

Abstract
The adenosine deaminase acting on RNA (ADAR) enzymes are essential for neuronal function and innate immune control. ADAR1 RNA editing prevents aberrant activation of antiviral dsRNA sensors through editing of long, double-stranded RNAs (dsRNAs). In this review, we focus on the ADAR2 proteins involved in the efficient, highly site-specific RNA editing to recode open reading frames first discovered in the GRIA2 transcript encoding the key GLUA2 subunit of AMPA receptors; ADAR1 proteins also edit many of these sites. We summarize the history of ADAR2 protein research and give an up-to-date review of ADAR2 structural studies, human ADARBI (ADAR2) mutants causing severe infant seizures, and mouse disease models. Structural studies on ADARs and their RNA substrates facilitate current efforts to develop ADAR RNA editing gene therapy to edit disease-causing single nucleotide polymorphisms (SNPs). Artificial ADAR guide RNAs are being developed to retarget ADAR RNA editing to new target transcripts in order to correct SNP mutations in them at the RNA level. Site-specific RNA editing has been expanded to recode hundreds of sites in CNS transcripts in Drosophila and cephalopods. In Drosophila and C. elegans, ADAR RNA editing also suppresses responses to self dsRNA.
Keywords: ADAR, ADARB1, dsRNA, recoding RNA editing, neurons
INTRODUCTION
The adenosine deaminase acting on RNA (ADAR) enzymes convert adenosine to inosine in double-stranded (ds)RNA (for reviews, see Heraud-Farlow and Walkley 2020; Erdmann et al. 2021). In recent years, ADAR1, which is one of the two enzymatically active members of the ADAR family in mammals, has received the lion's share of attention (for reviews, see Quin et al. 2021; Baker and Slack 2022; Song et al. 2022). This is understandable considering that ADAR1 has been demonstrated to be essential for the innate immune response by editing endogenous dsRNA (Mannion et al. 2014; Liddicoat et al. 2015). The innate immune sensors recognize the edited, inosine-containing dsRNAs as being of cellular origin, and not from a pathogen, thereby preventing aberrant activation of innate immune responses. This property of ADAR1 has attracted attention, especially in the field of cancer therapeutics, as knocking down ADAR1 in cell lines has been shown to increase the efficacy of immune checkpoint inhibitors such as α-PD1 antibodies (Ishizuka et al. 2019). ADAR1 can also perform site-specific RNA editing, and many specific sites are edited by both ADAR1 and ADAR2. However, unlike ADAR2, there is no site-specific position where editing by ADAR1 is essential; ADAR1 appears to play a backup role to ADAR2. ADAR2 is an essential RNA editing enzyme whose biological function is vital and is the focus of this review. We will highlight the structure of ADAR2, its cellular localization, the proteins that have been identified that interact with it, human mutations in the ADARB1 gene encoding ADAR2, transcripts that are edited by ADAR2, the evolutionarily conserved function of ADAR2 in vertebrates and invertebrates and progress toward ADAR RNA editing gene therapy.
HISTORICAL PERSPECTIVE
5-methylcytosine, discovered in Mycobacterium tuberculosis DNA hydrolysates in 1925 and confirmed by paper chromatography in 1950 (Wyatt 1950), was the first example of a modified canonical nucleoside (Johnson and Coghill 1925). The first modified canonical RNA nucleoside was detected in 1951 as a novel component in calf liver RNA hydrolysate (Cohn and Volkin 1951) and was later identified as 5-ribosyluracil, which is also known as pseudouridine (Cohn 1959).
In the late 1980s, an “unwinding” activity was observed in RNA antisense experiments performed in Xenopus oocytes and extracts; the dsRNA generated by pairing of the antisense RNA to its target transcript appeared to be unwound when electrophoresed on a native DNA gel (Bass and Weintraub 1987; Rebagliati and Melton 1987). At this time many groups were interested in identifying RNA helicases involved in splicing, so this “unwinding activity” was thought to be a candidate. However, further research demonstrated that this unwinding activity resulted from the deamination of adenosine to inosine in dsRNA (Bass and Weintraub 1988). Inosine-uracil base pairs are weaker than adenosine-uracil base pairs; thus, when edited dsRNA is electrophoresed on a native gel, the base-pairing is no longer perfect and it appears unwound.
In the early 1990s, ADAR1 was initially purified to homogeneity (Hough and Bass 1994; Kim et al. 1994a; O'Connell and Keller 1994) and the encoding ADAR gene was subsequently cloned by different groups (Kim et al. 1994b; O'Connell et al. 1995; Patterson and Samuel 1995). The assay for the purification was the in vitro conversion of adenosine to inosine in long dsRNA which had been transcribed in vitro with α-p32-ATP, so that adenosine phosphates were radioactively labeled (Hough and Bass 1994; Kim et al. 1994a; O'Connell and Keller 1994). The protein extract was incubated with dsRNA, and afterwards the dsRNA was digested with P1 nuclease and the products separated by thin layer chromatography (TLC). Adenosine phosphate can easily be distinguished from inosine phosphate by this method. However, there is one caveat: this editing activity assay used to purify ADAR1 is not specific for ADAR1.
At the time there was only one transcript that was known to be edited; the Seeburg group in Heidelberg had found site-specific editing in the GRIA2 transcript encoding the glutamate receptor subunit 2 (GLUA2) at a position where a glutamine (Q) codon was converted to an arginine (R) codon by RNA editing (Fig. 1; Higuchi et al. 1993). The amino acid at this position in GLUA2 is critical, as it is located in the channel pore of the tetrameric α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, and the Q to R conversion greatly reduces the calcium permeability of AMPA receptors containing the GLUA2 subunit. Arginine at this position inserts a polar group into the central ion channel of the AMPA receptor, thus preventing calcium ions from entering (this editing event is discussed in greater detail later). Thus, when ADAR1 was purified it was assumed it would be trivial to demonstrate its ability to edit the Q/R site in the GRIA2 transcript. When it did not, it was presumed that an essential cofactor was required. This hypothesis was proven incorrect because the activity required to edit the GRIA2 Q/R site had a smaller molecular weight than ADAR1, implying that there was another enzyme responsible (Maas et al. 1996). The ADARB1 gene encoding ADAR2 was cloned after a short expressed-sequence tag similar to, but not identical to, ADAR1 was identified and used to screen a cDNA library (Melcher et al. 1996b). ADAR2 was also subsequently purified from HeLa nuclear extract (O'Connell et al. 1997). An overlooked detail is that ADAR1 and ADAR2 purified from tissues behave differently from when they are overexpressed. ADAR2 is relatively easy to overexpress and stable when purified; however, when endogenous ADAR2 is purified the activity is highly labile (O'Connell et al. 1997). ADAR1 on the other hand is stable when purified from tissue but more difficult to overexpress in sufficient quantity, in particular the ADAR1 p150 isoform.
FIGURE 1.

The consequences of GRIA2 Q/R site editing by ADAR2. The editing of GRIA2 pre-mRNA at the Q/R site affects the properties of the resultant GLUA2 protein, which assembles with other subunits to form AMPA receptors. The edited GRIA2 codes for GLUA2 protein with arginine in the pore-forming region, which regulates receptor assembly and blocks Ca2+ entry through AMPA receptors. Unedited GLUA2 protein has glutamine in the pore-forming region, and it assembles to form Ca2+-permeable AMPA receptors (CP-AMPARs). Aberrantly increased production of unedited GLUA2 and the subsequent increase in Ca2+-permeable CP-AMPARs leads to epilepsy in mice and humans.
Thus, it was partly due to the ease of expressing and working with ADAR2 and its ability to perform site-specific editing of transcripts encoding proteins expressed in the nervous system that, originally, most of the research on ADARs focused on ADAR2. Many researchers could not see the relevance of promiscuous ADAR1 editing of Alu-length dsRNA hairpins in transcripts, despite the great abundance of this editing (Ulbricht and Emeson 2014). This editing occurs when two Alu elements are embedded close to each other in transcribed regions, usually in introns or 3′-UTRs, in opposite orientations so that they form Alu-length dsRNA hairpins within the longer transcripts. It was incorrectly assumed that this editing of Alu dsRNA hairpin-containing transcripts was simply a by-product of site-specific editing, despite the extensive work by the groups of Levanon and Eisenberg who demonstrated how prevalent it is (for review, see Eisenberg and Levanon 2018).
ISOFORMS OF ADAR2 PROTEINS EXPRESSED IN VERTEBRATES AND DROSOPHILA
The human ADARB1 gene encoding ADAR2 is located at chromosome 21 band q22.3 and spans approximately 152 kb (Melcher et al. 1996a). There are two major isoforms of ADAR2 protein: ADAR2S (or ADAR2a) and ADAR2L (or ADAR2b) (Gerber et al. 1997). These two isoforms have the same domain structure that is found in almost all ADAR proteins, with two or more amino-terminal dsRNA binding domains (dsRBDs) and a carboxy-terminal deaminase domain (Fig. 2). The longer human ADAR2L isoform is generated by inclusion of an alternatively spliced exon of 40 amino acids that evolved from an Alu J subfamily retrotransposon exonized in the reverse orientation within the deaminase domain (Gerber et al. 1997; Lai et al. 1997). It is uncertain if this insertion in the deaminase domain of ADAR2L has any major biological role; however, it is situated in the 5′ RNA-binding loop of the deaminase domain that is disordered in the RNA-free state (Macbeth et al. 2005) and which becomes ordered upon binding to dsRNA substrates (Matthews et al. 2016). This 5′ RNA-binding loop partly determines the slightly different substrate specificities of ADAR1 and ADAR2 deaminase domains; both have different 5′ binding loops that interact with the edited strand of the dsRNA substrate at phosphates one turn 5′ of the edited adenosine. The 5′ binding loop of ADAR2 creates more interactions with the dsRNA, hence favoring editing sites with longer duplexes 5′ of the edited A (Wang and Beal 2016; Wang et al. 2018). Thus, this insertion may influence the editing activity and possibly even the substrate specificity of ADAR2L, and it has been reported that ADAR2L has lower enzymatic activity than ADAR2S (Gerber et al. 1997; Lai et al. 1997).
FIGURE 2.

The domain structure of major ADAR2 protein isoforms. Human ADAR2 has two major isoforms: ADAR2a (ADAR2S) and ADAR2b (ADAR2L). Both isoforms have two amino-terminal dsRBDs (pink) and a carboxy-terminal deaminase domain (light yellow), as well as an NLS (lilac) at the amino terminus. The longer isoform ADAR2b (ADAR2L) has an extra Alu-derived insert (light blue) in the deaminase domain. ADAR2 has further isoforms differing at the carboxyl terminus, with either a long (blue) or short (yellow; C-ter. S) terminus that is generated by alternative splicing. The adult Drosophila ADAR is the ortholog of mammalian ADAR2, with a slightly shorter deaminase domain.
Rats similarly generate two Adar2 isoforms. In contrast, the two rat isoforms of Adar2 protein display the same enzymatic activities on some substrates, whereas Adar2L was slightly less active on other substrates (Filippini et al. 2018). The longer rat Adar2 isoform does not have the same evolutionary origin as the primate ADAR2L (Slavov and Gardiner 2002), and it adds only ten additional amino acids, but it occurs at the same position within the deaminase domain 5′ RNA-binding loop, implying some functional significance. There is no sequence homology at the RNA or protein level between the rodent insertion, which is generated from an alternative 5′ splice site resulting in an extension of an exon (Slavov and Gardiner 2002; Filippini et al. 2018), and that in primates.
There is an additional human isoform with a shorter deaminase domain carboxyl terminus due to alternative splicing of the last exon (either with or without the inserted Alu J sequence) (Lai et al. 1997). The shorter carboxyl terminus isoform lacks the final 29 amino acids, has very low editing activity on dsRNA, and is unable to edit the GRIA2 Q/R site. Surprisingly, this isoform is expressed at a 3:2 ratio to the longer isoform in both HeLa and HEK 293 cell lines, and this may help explain the anomaly whereby ADAR2 is expressed but not very active in these cell lines.
Another ADAR2 isoform conserved across vertebrate species has an extended amino terminus of 49 amino acids (Maas and Gommans 2009b). In humans, this isoform has the highest expression in the cerebellum and likely arose due to alternative promoter usage. It encodes a sequence motif related to the R-domain of ADAR3, which is a single-stranded RNA binding domain. When comparing its editing activity in HeLa and HEK293 cells to the major ADAR2 isoform, no difference in editing activity was observed.
A very short amino-terminally truncated Adar2 protein is generated in rats by Adar2 editing of the Adarb1 transcript to create a new splice 3′ acceptor site leading to an alternative splicing event resulting in the insertion of 47 nt. This self-editing of Adarb1 pre-mRNA does not lead to production of a functional Adar2 protein isoform, and it may be a mechanism for auto-regulatory negative feedback on Adar2 levels in rodents (Rueter et al. 1999; Feng et al. 2006). As the level of this self-editing event is much lower in humans than in rodents, it is not clear how relevant this auto-regulation is in humans (Rueter et al. 1999; Slavov and Gardiner 2002).
Drosophila Adar shows an interesting parallel to the alternative amino acid sequences in the 5′ RNA-binding loops of the vertebrate proteins. The Drosophila Adar transcript undergoes editing to change a residue in the 5′ RNA-binding loop from a conserved serine (S) to a glycine (G) (Palladino et al. 2000a; Keegan et al. 2005). The residue changed by editing in Drosophila Adar is located at the start of the unstructured residues of the 5′ RNA-binding loop where the insertions occur in the vertebrate ADAR2 proteins. Editing at this Adar S/G site increases at metamorphosis when the larval brain grows to form the adult brain; editing at this site is absent in the embryo and reaches approximately 40% in adult flies. The genome-encoded Adar S isoform is more active than the edited Adar G isoform; therefore, self-editing regulates the enzymatic activity of the Adar protein (Keegan et al. 2005), or retargets it (Savva et al. 2012b, 2013).
ADAR2 BINDS TO dsRNA AS A SURPRISING ASYMMETRIC DEAMINASE DOMAIN DIMER
When the crystal structure of the ADAR2 adenosine deaminase domain was originally determined, a highly negatively charged inositol-hexakisphosphate (IP6) was discovered buried within a large protein cavity lined with positively charged lysines (Macbeth et al. 2005). The bound IP6 molecule communicates allosterically with the deaminase active site and IP6 is required for enzymatic activity. The IP6 is not exchangeable in ADAR2 and is inserted during protein synthesis. The presence of IP6 remains mysterious, and it is due to the requirement for IP6 that active ADAR proteins cannot be obtained by expression in E. coli, which does not produce IP6; initially active ADARs were expressed in and purified from yeasts.
The structures of ADAR2 dsRNA-binding domains and their complexes with dsRNA were determined by NMR analyses (Stefl et al. 2010). The main remaining requirement was to determine the orientation of the deaminase domain on dsRNA, how it accesses the edited adenosine and how it interacts with the two dsRBDs, especially dsRBDII, which also binds close to the edited adenosine. It was known that ADARs position a dsRBD on the 3′ side of where the deaminase domain binds and it could have been expected that this would be dsRBDII of the same ADAR2 monomer that edits the target adenosine (Yi-Brunozzi et al. 2001).
Recently, a structure of an ADAR2 deaminase domain-dsRBD 2 complex has been obtained which shows that the asymmetric footprint of ADAR2 covering the edited A and extending 3′ of it is due to binding of an asymmetric ADAR2 deaminase domain dimer to dsRNA (Fig. 3; Thuy-Boun et al. 2020). The first deaminase domain (shown in red in Fig. 3) binds dsRNA and performs base flipping and adenosine deamination; surprisingly, the dsRBD 2 of this monomer is not resolved in the structure. The noncatalytic deaminase domain from the second ADAR2 monomer (in green), interacts with a dimerization α helix on the side of the catalytic deaminase domain. The dsRNA binding face of the second deaminase domain is blocked by dimerization; another part of this deaminase domain is still near the dsRNA but just barely contacts it. Instead, the second deaminase domain holds its own dsRBD 2 (shown in orange) by the side, perfectly positioning it to contact dsRNA in the way dsRBDs usually do, without much or any interaction of this dsRBD 2 with the catalytic deaminase domain.
FIGURE 3.
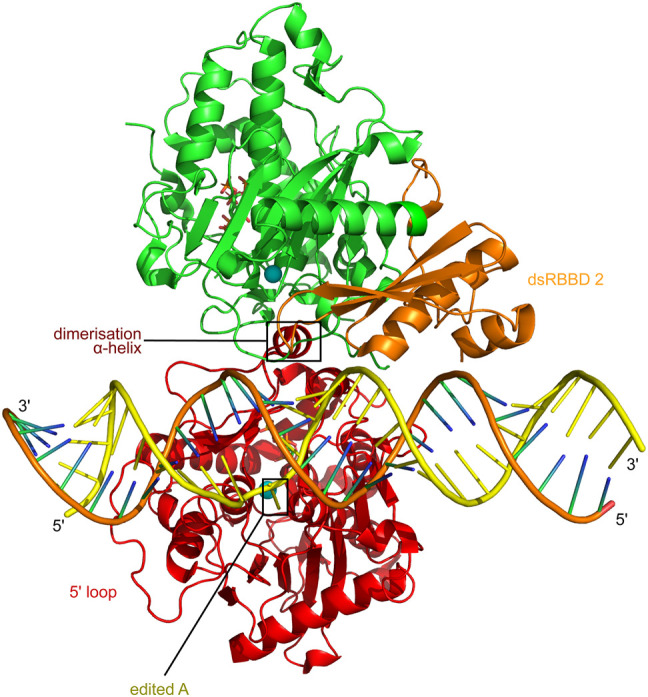
ADAR2 recognizes the editing site as an asymmetric deaminase domain dimer with one positioned dsRBD 2. The ADAR2 catalytic deaminase domain in red is shown behind the dsRNA in this view of the ADAR2 deaminase domain plus dsRBD 2 protein complex with a GLI1 substrate RNA containing 8-azanebularine (8-AZ) at the edited A (Thuy-Boun et al. 2020). The dsRBD 2 of the catalytic deaminase monomer is not resolved in the structure. The adenosine-analog, 8-AN, editing target base, is on the yellow edited strand where the phosphate backbone is slightly kinked, and the 8-AN base is seen flipped back out through the dsRNA minor groove and down towards the blue sphere of the catalytic site zinc atom. A short dimerization helix on the catalytic deaminase domain holds the second, noncatalytic deaminase domain, in green. This second, noncatalytic deaminase domain then positions its associated dsRBDII, shown in orange, for normal dsRBD binding to the dsRNA, without contacting the catalytic deaminase domain.
The final asymmetric dimer structure suggests that expressing a catalytically active ADAR2 deaminase domain without dsRBDs in the presence of full-length catalytically inactive ADAR2 would allow editing. If this is not the case, then the dsRBDs on both monomers may participate in steps involved in forming the final complex. Studies on the GRIA2 R/G RNA substrate showed that dsRBDs alone alter dsRNA structure at the edited A and 3′ to it (Yi-Brunozzi et al. 2001), before deaminase domain binding. DsRBDs could alter dsRNA structure at the target adenosine to facilitate deaminase domain recruitment. The dsRBD2 from the catalytic monomer of ADAR2 (Thuy-Boun et al. 2020) may also have been held by the side to its deaminase domain before dsRNA binding and may depart to allow the catalytic deaminase domain to bind to the dsRNA. Drosophila Adar requires the amino terminus and two subunits with functional dsRBDs for binding to a dsRNA substrate and then for editing activity to occur in S2 cells (Gallo et al. 2003), interpreted as reflecting dimer formation. On the other hand, interactions of the amino terminus of ADAR2 with other parts of the protein were interpreted as intramolecular dynamic control of ADAR activity without any dimer formation (Macbeth et al. 2004); ultimately, both types of process may occur; dsRBDs may act partly as grapples to grab and hold dsRNAs to allow other protein domains to engage with them more accurately and with somewhat more sequence-specificity.
ADAR2 ACTIVITY IS GENERALLY HIGHER IN PRIMATES THAN IN MICE AND IS MUCH HIGHER IN SOME TISSUES IN HUMANS
Human ADAR2 performs site-specific editing, recoding codons to modulate the properties of the encoded proteins (Tan et al. 2017). This site-specific editing can attain a high level and can be even 100% efficient (for reviews, see Pullirsch and Jantsch 2010; Holmgren and Rosenthal 2015), whereas ADAR1 performs promiscuous editing of Alu dsRNA hairpins embedded in longer transcripts, which is generally low efficiency, <1% (Bazak et al. 2014). The first important factor affecting Adar2 expression is strong differences between mammalian species. Comparisons of editing efficiencies at conserved RNA editing sites demonstrated that the primary factor determining RNA editing levels is species rather than the type of tissue (Tan et al. 2017), that is, primates and most mammals have higher editing levels than rodents in many tissues. This difference correlates with editing that is also more widespread across the transcriptome in primates, largely due to ongoing Alu and other repetitive element expansions; rodents are exceptional among mammals in having lower Adar editing levels and much less ongoing expansion of repetitive elements.
There are also differences in tissue-specific expression between rodents and humans; mouse Adar2 has the highest expression in the brain, whereas in humans, expression is also high in arteries, lungs and bladder as well as in the brain. As most studies were performed in mice, it came as a surprise when it was found that in humans ADAR2 expression and activity is ten times higher in arteries than in the brain. A major transcript that is edited in arteries is Flna encoding the actin crosslinking protein Filamin A (FLNA), which is expressed mainly in the vascular system (Jain et al. 2018). Cardiovascular tissues from patients with cardiovascular disorders showed reduced FLNA editing. Flna transgenic mice unable to edit the Flna transcript have increased vascular contraction, elevated blood pressure, arterial remodeling, and left ventricular wall thickening that develops into left ventricular hypertrophy and cardiac remodeling.
Various transcription factors have been shown to regulate Adarb1 expression in rats and mice. Editing of the Gria2 Q/R site is disrupted by transient forebrain ischemia in rats, and Adar2 protein levels in the ischemic rat brain can be restored by expression of constitutively active Creb1 protein (Peng et al. 2006). In the mouse liver, the Clock-Arntl complex involved in circadian transcription of genes can increase Adarb1 mRNA expression. The first intron of the mouse Adarb1 gene contains two E-box sequences that are recognized by the Clock-Arntl complex, which regulates the circadian changes in the level of the Adar2 protein (Terajima et al. 2017). Nutritional and metabolic cues regulate Adar2 expression in mouse pancreatic islet cells by activation of the JNK1-c-Jun pathway acting through an AP-1 binding site in the Adarb1 promoter (Gan et al. 2006; Yang et al. 2012). In humans, an androgen response element is present in the promoter of the ADARB1 gene, and mRNA expression was repressed when the androgen receptor bound to this response element in bladder cancer cell lines (Chen et al. 2020). Induction of DNA damage in human osteosarcoma and lymphoblastoid cell lines revealed that p53 induces ADAR2 expression by binding to a response element near the ADARB1 transcription start site (Bandele et al. 2011).
SUBCELLULAR LOCALIZATION AND POST-TRANSLATIONAL MODIFICATIONS OF ADAR2
ADAR2 is a nuclear localized protein; two amino-terminal NLS sequences are recognized by importin α4 (KPNA3) (Maas and Gommans 2009a; Behm et al. 2017). ADAR2 localizes to both the nucleoplasm and to nucleoli, where it binds to rRNAs (Desterro et al. 2003; Sansam et al. 2003). ADAR2 mutant proteins delocalize to the nucleoplasm from the nucleoli if they lack the ability to bind dsRNA. Inosine has not been found in rRNA despite ADAR2 having editing activity in the nucleoli (Vitali et al. 2005). ADAR2 migrates from the nucleoli into the nucleoplasm when a dsRNA substrate is overexpressed, suggesting that ADAR2 is sequestered in the nucleoli by binding rRNA, thus regulating its editing activity by decreasing its access to dsRNA substrates (Desterro et al. 2003).
ADAR2 is regulated by post-translational modifications. Pin1, the peptidyl-prolyl cis/trans isomerase, isomerizes proline at position 33; this is required for the nuclear localization of ADAR2 and for efficient editing activity (Marcucci et al. 2011). Pin1 binds to a proline residue only if it is preceded by a phosphorylated serine/threonine residue. In ADAR2 this phosphorylation is performed by an unknown kinase. If the isomerization of Pro33 by Pin1 is inhibited, then ADAR2 localizes to the cytoplasm where it is ubiquitinated by an E3 ubiquitin ligase, WWP2, and degraded (Marcucci et al. 2011). Between dsRBD1 and dsRBD2 there is phosphorylation of Ser211 and Ser216 by PKCζ, and ADAR2 editing activity is increased after this phosphorylation event (Shelton et al. 2018). However, ADAR2 editing activity is decreased after phosphorylation of residue Thr553 within the deaminase domain by AKT family kinases (Bavelloni et al. 2019).
THE ADAR2 PROTEIN INTERACTOME
A number of protein–protein interactions regulate ADAR2 editing activity. A study in primate and mouse tissues to identify proteins interacting with ADAR1 and ADAR2 identified; FAST kinase domain-containing protein 5 (FASTKD5), 39S ribosomal protein L15, mitochondrial (MRPL15), and NEDD4-binding protein 2-like 1 (N4BP2L1) (Tan et al. 2017). N4BP2L1 is a putative positive regulator; however, two other proteins have mitochondrial localization, and it is unclear how they can negatively regulate ADAR editing. This study also reported that Aminoacyl tRNA synthase complex-interacting multifunctional protein 2 (AIMP2) interacts with ADAR2 and regulates its protein levels (Tan et al. 2017).
Proteins reported to be negative regulators of ADAR2 editing activity without affecting ADARB1 mRNA levels include two ribosomal proteins, 40S ribosomal protein S14 (RPS14), and serine/arginine-rich splicing factor 9 (SFRS9) (Tariq et al. 2013). These two proteins colocalize with ADAR2 and interact with it in an RNA-independent manner. Fragile X mental retardation protein 1 (FMRP1), which binds single-stranded RNA, interacts with and colocalizes with ADAR2, down-regulating its editing activity without affecting the transcription or translation of ADAR2 (Bhogal et al. 2011; Filippini et al. 2017).
Finally, proteins with a DZF (domain associated with zinc fingers), such as interleukin enhancer-binding factor 2 (ILF2 or NF45), interleukin enhancer-binding factor 3 (ILF3 or NF90), spermatid perinuclear RNA-binding protein (STRBP), and zinc finger RNA-binding protein (ZFR), can interact with ADAR1 and ADAR2 and are site-specific regulators of editing (Freund et al. 2020). The DZF domain, which allows NF90 and NF45 to dimerize with other members of the family, encodes an inactive homolog of template-free nucleotidyl transferases such as poly(A) polymerase and tRNA CCA-adding enzymes.
TRANSCRIPTS SITE-SPECIFICALLY EDITED BY ADAR2
As previously mentioned, the first example of editing by ADAR2 resulting in recoding was found in the GRIA2 transcript (Sommer et al. 1991; Higuchi et al. 1993; for review, see Pullirsch and Jantsch 2010; Holmgren and Rosenthal 2015). This type of editing at coding positions is primarily performed by ADAR2 rather than ADAR1 (Table 1; Tan et al. 2017). There is an additional editing site in the GRIA transcripts, the R/G site which results in the conversion of an arginine codon to a glycine codon, near an exon/intron boundary in transcripts encoding GluA2, GluA3, and GluA4 subunits of AMPAR. The R/G site in the GRIA2 transcript is edited by both ADAR1 and ADAR2. RNA editing at this site can affect splicing of two mutually exclusive downstream exons, resulting in the generation of alternative flip and flop isoforms of GluA2–4, which in turn affect the gating, desensitization, and resensitization kinetics of the AMPARs (Lomeli et al. 1994; Koike et al. 2000; Krampfl et al. 2002; Grosskreutz et al. 2003).
TABLE 1.
Human ADAR2-edited transcripts

Transcripts encoding the kainate receptor subunits GluK1 and GluK2 also undergo editing of a Q/R site by both ADAR1 and ADAR2 (for review, see Nishikura 2016); however, the editing in kainite receptor subunit transcripts never reaches 100%. The Q/R site editing events in GRIK1 and GRIK2 transcripts are developmentally and regionally regulated (Bernard and Khrestchatisky 1994; Bernard et al. 1999). Q/R site editing in GRIK1 transcripts determines the Ca2+ permeability of the kainate receptor (Lee et al. 2001). However, in the transcript encoding GRIK2, two additional editing events, together with the Q/R site, all contribute to the cation selectivity of the kainate receptor channel (Egebjerg and Heinemann 1993; Köhler et al. 1993). GRIK2 editing may play a role in synaptic plasticity via induction of long-term potentiation as observed in studies in mice (Vissel et al. 2001).
Another important neuronal transcript that undergoes editing by both ADAR1 and ADAR2 in a site-specific manner at five sites, resulting in the recoding of three codons, is the serotonin receptor subtype 2C (5-HT2C) receptor (Burns et al. 1997). Editing of the 5-HT2C receptor was found to regulate serotonergic neurotransmission by decreasing the G protein coupling efficiency (Burns et al. 1997; Niswender et al. 1999; Price et al. 2001) as well as by altering desensitization and trafficking of the receptor (Marion et al. 2004). A link between 5-HT2C receptor editing and energy homeostasis was established in mice (Kawahara et al. 2008). Environmental cues such as stress can affect editing of the transcript encoding the 5-HT2C receptor (Zaidan et al. 2018), and editing efficiency also shows ligand-dependent regulation by serotonin (Gurevich et al. 2002).
When new methods such as comparative genomics were applied, the number of known recoding events increased (Hoopengardner et al. 2003; Levanon et al. 2005). However, it was the widespread use of RNA-seq on RNA from human tissue and cell line samples that identified the most recoding editing events. Site-specific editing that results in recoding is abundant in brain transcripts and has been evolutionarily conserved (Ramaswami et al. 2013; Picardi et al. 2015). Despite further recoding events having been found in other transcripts by transcriptome analyses, very few of these have been functionally characterized. In mammals, these include GABRA3 transcripts encoding the GABAA receptor subunit α3 (Ohlson et al. 2007; Rula et al. 2008; Daniel et al. 2011), and transcripts encoding voltage-gated potassium channel subunit Kv1.1 (Bhalla et al. 2004; Streit et al. 2014), voltage-gated calcium channel subunit Cav1.3 (Huang et al. 2012; Bazzazi et al. 2013), brain-specific alternative splicing factor NOVA1 (Irimia et al. 2012), and CAPS1, a protein involved in vesicle exocytosis (Miyake et al. 2016). As the editing of these transcripts does not reach 100%, there is a mixture of unedited and edited isoforms in cells. However, this RNA recoding may be important for brain function, as it increases diversity in the proteins encoded by edited transcripts.
Evidence suggests that there is sometimes convergent evolution of editing in neuronal transcripts (Jin et al. 2009). The paralogous transcripts encoding GluA2, GluK1, and GluK2 all have a similar Q/R editing event; however, it is likely they evolved, at least partly, separately, as the dsRNA structure required for editing of the GRIA2 Q/R site differs from those present in GRIK1 and GRIK2 transcripts (Higuchi et al. 1993; Herb et al. 1996). The same editing site emerged much more clearly independently in the evolutionarily distant voltage-gated K+ channel subunits from mammals, flies, and cephalopods (Bhalla et al. 2004). This suggests that RNA recoding at some sites is physiologically important in the nervous system and perhaps indicates that the regulation of ion channel properties via editing is evolutionarily advantageous.
EDITING OF THE Gria2 TRANSCRIPT IN MICE AND GRIA2 IN HUMANS
In studies performed in mice, it was demonstrated that although ADAR2 recoding is essential for mammalian brain physiology, there is only a single editing event in the Gria2 transcript that is absolutely required for survival (Higuchi et al. 1993, 2000). Mice that have a homozygous Adarb1 null mutation due to deletion of all the exons encoding the deaminase domain develop epileptic seizures and die within three weeks of birth. The Adarb1 null mutant mouse pup lethality is rescued by a concomitant homozygous knock-in of a preedited allele of Gria2R, which has the genomic glutamine codon at the Q/R site already mutated to an arginine codon. Only very subtle abnormalities in general behavior were observed in the rescued Adarb1, Gria2R double-mutant mice (Higuchi et al. 2000). In the Adarb1 null mice, Gria2 transcripts are still ∼40% edited at the Q/R site, probably catalyzed by Adar1 (Higuchi et al. 2000; Horsch et al. 2011), suggesting that having 60% of transcripts unedited at the Q/R site is too high for viability; other experiments indicate that no more than 25% of unedited transcripts can be tolerated (Horsch et al. 2011). This establishes in rodents the importance of editing of the Gria2 Q/R site and shows that recoding in other neuronal transcripts is not essential for viability. However, it is important to remember that there are significant differences in RNA editing between mouse and human; in humans, editing of FLNA transcripts in arteries is very high and may be physiologically significant (Tan et al. 2017; Jain et al. 2018).
Transgenic mice were generated that were Gria2 Q/R site editing-incompetent, as they lack a region in Gria2 intron 11 that is required for dsRNA formation with the exon and subsequently for editing (Brusa et al. 1995). Mice heterozygous for this editing-incompetent allele have seizures and die at or before weaning about postnatal day P21. Except for the death of some pyramidal neurons in the CA3 region of the hippocampus in pups that suffer from prolonged seizures, there are no signs of excessive neurodegeneration. The pups die after just a few days of seizures and the authors assumed that aberrant excitatory signaling rather than any morphological changes in the mouse brain was likely the cause of the lethal phenotype (Brusa et al. 1995). Mouse strains with different levels of unedited Gria2 expression were generated (Feldmeyer et al. 1999). Ca2+-triggered neuronal cell death was not detected, but mutant mice had mild to severe neurological dysfunctions, as well as epilepsy and abnormal dendritic architecture. The functional study of AMPARs from the mutant mice elucidated that the seizure-prone lethal phenotype is likely associated with an increase in macroscopic conductance of AMPARs, which is determined by the number of AMPA receptors as well as by their Ca2+ permeabilities (Feldmeyer et al. 1999).
Surprisingly, mice with a homozygous deletion of Gria2 are viable (Jia et al. 1996). The mice are smaller and weigh less at around 2–3 wk after birth and there is high mortality at this stage. However, if siblings are removed and the mutants are left with the mothers, they catch up in growth later and have a normal lifespan. Surprisingly, there are no signs of seizure activity in these mice despite having increased AMPAR Ca2+ permeability, and they exhibit mild behavioral defects (Jia et al. 1996). It was later shown that the Gria2 null mutant mice have aberrant heteromeric AMPA receptor complexes containing GluA1 and GluA3 subunits and also have an increased number of homomeric GluA1 and GluA3 receptors that are less efficiently delivered from the ER to the synapse (Sans et al. 2003). Therefore, GluA2 is essential for proper assembly and trafficking to give normal AMPA receptor types and levels at synapses. It was later shown that, in addition to having the GluA2 subunit present, editing of the Gria2 transcript to generate GluA2 R subunits is essential for correct assembly and trafficking of AMPA receptors (Greger et al. 2002, 2003). Transgenic mice with the preedited allele, Gria2R, are healthy with no observable defects in brain development or physiology (Kask et al. 1998), therefore the functional relevance of unedited GluA2 is unknown.
It was inferred from the above mouse studies that absence of GRIA2 Q/R site RNA editing in humans would cause various neurological and psychiatric disorders (Akbarian et al. 1995; Kawahara et al. 2004; Peng et al. 2006; Iwamoto et al. 2009; Khermesh et al. 2016). Recently, patients with de novo heterozygous mutations in GRIA2 were reported to have a range of severe neurodevelopmental disorders (Salpietro et al. 2019). One patient had a mutation at the Q/R site in GRIA2 and presented with intractable epilepsy and global developmental and intellectual disability. This confirmed the importance of editing the Q/R site in humans as it has a profound effect on the human brain.
CONSEQUENCES OF DYSREGULATION OF ADAR2 EDITING AND MUTATIONS IN ADARB1 IN HUMAN DISORDERS
Transcriptome-wide A-to-I editing analysis has shown that the majority of mammalian conserved recoding sites reside in transcripts expressed in the central nervous system (Pinto et al. 2014). This underlines the importance of A-to-I editing for correct functioning of the brain. Thus, the role of ADAR2 in various brain disorders has been investigated (Akbarian et al. 1995; Peng et al. 2006; Iwamoto et al. 2009). One focus has been on patients with neurological and psychiatric disorders to determine if they have aberrant A-to-I editing (Akbarian et al. 1995; Iwamoto and Kato 2003; Gaisler-Salomon et al. 2014; Khermesh et al. 2016; Kitaura et al. 2017; Tran et al. 2019). A-to-I editing levels were found to be altered in patients with Alzheimers, epilepsy, or autism compared to controls; however, further evidence is necessary to prove that the detected dysregulation of editing has a role in the studied diseases.
Sporadic amyotrophic lateral sclerosis (ALS), which is a neurodegenerative disorder with progressive loss of motor neurons, has been reported to involve down-reregulation of ADAR2 in motor neurons and a subsequent decrease in editing of the Q/R site in the GRIA2 transcript (Kawahara et al. 2004). Decreased levels of ADAR2 protein and reduced editing at the GRIA2 Q/R site were found in spinal motor neurons from patients with sporadic ALS (Aizawa et al. 2010; Hideyama et al. 2012). To model the motor neuron death associated with sporadic ALS, a conditional knockout of Adarb1 in motor neurons of mice was generated. This mouse model showed slow motor neuron death (Hideyama et al. 2010; Yamashita et al. 2012), which was rescued either by a concomitant knock-in of preedited Gria2R gene or by the administration of perampanel, which is a selective antagonist of AMPARs (Hideyama et al. 2010; Akamatsu et al. 2016). These results suggest that decreased ADAR2 editing at the GRIA2 Q/R site is involved in the pathogenesis of ALS. However, the full mechanism by which the reduction in ADAR2 editing causes motor neuron death needs to be elucidated. Loss of GRIA2 Q/R site editing by ADAR2 results in excessive calcium influx through AMPARs, and one possible way this could lead to cell death is by aberrant activation of calpain, a calcium-dependent protease (Yamashita and Kwak 2019); there may also be other ways calcium influx precipitates cell death. Why there is a decrease in ADAR2 activity in motor neurons of ALS patients is unknown; however, patients with mutations in FUS or C9orf72, genes associated with familial ALS, also have dysregulated ADAR2 activity (Aizawa et al. 2016; Moore et al. 2019). Therefore, mutations in some other genes may indirectly contribute to ALS by disrupting normal ADAR2 activity.
Only recently, seven human ADARB1 mutations were reported in six individuals with microcephaly, intellectual disability, and intractable seizures (Tan et al. 2020; Maroofian et al. 2021). Similar to mutations found in ADAR (Rice et al. 2012), most occurred in the deaminase domain, six were missense SNPs, while one was a small deletion at an exon–intron boundary. These mutations were recessive (either homozygous or bi-allelic in patients), and functional testing of mutated proteins expressed in cultured cells revealed that they are deleterious at either RNA or protein levels. All but one of the seven reported ADARB1 mutations resulted in either complete loss of or decrease in ADAR2 editing activity (Tan et al. 2020; Maroofian et al. 2021). The seizure-prone state observed in Adarb1 knockout mice with defective editing at the Gria2 Q/R site is a good model for the human disorder, as patients with GRIA2 mutations also have seizures (Brusa et al. 1995; Higuchi et al. 2000; Salpietro et al. 2019). Thus, insufficient editing of the GRIA2 Q/R site is likely to be the main cause of the neurodevelopmental disorders in individuals with ADARB1 mutations. However, when compared to mice with deficient GRIA2 Q/R site editing, the situation may be more complicated in humans, as patients with ADARB1 mutations presented with cerebral atrophy and white matter loss (Maroofian et al. 2021) after a much longer time with seizures, whereas brain lesions in Gria2 mice were restricted to hippocampal CA1 (Konen et al. 2020) or CA3 (Brusa et al. 1995) regions. Also, seizures in two patients with GRIA2 mutations were unsuccessfully treated with perampanel (Salpietro et al. 2019), unlike the success with treatment of neuronal death in the motor neurons of conditional knockout of Adarb1 mice (Akamatsu et al. 2016), suggesting that brain plasticity in humans leads to changes in other receptors which are not corrected by perampanel. Zebrafish deficient in gria2a have defects in brain morphology, including developmental abnormalities in neural crest and cranial cartilage, but are not seizure-prone (Ali et al. 2014). Thus, further studies are required to elucidate the pathological mechanisms underlying the neurodevelopmental disorder in patients with ADARB1 mutations.
ADAR2 PROTEINS IN INVERTEBRATES; DROSOPHILA ADAR IS INVOLVED IN BOTH RECODING EDITING AND INNATE IMMUNE RESPONSE SUPPRESSION
Drosophila has only one Adar gene (Palladino et al. 2000b; Keegan et al. 2011; Savva et al. 2012a). In arthropods such as Drosophila, ADAR1 has been lost (Keegan et al. 2011; Grice and Degnan 2015), and the annotated Adar gene is the sequence ortholog of the mammalian ADARB1 gene encoding ADAR2. Drosophila Adar is mainly expressed in the nervous system (Jepson et al. 2011), and ADAR2-type site-specific RNA editing in CNS has been greatly expanded in Drosophila and other insects (Graveley et al. 2011). The target transcripts for A-to-I editing are enriched in ion channels, neurotransmitter receptor subunits, and calcium- and synapse-associated neuronal proteins (Duan et al. 2017). In Drosophila, over 1300 site-specific RNA editing events have been detected in mRNAs. A subset of 50–100 sites is edited with high efficiency, and editing at these sites is also well-conserved across different insect groups, suggesting that editing at these sites is functionally significant. In bumblebees, paralleling the mouse-human difference in mammals, there is a higher level of ADAR-mediated RNA editing despite the lack of an ADAR1-homolog (Porath et al. 2019). There are 1.15 million identified unique genomic sites, and 164 recoding sites residing in 100 protein coding genes, including ion channels, transporters, and receptors predicted to affect brain function and behavior. Furthermore, in the honeybee Apis mellifera, 464 A-to-I editing sites have been identified, especially in heads (Porath et al. 2019). Four recoding sites and one synonymous editing site are highly conserved between honeybee, bumblebee, and Drosophila. There is a diverged auto-editing site in Adar mRNA in bees and flies, which might play an auto-regulatory role in the two clades (Porath et al. 2019).
Drosophila Adar5G1 null mutant flies show reduced viability in terms of numbers of individuals surviving through earlier stages to eclose from pupae as live flies (Khan et al. 2020), and mutant larvae show increased neuronal excitability (Li et al. 2014) and aberrant accumulation of neurotransmitter synaptic vesicles in neurons (Maldonado et al. 2013). Adar5G1 mutant flies show locomotion defects from the time of pupal eclosion, still have aberrant accumulation of neurotransmitter synaptic vesicle proteins (Robinson et al. 2016), and, after 20–30 d, show age-related neurodegeneration with large vacuoles in the brain (Palladino et al. 2000b). All Adar5G1 mutant defects are suppressed by reduced Tor or by overexpression of Atg5, both of which increase canonical autophagy initiation and reduce aberrant accumulation of synaptic vesicle proteins (Khan et al. 2020). Moreover, increased expression of Hsc70–4, which mediates endosomal microautophagy, also reduces aberrant accumulation of synaptic vesicle proteins and suppresses all Adar5G1 mutant defects (Khan et al. 2020). The cause of the underlying aberrant autophagy in Adar5G1 is unknown, but it might be part of an aberrant antiviral response.
Interestingly, a new study has shown that the loss of Adar RNA editing activity leads to innate immune induction, indicating that Drosophila Adar, despite being the homolog of mammalian ADAR2, also has functions similar to mammalian ADAR1 (Deng et al. 2020). The innate immune induction in fly Adar mutants is suppressed by silencing of Dicer-2 in heads. Dicer-2 has an RNA helicase domain similar to that of MDA5, which senses unedited dsRNAs in mammalian Adar mutants; insects have also lost Mda5-like helicase domains during evolution. Studying effects of restoring expression of catalytically inactive Adar protein in Adar mutants allows us to determine which Adar5G1 mutant defects depend entirely on editing of target transcripts. When Adar are expressed at physiological levels, adenosine deamination activity is required to restore normal locomotion and to prevent neurodegeneration, indicating a requirement for the edited isoforms of CNS proteins. However, the Adar E347A catalytically inactive protein, when expressed at a higher than physiological level, can rescue neurodegeneration and block aberrant Dicer-2 activation in Adar mutants, though it still does not rescue locomotion defects, suggesting the existence of editing-independent effects on neurodegeneration and innate immune responses (Deng et al. 2020).
The involvement of Drosophila Adar in genome defense and antiviral dsRNA responses raises the question of whether this role is conserved in other invertebrate ADAR2 proteins or if it might have escaped detection even in vertebrate ADAR2 proteins. In the planarian Schmidtea mediterranea, it has been shown that ADAR2, but not ADAR1, mediates mRNA editing (Bar Yaacov 2022). However, knockdown of either planarian adar1 or adar2 by RNA interference (RNAi) resulted in up-regulation of dsRNA-response genes, including three planarian rig-I-like receptor (prlr) homologs. Furthermore, independent knockdown of either adar1 or adar2 reduced the number of cells infected with a dsRNA virus, suggesting they both suppress an antiviral dsRNA-response activity (Bar Yaacov 2022). The possibility exists that ADAR2 has a role in innate immunity similar to ADAR1; however, due to the nuclear localization, ADAR2 might have limited impact (Desterro et al. 2003). However, in cell lines when ADAR2 is mislocalized to the cytoplasm, then it can reduce the expression of interferon response genes (Mannion et al. 2014), suggesting that the nuclear localization of ADAR2 limits its involvement.
Caenorhabditis elegans, on the other hand, has two ADAR-like proteins: ADR-1 (catalytically inactive) and ADR-2 (catalytically active) (Knight and Bass 2002). The binding of ADR-1 to target dsRNAs promotes the editing by ADR-2 (Rajendren et al. 2018). It has been reported that transcribed regions overlapping ADR-modulated loci in C. elegans exhibit significant up-regulation of secondary siRNAs. This suggests an effective engagement of the RNAi machinery that arises as a consequence of the production of an aberrant population of primary siRNAs in the absence of ADAR activity, demonstrating the competitive relationship between ADARs and the RNAi pathways (Wu et al. 2011). Nevertheless, ADR-1 and ADR-2 do not correspond to mammalian ADAR1 and ADAR2. C. elegans adr-2 is similar in structure to ADAR1, with multiple dsRBDs, and adr-1 has only a deaminase domain and no dsRBDs. Loss of Adr1 function also leads to increased RNAi responses in nematodes (Tonkin and Bass 2003).
Some Drosophila melanogaster Adar RNA editing events can appear and disappear rapidly during evolution and can differ even between different geographic populations of D. melanogaster, even though other editing events are evolutionarily conserved and maintained widely across insect species (Popitsch et al. 2020). A significant proportion of D. melanogaster Adar RNA editing events may be present in a limited range of species; unexpected absences of some editing events and presences of different editing events had already been observed in closely related species. A recent study using the extensive genomic and transcriptomic data available for drosophilids suggests that a large proportion of ADAR editing events have evolved because they correct, at the RNA level, genomic G to A mutations (Popitsch et al. 2020); perhaps ADARs have always been engaged in gene therapy to some extent. The evidence for this is that the G residue is present and conserved in a wider range of drosophilids and dipteran insects, suggesting that G is selectively advantageous. However, Drosophila melanogaster has acquired a deleterious G to A mutation which can be maintained because of ADAR RNA editing. Similar evidence has been presented for ADAR RNA editing in cephalopods and even for vertebrates (Popitsch et al. 2020). It is tempting to suggest also that the evolution of complete metamorphosis in dipteran insects has permitted accumulation of G to A mutations in CNS-expressed genes in which the mutations can be tolerated during the simplified, headless, larval feeding stages. Adar RNA editing increases dramatically downstream from ecdysome signaling at metamorphosis (Palladino et al. 2000a), in time to correct these mutations in CNS transcripts at the RNA level before they affect the adult brain and capacities for critical adult behaviors and choices.
Reversion of G to A mutations back to A is detected variably in different populations of the Drosophila melanogaster species (Popitsch et al. 2020). Surprisingly, such revertant genomic sites appear to be common in Drosophila melanogaster populations (Popitsch et al. 2020). Drosophila species have large effective population sizes in the millions, meaning that genomic sequence variation within the species is greater than it is within larger, less fecund species such as mammals. In D. melanogaster populations, the mutant genomic A base at an edited site more frequently reverts back to G again than a similar A that is not at an edited site. It is interesting to consider how ADAR editing could facilitate these genomic reversion mutations. In male germline cells where many genes are transcribed, any expression of Adar might permit an edited transcript-facilitated DNA mutagenesis if RNA strands can participate as templates in DNA repair, as has been proposed (Keskin et al. 2014); however, more evidence for the existence of this process in Drosophila is required.
APPLICATIONS OF ADARs IN GENE THERAPY
Retargeting ADAR RNA editing to correct disease mutations at the transcript level is a very exciting prospect for gene therapy. CRISPR–Cas9 DNA mutagenesis faces difficulties in human application because off-target genome editing is common, irreversible, and potentially very dangerous. An additional problem is that the DNA repair pathways required are active in proliferating human cells in culture but not in postmitotic cells that make up the bulk of tissues and the majority of target cell types for gene therapy (Montiel-Gonzalez et al. 2019). Also, expression of bacterial proteins is likely to be immunogenic. For these reasons and others, including the successes of Covid mRNA vaccines and the Spinrada antisense RNA treatment against SMA, there is now increased interest in using ADAR RNA editing to correct disease mutations in RNA transcripts rather than in DNA. ADAR RNA editing gene therapy might be achieved without expressing foreign proteins and, even if off-target editing during therapy cannot be entirely eliminated, the large number of editing events known to occur at low levels in the human transcriptome suggest that some off-target RNA editing might be well tolerated in patients. It is estimated that about 60% of human disease mutations are due to SNPs and, conveniently, ADAR editing potentially directly corrects the most common type; G-to-A changes making up nearly fifty percent of SNPs. Of these G-to-A mutations, some will be directly ADAR targetable when a mutant adenosine is in the C/U-A-G/A context favored by ADARs. For open reading frame mutations where the mutant A is not in a good context for editing, codon redundancy might allow coding correction by targeting another adenosine in the codon.
Some early approaches to retargeting ADAR RNA editing used fusions of other RNA-binding proteins to an ADAR deaminase domain, and some of these gave efficient target editing; however, they also identified a critical problem arising from high off-target editing (Montiel-Gonzalez et al. 2019). This, and difficulties expected with expression of proteins and possible immunogenicity of foreign proteins in humans, means that current approaches concentrate on retargeting endogenous ADAR proteins by providing an ADAR-recruiting “guide” RNA that will also base-pair across the mutation site and make the target adenosine available for editing (Katrekar et al. 2022; Reautschnig et al. 2022; Yi et al. 2022). Studies on ADAR2 have been the predominant source of information on how ADARs recognize RNA substrates for highly efficient site-specific RNA editing and on how we might design guide RNAs to retarget ADARs for gene therapy. There is still no direct structural information on the ADAR1 protein which is more highly expressed than ADAR2 in many human tissues, and for ADAR1 we depend on inferences from, and comparisons to, ADAR2. The structures of highly edited substrate RNAs and their ADAR complexes have been essential to design effective artificial guide RNAs. Examples include guide RNAs that place the target adenosine opposite a C or U for more efficient base flipping, RNA guides that incorporate base mismatches to reduce off target editing, RNA guides that use the four base loop structure of the Gria2 R/G site to recruit ADAR2 dsRBD1, and RNA guides that incorporate bulges at certain positions, most interestingly perhaps between the deaminase domain and the 5′ RNA binding loop. Guide RNAs that are expressed in cells from a plasmid may be able to be delivered as Covid mRNA vaccines were, in nanoparticles or in engineered viruses, though the particles may usually need to be targeted to infect particular cell types.
There are of course still unanswered questions regarding ADAR2 protein RNA substrate specificity, in addition to possible ADAR2 domain dynamics already discussed. Only a limited set of RNA substrates have been studied in detail and, in organisms like Drosophila with many site-specifically edited substrates, correct structures for more efficiently edited RNA substrates are likely to improve understanding; human ADAR2 rescues Drosophila Adar mutant defects and edits these substrates (Keegan et al. 2011). For example, some Drosophila guide RNAs (previously called editing site complementary sequences [ECSs]) appear to be composed of short sections that may be better able to find their correct editing target because together they establish continuous base stacking along the target sequence (Reenan 2005). Contiguous base stacking of shorter guide RNA sections along the target sequence and base stacking continued over the ends of the target sequence by guide RNA hairpins might aid the specificity of the intermolecular pairing of artificial ADAR guide RNAs to target sites and reduce off-target editing. Artificial guide RNAs often attempt to have ADARs recruit to intramolecular hairpins in the guide RNA somewhat separately from the intermolecular pairing of the guide to the target RNA. Some vertebrate RNA substrates have bulges at positions where they do not interfere with ADAR binding, and structures for a wider range of Drosophila RNA substrates may identify examples of how additional ADAR interactions could be engineered into the guide RNA by adding bulges or even additional dsRNA hairpins. The catalytically inactive deaminase domain in the asymmetric dimer probably does not have all of its possible dsRNA contacts blocked by the dimerization so additional dsRNA interactions may be possible.
CONCLUSION
We believe it is fair to say that the ADAR enzymes have surpassed all expectations. Not only are they essential for different biological processes such as brain and innate immune function, but also, they may be instrumental to new and innovative therapies for an assortment of diseases (Montiel-Gonzalez et al. 2019). There is huge interest in ADARs themselves from the pharmaceutical industry, as an inhibitor of ADAR1 would likely increase the efficacy of cancer cell-killing and of checkpoint inhibitors in the treatment of cancer (Ishizuka et al. 2019). This success of ADAR proteins highlights the advantages of basic scientific research, where the persistence and curiosity to study an “odd” enzymatic activity reaped unforeseeable rewards.
There is also room to improve understanding of ADAR2 biology. In the CNS, development of glutamate excitotoxic neuron death through AMPA receptors may be mainly related to the ADAR editing process and the possibility to produce a toxic proteoform of GluA2 that confers excessive calcium entry and channel responsiveness. Some similar effects might also contribute to kainite receptor mediated toxicity. How aberrantly responsive CP-AMPAs lead to neuron death or to more widespread changes in other receptors in the seizure-prone state in mutants that fail to edit the Gria2 Q/R site needs to be better understood. Outside the CNS, ADAR2 in humans may have roles in vascular endothelium, in the pancreas, and possibly in other organs such as lungs that are less prominent in mice. Other questions include whether any functions of mammalian ADAR2 are substantially editing-independent and whether ADAR2 also affects innate immunity, either inside the CNS or elsewhere.
ACKNOWLEDGMENTS
This work was supported by a grant from the Czech Science Foundation, project no. GAČR 21-27329X to M.A.O'C. J.S. was funded by GAČR 20-11101S. D.A. is funded by the European Union's Horizon 2020 research and innovation program under Marie Skłodowska-Curie grant agreement no. 956810.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079266.122.
Freely available online through the RNA Open Access option.
REFERENCES
- Aizawa H, Sawada J, Hideyama T, Yamashita T, Katayama T, Hasebe N, Kimura T, Yahara O, Kwak S. 2010. TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol 120: 75–84. 10.1007/s00401-010-0678-x [DOI] [PubMed] [Google Scholar]
- Aizawa H, Hideyama T, Yamashita T, Kimura T, Suzuki N, Aoki M, Kwak S. 2016. Deficient RNA-editing enzyme ADAR2 in an amyotrophic lateral sclerosis patient with a FUSP525L mutation. J Clin Neurosci 32: 128–129. 10.1016/j.jocn.2015.12.039 [DOI] [PubMed] [Google Scholar]
- Akamatsu M, Yamashita T, Hirose N, Teramoto S, Kwak S. 2016. The AMPA receptor antagonist perampanel robustly rescues amyotrophic lateral sclerosis (ALS) pathology in sporadic ALS model mice. Sci Rep 6: 28649. 10.1038/srep28649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarian S, Smith MA, Jones EG. 1995. Editing for an AMPA receptor subunit RNA in prefrontal cortex and striatum in Alzheimer's disease, Huntington's disease and schizophrenia. Brain Res 699: 297–304. 10.1016/0006-8993(95)00922-D [DOI] [PubMed] [Google Scholar]
- Ali IM, Evehe MSB, Netongo PM, Atogho-Tiedeu B, Akindeh-Nji M, Ngora H, Domkam IK, Diakite M, Baldip K, Ranford-Cartwright L, et al. 2014. Host candidate gene polymorphisms and associated clearance of P. falciparum amodiaquine and fansidar resistance mutants in children less than 5 years in Cameroon. Pathog Glob Health 108: 323–333. 10.1179/2047773214Y.0000000159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AR, Slack FJ. 2022. ADAR1 and its implications in cancer development and treatment. Trends Genet 38: 821–830. 10.1016/j.tig.2022.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandele OJ, Wang X, Campbell MR, Pittman GS, Bell DA. 2011. Human single-nucleotide polymorphisms alter p53 sequence-specific binding at gene regulatory elements. Nucleic Acids Res 39: 178–189. 10.1093/nar/gkq764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar Yaacov D. 2022. Functional analysis of ADARs in planarians supports a bilaterian ancestral role in suppressing double-stranded RNA-response. PLoS Pathog 18: e1010250. 10.1371/journal.ppat.1010250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Weintraub H. 1987. A developmental regulated activity that unwinds RNA duplexes. Cell 48: 607–613. 10.1016/0092-8674(87)90239-X [DOI] [PubMed] [Google Scholar]
- Bass BL, Weintraub H. 1988. An unwinding activity that covalently modifies its double-strand RNA substrate. Cell 55: 1089–1098. 10.1016/0092-8674(88)90253-X [DOI] [PubMed] [Google Scholar]
- Bavelloni A, Focaccia E, Piazzi M, Raffini M, Cesarini V, Tomaselli S, Orsini A, Ratti S, Faenza I, Cocco L, et al. 2019. AKT-dependent phosphorylation of the adenosine deaminases ADAR-1 and -2 inhibits deaminase activity. FASEB J 33: 9044–9061. 10.1096/fj.201800490RR [DOI] [PubMed] [Google Scholar]
- Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, et al. 2014. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24: 365–376. 10.1101/gr.164749.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzazi H, Ben Johny M, Adams PJ, Soong TW, Yue DT. 2013. Continuously tunable Ca2+ regulation of RNA-edited CaV1.3 channels. Cell Rep 5: 367–377. 10.1016/j.celrep.2013.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm M, Wahlstedt H, Widmark A, Eriksson M, Ohman M. 2017. Accumulation of nuclear ADAR2 regulates adenosine-to-inosine RNA editing during neuronal development. J Cell Sci 130: 745–753. [DOI] [PubMed] [Google Scholar]
- Bernard A, Khrestchatisky M. 1994. Assessing the extent of RNA editing in the TMII region of GluR5 and GluR6 kainate receptors during rat brain development. J Neurochem 62: 2057–2060. 10.1046/j.1471-4159.1994.62052057.x [DOI] [PubMed] [Google Scholar]
- Bernard A, Ferhat L, Dessi F, Charton G, Represa A, Ben-Ari Y, Khrestchatisky M. 1999. Q/R editing of the rat GluR5 and GluR6 kainate receptors in vivo and in vitro: evidence for independent developmental, pathological and cellular regulation. Eur J Neurosci 11: 604–616. 10.1046/j.1460-9568.1999.00479.x [DOI] [PubMed] [Google Scholar]
- Bhalla T, Rosenthal JJC, Holmgren M, Reenan R. 2004. Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat Struct Mol Biol 11: 950–956. 10.1038/nsmb825 [DOI] [PubMed] [Google Scholar]
- Bhogal B, Jepson JE, Savva YA, Pepper ASR, Reenan RA, Jongens TA. 2011. Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat Neurosci 14: 1517–1524. 10.1038/nn.2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusa R, Zimmermann F, Koh D-S, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. 1995. Early-onset epilepsy and postnatal lethality associated with editing-deficient GluR-B allele in mice. Science 270: 1677–1680. 10.1126/science.270.5242.1677 [DOI] [PubMed] [Google Scholar]
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. 1997. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387: 303–308. 10.1038/387303a0 [DOI] [PubMed] [Google Scholar]
- Chen J, Sun Y, Ou Z, Yeh S, Huang C-P, You B, Tsai Y-C, Sheu T-J, Zu X, Chang C. 2020. Androgen receptor-regulated circFNTA activates KRAS signaling to promote bladder cancer invasion. EMBO Rep 21: e48467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn WE. 1959. 5-Ribosyl uracil, a carbon-carbon ribofuranosyl nucleoside in ribonucleic acids. Biochim Biophys Acta 32: 569–571. 10.1016/0006-3002(59)90644-4 [DOI] [PubMed] [Google Scholar]
- Cohn WE, Volkin E. 1951. Nucleoside-5′-phosphates from ribonucleic acids. Nature 169: 483–484. 10.1038/167483a0 [DOI] [Google Scholar]
- Daniel C, Wahlstedt H, Ohlson J, Björk P, Öhman M. 2011. Adenosine-to-inosine RNA editing affects trafficking of the γ-aminobutyric acid type A (GABAA) receptor. J Biol Chem 286: 2031–2040. 10.1074/jbc.M110.130096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng P, Khan A, Jacobson D, Sambrani N, McGurk L, Li X, Jayasree A, Hejatko J, Shohat-Ophir G, O'Connell MA, et al. 2020. Adar RNA editing-dependent and -independent effects are required for brain and innate immune functions in Drosophila. Nat Commun 11: 1580. 10.1038/s41467-020-15435-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desterro JMP, Keegan LP, Lafarga M, Berciano MT, O'Connell M, Carmo-Fonseca M. 2003. Dynamic association of RNA-editing enzymes with the nucleolus. J Cell Sci 116: 1805–1818. 10.1242/jcs.00371 [DOI] [PubMed] [Google Scholar]
- Duan Y, Dou S, Luo S, Zhang H, Lu J. 2017. Adaptation of A-to-I RNA editing in Drosophila. PLoS Genet 13: e1006648. 10.1371/journal.pgen.1006648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egebjerg J, Heinemann SF. 1993. Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proc Natl Acad Sci 90: 755–759. 10.1073/pnas.90.2.755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Levanon EY. 2018. A-to-I RNA editing—immune protector and transcriptome diversifier. Nat Rev Genet 19: 473–490. 10.1038/s41576-018-0006-1 [DOI] [PubMed] [Google Scholar]
- Erdmann EA, Mahapatra A, Mukherjee P, Yang B, Hundley HA. 2021. To protect and modify double-stranded RNA - the critical roles of ADARs in development, immunity and oncogenesis. Crit Rev Biochem Mol Biol 56: 54–87. 10.1080/10409238.2020.1856768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Kask K, Brusa R, Kornau HC, Kolhekar R, Rozov A, Burnashev N, Jensen V, Hvalby O, Sprengel R, et al. 1999. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci 2: 57–64. 10.1038/4561 [DOI] [PubMed] [Google Scholar]
- Feng Y, Sansam CL, Singh M, Emeson RB. 2006. Altered RNA editing in mice lacking ADAR2 autoregulation. Mol Cell Biol 26: 480–488. 10.1128/MCB.26.2.480-488.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini A, Bonini D, Lacoux C, Pacini L, Zingariello M, Sancillo L, Bosisio D, Salvi V, Mingardi J, La Via L, et al. 2017. Absence of the Fragile X Mental Retardation Protein results in defects of RNA editing of neuronal mRNAs in mouse. RNA Biol 14: 1580–1591. 10.1080/15476286.2017.1338232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini A, Bonini D, Giacopuzzi E, La Via L, Gangemi F, Colombi M, Barbon A. 2018. Differential enzymatic activity of rat ADAR2 splicing variants is due to altered capability to interact with RNA in the deaminase domain. Genes (Basel) 9: 79. 10.3390/genes9020079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzen O, Ermel R, Sukhavasi K, Jain R, Jain A, Betsholtz C, Giannarelli C, Kovacic JC, Ruusalepp A, Skogsberg J, et al. 2018. Global analysis of A-to-I RNA editing reveals association with common disease variants. PeerJ 6: e4466. 10.7717/peerj.4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund EC, Sapiro AL, Li Q, Linder S, Moresco JJ, Yates JR III, Li JB. 2020. Unbiased identification of trans regulators of ADAR and A-to-I RNA editing. Cell Rep 31: 107656. 10.1016/j.celrep.2020.107656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisler-Salomon I, Kravitz E, Feiler Y, Safran M, Biegon A, Amariglio N, Rechavi G. 2014. Hippocampus-specific deficiency in RNA editing of GluA2 in Alzheimer's disease. Neurobiol Aging 35: 1785–1791. 10.1016/j.neurobiolaging.2014.02.018 [DOI] [PubMed] [Google Scholar]
- Gallo A, Keegan LP, Ring GM, O'Connell MA. 2003. An ADAR that edits transcripts encoding ion channel subunits functions as a dimer. Embo J 22: 3421–3430. 10.1093/emboj/cdg327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Z, Zhao L, Yang L, Huang P, Zhao F, Li W, Liu Y. 2006. RNA editing by ADAR2 is metabolically regulated in pancreatic islets and β-cells. J Biol Chem 281: 33386–33394. 10.1074/jbc.M604484200 [DOI] [PubMed] [Google Scholar]
- Gerber A, O'Connell MA, Keller W. 1997. Two forms of human double-stranded RNA-specific editase 1 (hRED1) generated by the insertion of an Alu cassette. RNA 3: 453–463. [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479. 10.1038/nature09715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Ziff EB. 2002. RNA editing at Arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 34: 759–772. 10.1016/S0896-6273(02)00693-1 [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Kong X, Ziff EB. 2003. AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40: 763–774. 10.1016/S0896-6273(03)00668-8 [DOI] [PubMed] [Google Scholar]
- Grice LF, Degnan BM. 2015. The origin of the ADAR gene family and animal RNA editing. BMC Evol Biol 15: 4. 10.1186/s12862-015-0279-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskreutz J, Zoerner A, Schlesinger F, Krampfl K, Dengler R, Bufler J. 2003. Kinetic properties of human AMPA-type glutamate receptors expressed in HEK293 cells. Eur J Neurosci 17: 1173–1178. 10.1046/j.1460-9568.2003.02531.x [DOI] [PubMed] [Google Scholar]
- Gurevich I, Englander MT, Adlersberg M, Siegal NB, Schmauss C. 2002. Modulation of serotonin 2C receptor editing by sustained changes in serotonergic neurotransmission. J Neurosci 22: 10529–10532. 10.1523/JNEUROSCI.22-24-10529.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heraud-Farlow JE, Walkley CR. 2020. What do editors do? Understanding the physiological functions of A-to-I RNA editing by adenosine deaminase acting on RNAs. Open Biol 10: 200085. 10.1098/rsob.200085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herb A, Higuchi M, Sprengel R, Seeburg PH. 1996. Q/R site editing in kainate receptor GluR5 and GluR6 pre-mRNAs requires distant intronic sequences. Proc Natl Acad Sci 93: 1875–1880. 10.1073/pnas.93.5.1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideyama T, Yamashita T, Suzuki T, Tsuji S, Higuchi M, Seeburg PH, Takahashi R, Misawa H, Kwak S. 2010. Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J Neurosci 30: 11917–11925. 10.1523/JNEUROSCI.2021-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideyama T, Yamashita T, Aizawa H, Tsuji S, Kakita A, Takahashi H, Kwak S. 2012. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol Dis 45: 1121–1128. 10.1016/j.nbd.2011.12.033 [DOI] [PubMed] [Google Scholar]
- Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, Seeburg PH. 1993. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell 75: 1361–1370. 10.1016/0092-8674(93)90622-W [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single F, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg P. 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78–81. 10.1038/35017558 [DOI] [PubMed] [Google Scholar]
- Holmgren M, Rosenthal JJ. 2015. Regulation of ion channel and transporter function through RNA editing. Curr Issues Mol Biol 17: 23–36. [PMC free article] [PubMed] [Google Scholar]
- Hoopengardner B, Bhalla T, Staber C, Reenan R. 2003. Nervous system targets of RNA editing identified by comparative genomics. Science 301: 832–836. 10.1126/science.1086763 [DOI] [PubMed] [Google Scholar]
- Horsch M, Seeburg PH, Adler T, Aguilar-Pimentel JA, Becker L, Calzada-Wack J, Garrett L, Gotz A, Hans W, Higuchi M, et al. 2011. Requirement of the RNA-editing enzyme ADAR2 for normal physiology in mice. J Biol Chem 286: 18614–18622. 10.1074/jbc.M110.200881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough RF, Bass BL. 1994. Purification of the Xenopus laevis dsRNA adenosine deaminase. J Biol Chem 269: 9933–9939. 10.1016/S0021-9258(17)36972-7 [DOI] [PubMed] [Google Scholar]
- Huang H, Tan BZ, Shen Y, Tao J, Jiang F, Sung YY, Ng CK, Raida M, Köhr G, Higuchi M, et al. 2012. RNA editing of the IQ domain in Cav1.3 channels modulates their Ca2+-dependent inactivation. Neuron 73: 304–316. 10.1016/j.neuron.2011.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, Denuc A, Ferran JL, Pernaute B, Puelles L, Roy SW, Garcia-Fernàndez J, Marfany G. 2012. Evolutionarily conserved A-to-I editing increases protein stability of the alternative splicing factor Nova1. RNA Biol 9: 12–21. 10.4161/rna.9.1.18387 [DOI] [PubMed] [Google Scholar]
- Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. 2019. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565: 43–48. 10.1038/s41586-018-0768-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto K, Kato T. 2003. RNA editing of serotonin 2C receptor in human postmortem brains of major mental disorders. Neurosci Lett 346: 169–172. 10.1016/S0304-3940(03)00608-6 [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, Kato T. 2009. Serotonin receptor 2C and mental disorders: genetic, expression and RNA editing studies. RNA Biol 6: 248–253. 10.4161/rna.6.3.8370 [DOI] [PubMed] [Google Scholar]
- Jain M, Mann TD, Stulic M, Rao SP, Kirsch A, Pullirsch D, Strobl X, Rath C, Reissig L, Moreth K, et al. 2018. RNA editing of Filamin A pre-mRNA regulates vascular contraction and diastolic blood pressure. EMBO J 37: e94813. 10.15252/embj.201694813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson JE, Savva YA, Yokose C, Sugden AU, Sahin A, Reenan RA. 2011. Engineered alterations in RNA editing modulate complex behavior in Drosophila: regulatory diversity of adenosine deaminase acting on RNA (ADAR) targets. J Biol Chem 286: 8325–8337. 10.1074/jbc.M110.186817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, McDonald J, Carlen P, et al. 1996. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 17: 945–956. 10.1016/S0896-6273(00)80225-1 [DOI] [PubMed] [Google Scholar]
- Jin Y, Zhang W, Li Q. 2009. Origins and evolution of ADAR-mediated RNA editing. IUBMB Life 61: 572–578. 10.1002/iub.207 [DOI] [PubMed] [Google Scholar]
- Johnson TB, Coghill RD. 1925. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the tubercle Bacillus. J Am Chem Soc 47: 2838–2844. 10.1021/ja01688a030 [DOI] [Google Scholar]
- Kask K, Zamanillo D, Rozov A, Burnashev N, Sprengel R, Seeburg PH. 1998. The AMPA receptor subunit GluR-B in its Q/R site-unedited form is not essential for brain development and function. Proc Natl Acad Sci 95: 13777–13782. 10.1073/pnas.95.23.13777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katrekar D, Yen J, Xiang Y, Saha A, Meluzzi D, Savva Y, Mali P. 2022. Efficient in vitro and in vivo RNA editing via recruitment of endogenous ADARs using circular guide RNAs. Nat Biotechnol 40: 938–945. 10.1038/s41587-021-01171-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. 2004. Glutamate receptors: RNA editing and death of motor neurons. Nature 427: 801. 10.1038/427801a [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Grimberg A, Teegarden S, Mombereau C, Liu S, Bale TL, Blendy JA, Nishikura K. 2008. Dysregulated editing of serotonin 2C receptor mRNAs results in energy dissipation and loss of fat mass. J Neurosci 28: 12834–12844. 10.1523/JNEUROSCI.3896-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan LP, Brindle J, Gallo A, Leroy A, Reenan RA, O'Connell MA. 2005. Tuning of RNA editing by ADAR is required in Drosophila. EMBO J 24: 2183–2193. 10.1038/sj.emboj.7600691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan LP, McGurk L, Palavicini JP, Brindle J, Paro S, Li X, Rosenthal JJ, O'Connell MA. 2011. Functional conservation in human and Drosophila of Metazoan ADAR2 involved in RNA editing: loss of ADAR1 in insects. Nucleic Acids Res 39: 7249–7262. 10.1093/nar/gkr423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, Storici F. 2014. Transcript-RNA-templated DNA recombination and repair. Nature 515: 436–439. 10.1038/nature13682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Paro S, McGurk L, Sambrani N, Hogg MC, Brindle J, Pennetta G, Keegan LP, O'Connell MA. 2020. Membrane and synaptic defects leading to neurodegeneration in Adar mutant Drosophila are rescued by increased autophagy. BMC Biol 18: 15. 10.1186/s12915-020-0747-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khermesh K, D'Erchia AM, Barak M, Annese A, Wachtel C, Levanon EY, Picardi E, Eisenberg E. 2016. Reduced levels of protein recoding by A-to-I RNA editing in Alzheimer's disease. RNA 22: 290–302. 10.1261/rna.054627.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U, Garner TL, Sanford T, Speicher D, Murray JM, Nishikura K. 1994a. Purification and characterization of double-stranded RNA adenosine deaminase from bovine nuclear extracts. J Biol Chem 269: 13480–13489. 10.1016/S0021-9258(17)36857-6 [DOI] [PubMed] [Google Scholar]
- Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. 1994b. Molecular cloning of cDNAs for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc Natl Acad Sci 91: 11457–11461. 10.1073/pnas.91.24.11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaura H, Sonoda M, Teramoto S, Shirozu H, Shimizu H, Kimura T, Masuda H, Ito Y, Takahashi H, Kwak S, et al. 2017. Ca2+-permeable AMPA receptors associated with epileptogenesis of hypothalamic hamartoma. Epilepsia 58: e59–e63. 10.1111/epi.13700 [DOI] [PubMed] [Google Scholar]
- Knight SW, Bass BL. 2002. The role of RNA editing by ADARs in RNAi. Mol Cell 10: 809–817. 10.1016/S1097-2765(02)00649-4 [DOI] [PubMed] [Google Scholar]
- Köhler M, Burnashev N, Sakmann B, Seeburg PH. 1993. Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron 10: 491–500. 10.1016/0896-6273(93)90336-P [DOI] [PubMed] [Google Scholar]
- Koike M, Tsukada S, Tsuzuki K, Kijima H, Ozawa S. 2000. Regulation of kinetic properties of GluR2 AMPA receptor channels by alternative splicing. J Neurosci 20: 2166–2174. 10.1523/JNEUROSCI.20-06-02166.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konen LM, Wright AL, Royle GA, Morris GP, Lau BK, Seow PW, Zinn R, Milham LT, Vaughan CW, Vissel B. 2020. A new mouse line with reduced GluA2 Q/R site RNA editing exhibits loss of dendritic spines, hippocampal CA1-neuron loss, learning and memory impairments and NMDA receptor-independent seizure vulnerability. Mol Brain 13: 27. 10.1186/s13041-020-0545-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krampfl K, Schlesinger F, Zörner A, Kappler M, Dengler R, Bufler J. 2002. Control of kinetic properties of GluR2 flop AMPA-type channels: impact of R/G nuclear editing. Eur J Neurosci 15: 51–62. 10.1046/j.0953-816x.2001.01841.x [DOI] [PubMed] [Google Scholar]
- Lai F, Chen C-X, Carter KC, Nishikura K. 1997. Editing of glutamate receptor B subunit ion channel RNAs by four alternatively spliced DRADA2 double-stranded RNA adenosine deaminases. Mol Cell Biol 17: 2413–2424. 10.1128/MCB.17.5.2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CJ, Kong H, Manzini MC, Albuquerque C, Chao MV, MacDermott AB. 2001. Kainate receptors expressed by a subpopulation of developing nociceptors rapidly switch from high to low Ca2+ permeability. J Neurosci 21: 4572–4581. 10.1523/JNEUROSCI.21-13-04572.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon EY, Hallegger M, Kinar Y, Shemesh R, Djinovic-Carugo K, Rechavi G, Jantsch MF, Eisenberg E. 2005. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res 33: 1162–1168. 10.1093/nar/gki239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Overton IM, Baines RA, Keegan LP, O'Connell MA. 2014. The ADAR RNA editing enzyme controls neuronal excitability in Drosophila melanogaster. Nucleic Acids Res 42: 1139–1151. 10.1093/nar/gkt909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. 10.1126/science.aac7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli H, Mosbacher J, Melcher T, Höger T, Geiger JR, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg PH. 1994. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 266: 1709–1713. 10.1126/science.7992055 [DOI] [PubMed] [Google Scholar]
- Maas S, Gommans WM. 2009a. Identification of a selective nuclear import signal in adenosine deaminases acting on RNA. Nucleic Acids Res 37: 5822–5829. 10.1093/nar/gkp599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S, Gommans WM. 2009b. Novel exon of mammalian ADAR2 extends open reading frame. PLoS One 4: e4225. 10.1371/journal.pone.0004225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S, Melcher T, Herb A, Seeburg PH, Keller W, Krause S, Higuchi M, O'Connell MA. 1996. Structural requirements for RNA editing in glutamate receptor pre-mRNAs by recombinant double-stranded RNA adenosine deaminase. J Biol Chem 271: 12221–12226. 10.1074/jbc.271.21.12221 [DOI] [PubMed] [Google Scholar]
- Macbeth MR, Lingam AT, Bass BL. 2004. Evidence for auto-inhibition by the N terminus of hADAR2 and activation by dsRNA binding. RNA 10: 1563–1571. 10.1261/rna.7920904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macbeth MR, Schubert HL, VanDemark AF, Lingam AT, Hill CP, Bass BL. 2005. Structural biology: inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 309: 1534–1539. 10.1126/science.1113150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado C, Alicea D, Gonzalez M, Bykhovskaia M, Marie B. 2013. Adar is essential for optimal presynaptic function. Mol Cell Neurosci 52: 173–180. 10.1016/j.mcn.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. 10.1016/j.celrep.2014.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci R, Brindle J, Paro S, Casadio A, Hempel S, Morrice N, Bisso A, Keegan LP, Del Sal G, O'Connell MA. 2011. Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J 30: 4211–4222. 10.1038/emboj.2011.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion S, Weiner DM, Caron MG. 2004. RNA editing induces variation in desensitization and trafficking of 5-hydroxytryptamine 2c receptor isoforms. J Biol Chem 279: 2945–2954. 10.1074/jbc.M308742200 [DOI] [PubMed] [Google Scholar]
- Maroofian R, Sedmik J, Mazaheri N, Scala M, Zaki MS, Keegan LP, Azizimalamiri R, Issa M, Shariati G, Sedaghat A, et al. 2021. Biallelic variants in ADARB1, encoding a dsRNA-specific adenosine deaminase, cause a severe developmental and epileptic encephalopathy. J Med Genet 58: 495–504. 10.1136/jmedgenet-2020-107048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, Scott AI, Havel J, Fisher AJ, Beal PA. 2016. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol 23: 426–433. 10.1038/nsmb.3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH. 1996a. RED2, a brain specific member of the RNA-specific adenosine deaminase family. J Biol Chem 271: 31795–31798. 10.1074/jbc.271.50.31795 [DOI] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. 1996b. A mammalian RNA editing enzyme. Nature 379: 460–464. 10.1038/379460a0 [DOI] [PubMed] [Google Scholar]
- Miyake K, Ohta T, Nakayama H, Doe N, Terao Y, Oiki E, Nagatomo I, Yamashita Y, Abe T, Nishikura K, et al. 2016. CAPS1 RNA editing promotes dense core vesicle exocytosis. Cell Rep 17: 2004–2014. 10.1016/j.celrep.2016.10.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-Gonzalez MF, Diaz Quiroz JF, Rosenthal JJC. 2019. Current strategies for site-directed RNA editing using ADARs. Methods 156: 16–24. 10.1016/j.ymeth.2018.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore S, Alsop E, Lorenzini I, Starr A, Rabichow BE, Mendez E, Levy JL, Burciu C, Reiman R, Chew J, et al. 2019. ADAR2 mislocalization and widespread RNA editing aberrations in C9orf72-mediated ALS/FTD. Acta Neuropathol 138: 49–65. 10.1007/s00401-019-01999-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. 2016. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17: 83–96. 10.1038/nrm.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Copeland SC, Herrick-Davis K, Emeson RB, Sanders-Bush E. 1999. RNA editing of the human serotonin 5-hydroxytryptamine 2C receptor silences constitutive activity. J Biol Chem 274: 9472–9478. 10.1074/jbc.274.14.9472 [DOI] [PubMed] [Google Scholar]
- O'Connell MA, Keller W. 1994. Purification and properties of double-stranded RNA-specific adenosine deaminase from calf thymus. Proc Natl Acad Sci 91: 10596–10600. 10.1073/pnas.91.22.10596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell MA, Krause S, Higuchi M, Hsuan JJ, Totty NF, Jenny A, Keller W. 1995. Cloning of cDNAs encoding mammalian double-stranded RNA-specific adenosine deaminase. Mol Cell Biol 15: 1389–1397. 10.1128/MCB.15.3.1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell MA, Gerber A, Keller W. 1997. Purification of human double-stranded RNA-specific editase 1 (hRED1) involved in editing of brain glutamate receptor B pre-mRNA. J Biol Chem 272: 473–478. 10.1074/jbc.272.1.473 [DOI] [PubMed] [Google Scholar]
- Ohlson J, Pedersen JS, Haussler D, Ohman M. 2007. Editing modifies the GABAA receptor subunit α3. RNA 13: 698–703. 10.1261/rna.349107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. 2000a. dADAR, a Drosophila double-stranded RNA-specific adenosine deaminase is highly developmentally regulated and is itself a target for RNA editing. RNA 6: 1004–1018. 10.1017/S1355838200000248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. 2000b. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 102: 437–449. 10.1016/S0092-8674(00)00049-0 [DOI] [PubMed] [Google Scholar]
- Patterson JB, Samuel CE. 1995. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol 15: 5376–5388. 10.1128/MCB.15.10.5376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng PL, Zhong X, Tu W, Soundarapandian MM, Molner P, Zhu D, Lau L, Liu S, Liu F, Lu Y. 2006. ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron 49: 719–733. 10.1016/j.neuron.2006.01.025 [DOI] [PubMed] [Google Scholar]
- Picardi E, Manzari C, Mastropasqua F, Aiello I, D'Erchia AM, Pesole G. 2015. Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci Rep 5: 14941. 10.1038/srep14941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto Y, Cohen H, Levanon E. 2014. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol 15: R5. 10.1186/gb-2014-15-1-r5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popitsch N, Huber CD, Buchumenski I, Eisenberg E, Jantsch M, von Haeseler A, Gallach M. 2020. A-to-I RNA editing uncovers hidden signals of adaptive genome evolution in animals. Genome Biol Evol 12: 345–357. 10.1093/gbe/evaa046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath HT, Hazan E, Shpigler H, Cohen M, Band M, Ben-Shahar Y, Levanon EY, Eisenberg E, Bloch G. 2019. RNA editing is abundant and correlates with task performance in a social bumblebee. Nat Commun 10: 1605. 10.1038/s41467-019-09543-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RD, Weiner DM, Chang MS, Sanders-Bush E. 2001. RNA editing of the human serotonin 5-HT2C receptor alters receptor-mediated activation of G13 protein. J Biol Chem 276: 44663–44668. 10.1074/jbc.M106745200 [DOI] [PubMed] [Google Scholar]
- Pullirsch D, Jantsch MF. 2010. Proteome diversification by adenosine to inosine RNA-editing. RNA Biol 7: 205–212. 10.4161/rna.7.2.11286 [DOI] [PubMed] [Google Scholar]
- Quin J, Sedmík J, Vukić D, Khan A, Keegan LP, O'Connell MA. 2021. ADAR RNA modifications, the epitranscriptome and innate immunity. Trends Biochem Sci 46: 758–771. 10.1016/j.tibs.2021.02.002 [DOI] [PubMed] [Google Scholar]
- Rajendren S, Manning AC, Al-Awadi H, Yamada K, Takagi Y, Hundley HA. 2018. A protein–protein interaction underlies the molecular basis for substrate recognition by an adenosine-to-inosine RNA-editing enzyme. Nucleic Acids Res 46: 9647–9659. 10.1093/nar/gky800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. 2013. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128–132. 10.1038/nmeth.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reautschnig P, Wahn N, Wettengel J, Schulz AE, Latifi N, Vogel P, Kang T-W, Pfeiffer LS, Zarges C, Naumann U. 2022. CLUSTER guide RNAs enable precise and efficient RNA editing with endogenous ADAR enzymes in vivo. Nat Biotechnol 40: 759–768. 10.1038/s41587-021-01105-0 [DOI] [PubMed] [Google Scholar]
- Rebagliati MR, Melton DA. 1987. Antisense RNA injections in fertilized frog eggs reveal an RNA duplex unwinding activity. Cell 48: 599–605. 10.1016/0092-8674(87)90238-8 [DOI] [PubMed] [Google Scholar]
- Reenan RA. 2005. Molecular determinants and guided evolution of species-specific RNA editing. Nature 434: 409–413. 10.1038/nature03364 [DOI] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA. 2012. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248. 10.1038/ng.2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J, Paluch J, Dickman D, Joiner W. 2016. ADAR-mediated RNA editing suppresses sleep by acting as a brake on glutamatergic synaptic plasticity. Nat Commun 7: 10512. 10.1038/ncomms10512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB. 1999. Regulation of alternative splicing by RNA editing. Nature 399: 75–80. 10.1038/19992 [DOI] [PubMed] [Google Scholar]
- Rula EY, Lagrange AH, Jacobs MM, Hu N, Macdonald RL, Emeson RB. 2008. Developmental modulation of GABAA receptor function by RNA editing. J Neurosci 28: 6196–6201. 10.1523/JNEUROSCI.0443-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpietro V, Dixon CL, Guo H, Bello OD, Vandrovcova J, Efthymiou S, Maroofian R, Heimer G, Burglen L, Valence S, et al. 2019. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat Commun 10: 3094. 10.1038/s41467-019-10910-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Vissel B, Petralia RS, Wang Y-X, Chang K, Royle GA, Wang C-Y, O'Gorman S, Heinemann SF, Wenthold RJ. 2003. Aberrant formation of glutamate receptor complexes in hippocampal neurons of mice lacking the GluR2 AMPA receptor subunit. J Neurosci 23: 9367–9373. 10.1523/JNEUROSCI.23-28-09367.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansam CL, Wells KS, Emeson RB. 2003. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci 100: 14018–14023. 10.1073/pnas.2336131100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva Y, Rieder L, Reenan R. 2012a. The ADAR protein family. Genome Biol 13: 252. 10.1186/gb-2012-13-12-252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva YA, Jepson JEC, Sahin A, Sugden AU, Dorsky JS, Alpert L, Lawrence C, Reenan RA. 2012b. Auto-regulatory RNA editing fine-tunes mRNA re-coding and complex behaviour in Drosophila. Nat Commun 3: 790. 10.1038/ncomms1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva YA, Jepson JE, Chang Y-J, Whitaker R, Jones BC, St Laurent G, Tackett MR, Kapranov P, Jiang N, Du G. 2013. RNA editing regulates transposon-mediated heterochromatic gene silencing. Nat Commun 4: 2745. 10.1038/ncomms3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton PM, Duran A, Nakanishi Y, Reina-Campos M, Kasashima H, Llado V, Ma L, Campos A, García-Olmo D, García-Arranz M, et al. 2018. The secretion of miR-200s by a PKCζ/ADAR2 signaling axis promotes liver metastasis in colorectal cancer. Cell Rep 23: 1178–1191. 10.1016/j.celrep.2018.03.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov D, Gardiner K. 2002. Phylogenetic comparison of the pre-mRNA adenosine deaminase ADAR2 genes and transcripts: conservation and diversity in editing site sequence and alternative splicing patterns. Gene 299: 83–94. 10.1016/S0378-1119(02)01016-8 [DOI] [PubMed] [Google Scholar]
- Sommer B, Köhler M, Sprengel R, Seeburg PH. 1991. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67: 11–19. 10.1016/0092-8674(91)90568-J [DOI] [PubMed] [Google Scholar]
- Song B, Shiromoto Y, Minakuchi M, Nishikura K. 2022. The role of RNA editing enzyme ADAR1 in human disease. Wiley Interdiscip Rev RNA 13: e1665. 10.1002/wrna.1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefl R, Oberstrass FC, Hood JL, Jourdan M, Zimmermann M, Skrisovska L, Maris C, Peng L, Hofr C, Emeson RB, et al. 2010. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell 143: 225–237. 10.1016/j.cell.2010.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit AK, Matschke LA, Dolga AM, Rinné S, Decher N. 2014. RNA editing in the central cavity as a mechanism to regulate surface expression of the voltage-gated potassium channel Kv1.1. J Biol Chem 289: 26762–26771. 10.1074/jbc.M113.545731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. 2017. Dynamic landscape and regulation of RNA editing in mammals. Nature 550: 249–254. 10.1038/nature24041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TY, Sedmik J, Fitzgerald MP, Halevy RS, Keegan LP, Helbig I, Basel-Salmon L, Cohen L, Straussberg R, Chung WK, et al. 2020. Bi-allelic ADARB1 variants associated with microcephaly, intellectual disability, and seizures. Am J Hum Genet 106: 467–483. 10.1016/j.ajhg.2020.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariq A, Garncarz W, Handl C, Balik A, Pusch O, Jantsch MF. 2013. RNA-interacting proteins act as site-specific repressors of ADAR2-mediated RNA editing and fluctuate upon neuronal stimulation. Nucleic Acids Res 41: 2581–2593. 10.1093/nar/gks1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terajima H, Yoshitane H, Ozaki H, Suzuki Y, Shimba S, Kuroda S, Iwasaki W, Fukada Y. 2017. ADARB1 catalyzes circadian A-to-I editing and regulates RNA rhythm. Nat Genet 49: 146–151. 10.1038/ng.3731 [DOI] [PubMed] [Google Scholar]
- Thuy-Boun AS, Thomas JM, Grajo HL, Palumbo CM, Park S, Nguyen LT, Fisher AJ, Beal PA. 2020. Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res 48: 7958–7972. 10.1093/nar/gkaa532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonkin LA, Bass BL. 2003. Mutations in RNAi rescue aberrant chemotaxis of ADAR mutants. Science 302: 1725. 10.1126/science.1091340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran SS, Jun HI, Bahn JH, Azghadi A, Ramaswami G, Van Nostrand EL, Nguyen TB, Hsiao YE, Lee C, Pratt GA, et al. 2019. Widespread RNA editing dysregulation in brains from autistic individuals. Nat Neurosci 22: 25–36. 10.1038/s41593-018-0287-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbricht RJ, Emeson RB. 2014. One hundred million adenosine-to-inosine RNA editing sites: hearing through the noise. Bioessays 36: 730–735. 10.1002/bies.201400055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissel B, Royle GA, Christie BR, Schiffer HH, Ghetti A, Tritto T, Perez-Otano I, Radcliffe RA, Seamans J, Sejnowski T, et al. 2001. The role of RNA editing of kainate receptors in synaptic plasticity and seizures. Neuron 29: 217–227. 10.1016/S0896-6273(01)00192-1 [DOI] [PubMed] [Google Scholar]
- Vitali P, Basyuk E, Le Meur E, Bertrand E, Muscatelli F, Cavaille J, Huttenhofer A. 2005. ADAR2-mediated editing of RNA substrates in the nucleolus is inhibited by C/D small nucleolar RNAs. J Cell Biol 169: 745–753. 10.1083/jcb.200411129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Beal PA. 2016. Probing RNA recognition by human ADAR2 using a high-throughput mutagenesis method. Nucleic Acids Res 44: 9872–9880. 10.1093/nar/gkw799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Park S, Beal PA. 2018. Selective recognition of RNA substrates by ADAR deaminase domains. Biochemistry 57: 1640–1651. 10.1021/acs.biochem.7b01100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Lamm AT, Fire AZ. 2011. Competition between ADAR and RNAi pathways for an extensive class of RNA targets. Nat Struct Mol Biol 18: 1094–1101. 10.1038/nsmb.2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt GR. 1950. Occurrence of 5-methylcytosine in nucleic acids. Nature 166: 237–238. 10.1038/166237b0 [DOI] [PubMed] [Google Scholar]
- Yamashita T, Kwak S. 2019. Cell death cascade and molecular therapy in ADAR2-deficient motor neurons of ALS. Neurosci Res 144: 4–13. 10.1016/j.neures.2018.06.004 [DOI] [PubMed] [Google Scholar]
- Yamashita T, Hideyama T, Hachiga K, Teramoto S, Takano J, Iwata N, Saido TC, Kwak S. 2012. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat Commun 3: 1307. 10.1038/ncomms2303 [DOI] [PubMed] [Google Scholar]
- Yang L, Huang P, Li F, Zhao L, Zhang Y, Li S, Gan Z, Lin A, Li W, Liu Y. 2012. c-Jun amino-terminal kinase-1 mediates glucose-responsive upregulation of the RNA editing enzyme ADAR2 in pancreatic beta-cells. PLoS One 7: e48611. 10.1371/journal.pone.0048611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Z, Qu L, Tang H, Liu Z, Liu Y, Tian F, Wang C, Zhang X, Feng Z, Yu Y. 2022. Engineered circular ADAR-recruiting RNAs increase the efficiency and fidelity of RNA editing in vitro and in vivo. Nat Biotechnol 40: 946–955. 10.1038/s41587-021-01180-3 [DOI] [PubMed] [Google Scholar]
- Yi-Brunozzi HY, Stephens OM, Beal PA. 2001. Conformational changes that occur during an RNA editing adenosine deamination reaction. J Biol Chem 276: 37827–37833. 10.1074/jbc.M106299200 [DOI] [PubMed] [Google Scholar]
- Zaidan H, Ramaswami G, Golumbic YN, Sher N, Malik A, Barak M, Galiani D, Dekel N, Li JB, Gaisler-Salomon I. 2018. A-to-I RNA editing in the rat brain is age-dependent, region-specific and sensitive to environmental stress across generations. BMC Genomics 19: 28. 10.1186/s12864-017-4409-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Qian H, Xu J, Gao W. 2019. ADAR, the carcinogenesis mechanisms of ADAR and related clinical applications. Ann Transl Med 7: 686. 10.21037/atm.2019.11.06 [DOI] [PMC free article] [PubMed] [Google Scholar]