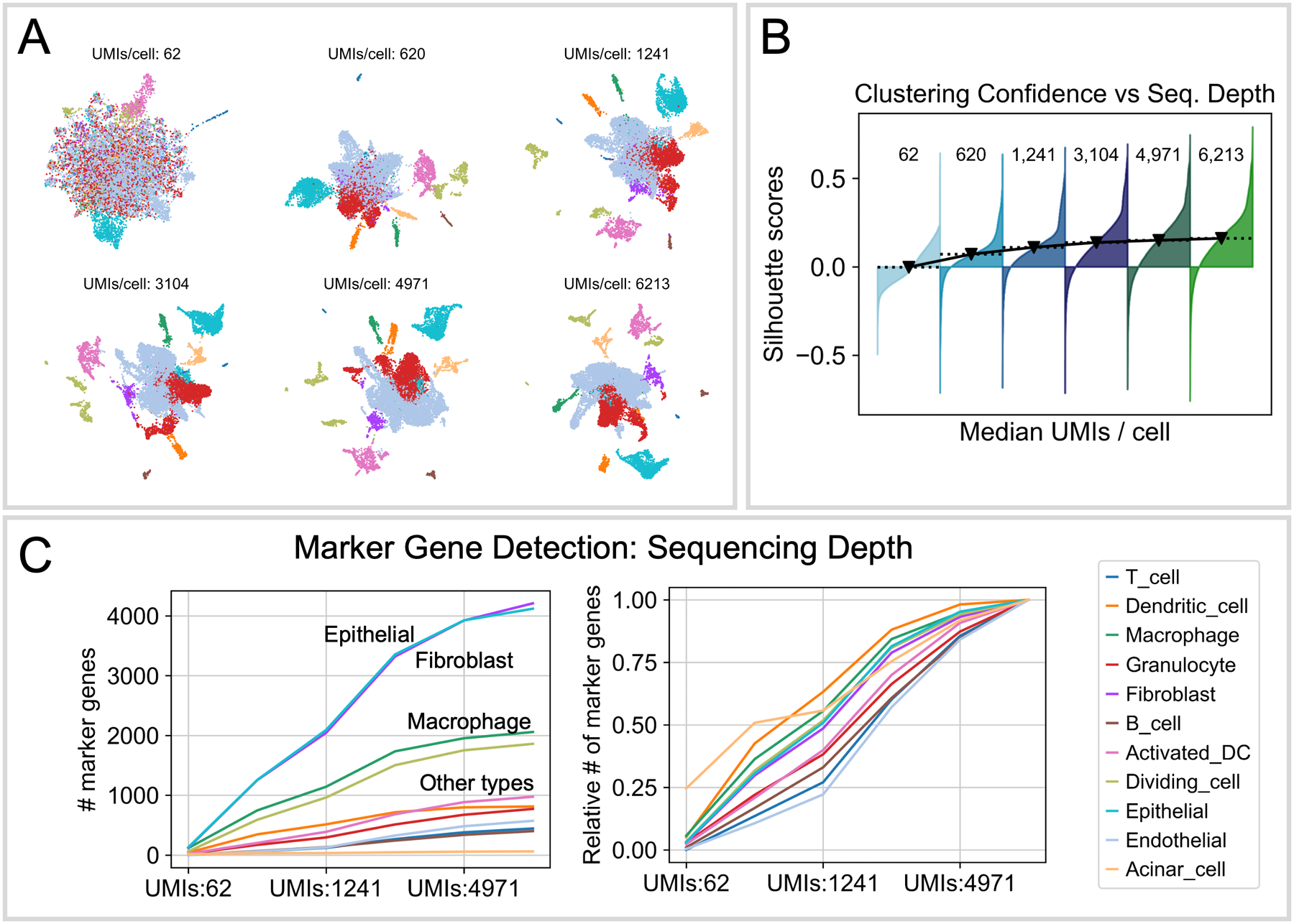
Ideal sequencing depth is dependent upon the project’s goals. For example, clustering and cell type identification tasks in scRNA-seq are relatively robust to shallower sequencing, modeled here by in silico downsampling of a mouse pancreatic tumor dataset (Elyada et al., 2019) to different median numbers of UMIs per cell. Dimensionality reduction by UMAP (A) and clustering confidence (B) demonstrate the impact of sequencing depth on resolving cell types. Distinct clusters (A, colored points) become resolvable with only a few hundred UMIs per cell, with rarer cell types emerging as separate clusters only at higher depths. (B) Unsupervised clustering was run on the downsampled datasets, and inter-cluster silhouette score was calculated as a measure of confidence in cluster assignments. Higher sequencing depth returns only a modest improvement in unsupervised clustering confidence as median UMI counts increase beyond ~2,000/cell. (C) Marker gene detection scales roughly linearly over the full range of subsampled depths. Cell type labels were fixed, and differential expression was performed to detect marker genes across cell types at each subsampled depth. Cell types with high mRNA content, such as epithelial and fibroblast cells, yield more marker genes at low sequencing depth compared with other types (left panel). This phenomenon is typically the result of the fact that a greater proportion of the total UMIs in the dataset come from mRNA-rich cells. Nonetheless, marker gene detection as a function of depth is roughly linear for all cell types, as visualized by normalizing the trend to the maximum number of recovered markers for each type (right panel). This illustrates how sequencing more deeply can help recover more marker genes in cell types with low mRNA content or which comprise only a small proportion of the total library.
If the goal is to capture and quantify weakly expressed genes, then an alternative sequencing approach is required, to increase the sensitivity to these low-abundance molecules. One option could be to simply expend more resources to sequence the entire library as deeply as possible in the hopes of detecting rare transcripts. This may be, however, a doomed endeavor for some experiments, if the genes of interest are below the detection limits of the method being used. Estimates of the mRNA detection efficiency of scRNA-seq platforms vary. For example, the current 10X Genomics chemistry is able to recover only in the range of 10–15% of the mRNA molecules within a given cell (M. J. Zhang et al., 2020), whereas the plate-based Quartz-Seq2 protocol can detect 30% or more (Sasagawa et al., 2018). Given that some mRNAs are present at only a few copies per cell, they may often escape reverse transcription altogether, and no amount of sequencing can be expected to uncover them. Similarly, rare mRNAs could be initially barcoded but subsequently lost or swamped out during PCR and library clean-up steps, making them exceedingly difficult to detect during sequencing.
Rather than wasting resources trying to quantify rare transcripts by brute force using ultra-deep sequencing, it may be preferable to enrich the library for a subset of genes of interest. Reduced-complexity libraries enriched for target genes can be fully saturated with shallower sequencing and still provide valuable, curated information (see below). Targeted gene enrichment can also improve the dynamic range and limit of detection of certain digital counting methods by capturing molecules at an early step in library construction, where fewer unique molecules have been lost due to stochastic processes in the enzymatic workflow. Target gene panel enrichment is emerging as a useful strategy in a growing number of single-cell applications and is discussed in more detail below.
9.1.3. Targeted panel sequencing.
The growing scale of single-cell experiments is a double-edged sword: larger cell numbers fuel more robust discoveries but simultaneously drive up costs associated with sequencing. Unbiased whole-transcriptome libraries include data from tens of thousands of genes, but for many applications, a focused panel of relevant, highly informative genes can effectively capture cell type heterogeneity and changes in gene expression for select pathways of interest. Targeted panels can reduce sequencing costs by as much as 90% and simplify downstream bioinformatic analysis, and have become an important part of the forward-looking strategy for commercial vendors such as 10X Genomics and BD Biosciences.
In scRNA-seq, targeted panel enrichment is generally achieved by one of two methods: gene-specific priming during the RT step, or hybridization capture of amplified cDNA. The BD Rhapsody platform has standardized the gene-specific priming approach, and offers a set of pre-designed panels as well as a design tool for custom gene sets. Oligo-dT priming followed by two rounds of nested PCR with gene-specific panels are used to generate low-background libraries (Fan, Fu, & Fodor, 2015). In contrast, the 10X Genomics solution first carries out unbiased cDNA amplification and standard whole-transcriptome library generation. Biotinylated bait oligos tiling the genes of interest are then hybridized to the library and enriched on streptavidin-coated beads, and the target genes are then amplified with a final PCR step prior to sequencing. A key advantage of this protocol is that since a standard whole-transcriptome library is produced, it also has the option to be fully sequenced or queried with alternative panels at a later date. In contrast, the gene-specific priming approach may offer increased sensitivity and dynamic range, but the practical benefit remains to be rigorously demonstrated.
Reducing sequencing costs and analytical complexity is especially important for functional genomics applications, which often aim to profile phenotypic changes in hundreds or even thousands of genetically perturbed populations in a single experiment (Replogle et al., 2020). Likewise, atlas-making efforts aspiring to the million-cell scale and beyond can leverage targeted panel sequencing to keep costs under control while providing the flexibility for deeper exploration in the future.
9.2. Read Format
Many single-cell libraries have strict length requirements for the barcode and UMI portions of the read, but somewhat looser recommendations about the length of the genomic insert read, provided some minimal mappable length is met. Exploiting knowledge of the sequencing platform and bioinformatic requirements can help squeeze the most out of your sequencing budget.
9.2.1. Kit “Hacking”.
Every Illumina sequencing kit with a given advertised cycle number also contains “extra” reagents to accommodate for different kinds of paired-end barcoding strategies. For example, the NextSeq 500 75-cycle kit is actually packaged with 92 cycles worth of consumables: these can be assigned freely to the cell barcode/UMI and sample index reads as per the required structure of the library (e.g. 28bp and 8bp for current 10X Genomics 3’ Gene Expression chemistries), reserving the remaining cycles (56bp) for the genomic insert read(s). Maximizing reagent utilization in this way can dramatically improve the cost-effectiveness of sequencing kits, provided a suitable run type can be constructed (see below for a discussion of read length considerations).
Table 2 summarizes some common Illumina kits and their actual packaged reagent content, according to the manufacturer’s website: (https://support.illumina.com/bulletins/2016/10/how-many-cycles-of-sbs-chemistry-are-in-my-kit.html):
Table 2. Usable cycle numbers in common Illumina sequencing kits.
The combined lengths of the barcode and genomic portions of a given single-cell library type must fit within the reagent constraints of available sequencing kits. Most Illumina kits include additional cycles to allow for reading barcodes, including the common kit types listed here. Use this information to maximize kit utilization: for instance, a NextSeq2000 “100-cycle” kit can be used to sequence a 10X Genomics 3’ Gene Expression library in the following format: 28-bp UMI/barcode + 10-bp i5 index + 10-bp i7 index + 90-bp gene body = 138 total bases. Exploiting this information can improve mapping rates, variant calling, transcript assembly, or other applications.
| Kit Type | Billion Read Pairs | Kit Size | Max Cycle Num. | |
|---|---|---|---|---|
| NextSeq 500/550 | High Output | 0.4 | 75 | 92 |
| 0.4 | 150 | 168 | ||
| NextSeq 2000 | P2 v3 | 0.4 | 100 | 138 |
| 0.4 | 200 | 238 | ||
| P3 v3 | 1.2 | 50 | 88 | |
| 1.2 | 100 | 138 | ||
| 1.2 | 200 | 238 | ||
| NovaSeq 6000 | SP v1.5 | 0.8 | 100 | 138 |
| 0.8 | 200 | 238 | ||
| S1 v1.5 | 1.6 | 100 | 138 | |
| 1.6 | 200 | 238 | ||
| S2 v1.5 | 4.1 | 100 | 138 | |
| 4.1 | 200 | 238 | ||
| S4 v1.5 | 20 | 200 | 238 | |
9.2.2. Genomic Insert Read Length.
The minimum required read length will depend on the modality, organism, and analysis goals. Longer reads will generally improve mappability, but for counting-based applications, the return on investment may not be justified beyond a certain length. For example, standard digital gene expression libraries are heavily enriched for non-degenerate, easily mappable sequences, and a cost-saving optimum of 50 bp has been recommended for differential expression applications (Chhangawala, Rudy, Mason, & Rosenfeld, 2015). This contrasts with the official recommendation from 10X Genomics of 90 bp for their scRNA-seq libraries. Judiciously opting for a shorter kit size can dramatically save on overall experiment costs, in the right circumstances.
Optimal read lengths can be determined empirically by carrying out a pilot experiment or finding a relevant public dataset that employs longer reads, then trimming these reads in silico to observe the tradeoff in mapping rates for using less expensive run types. For example, Figure 9A (top) illustrates the modest 10% reduction in mapping rate when simulating the step down from longer reads to 50 bp for a publicly available human PBMC dataset. Considering that shorter reads could generate a 50% or greater cost savings, the protocol modification seems justified. Here, the best use of resources may come from investing in additional lanes of shorter-read sequencing to maximize the recovery of unique molecules sequenced per dollar spent. Similarly, scATAC-seq libraries can be very forgiving, suffering virtually no loss in mapping rate when downsampling to paired-end 34bp reads (Figure 9A, bottom).
Figure 9. Read length requirements.
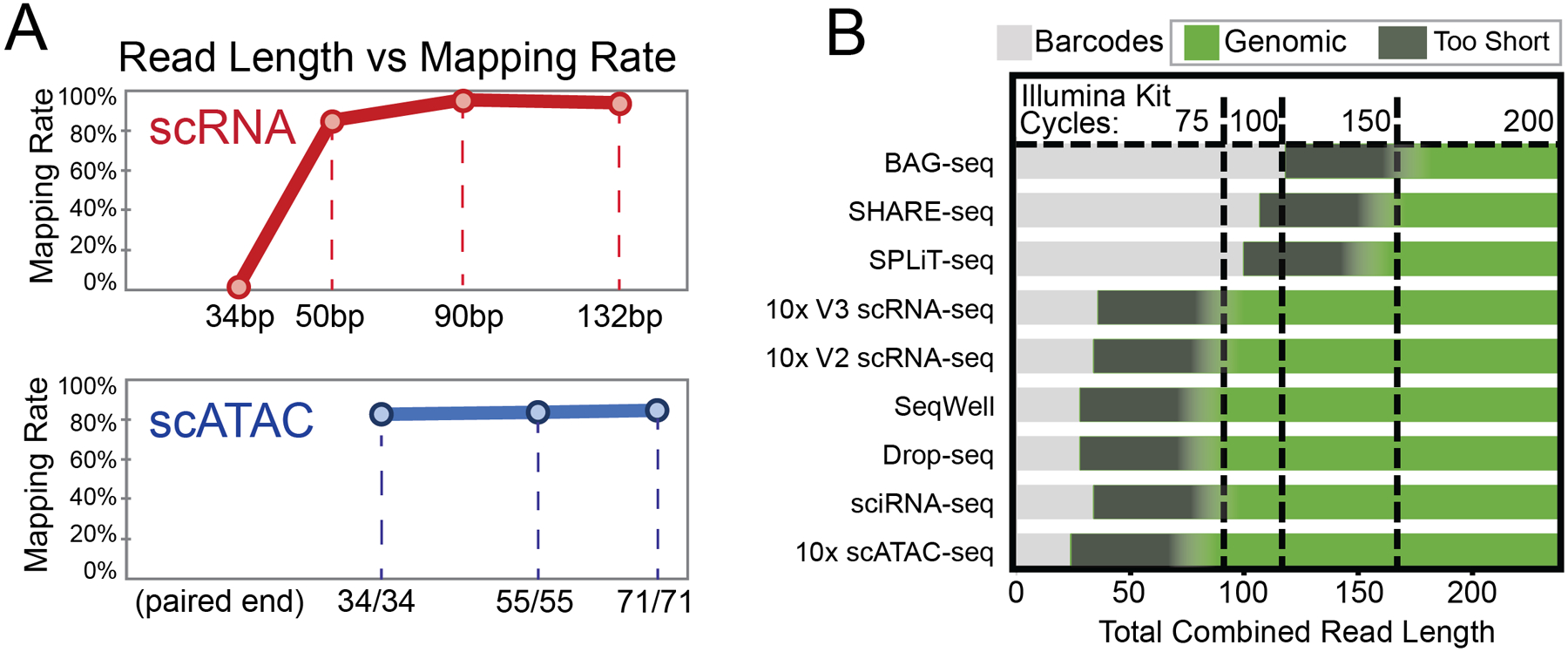
Single-cell modalities require different read lengths for optimal performance. For example, using the 10X Genomics scRNA-seq platform, longer reads return modestly higher mapping rates above a certain minimum threshold length of the gene body read (A, top). The example shown represents human peripheral blood mononuclear cells prepared using 10X Genomics Single Cell 3’ Gene Expression version 3 chemistry and sequenced on an Illumina NextSeq 500 with a 132 bp gene-body read (unpublished data). Reads were trimmed in silico and remapped to assess overall mappability as a function of gene-body read length. Similarly, commonly used read lengths for scATAC-seq are largely indistinguishable in mapping rate (A, bottom). Here, 10X Genomics Single Cell ATAC libraries were created from dissociated mouse pancreatic tumors and sequenced on an Illumina NextSeq 500 with symmetric paired-end reads, informatically trimmed to various lengths (unpublished data). (B) Required read lengths also differ by technology platform as a consequence of their barcode design. Single-cell protocols use a variety of library design strategies, resulting in different required read lengths to cover key library features. For instance, split/pool methods such as BAG-seq, SHARE-seq, and SPLiT-seq generally require 100 or more bases to cover all barcode regions, while droplet methods employ compact barcodes requiring fewer bases, leaving more sequencing reagents available to dedicate to the gene-body or other features in the library amplicon. Combined barcode and minimum mappable genomic read lengths help determine the most efficient kit size to select for sequencing.
9.2.3. Barcode Length Efficiency.
The proportion of sequencing dedicated to barcode regions can vary dramatically depending on the protocol and how the barcodes are synthesized. Split-pool methods that build barcodes by serial addition of hybridizing oligos can have barcodes extending up to 100 bp, while other methods can have barcodes as short as 12–16 bp. Since sequencing cost scales proportionally with read lengths, longer barcodes can significantly drive up the total cost of a single-cell project, which should be accounted for in advance. In order to achieve a minimal mappable genomic read length (e.g. 50 or more high-quality bases), some protocols require kit sizes of 150 cycles or more, while others can fit mappable reads into a 75 cycle kit (Figure 9B).
9.3. Long Read Sequencing for scRNA-seq
The majority of single-cell genomics protocols have been designed to be compatible with and sequenced on an Illumina platform, because of its unmatched throughput and low error rate, which facilitates counting of short barcodes. Digital gene expression methods necessarily sacrifice transcript coverage for quantifiability, but recent advances in long-read sequencing have made full-length, isoform-discriminating cDNA sequencing in single-cell workflows a possibility. Adapting a scRNA-seq library for long-read sequencing is generally trivial: in the case of 10X Genomics, full-length, amplified cDNA is already produced during the workflow, so it is simply a matter of reserving a portion of this material as input for a standard commercial long-read library kit. In some cases, it may be beneficial to lengthen the extension time during cDNA amplification to favor longer cDNAs (Long et al., 2021).
The Oxford Nanopore (ONT) Promethion platform has the capability of sequencing tens of millions of unique, multi-kilobase long molecules per run at a cost comparable to an Illumina flow cell, but with a much higher per-base error rate (Sahlin & Medvedev, 2021). These high error rates greatly hinder the unambiguous assignment of cell barcodes and UMIs, but can be compensated for by sequencing the same library in parallel using high-fidelity Illumina chemistry in order to build a restricted ‘whitelist’ of observed cell barcodes in the experiment. Computational strategies to jointly assign long and short reads to shared cell barcodes have been developed by several groups and used to profile gene expression with isoform specificity from droplet-based single-cell platforms (Gupta et al., 2018; Lebrigand, Magnone, Barbry, & Waldmann, 2020; Long et al., 2021; Singh et al., 2019).
In contrast to ONT instruments, the Pacific Biosciences (PacBio) Single Molecule Real-Time (SMRT) platform can achieve extremely low error rates, but generates 5- to 10-fold fewer unique reads. As with ONT, PacBio-based sparse isoform sequencing can be used to augment Illumina-based gene expression from the same sample (Gupta et al., 2018). The accuracy of PacBio has been leveraged for other single-cell applications such as characterizing genetic and transcriptional variation in influenza (Russell, Elshina, Kowalsky, Velthuis, & Bloom, 2019), and for lineage tracing of hematopoietic stem cells (Pei et al., 2020). Recently, a novel library concatemerization strategy has been reported that enables multiple cDNA molecules to be linked together and sequenced with high fidelity from one long molecule, increasing the throughput of PacBio-based scRNA isoform sequencing severalfold (Zheng et al., 2020).
10. Concluding Remarks
The landscape of technology platforms for single-cell genomics is evolving rapidly. Continuous improvements in the scalability, sensitivity, and costs of easy-to-use commercial products have lowered the barrier to entry for many laboratories. While it is easier than ever before to generate complex single-cell datasets, it is critical for inexperienced investigators to research and adhere to the current best practices to avoid common pitfalls, unnecessary failures, and confounded data. Each technology platform will have its own unique set of strengths and limitations, but we have attempted to highlight here the general principles of good experimental design that apply to the broadest possible set of study types. Considering the current trend toward multi-modal workflows, new guidelines will undoubtedly emerge to address the unique challenges of combining different chemistries. Before embarking on any expensive single-cell project, it is critical to adopt a ‘measure twice, cut once’ mindset and carefully plan each stage of the study, from statistical power analysis and batch design to sample preparation and sequencing strategy, to best ensure high quality data at its conclusion.
ACKNOWLEDGEMENTS:
The authors are supported in part by the National Institutes of Health Cancer Center Support Grant (CCSG) 5P30CA045508 and NIH grant 1R01CA249002. Figures 1, 2, 3, and 4 were created with Biorender.com.
Footnotes
CONFLICT OF INTEREST STATEMENT:
The authors declare no conflicts of interest.
Contributor Information
Claire Regan, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, USA..
Jonathan Preall, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, USA..
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study
LITERATURE CITED:
- Adam M, Potter AS, & Potter SS (2017). Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: A molecular atlas of kidney development. Development (Cambridge, England), 144(19), 3625–3632. 10.1242/dev.151142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge S, & Teichmann SA (2020). Single cell transcriptomics comes of age. Nature Communications, 11(1), 4307. 10.1038/s41467-020-18158-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaway KC, Gabitto MI, Wapinski O, Saldi G, Wang C-Y, Bandler RC, … Fishell G (2021). Genetic and epigenetic coordination of cortical interneuron development. Nature, 597(7878), 693–697. 10.1038/s41586-021-03933-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alles J, Karaiskos N, Praktiknjo SD, Grosswendt S, Wahle P, Ruffault P-L, … Rajewsky N (2017a). Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC Biology, 15(1), 44. 10.1186/s12915-017-0383-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alles J, Karaiskos N, Praktiknjo SD, Grosswendt S, Wahle P, Ruffault P-L, … Rajewsky N (2017b). Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC Biology, 15(1), 44. 10.1186/s12915-017-0383-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar M, Sharma E, Li S, Bryer C, Cubitt L, Broxholme J, … Bowden R (2018). A practical solution for preserving single cells for RNA sequencing. Scientific Reports, 8(1), 2151. 10.1038/s41598-018-20372-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayhan F, Douglas C, Lega BC, & Konopka G (2021). Nuclei isolation from surgically resected human hippocampus. STAR Protocols, 2(4), 100844. 10.1016/j.xpro.2021.100844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bageritz J, Krausse N, Yousefian S, Leible S, Valentini E, & Boutros M (2021). Glyoxal as alternative fixative for single cell RNA sequencing (preprint). Genomics. 10.1101/2021.06.06.447272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, … Tasic B (2018). Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE, 13(12), e0209648. 10.1371/journal.pone.0209648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baslan T, Kendall J, Rodgers L, Cox H, Riggs M, Stepansky A, … Hicks J (2012). Genome-wide copy number analysis of single cells. Nature Protocols, 7(6), 1024–1041. 10.1038/nprot.2012.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute. (Accessed: 2022/04/21; version 2.27.1). “Picard Tools.” Broad Institute, GitHub repository. http://broadinstitute.github.io/picard/. [Google Scholar]
- Broseus L, & Ritchie W (2020). Challenges in detecting and quantifying intron retention from next generation sequencing data. Computational and Structural Biotechnology Journal, 18, 501–508. 10.1016/j.csbj.2020.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, … Shendure J (2017). Comprehensive single cell transcriptional profiling of a multicellular organism. Science (New York, N.Y.), 357(6352), 661–667. 10.1126/science.aam8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Abu Zaid MI, Reiter JL, Czader M, Wang L, McGuire P, … Liu Y (2021). Cryopreservation Preserves Cell-Type Composition and Gene Expression Profiles in Bone Marrow Aspirates From Multiple Myeloma Patients. Frontiers in Genetics, 12, 583. 10.3389/fgene.2021.663487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Liao Y, Zhang G, Sun Z, Yang L, Fang X, … Guo G (2021). High-throughput Microwell-seq 2.0 profiles massively multiplexed chemical perturbation. Cell Discovery, 7(1), 1–4. 10.1038/s41421-021-00333-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Pei Y, He L, Peng G, Reinius B, Tam PPL, … Deng Q (2019). Single-Cell RNA-Seq Reveals Cellular Heterogeneity of Pluripotency Transition and X Chromosome Dynamics during Early Mouse Development. Cell Reports, 26(10), 2593–2607.e3. 10.1016/j.celrep.2019.02.031 [DOI] [PubMed] [Google Scholar]
- Chhangawala S, Rudy G, Mason CE, & Rosenfeld JA (2015). The impact of read length on quantification of differentially expressed genes and splice junction detection. Genome Biology, 16(1), 131. 10.1186/s13059-015-0697-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium TTS, & Quake SR (2021). The Tabula Sapiens: A single cell transcriptomic atlas of multiple organs from individual human donors (p. 2021.07.19.452956). 10.1101/2021.07.19.452956 [DOI]
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, … Shendure J (2015). Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science, 348(6237), 910–914. 10.1126/science.aab1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley T, & Smith AD (2013). Predicting the molecular complexity of sequencing libraries. Nature Methods, 10(4), 325–327. 10.1038/nmeth.2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammer EB, Duong DM, Diner I, Gearing M, Feng Y, Lah JJ, … Seyfried NT (2013). A Neuron Enriched Nuclear Proteome Isolated from Human Brain. Journal of Proteome Research, 12(7), 3193–3206. 10.1021/pr400246t [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rop FV, Ismail JN, Bravo González-Blas C, Hulselmans GJ, Flerin CC, Janssens J, … Aerts S (2022). Hydrop enables droplet-based single-cell ATAC-seq and single-cell RNA-seq using dissolvable hydrogel beads. ELife, 11, e73971. 10.7554/eLife.73971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deerhake ME, Reyes EY, Xu-Vanpala S, & Shinohara ML (2021). Single-Cell Transcriptional Heterogeneity of Neutrophils During Acute Pulmonary Cryptococcus neoformans Infection. Frontiers in Immunology, 12. Retrieved from https://www.frontiersin.org/article/10.3389/fimmu.2021.670574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del-Aguila JL, Li Z, Dube U, Mihindukulasuriya KA, Budde JP, Fernandez MV, … Harari O (2019). A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimer’s Research & Therapy, 11(1), 71. 10.1186/s13195-019-0524-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delley CL, & Abate AR (2021). Modular barcode beads for microfluidic single cell genomics. Scientific Reports, 11(1), 10857. 10.1038/s41598-021-90255-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisenko E, Guo BB, Jones M, Hou R, de Kock L, Lassmann T, … Forrest ARR (2020). Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biology, 21(1), 130. 10.1186/s13059-020-02048-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, Hill AJ, Daza RM, Cao J, O’Day DR, Pliner HA, … Shendure J (2020). A human cell atlas of fetal chromatin accessibility. Science (New York, N.Y.), 370(6518), eaba7612. 10.1126/science.aba7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, … Turley SJ (2020). Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discovery, 10(2), 232–253. 10.1158/2159-8290.CD-19-0644 [DOI] [PubMed] [Google Scholar]
- Dong J, Hu Y, Fan X, Wu X, Mao Y, Hu B, … Tang F (2018). Single-cell RNA-seq analysis unveils a prevalent epithelial/mesenchymal hybrid state during mouse organogenesis. Genome Biology, 19(1), 31. 10.1186/s13059-018-1416-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrity MW, Alexandre CM, Hamm MO, Vigil A-L, Fields S, Queitsch C, & Cuperus JT (2021). The regulatory landscape of Arabidopsis thaliana roots at single-cell resolution. Nature Communications, 12(1), 3334. 10.1038/s41467-021-23675-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drokhlyansky E, Smillie CS, Van Wittenberghe N, Ericsson M, Griffin GK, Eraslan G, … Regev A (2020). The Human and Mouse Enteric Nervous System at Single-Cell Resolution. Cell, 182(6), 1606–1622.e23. 10.1016/j.cell.2020.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, … Tuveson DA (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discovery, 9(8), 1102–1123. 10.1158/2159-8290.CD-19-0094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, … Coleman P (1992). Analysis of gene expression in single live neurons. Proceedings of the National Academy of Sciences of the United States of America, 89(7), 3010–3014. 10.1073/pnas.89.7.3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eraslan G, Drokhlyansky E, Anand S, Subramanian A, Fiskin E, Slyper M, … Regev A (2021). Single-nucleus cross-tissue molecular reference maps to decipher disease gene function (p. 2021.07.19.452954). 10.1101/2021.07.19.452954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Savill JS, Haslett C, Bratton DL, Doherty DE, Campbell PA, & Henson PM (1992). Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. Journal of Immunology (Baltimore, Md.: 1950), 149(12), 4029–4035. [PubMed] [Google Scholar]
- Fan HC, Fu GK, & Fodor SPA (2015). Combinatorial labeling of single cells for gene expression cytometry. Science, 347(6222), 1258367. 10.1126/science.1258367 [DOI] [PubMed] [Google Scholar]
- Fan X, Tang D, Liao Y, Li P, Zhang Y, Wang M, … Tang F (2020). Single-cell RNA-seq analysis of mouse preimplantation embryos by third-generation sequencing. PLoS Biology, 18(12), e3001017. 10.1371/journal.pbio.3001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, … Ren B (2021). Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nature Communications, 12(1), 1337. 10.1038/s41467-021-21583-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B, Meier A, Dehne A, Salhotra A, Tran TA, Neumann S, … Gentile L (2018). A complete workflow for the differentiation and the dissociation of hiPSC-derived cardiospheres. Stem Cell Research, 32, 65–72. 10.1016/j.scr.2018.08.015 [DOI] [PubMed] [Google Scholar]
- Fleming SJ, Marioni JC, & Babadi M (2019). CellBender remove-background: A deep generative model for unsupervised removal of background noise from scRNA-seq datasets (p. 791699). 10.1101/791699 [DOI] [Google Scholar]
- Frantz C, Stewart KM, & Weaver VM (2010). The extracellular matrix at a glance. Journal of Cell Science, 123(Pt 24), 4195–4200. 10.1242/jcs.023820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu GK, Hu J, Wang P-H, & Fodor SPA (2011). Counting individual DNA molecules by the stochastic attachment of diverse labels. Proceedings of the National Academy of Sciences of the United States of America, 108(22), 9026–9031. 10.1073/pnas.1017621108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Wu P-H, Beane T, Zamore PD, & Weng Z (2018). Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genomics, 19(1), 531. 10.1186/s12864-018-4933-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara S, Baek S, Varticovski L, Kim S, & Hager GL (2019). High Quality ATAC-Seq Data Recovered from Cryopreserved Breast Cell Lines and Tissue. Scientific Reports, 9(1), 516. 10.1038/s41598-018-36927-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granja JM, Klemm S, McGinnis LM, Kathiria AS, Mezger A, Corces MR, … Greenleaf WJ (2019). Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nature Biotechnology, 37(12), 1458–1465. 10.1038/s41587-019-0332-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaumet-Adkins A, Rodríguez-Esteban G, Mereu E, Mendez-Lago M, Jaitin DA, Villanueva A, … Heyn H (2017). Single-cell transcriptome conservation in cryopreserved cells and tissues. Genome Biology, 18(1), 45. 10.1186/s13059-017-1171-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldner IH, Golomb SM, Wang Q, Wang E, & Zhang S (2021). Isolation of mouse brain-infiltrating leukocytes for single cell profiling of epitopes and transcriptomes. STAR Protocols, 2(2), 100537. 10.1016/j.xpro.2021.100537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta I, Collier PG, Haase B, Mahfouz A, Joglekar A, Floyd T, … Tilgner HU (2018). Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nature Biotechnology, 36(12), 1197–1202. 10.1038/nbt.4259 [DOI] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, … Regev A (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods, 14(10), 955–958. 10.1038/nmeth.4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, & Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biology, 20(1), 296. 10.1186/s13059-019-1874-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, … Guo G (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell, 172(5), 1091–1107.e17. 10.1016/j.cell.2018.02.001 [DOI] [PubMed] [Google Scholar]
- Hanamsagar R, Reizis T, Chamberlain M, Marcus R, Nestle FO, de Rinaldis E, & Savova V (2020). An optimized workflow for single-cell transcriptomics and repertoire profiling of purified lymphocytes from clinical samples. Scientific Reports, 10(1), 2219. 10.1038/s41598-020-58939-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton H, Talman AM, Knights A, Imaz M, Gaffney DJ, Durbin R, … Lawniczak MKN (2020). Souporcell: Robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nature Methods, 17(6), 615–620. 10.1038/s41592-020-0820-1 [DOI] [PubMed] [Google Scholar]
- How many cycles of SBS chemistry are in my kit? (n.d.). Retrieved December 1, 2021, from https://support.illumina.com/bulletins/2016/10/how-many-cycles-of-sbs-chemistry-are-in-my-kit.html [Google Scholar]
- Huang Y, McCarthy DJ, & Stegle O (2019). Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biology, 20(1), 273. 10.1186/s13059-019-1865-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jariani A, Vermeersch L, Cerulus B, Perez-Samper G, Voordeckers K, Van Brussel T, … Verstrepen KJ (2020). A new protocol for single-cell RNA-seq reveals stochastic gene expression during lag phase in budding yeast. ELife, 9, e55320. 10.7554/eLife.55320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Baptiste K, McFaline-Figueroa JL, Alexandre CM, Dorrity MW, Saunders L, Bubb KL, … Cuperus JT (2019). Dynamics of Gene Expression in Single Root Cells of Arabidopsis thaliana. The Plant Cell, 31(5), 993–1011. 10.1105/tpc.18.00785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- kb.10xgenomics.com - a. Can I sort nuclei for Single Cell ATAC sequencing or Single Cell Multiome ATAC + GEX?[Internet]. 10X Genomics, Inc.; [Accessed 2022 May 17]. Available from: https://kb.10xgenomics.com/hc/en-us/articles/360027640311 [Google Scholar]
- kb.10xgenomics.com - b. What are the best practices for flow sorting cells for 10x Genomics assays? [Internet]. 10X Genomics, Inc.; [Accessed 2022 May 17]. Available from: https://kb.10xgenomics.com/hc/en-us/articles/360048826911 [Google Scholar]
- Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, … Ye CJ (2018). Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nature Biotechnology, 36(1), 89–94. 10.1038/nbt.4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís-Calleja F, … Camp JG (2019). Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature, 574(7778), 418–422. 10.1038/s41586-019-1654-9 [DOI] [PubMed] [Google Scholar]
- Kaplan N, Wang J, Wray B, Patel P, Yang W, Peng H, & Lavker RM (2019). Single-Cell RNA Transcriptome Helps Define the Limbal/Corneal Epithelial Stem/Early Transit Amplifying Cells and How Autophagy Affects This Population. Investigative Ophthalmology & Visual Science, 60(10), 3570–3583. 10.1167/iovs.19-27656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenelenbogen Y, Sheban F, Yalin A, Yofe I, Svetlichnyy D, Jaitin DA, … Amit I (2020). Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell, 182(4), 872–885.e19. 10.1016/j.cell.2020.06.032 [DOI] [PubMed] [Google Scholar]
- Kielberg V fishersci.com [Internet] Thermo Fisher, Inc.; [Updated 2010; Accessed 2022 May 17]. Available from: https://assets.fishersci.com/TFS-Assets/LSG/Application-Notes/D19575.pdf [Google Scholar]
- Koopman G, Reutelingsperger C, Kuijten G, Keehnen R, Pals S, & van Oers M (1994). Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood, 84(5), 1415–1420. 10.1182/blood.V84.5.1415.1415 [DOI] [PubMed] [Google Scholar]
- Krishnaswami SR, Grindberg RV, Novotny M, Venepally P, Lacar B, Bhutani K, … Lasken RS (2016). Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nature Protocols, 11(3), 499–524. 10.1038/nprot.2016.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, … Zhang K (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nature Biotechnology, 36(1), 70–80. 10.1038/nbt.4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrigand K, Magnone V, Barbry P, & Waldmann R (2020). High throughput error corrected Nanopore single cell transcriptome sequencing. Nature Communications, 11(1), 4025. 10.1038/s41467-020-17800-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK (2018). Sex as an important biological variable in biomedical research. BMB Reports, 51(4), 167–173. 10.5483/BMBRep.2018.51.4.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, & Wigler M (2014). Facilitated sequence counting and assembly by template mutagenesis. Proceedings of the National Academy of Sciences of the United States of America, 111(43), E4632–E4637. 10.1073/pnas.1416204111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Janssens J, Waegeneer MD, Kolluru SS, Davie K, Gardeux V, … Aerts S (2021). Fly Cell Atlas: A single-cell transcriptomic atlas of the adult fruit fly (p. 2021.07.04.451050). 10.1101/2021.07.04.451050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Kendall J, Park S, Wang Z, Alexander J, Moffitt A, … Wigler M (2020). Copolymerization of single-cell nucleic acids into balls of acrylamide gel. Genome Research, 30(1), 49–61. 10.1101/gr.253047.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim B, Lin Y, & Navin N (2020). Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell, 37(4), 456–470. 10.1016/j.ccell.2020.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y, Liu Z, Jia J, Mo W, Fang L, Lu D, … Zhai J (2021). FlsnRNA-seq: Protoplasting-free full-length single-nucleus RNA profiling in plants. Genome Biology, 22(1), 66. 10.1186/s13059-021-02288-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Ramos CM, Chen C-Y, Kuijjer ML, Paulson JN, Sonawane AR, Fagny M, … DeMeo DL (2020). Sex Differences in Gene Expression and Regulatory Networks across 29 Human Tissues. Cell Reports, 31(12), 107795. 10.1016/j.celrep.2020.107795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Anido CB, Vatén A, Smoot NK, Sharma N, Guo V, Gong Y, … Bergmann DC (2021). Single-cell resolution of lineage trajectories in the Arabidopsis stomatal lineage and developing leaf. Developmental Cell, 56(7), 1043–1055.e4. 10.1016/j.devcel.2021.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, & Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15(12), 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, … Buenrostro JD (2020). Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell, 183(4), 1103–1116.e20. 10.1016/j.cell.2020.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado L, Relaix F, & Mourikis P (2021). Stress relief: Emerging methods to mitigate dissociation-induced artefacts. Trends in Cell Biology, S0962–8924(21)00096–9. 10.1016/j.tcb.2021.05.004 [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, … McCarroll SA (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell, 161(5), 1202–1214. 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madissoon E, Wilbrey-Clark A, Miragaia RJ, Saeb-Parsy K, Mahbubani KT, Georgakopoulos N, … Meyer KB (2019). ScRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biology, 21, 1. 10.1186/s13059-019-1906-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra M, Nagy C, Chawla A, Wang YC, Nascimento C, Suderman M, … Turecki G (2021). Extraction of nuclei from archived postmortem tissues for single-nucleus sequencing applications. Nature Protocols, 16(6), 2788–2801. 10.1038/s41596-021-00514-4 [DOI] [PubMed] [Google Scholar]
- Marand AP, Chen Z, Gallavotti A, & Schmitz RJ (2021). A cis-regulatory atlas in maize at single-cell resolution. Cell, 184(11), 3041–3055.e21. 10.1016/j.cell.2021.04.014 [DOI] [PubMed] [Google Scholar]
- Mascotti K, McCullough J, & Burger SR (2000). HPC viability measurement: Trypan blue versus acridine orange and propidium iodide. Transfusion, 40(6), 693–696. 10.1046/j.1537-2995.2000.40060693.x [DOI] [PubMed] [Google Scholar]
- McGinnis CS, Patterson DM, Winkler J, Conrad DN, Hein MY, Srivastava V, … Gartner ZJ (2019). MULTI-seq: Sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nature Methods, 16(7), 619–626. 10.1038/s41592-019-0433-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, … Izar B (2021). A molecular single-cell lung atlas of lethal COVID-19. Nature, 595(7865), 114–119. 10.1038/s41586-021-03569-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu E, Lafzi A, Moutinho C, Ziegenhain C, McCarthy DJ, Álvarez-Varela A, … Heyn H (2020). Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nature Biotechnology, 38(6), 747–755. 10.1038/s41587-020-0469-4 [DOI] [PubMed] [Google Scholar]
- Miller K (2016, June 6). Red Blood Cell Lysis Protocols. Retrieved January 7, 2022, from https://www.protocols.io/view/Red-Blood-Cell-Lysis-Protocols-e3dbgi6 [Google Scholar]
- Mimitou EP, Lareau CA, Chen KY, Zorzetto-Fernandes AL, Hao Y, Takeshima Y, … Smibert P (2021). Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nature Biotechnology, 39(10), 1246–1258. 10.1038/s41587-021-00927-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minussi DC, Nicholson MD, Ye H, Davis A, Wang K, Baker T, … Navin NE (2021). Breast tumours maintain a reservoir of subclonal diversity during expansion. Nature, 592(7853), 302–308. 10.1038/s41586-021-03357-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsey B, Niu M, Dyavar SR, Fletcher CV, Lamberty BG, Emanuel K, … Fox HS (2021). Cryopreservation of microglia enables single-cell RNA sequencing with minimal effects on disease-related gene expression patterns. IScience, 24(4), 102357. 10.1016/j.isci.2021.102357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadelmann ER, Gorham JM, Reichart D, Delaughter DM, Wakimoto H, Lindberg EL, … Seidman JG (2021). Isolation of Nuclei from Mammalian Cells and Tissues for Single-Nucleus Molecular Profiling. Current Protocols, 1(5), e132. 10.1002/cpz1.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, … Wigler M (2011). Tumour evolution inferred by single-cell sequencing. Nature, 472(7341), 90–94. 10.1038/nature09807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noland TL, & Mohammed GH (1997). Fluorescein diacetate as a viability stain for tree roots and seeds. New Forests, 14(3), 221–232. 10.1023/A:1006561829931 [DOI] [Google Scholar]
- Nott A, Schlachetzki JCM, Fixsen BR, & Glass CK (2021). Nuclei isolation of multiple brain cell types for omics interrogation. Nature Protocols, 16(3), 1629–1646. 10.1038/s41596-020-00472-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Flanagan CH, Campbell KR, Zhang AW, Kabeer F, Lim JLP, Biele J, … Aparicio S (2019). Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biology, 20(1), 210. 10.1186/s13059-019-1830-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavel A, Sandra L, Jaroslav T, Mikael K, & Radek S (2019). Preparation of single-cell suspension from mouse breast cancer focusing on preservation of original cell state information and cell type composition (p. 824714). 10.1101/824714 [DOI] [Google Scholar]
- Pei W, Shang F, Wang X, Fanti A-K, Greco A, Busch K, … Rodewald H-R (2020). Resolving Fates and Single-Cell Transcriptomes of Hematopoietic Stem Cell Clones by PolyloxExpress Barcoding. Cell Stem Cell, 27(3), 383–395.e8. 10.1016/j.stem.2020.07.018 [DOI] [PubMed] [Google Scholar]
- Pennartz S, Reiss S, Biloune R, Hasselmann D, & Bosio A (2009). Generation of Single-Cell Suspensions from Mouse Neural Tissue. Journal of Visualized Experiments : JoVE, (29), 1267. 10.3791/1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, … Klappenbach JA (2017). Multiplexed quantification of proteins and transcripts in single cells. Nature Biotechnology, 35(10), 936–939. 10.1038/nbt.3973 [DOI] [PubMed] [Google Scholar]
- Phan HV, van Gent M, Drayman N, Basu A, Gack MU, & Tay S (2021). High-throughput RNA sequencing of paraformaldehyde-fixed single cells. Nature Communications, 12(1), 5636. 10.1038/s41467-021-25871-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, & Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nature Protocols, 9(1), 171–181. 10.1038/nprot.2014.006 [DOI] [PubMed] [Google Scholar]
- Potter AS, & Steven Potter S (2019). Dissociation of Tissues for Single-Cell Analysis. Methods in Molecular Biology (Clifton, N.J.), 1926, 55–62. 10.1007/978-1-4939-9021-4_5 [DOI] [PubMed] [Google Scholar]
- Qi X, Yu Y, Sun R, Huang J, Liu L, Yang Y, … Sun B (2021). Identification and characterization of neutrophil heterogeneity in sepsis. Critical Care, 25(1), 50. 10.1186/s13054-021-03481-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, … Human Cell Atlas Meeting Participants. (2017). The Human Cell Atlas. ELife, 6, e27041. 10.7554/eLife.27041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard A, & Asosingh K (2019). Best Practices for Preparing a Single Cell Suspension from Solid Tissues for Flow Cytometry. Cytometry. Part A: The Journal of the International Society for Analytical Cytology, 95(2), 219–226. 10.1002/cyto.a.23690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Replogle JM, Norman TM, Xu A, Hussmann JA, Chen J, Cogan JZ, … Adamson B (2020). Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nature Biotechnology, 38(8), 954–961. 10.1038/s41587-020-0470-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, & Smyth GK (2010). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26(1), 139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocks D, Jaric I, Tesfa L, Greally JM, Suzuki M, & Kundakovic M (2021). Cell type-specific chromatin accessibility analysis in the mouse and human brain. Epigenetics, 0(0), 1–18. 10.1080/15592294.2021.1896983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozhnikov A, Ramkumar P, Shah K, Bedi R, Kato S, & Escola GS (2021). Demuxalot: Scaled up genetic demultiplexing for single-cell sequencing (p. 2021.05.22.443646). 10.1101/2021.05.22.443646 [DOI] [Google Scholar]
- Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, … Seelig G (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science (New York, N.Y.), 360(6385), 176–182. 10.1126/science.aam8999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AB, Elshina E, Kowalsky JR, Velthuis A. J. W. te, & Bloom JD (2019). Single-Cell Virus Sequencing of Influenza Infections That Trigger Innate Immunity. Journal of Virology, 93(14). 10.1128/JVI.00500-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KH, Huang L, Kang HM, & Schiefelbein J (2019). Single-Cell RNA Sequencing Resolves Molecular Relationships Among Individual Plant Cells. Plant Physiology, 179(4), 1444–1456. 10.1104/pp.18.01482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlin K, & Medvedev P (2021). Error correction enables use of Oxford Nanopore technology for reference-free transcriptome analysis. Nature Communications, 12(1), 2. 10.1038/s41467-020-20340-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MD, Gioftsidi S, Backer S, Machado L, Relaix F, Maire P, & Mourikis P (2021). Extraction and sequencing of single nuclei from murine skeletal muscles. STAR Protocols, 2(3), 100694. 10.1016/j.xpro.2021.100694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasagawa Y, Danno H, Takada H, Ebisawa M, Tanaka K, Hayashi T, … Nikaido I (2018). Quartz-Seq2: A high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads. Genome Biology, 19(1), 29. 10.1186/s13059-018-1407-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid KT, Höllbacher B, Cruceanu C, Böttcher A, Lickert H, Binder EB, … Heinig M (2021). ScPower accelerates and optimizes the design of multi-sample single cell transcriptomic studies. Nature Communications, 12(1), 6625. 10.1038/s41467-021-26779-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan BS, & Lefrançois L (2012). Isolation of Mouse Lymphocytes from Small Intestine Tissues. Current Protocols in Immunology, 99(1), 3.19.1–3.19.11. 10.1002/0471142735.im0319s99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Al-Eryani G, Carswell S, Ferguson JM, Blackburn J, Barton K, … Swarbrick A (2019). High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nature Communications, 10(1), 3120. 10.1038/s41467-019-11049-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, … Regev A (2020). A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nature Medicine, 26(5), 792–802. 10.1038/s41591-020-0844-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaethling JM, Na Y-J, Lee J, Ulyanova AV, Baltuch GH, Bell TJ, … Eberwine JH (2017). Primary Cell Culture of Live Neurosurgically Resected Aged Adult Human Brain Cells and Single Cell Transcriptomics. Cell Reports, 18(3), 791–803. 10.1016/j.celrep.2016.12.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squair JW, Gautier M, Kathe C, Anderson MA, James ND, Hutson TH, … Courtine G (2021). Confronting false discoveries in single-cell differential expression. Nature Communications, 12(1), 5692. 10.1038/s41467-021-25960-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenn KS, Link R, Moellmann G, Madri J, & Kuklinska E (1989). Dispase, a neutral protease from Bacillus polymyxa, is a powerful fibronectinase and type IV collagenase. The Journal of Investigative Dermatology, 93(2), 287–290. 10.1111/1523-1747.ep12277593 [DOI] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, … Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nature Methods, 14(9), 865–868. 10.1038/nmeth.4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM, … Satija R (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biology, 19(1), 224. 10.1186/s13059-018-1603-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su K, Wu Z, & Wu H (2020). Simulation, power evaluation and sample size recommendation for single-cell RNA-seq. Bioinformatics, 36(19), 4860–4868. 10.1093/bioinformatics/btaa607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Song D, Li WV, & Li JJ (2021). scDesign2: A transparent simulator that generates high-fidelity single-cell gene expression count data with gene correlations captured. Genome Biology, 22(1), 163. 10.1186/s13059-021-02367-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- support.illumina.com. How many cycles of SBS chemistry are in my kit? [Internet]. Illumina, Inc.; [Updated 2021 Mar 17; Accessed 2022 May 17]. Available from: https://support.illumina.com/bulletins/2016/10/how-many-cycles-of-sbs-chemistry-are-in-my-kit.html [Google Scholar]
- Svensson V, Beltrame E. da V., & Pachter L (2019). Quantifying the tradeoff between sequencing depth and cell number in single-cell RNA-seq (p. 762773). 10.1101/762773 [DOI] [Google Scholar]
- Swann JC, Reynolds JJ, & Galloway WA (1981). Zinc metalloenzyme properties of active and latent collagenase from rabbit bone. Biochemical Journal, 195(1), 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson E, Lord C, Reading J, Heubeck AT, Genge PC, Thomson Z, … Skene PJ (2021). Simultaneous trimodal single-cell measurement of transcripts, epitopes, and chromatin accessibility using TEA-seq. ELife, 10, e63632. 10.7554/eLife.63632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabula Muris Consortium. (2020). A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature, 583(7817), 590–595. 10.1038/s41586-020-2496-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis, … Principal investigators. (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature, 562(7727), 367–372. 10.1038/s41586-018-0590-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, & Regev A (2017). Single cell genomics: From phenomenology to mechanism. Nature, 541(7637), 331–338. 10.1038/nature21350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, … Surani MA (2009). MRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods, 6(5), 377–382. 10.1038/nmeth.1315 [DOI] [PubMed] [Google Scholar]
- Tatsuoka H, Sakamoto S, Yabe D, Kabai R, Kato U, Okumura T, … Inagaki N (2020). Single-Cell Transcriptome Analysis Dissects the Replicating Process of Pancreatic Beta Cells in Partial Pancreatectomy Model. IScience, 23(12), 101774. 10.1016/j.isci.2020.101774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattikota SG, Cho B, Liu Y, Hu Y, Barrera V, Steinbaugh MJ, … Perrimon N (2020). A single-cell survey of Drosophila blood. ELife, 9, e54818. 10.7554/eLife.54818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibivilliers S, Anderson D, & Libault M (2020). Isolation of Plant Root Nuclei for Single Cell RNA Sequencing. Current Protocols in Plant Biology, 5(4), e20120. 10.1002/cppb.20120 [DOI] [PubMed] [Google Scholar]
- Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, … Fiers M (2020). Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Reports, 32(13), 108189. 10.1016/j.celrep.2020.108189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevino AE, Müller F, Andersen J, Sundaram L, Kathiria A, Shcherbina A, … Greenleaf WJ (2021). Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell, 184(19), 5053–5069.e23. 10.1016/j.cell.2021.07.039 [DOI] [PubMed] [Google Scholar]
- Troiano NW, Ciovacco WA, & Kacena MA (2009). The Effects of Fixation and Dehydration on the Histological Quality of Undecalcified Murine Bone Specimens Embedded in Methylmethacrylate. Journal of Histotechnology, 32(1), 27–31. 10.1179/his.2009.32.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji K, Ojima M, Otabe K, Horie M, Koga H, Sekiya I, & Muneta T (2017). Effects of Different Cell-Detaching Methods on the Viability and Cell Surface Antigen Expression of Synovial Mesenchymal Stem Cells. Cell Transplantation, 26(6), 1089–1102. 10.3727/096368917X694831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung P-Y, Blischak JD, Hsiao CJ, Knowles DA, Burnett JE, Pritchard JK, & Gilad Y (2017). Batch effects and the effective design of single-cell gene expression studies. Scientific Reports, 7, 39921. 10.1038/srep39921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson-Maduro J, Baron CS, … van Oudenaarden A (2017). Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nature Methods, 14(10), 935–936. 10.1038/nmeth.4437 [DOI] [PubMed] [Google Scholar]
- Vieth B, Ziegenhain C, Parekh S, Enard W, & Hellmann I (2017). powsimR: Power analysis for bulk and single cell RNA-seq experiments. Bioinformatics (Oxford, England), 33(21), 3486–3488. 10.1093/bioinformatics/btx435 [DOI] [PubMed] [Google Scholar]
- Volovitz I, Shapira N, Ezer H, Gafni A, Lustgarten M, Alter T, … Ram Z (2016). A non-aggressive, highly efficient, enzymatic method for dissociation of human brain-tumors and brain-tissues to viable single-cells. BMC Neuroscience, 17(1), 30. 10.1186/s12868-016-0262-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waise S, Parker R, Rose-Zerilli MJJ, Layfield DM, Wood O, West J, … Hanley CJ (2019). An optimised tissue disaggregation and data processing pipeline for characterising fibroblast phenotypes using single-cell RNA sequencing. Scientific Reports, 9(1), 9580. 10.1038/s41598-019-45842-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Xiao Z, Yan Y, Ye R, Hu M, Bai S, … Navin NE (2021). Simple oligonucleotide-based multiplexing of single-cell chromatin accessibility. Molecular Cell, 81(20), 4319–4332.e10. 10.1016/j.molcel.2021.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Xiong H, Ai S, Yu X, Liu Y, Zhang J, & He A (2019). CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Molecular Cell, 76(1), 206–216.e7. 10.1016/j.molcel.2019.07.015 [DOI] [PubMed] [Google Scholar]
- Wang W, Penland L, Gokce O, Croote D, & Quake SR (2018). High fidelity hypothermic preservation of primary tissues in organ transplant preservative for single cell transcriptome analysis. BMC Genomics, 19, 140. 10.1186/s12864-018-4512-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, & Macosko EZ (2019). Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell, 177(7), 1873–1887.e17. 10.1016/j.cell.2019.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohnhaas CT, Leparc GG, Fernandez-Albert F, Kind D, Gantner F, Viollet C, … Baum P (2019). DMSO cryopreservation is the method of choice to preserve cells for droplet-based single-cell RNA sequencing. Scientific Reports, 9(1), 10699. 10.1038/s41598-019-46932-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woitowich NC, Beery A, & Woodruff T (2020). A 10-year follow-up study of sex inclusion in the biological sciences. ELife, 9, e56344. 10.7554/eLife.56344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Malone AF, Donnelly EL, Kirita Y, Uchimura K, Ramakrishnan SM, … Humphreys BD (2018). Single-Cell Transcriptomics of a Human Kidney Allograft Biopsy Specimen Defines a Diverse Inflammatory Response. Journal of the American Society of Nephrology: JASN, 29(8), 2069–2080. 10.1681/ASN.2018020125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SZ, Roden DL, Al-Eryani G, Bartonicek N, Harvey K, Cazet AS, … Swarbrick A (2021). Cryopreservation of human cancers conserves tumour heterogeneity for single-cell multi-omics analysis. Genome Medicine, 13(1), 1–17. 10.1186/s13073-021-00885-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Shi Q, Wu P, Zhang X, Kambara H, Su J, … Luo HR (2020). Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nature Immunology, 21(9), 1119–1133. 10.1038/s41590-020-0736-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Falconer C, Nguyen Q, Crawford J, McKinnon BD, Mortlock S, … Coin LJM (2019). Genotype-free demultiplexing of pooled single-cell RNA-seq. Genome Biology, 20(1), 290. 10.1186/s13059-019-1852-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Crow M, Rice BR, Li F, Harris B, Liu L, … Jackson D (2021). Single-cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery. Developmental Cell, 56(4), 557–568.e6. 10.1016/j.devcel.2020.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki A, Shue F, Yamazaki Y, Martens YA, Bu G, & Liu C-C (2021). Preparation of single cell suspensions enriched in mouse brain vascular cells for single-cell RNA sequencing. STAR Protocols, 2(3), 100715. 10.1016/j.xpro.2021.100715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Corbett SE, Koga Y, Wang Z, Johnson WE, Yajima M, & Campbell JD (2020). Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biology, 21(1), 57. 10.1186/s13059-020-1950-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Chen L, Zhang D, Zhao P, Chen Z, Qin E-Q, … Yang P (2021). Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nature Cell Biology, 23(6), 620–630. 10.1038/s41556-021-00690-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, & Behjati S (2020). SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience, 9(12). 10.1093/gigascience/giaa151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Hocker JD, Miller M, Hou X, Chiou J, Poirion OB, … Ren B (2021). A single-cell atlas of chromatin accessibility in the human genome. Cell, S0092–8674(21)01279–4. 10.1016/j.cell.2021.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MJ, Ntranos V, & Tse D (2020). Determining sequencing depth in a single-cell RNA-seq experiment. Nature Communications, 11(1), 774. 10.1038/s41467-020-14482-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y-F, Chen Z-C, Shi Z-X, Hu K-H, Zhong J-Y, Wang C-X, … Xiao C-L (2020). HIT-scISOseq: High-throughput and High-accuracy Single-cell Full-length Isoform Sequencing for Corneal Epithelium (preprint). Genomics. 10.1101/2020.07.27.222349 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study