

Summary
Mitochondrial energetics and respiration have emerged as important factors in how cancer cells respond to or evade apoptotic signals. The study of the functional connection between these two processes may provide insight into questions old and new, such as: Why are respiration and apoptotic regulation housed in the same organelle? How might we target respiration or downstream signaling pathways to amplify apoptotic stress in the context of cancer therapy? Here we briefly review mitochondrial respiration and apoptosis, and then focus on how the intersection of these two processes is regulated by cytoplasmic signaling pathways such as the integrated stress response.
eTOC paragraph
Aerobic respiration and apoptosis are two of the most characteristic activities performed by mitochondria. Here we review how they are functionally interconnected via stress signaling pathways and describe how these connections might be exploited in the context of cancer therapy.
Introduction
Mitochondria perform critical metabolic and signaling functions within nearly all eukaryotic cells. Cancer cells are no exception, and they make unique demands of their mitochondria to resource and power the neoplastic process. Our understanding of mitochondria in cancer has evolved to include key metabolic processes such as energy homeostasis, redox balance and macromolecular biosynthesis, which support cancer cell survival and proliferation, while also including apoptosis. The two most characteristic functions of mitochondria, respiration and “expiration,” are in constant conflict in cancer cells, but they remain intricately intertwined with one another.
Mitochondria and the electron transport chain in cancer
Mitochondria are the product of an evolutionary endosymbiotic event, leaving them bound by two membranes (Archibald, 2015). The outer and inner mitochondrial membranes (OMM and IMM) are separated by an intermembrane space (IMS), with the innermost compartment being known as the mitochondrial matrix (Friedman and Nunnari, 2014). Among many other functions, mitochondria transport high energy electrons along a conduit of protein complexes and electron carriers in the IMM to eventually harness fuel in the form of adenosine triphosphate (ATP). NADH and succinate donate electrons as hydride ions to respiratory complexes I and II, respectively. Transport of these electrons to their destination of molecular oxygen on complex IV is coupled to proton pumping from the mitochondrial matrix to the IMS. This process of “aerobic respiration” is how the majority of oxygen is consumed by eukaryotic cells, and generates an electrochemical gradient across the IMM that complex V, the ATP synthase, uses to make ATP.
Cancer cell metabolism
Understanding the contributions of aerobic respiration to cancer cell biology has not been straightforward (DeBerardinis and Chandel, 2016; Spinelli and Haigis, 2018). Nearly a century ago, prominent biochemists, particularly the Nobel laureate Otto Warburg, believed that “irreversible injuring of respiration” not only occurred in cancer cells, but in fact was the universal cause of cancer (Warburg, 1956). Today, “The Warburg Effect,” refers to the tendency of cancer cells to avidly uptake glucose, perform glycolysis, and ferment pyruvate to lactate despite the presence of sufficient oxygen for aerobic respiration (DeBerardinis and Chandel, 2020; Schell et al., 2014). Although the interpretation of Warburg’s findings has changed, “aerobic glycolysis” is still a dominant feature in the metabolic profile of cancer cells (Luengo et al., 2021). Aerobic glycolysis is associated with cell proliferation in many normal and disease contexts, and can promote tumor initiation and growth (Bensard et al., 2020; Luengo et al., 2021).
The scientific pendulum has swung decidedly away from Warburg’s original claim of injured respiration in cancer cells. The predominant view in the field today regarding the role that mitochondria fill in the cancer cell is more nuanced. This view posits that there are many interdependent functions of mitochondria that are important for the cancer cell phenotype, including but not limited to: Redox regulation, signaling, biosynthesis of essential molecules, energy capture in ATP, and regulation of apoptosis. The activity of the electron transport chain (ETC) is intimately tied to each of those functions.
What’s ATP got to do with it?
A surprising concept for cancer metabolism neophytes is that the ETC supports cancer cell proliferation in ways that extend beyond ATP generation alone. Over the past several years, it has become clear that the essential function of the ETC in cancer cells is the recycling of electron acceptors that are needed for the biosynthesis of crucial macromolecules. To illustrate this point, we will highlight two key discoveries.
First, let us consider how the ETC contributes to aspartate synthesis. In every redox reaction, one reactant is reduced while another is oxidized– which conversion is more important depends on the context. In an “ATP-centric” view of the ETC, the key function of the tricarboxylic acid (TCA) cycle is to generate NADH and succinate, which then donate electrons to complex I and II to enable the generation of a proton gradient. From this perspective, the “important” redox state of NAD/NADH is NADH, and NAD+ is a mere byproduct. But in its role as an electron acceptor, complex I extracts an electron to oxidize NADH and regenerate NAD+, which is itself an essential electron acceptor used by other dehydrogenases, including malate dehydrogenase (MDH2). Within the mitochondrial matrix, MDH2 generates oxaloacetate, which can be converted to aspartate. In contrast to the “ATP-centric” perspective of the ETC, free NAD+ is really what is important in the context of proliferating cells – the dominant “purpose” of complex I is simply to regenerate NAD+ from NADH in order to make NAD+ available for other reactions, like that catalyzed by MDH2 to enable aspartate synthesis. Indeed, supplying cells with an alternative electron acceptor in alpha-ketobutyrate rescued ETC inhibition with respect to cell proliferation, as did supplying exogenous aspartate (Birsoy et al., 2015; Sullivan et al., 2015). The surprising conclusion of these studies is that the essential role of ETC activity in the context of proliferating cells is generating aspartate (Van Vranken and Rutter, 2015).
Second, new insights into the role of complex III in cancer have similarly turned the “powerhouse-of-the-cell” thinking on its head (Martínez-Reyes et al., 2020). The recent work by Chandel and colleagues reveals that, in the context of cancer, what is important about complex III accepting electrons from ubiquinol is simply that it generates ubiquinone, an obligate electron acceptor for DHODH, complex I, and complex II. The authors used ectopic expression of alternative oxidase (AOX), a ubiquinol oxidase present in tunicates, to bypass complex III. Although AOX does not pump protons, it does oxidize ubiquinol to ubiquinone. Hence, ectopic expression of AOX in complex III-deficient cells enables one to separate the two main functions of complex III, proton pumping toward ATP synthesis and ubiquinone regeneration, with only the latter being rescued by AOX. Surprisingly, AOX expression was sufficient to rescue tumor growth defects caused by complex III deficiency in vivo. Inhibition of complex I, complex II, or DHODH prevented the rescue of tumor growth by AOX, demonstrating that all three of these activities are essential and are enabled by complex III function in cancer. Altogether, these intriguing findings demonstrate that ETC activity is important for proliferating cancer cells, at least in these experimental contexts, simply because it gives the cell a way to dispose of high energy electrons by donating them to oxygen, thereby regenerating cofactors like NAD+ and ubiquinone.
Metformin and related drugs reveal ETC dependency in cancer
As described above, complex I initiates the ETC by extracting high energy electrons from NADH and contributes to the mitochondrial membrane potential by pumping protons into the IMS. Despite the importance of complex I for normal cellular physiology, there appears to be a tractable therapeutic index for complex I inhibitors in the context of cancer. Metformin is used clinically to treat type 2 diabetes and is one of the most widely prescribed drugs in the world. The effects of metformin treatment on an organismal scale are complex, but a key effect of metformin on a cellular scale is the inhibition of complex I. For more information on metformin and complex I in cancer, the reader is referred to a recent and thorough review on this subject by Chandel and colleagues (Vasan et al., 2020).
How the cell senses and responds to ETC dysfunction
Since the ETC is essential for a wide variety of important cellular functions, it is not surprising that mechanisms have evolved to respond to ETC dysfunction and trigger adaptive changes. One important pathway activated by ETC dysfunction is the integrated stress response (ISR). Here we will briefly describe the ISR but the reader is referred to in-depth reviews on the topic for more information (Costa-Mattioli and Walter, 2020; Pakos-Zebrucka et al., 2016).
The Integrated Stress Response
The ISR is triggered by diverse stimuli, including heat stress, amino acid deprivation, and invasion by pathogens. The central feature of the ISR is the phosphorylation and inactivation of a translation initiation factor, eIF2α, which reduces the number of ternary complexes that can initiate cap-dependent translation (Harding et al., 2003; Hinnebusch, 2014; Pavitt et al., 1998). In mammals, four kinases are known to phosphorylate eIF2α and each does so in response to distinct stressors. For instance, uncharged tRNAs allosterically activate the kinase GCN2 to phosphorylate eIF2α. Regardless of the kinase responsible, eIF2α phosphorylation leads to a decreased rate of cap-dependent translation and a reduction in protein synthesis. In addition to a global decrease in translation rate, the reduction in ternary complexes also causes ribosomes to skip over inhibitory upstream open reading frames (uORFs) to increase translation of a subset of transcripts. One of these proteins induced by the ISR is the transcription factor ATF4, which initiates a transcriptional program with two temporal phases: Adaptive and apoptotic. The first phase of the ISR attempts to help the cell survive stress while the second phase kills cells that fail to successfully adapt. The pro-homeostatic output of ATF4 is complex but includes activation of autophagy (at least partially through suppressing mTORC1 via Sestrin2 and Redd1), induction of metabolic enzymes and transporters to alleviate nutrient deficiency, and production of chaperones (Condon et al., 2021). When adaptation fails nonetheless, the ISR promotes cell death, which we will discuss in detail in later sections.
It has been known for years that mitochondrial dysfunction –and ETC dysfunction in particular– activates the ISR (Mick et al., 2020; Quirós et al., 2017). For instance, the Mootha lab recently demonstrated that ETC inhibition can activate the ISR via depletion of aspartate and asparagine (Mick et al., 2020). As described above, amino acid depletion activates the ISR through the accumulation of uncharged cognate tRNAs, which bind to GCN2, an eIF2α kinase. In some contexts, mitochondrial dysfunction activates the ISR even when aspartate or other amino acids are replete (Mick et al., 2020), which indicates the presence of alternate pathways that communicate mitochondrial dysfunction to the ISR.
Mitochondrial dysfunction activates the ISR through an OMA1-DELE1-HRI pathway
Recently, two groups used complementary genetic screening approaches to decipher how mitochondrial stress is relayed to the ISR (Fessler et al., 2020; Guo et al., 2020). A clear signal emerged from both screens– the kinase heme-regulated inhibitor (HRI) was necessary for ISR activation in this context. Interestingly, HRI was required to activate the ISR in response to diverse stressors, such as oligomycin and CCCP. Oligomycin inhibits ATP Synthase and increases membrane potential across the IMM, while CCCP is a protonophore that dissipates IMM potential. The fact that two agents with opposing effects on membrane potential activate the same stress response pathway points to a shared sensing mechanism that could be fundamental to mitochondrial homeostasis (Eckl et al., 2021).
How does ETC dysfunction activate HRI? Another gene required for ISR activation in this context was OMA1, which encodes a stress-activated mitochondrial protease localized to the IMM (Anand et al., 2014; MacVicar and Langer, 2016). OMA1 becomes active in response to stresses as varied as heat shock, CCCP, oligomycin, hydrogen peroxide, and others. The canonical substrates of OMA1 are in the IMM/IMS, particularly the large GTPase OPA1 that is required for cristae junction formation and mitochondrial fusion (Frezza et al., 2006; Giacomello et al., 2020). In these studies, the authors discovered that OMA1 cleaves the poorly characterized IMS protein DELE1, generating DELE1-S, a shorter fragment that translocates to the cytoplasm. Here, DELE1-S binds and allosterically activates HRI to promote eIF2α phosphorylation and activate the ISR (Figure 1).
Figure 1: How mitochondria send and receive stress signals.
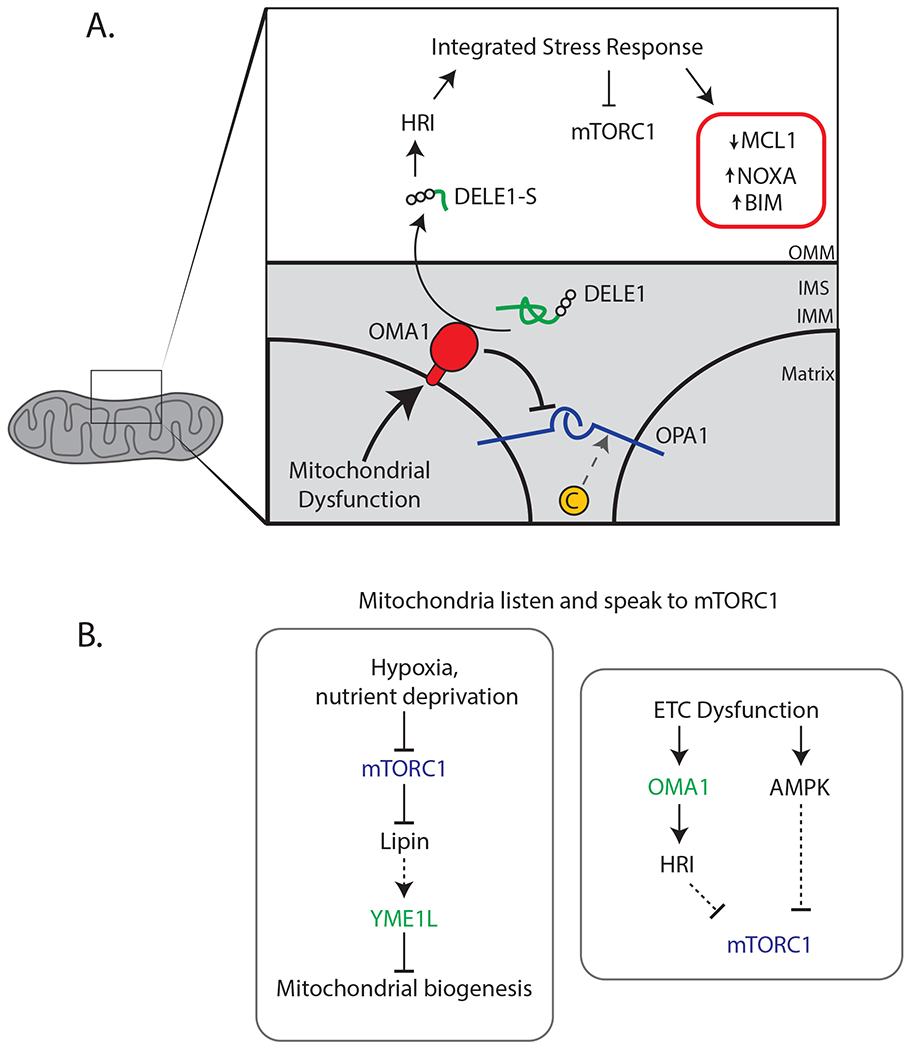
(A) Overview of a mito-nuclear signaling pathway that communicates mitochondrial dysfunction via the protein messenger DELE1. The protease OMA1 cleaves DELE1 to generate a short fragment (DELE1-S) that migrates to the cytoplasm and allosterically activates the eIF2α, kinase HRI. Phosphorylation of eIF2α, triggers the integrated stress response (ISR), which is a pleiotropic transcriptional program. The ISR inhibits mTORC1 via Sestrin2 and Redd1, leading to activation of autophagy and inhibition of translation. Other effects of the ISR include activation of the pro-apoptotic BH3-only proteins NOXA and BIM and a reduction in levels of the anti-apoptotic factor MCL1, which contribute to the pro-death phase of the ISR. (B) The mTORC1 pathway modulates mitochondrial function in part through the IMM protease YME1L. In cancer, this signaling pathway may serve to preserve anaplerotic capability during nutrient stress. Reciprocal communication is executed by a distinct but related IMM protease, OMA1, and also through AMPK.
OMA1 is somewhat unique among stress-activated proteins in that it responds to a wide variety of fundamentally different stressors (Baker et al., 2014). For instance, oligomycin, which inhibits ATP Synthase and hyperpolarizes the IMM, activates OMA1, but so does antimycin A, an inhibitor of complex III that depolarizes the IMM. Additionally, OMA1 is activated during apoptosis to trigger cristae remodeling and cytochrome C release from the IMS into the cytoplasm (Jiang et al., 2014). But, how OMA1 is activated by these stimuli remains an open question, the answer to which would have important implications in the treatment of malignancy.
The discovery of this OMA1-DELE1-HRI pathway raises interesting mechanistic questions. For instance, how does DELE1-S exit the IMS after cleavage and translocate to the cytoplasm? The efflux of proteins from the IMS to the cytoplasm is a tightly regulated process with life-or-death consequences. As we will explain below, cytoplasmic translocation of IMS factors such as cytochrome C directly activates apoptosis. The molecular weight of DELE1-S is approximately 30 kilodaltons (kD), while that of cytochrome C is ≈12 kD (Liu et al., 1996). If the larger DELE1-S can cross the OMM, what is to prevent cytochrome C from doing so, activating cytoplasmic APAF1, and thereby triggering apoptosis? One potential explanation is that cytochrome C is sequestered in cristae, which prevent its diffusion across the OMM. The membrane folds of cristae are clamped together at their opening by proteinaceous complexes known as cristae junctions, of which OPA1 is a key component. OPA1 is a large GTPase that mediates IMM fusion in addition to maintaining cristae junctions. However, in response to stress, OMA1 cleaves OPA1 and disrupts cristae junctions, so one might expect that cytochrome C would be “free” in the IMS under the same conditions in which DELE1 is cleaved (both depend on OMA1).
Another possibility is that DELE1 does not fully cross the OMM but stalls in the TOMM complex (Translocase of the Outer Mitochondrial Membrane), which is a translocation channel that imports proteins across the mitochondrial outer membrane (Schmidt et al., 2010). It could be that DELE1 stalls in the TOMM complex, and the N-terminus penetrates the IMS and is cleaved, which triggers the release of the C terminus back into the cytoplasm. There is precedent in mammals for similar mechanisms, illustrated by PINK1 stalling in the TOMM complex in response to IMM depolarization and recruiting the ubiquitin E3 ligase Parkin, which subsequently activates autophagic destruction of mitochondria (Harper et al., 2018; Pickrell and Youle, 2015). Further, in yeast there are multiple examples of TOMM substrates stalling during import and requiring active extraction by the AAA+ ATPases Cdc48 or Msp1 (Basch et al., 2020; Mårtensson et al., 2019; Weidberg and Amon, 2018). In yeast, similar mechanisms even signal via a retrograde pathway to the nucleus, which is reminiscent of the DELE1-ISR pathway (Boos et al., 2019).
Further work from the Jae lab provided additional insight into the molecular biology of DELE1, showing that DELE1 undergoes distinct processing events in response to distinct mitochondrial perturbations (Fessler et al., 2022). It was proposed that DELE1 acts as a sensor for mitochondrial protein import efficiency (Fessler et al., 2022), akin to a simpler mechanism mediated by ATFS-1 in C. elegans (Nargund et al., 2012). Thus, it appears that whether DELE1 is released from the IMS, or never imported in the first place, depends on the mitochondrial perturbation in question.
Mitochondria speak and listen to mTORC1
The mTORC1 complex regulates countless metabolic and signaling pathways and is activated in a variety of cancers (Okosun et al., 2016). Briefly, mTORC1 activity is subject to multiple inputs that convey the nutrient status of the cell. In the nutrient replete state, mTORC1 phosphorylates substrates such as 4E-BP1, which prevents its inhibitory binding of eIF4e preventing it from interacting with eIF4e and inhibiting cap-dependent translation (Saxton and Sabatini, 2017). It is intuitive that this key nutrient sensor would communicate with the metabolic hub of the cell, the mitochondrion.
A recent report demonstrated how the cell uses a protease, YME1L, to communicate nutrient status to mitochondria through the mTORC1 pathway (MacVicar et al., 2019). Briefly, nutrient deprivation inhibits mTORC1, relieving inhibition of the enzyme Lipin, and eventually decreasing phosphatidylethanolamine (PE) in the IMM. Low PE levels activate YME1L, which then cleaves a number of substrates to change mitochondrial behavior (MacVicar and Langer, 2016). For instance, YME1L directly cleaves components of the mitochondrial protein import channel, which decreases the rate of protein import. It is expected that decreased protein import would generally suppress mitochondrial activity, but it is also possible that this serves as a de facto filter to selectively import a subset of mitochondrial proteins. YME1L also cleaves phospholipid transfer proteins, resulting in altered distribution of phospholipids between the OMM and IMM (MacVicar et al., 2019).
This work reveals one mechanism by which cells sense poor nutrient status and respond by suppressing mitochondrial biogenesis. Precisely how YME1L “rewires” metabolism remains to be fleshed out, but the authors conducted insightful experiments using a protein import assay in isolated mitochondria. The authors treated cells with the mTORC1 inhibitor Torin, isolated mitochondria, and conducted protein import assays. Torin treatment decreased mitochondrial protein import efficiency in a YME1L-dependent manner, as did hypoxia treatment.
There is an interesting predilection of pancreatic ductal adenocarcinoma (PDAC) cells for this pathway connecting nutrient stress to YME1L, which has been proposed to sacrifice mitochondrial biogenesis for the sake of preserving anaplerosis. PDAC spheroids required YME1L for optimal growth, while hepatocellular carcinoma cell spheroids did not (MacVicar et al., 2019). PDAC tumors are some of the most nutrient and oxygen-starved malignancies, so adaptation to this context could explain why the regulation of mitochondrial activity via YME1L is so critical in that tumor type (Encarnación-Rosado and Kimmelman, 2021; Olivares et al., 2017). Since mTORC1 signaling is elevated in a large fraction of human tumors, and potent small molecule inhibitors of mTORC1 are already used clinically, it is important to understand the mTORC1 effects on mitochondria, including through YME1L. These findings likely extend beyond PDAC, and it will be important to understand how this signaling axis responds to other nutrient stressors and acts in other tumor contexts.
There is an intriguing inhibitory connection between YME1L and OMA1. In a physiological setting, it was shown that concomitant Oma1 knockout rescued the cardiac phenotype of Yme1l knockout mice, suggesting that unrestrained Oma1 activity is a consequence of Yme1l deficiency in mice (Wai et al., 2015). OMA1 becomes active in response to IMM depolarization, but YME1L quenches OMA1 activity via proteolysis when there is sufficient ATP available (Rainbolt et al., 2016). This is a fascinating case where a direct metabolic input, ATP, could change mitochondrial behavior and likely has relevance to cancer. For instance, it was shown that low expression of OMA1 predicted poor prognosis in breast cancer and silencing OMA1 in cell lines increased proliferation and migration (Daverey et al., 2019).
Just as mTORC1 inhibition inhibits mitochondrial biogenesis, ETC dysfunction can inhibit mTORC1. It was recently discovered that ETC dysfunction suppresses mTORC1 via a two-pronged response featuring the ISR and AMPK (Condon et al., 2021). Corroborating the work described above on DELE1, oligomycin required HRI to relay stress to the rest of the cell. HRI activated ATF4, which induced transcription of the mTORC1 inhibitors Redd1 and Sestrin2._(Figure 1). This study provides a helpful exhibit of how the HRI-dependent program is beneficial to cells. HRI-null cells hyperaccumulate adenosine monophosphate (AMP) in response to oligomycin, and fail to suppress AMP back to low levels within the time frame observed (six hours), in contrast to wildtype cells which quickly restored normal AMP levels.
The cell senses AMP concentration through an evolutionarily conserved pathway, wherein AMP directly binds and allosterically activates a subunit of the AMP kinase (AMPK) (Garcia and Shaw, 2017; Herzig and Shaw, 2018). Interpreting increased AMP as a sign of mitochondrial dysfunction is a direct and elegant way that the cell can respond to defective respiration, since this derives from reduced ATP synthase activity. This is one example of how metabolites can act as signaling molecules but there is a need for more work in this area. To understand how AMPK can influence mitochondrial form and function, consider work by Shaw and colleagues that discovered that AMPK phosphorylates mitochondrial fission factor (MFF) (Toyama et al., 2016). MFF is a transmembrane protein localized to the OMM that functions as a docking site for dynamin-related protein 1 (DRP1), a GTPase that catalyzes mitochondrial fission (Otera et al., 2010). Phosphorylation of MFF by AMPK activates it and promotes DRP1 recruitment. AMPK was necessary for oligomycin-induced fragmentation of the mitochondrial network in cultured cells, and AMPK mediated this response in a manner of minutes after ETC dysfunction. Fragmentation of mitochondria is thought to facilitate degradation of defective organelles by mitophagy. AMPK couples these two processes, mitochondrial fission and mitophagy, by phosphorylating and activating MFF and ULK1, the latter being the initial component of autophagy pathway. Additionally, mTORC1 phosphorylates ULK1 to inhibit it, so suppression of mTORC1 by ETC dysfunction further promotes autophagy (Kim et al., 2011).
Altogether these results indicate that mitochondria communicate with nutrient and stress-sensing pathways in the cell. The “language” of this communication can be protein or metabolite based, and both are activated by ETC dysfunction.
Apoptosis
Mitochondria also control apoptosis, with outcomes that diametrically oppose the function of generating electron acceptors to fuel proliferation. An elaborate “dance” of protein binding partners tightly regulates the initiation of apoptosis (Kale et al., 2018), which occurs principally on the OMM but also requires coordinated changes in the IMM.
MOMP and the BCL2 family
Mitochondrial outer membrane permeabilization (MOMP) is the process by which the proteins BAX and BAK oligomerize to form pores in the OMM. MOMP results in the release of IMS proteins into the cytosol, including cytochrome C, SMAC/Diablo, HTRA2/OMI, and others. These IMS factors activate the caspase cascade and initiate the apoptotic process (Letai, 2017). In a healthy cell, BAX and BAK exhibit low intrinsic activity and are also inhibited by direct binding to anti-apoptotic proteins of the BCL2 family, such as BCL2 itself, BCL-xL, and MCL1 (Figure 2 (Diepstraten et al., 2022)). Upstream BH3-only proteins stimulate MOMP induction by inhibiting anti-apoptotic family members (such as BCL2 and MCL1) and in some cases by directly activating BAX/BAK (Kale et al., 2018). In particular, the BH3-only proteins BIM and BID are direct activators of BAX/BAK. In many tumor cells, activated BH3-only proteins occupy the reservoir of anti-apoptotic proteins on the OMM. When cells lack free anti-apoptotic proteins such as MCL1 they are highly sensitive to further apoptotic stimuli and are said to be “primed” for apoptosis (Bhatt et al., 2020; Letai, 2017).
Figure 2). Processes throughout the mitochondrion can affect apoptotic priming.
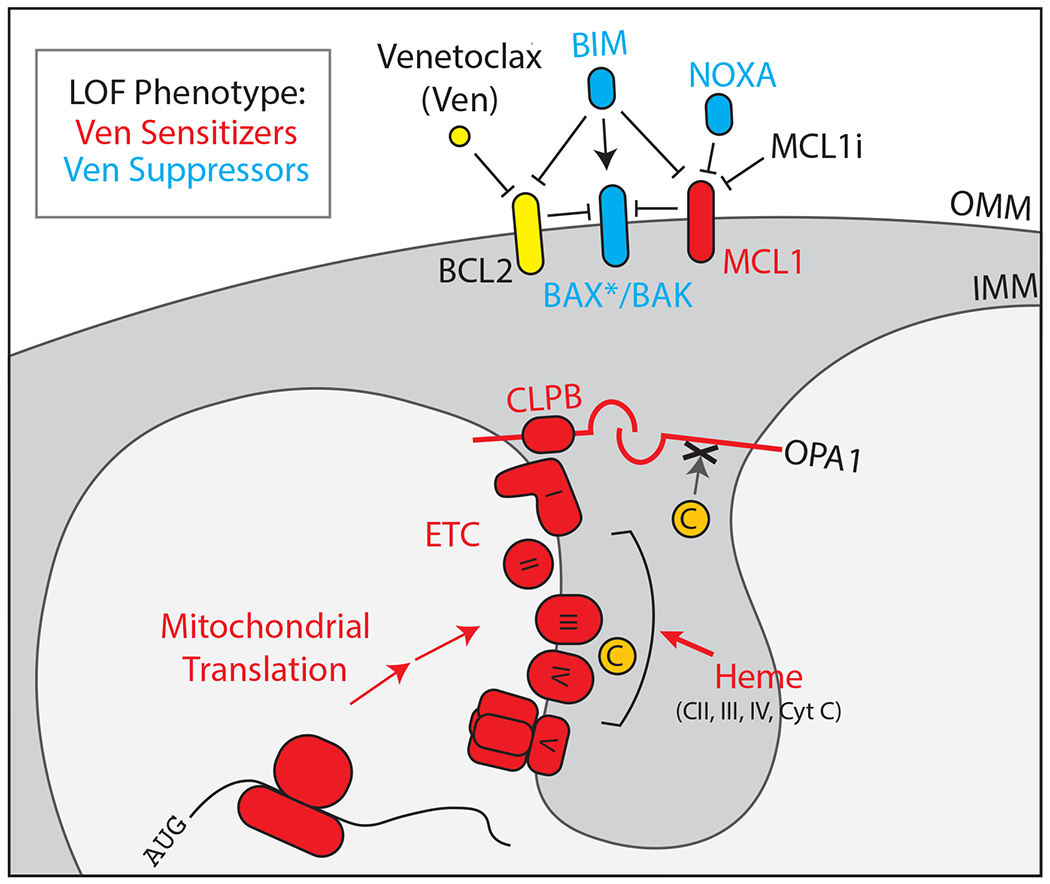
Venetoclax binds and inhibits BCL2, liberating pro-apoptotic factors that trigger the permeabilization of the OMM by BAX/BAK. Loss of function (e.g. genetic knockout) in proteins shown in cyan makes the cell resistant to Venetoclax. Loss of function in proteins shown in red causes the cell to become more sensitive to Venetoclax, and in some cases can reverse established Venetoclax resistance occurring in vivo. BAX* specifies active BAX that is bound to the OMM, as opposed to cytosolic BAX that exists under basal conditions. OPA1 and cristae junctions suppress cytochrome C efflux during MOMP, as indicated by the “X”.
Small molecule BH3 mimetics that inhibit anti-apoptotic proteins have yielded clinical success in particular cancers, such as the BCL2 inhibitor Venetoclax in CLL (Caenepeel et al., 2018; Greaves et al., 2018; Kotschy et al., 2016; Roberts et al., 2016). However, many malignancies have intrinsic or acquired resistance to these drugs. Understanding how to prime cells for apoptosis, thereby sensitizing them to BH3-mimetics, could translate into clinical benefit for many patients (Soderquist et al., 2018).
Circumventing Venetoclax
Many different processes have emerged as sensitizing or suppressing sensitivity to BH3 mimetics, suggesting that they functionally interact with apoptotic susceptibility. First, we describe work on mechanisms of apoptotic priming that localize to the OMM, where historically the focus has been. We then delve into novel and unexpected interactions occurring “below the surface” of the OMM. Understanding how these processes relate is more than an academic exercise: Many cancer patients succumb to their disease in spite of novel BH3 mimetic drugs, and unexpected mitochondrial functions may explain such resistance. Understanding how mitochondrial functions -including respiration- affect the response of cells to pro-apoptotic stimuli could help cancer patients live longer, healthier lives. The majority of the work we describe has been conducted in the context of leukemia, namely acute myeloid leukemia (AML) and chronic lymphocytic leukemia (CLL).
There are at least three established mechanisms whereby AML cells circumvent Venetoclax. First, point mutations in the Venetoclax binding pocket of BCL2 have been shown to cause drug resistance (Blombery et al., 2019). This finding demonstrates the on-target activity of Venetoclax in human cancer patients. However, we must acknowledge an important study by Tait and colleagues in which Venetoclax -at high concentrations- can have BCL2-independent effects on metabolism (Roca-Portoles et al., 2020). Even so, most studies we detail below use concentrations of Venetoclax that are up to 500-fold lower than what has been shown to have BCL2-independent effects (Lin et al., 2019). A second mechanism of resistance is the suppression of BAX/BAK downstream of BCL2 (Bhatt et al., 2020). Loss of BAX/BAK can make cells completely refractory to Venetoclax despite not affecting BCL2 itself. The third mechanism is the upregulation of other anti-apoptotic proteins that complement BCL2, such as MCL1 (Guièze et al., 2019). Upregulation of MCL1 appears to be a particularly widespread mechanism of resistance, and many signaling pathways converge on MCL1, such as MAPK/ERK, mTORC1, and AKT. All three of these examples occur on the OMM and fit nicely into current models of BCL2 family biology. New work in cell death research has uncovered how other processes affect apoptotic priming in unexpected ways.
What lies beneath: How the IMM affects apoptotic priming
CRISPR-based genetic screens enable robust interrogation of mammalian biology, and several groups have leveraged this technology to search for genes that either sensitize to or suppress apoptosis triggered by Venetoclax (Chen et al., 2019; Guièze et al., 2019; Lin et al., 2019; Sharon et al., 2019; Smith et al., 2020). It is important to point out that these screens tend to identify known genetic interactors with BCL2, which serves to validate the approach. For instance, deletion of NOXA (PMAIP1) and BAX protect cells from Venetoclax in multiple contexts, primarily in AML culture models. In addition to these conventional apoptosis-related genes, other genes that affect Venetoclax sensitivity function in mitochondrial translation, cristae structure (ClpB/Skd3), heme biosynthesis, and energy-sensing pathways like AMPK (Figure 2). All of these pathways converge on or respond to the ETC, and indeed many studies have found that ETC poisons synergize with Venetoclax (Bajpai et al., 2020; Chan et al., 2015). These findings raise interesting questions about intrinsic apoptosis. For instance, can IMM processes like cristae deconstruction act upstream of MOMP? Which steps in intrinsic apoptosis are rate-limiting in different contexts? And crucially, how does the cell respond to ETC dysfunction to modulate apoptotic priming?
The structure and shape of mitochondria modulate the cell’s response to apoptotic stimuli, and the regulation of cristae by OPA1 exemplifies this interplay. In response to a variety of stressors, the IMM protease OMA1 becomes active and cleaves OPA1 to disrupt cristae junctions. Cleavage of OPA1 generates a short form of the protein, OPA1-S, that is incompetent in forming cristae junctions or mediating IMM fusion. The disruption of cristae junctions is an important mechanistic step in apoptosis because it releases cytochrome C from the IMM to diffuse through BAX/BAK pores and activate APAF1 in the cytoplasm (Frezza et al., 2006). Beautiful genetic experiments have shown how crucial OMA1 and OPA1 are for apoptosis: Overexpression of OPA1 or deletion of OMA1 suppresses apoptosis while deletion of OPA1 sensitizes to apoptosis. Remarkably, OPA1 overexpression suppresses apoptosis despite the formation of BAX/BAK pores in the OMM, confirming that OPA1 acts downstream of MOMP in the conventional view (Frezza et al., 2006; MacVicar and Langer, 2016; Yamaguchi et al., 2008).
The maintenance of cristae junctions by OPA1 may be relevant to Venetoclax resistance. Chen, et al. conducted a genome-wide CRISPR screen for gene deletions that re-sensitized resistant AML cells to Venetoclax and identified CLPB, which encodes an IMS protein (Chen et al., 2019). Increased levels of ClpB were also identified in a subset of Venetoclax-resistant AML patient derived xenografts (PDXs) in an independent study (Bhatt et al., 2020). It was proposed that ClpB acts as a chaperone to stabilize OPA1 and maintain cristae junctions, which causes resistance to MOMP induced by Venetoclax. This model raises some questions that pertain to our discussion. For instance, since MOMP activates OMA1 (Jiang et al., 2014) to cleave OPA1, which step in that sequence does ClpB inhibit? ClpB, also known as Skd3, is a AAA+ ATPase that disassembles protein aggregates in vitro (Cupo and Shorter, 2020) – is this disaggregase activity relevant to its role in cristae junction maintenance?
The involvement of IMM lipids in cytochrome C release and consequent initiation of the apoptotic cascade is a significant mechanism through which the IMM modulates apoptotic priming. The IMM-resident lipid cardiolipin (CL) plays a major role in this process by virtue of its ability to bind cytochrome C, serving as an anchor that impedes the translocation of cytochrome C to the cytosol (Ott et al., 2007). CL is an interesting lipid in this regard as it is closely involved in the assembly and functional integrity of ETC supercomplexes (Arnarez et al., 2016). In fact, CL can directly bind these complexes and disrupting this interaction results in a near complete loss of mitochondrial respiratory activity, indicating that CL is indispensable for mitochondrial function (Musatov and Robinson, 2014). Further, CL is located adjacent to ETC complexes in the IMM- a major source of ROS- thus making it particularly prone to undergo oxidation. CL oxidation is highly important for cytochrome C release during apoptotic cascade, implying that ETC activity has a direct functional connection with apoptosis progression through the oxidative state of CL.
The onset of apoptosis in a cell is characterized by changes such as mitochondrial depolarization, membrane disruption and the accumulation of oxidizing agents such as H2O2. These changes in the mitochondrial membrane that make more and more CL available to bind cytochrome C, leading to the formation of the CL-cytochrome C complex (Bergstrom et al., 2013; Raemy and Martinou, 2014). Upon binding, hydrophobic interactions with CL change heme group coordination in cytochrome C and induce an increase in its peroxidase activity. Thus, as part of the complex, cytochrome C can now oxidize CL, a decisive step that signifies cytochrome C taking up a pro-apoptotic role (Belikova et al., 2006). CL oxidation by cytochrome C leads to the dissociation of the CL-cytochrome C complex, resulting in cytochrome C release and the onset of apoptosis (Kagan et al., 2005; Santucci et al., 2019). Two things therefore become clear from our discussion- one, CL oxidation might be a critical modality by which the mitochondrial redox biology informs apoptosis progression and second, the IMM integrates multiple cues from within and outside mitochondria and thus contributes extensively to the complexity of the apoptotic process. Understanding how mitochondria respond to different oxidative stresses or cues will be an important area for future research. Recent discoveries described a mechanism by which mitochondria import cytosolic gluthione (GSH), an important regulator of redox metabolism (Shi et al., 2022; Wang et al., 2021). This pathway will undoubtedly be relevant in cancer cells, since many type of cancer rely on a carefully curated balance of oxidative metabolism (Sullivan and Chandel, 2014).
Another trait of the IMM that is as characteristic as cristae is a membrane potential that results from pumping of protons into the IMS. Depolarization of the IMM is a cardinal feature of apoptosis and occurs downstream of MOMP. Work from Wood and colleagues proposed that IMM depolarization explained their finding that inhibiting heme biosynthesis sensitized cells to Venetoclax (Lin et al., 2019). Their genetic screening approach generated a “metabolic map of apoptosis,” and found that deletion of 7 out of 8 genes in the heme biosynthesis pathway sensitized cells to Venetoclax. ETC complexes II, III, and IV contain heme protein subunits and require heme to function; as expected, inhibition of heme biosynthesis by pharmacologic or genetic means decreased IMM potential. It is also possible that heme might act in a specific way to regulate apoptosis. IMM depolarization was shown to be sufficient for Venetoclax synergy, but necessity was not tested. It seems unlikely that IMM depolarization per se is required to sensitize to Venetoclax, since the Complex V inhibitor oligomycin also synergizes with Venetoclax despite hyperpolarizing IMM potential. How else might ETC dysfunction and resultant changes in IMM potential affect apoptotic priming? New discoveries on how ETC dysfunction signals to the rest of the cell may provide insight on this question. In particular, IMM depolarization is known to activate the integrated stress response (ISR), a complex signaling pathway with direct contributions to apoptotic priming. Loss of ClpB in the Chen, et al study described above also activated the ISR. As we alluded to, ISR activation is a prime candidate to explain how IMM processes like ETC dysfunction could influence apoptotic priming indirectly.
How do these new findings on mitochondrial stress signaling inform our discussion of apoptotic priming and cancer therapy?
Together, the inhibition of mTORC1 and activation of the ISR constitutes a dramatic shift in cellular behavior in response to ETC dysfunction. While this mechanism is typically acutely adaptive, prolonged activation pushes the cell toward apoptosis. Decreased translation rate and increased production of chaperones helps to alleviate proteotoxic stress in the short term. Indeed, this pathway protected cells from some mitochondrial stressors (such as deletion of MFN2, encoding Mitofusin2), while sensitizing to others (e.g. CCCP) (Fessler et al., 2020; Guo et al., 2020). Understanding how the duration and magnitude of these signals affects the net outcome on cell fitness will be important to translate these findings into the context of human disease.
The signaling output of mTORC1 inhibition and ISR activation is complex, but there are clear predictions on how it will affect apoptotic priming, and direct evidence of these effects is apparent in the literature._Decreased cap-dependent translation (from eIF2α phosphorylation or mTORC1 inhibition) promptly decreases steady state levels of short-lived proteins such as MCL1. ATF4-dependent activation of CHOP activates transcription of BH3 only proteins NOXA and BIM, which bind and inhibit MCL1 or all anti-apoptotic proteins, respectively._Indeed, the transcriptomics data provided by Fessler, et al reveal that depolarization of mitochondria with CCCP induced the transcription of NOXA (PMAIP1), but this was blocked by HRI deletion or treatment with ISRIB, a small-molecule inhibitor of the ISR. This finding fits with the observation that deletion of HRI conferred resistance to CCCP relative to wildtype, i.e. in the case of CCCP, HRI-mediated signaling promoted cell death.
Within the context of DELE1-HRI signaling, it is interesting to revisit the work by Lin et al, who used a metabolism-focused sgRNA library to describe an “apoptotic map of metabolism” in the context of AML. This study convincingly demonstrated that interfering with heme biosynthesis (through pharmacologic or genetic means) sensitizes AML cell lines to Venetoclax. The authors suggest that decreased heme inhibits complexes II, III, and IV, which decreases membrane potential across the IMM and accelerates the release of IMS factors (e.g. cytochrome C, Smac) through BAX/BAK pores and into the cytoplasm. An interesting conundrum regarding this mechanism is that cytochrome C is itself a hemeprotein and only in its heme-bound form can it bind APAF1 to activate the caspase cascade. Does the sensitization to Venetoclax conferred by heme-depletion occur independently of cytochrome C? Does cytochrome C bind heme with higher affinity than ETC complexes? Another question is whether inhibiting heme synthesis activates HRI, as it does in vivo, which would likely cause an induction of NOXA and BIM. Pharmacologic inhibition of heme synthesis with succinyl acetone did not appear to increase levels of NOXA and BIM by western blot (Lin et al., 2019), so whether this instance of heme depletion affects apoptosis through the ISR is unclear. Last, Lin et al demonstrate that oligomycin also sensitizes to Venetoclax treatment, despite it having the opposite effect on IMM potential (hyperpolarization) compared to succinyl acetone (depolarization). Perturbation of membrane potential in either direction is known to activate OMA1 and cause OPA1 cleavage, which would facilitate release of cytochrome C downstream of MOMP. Therefore, it could be that heme depletion does sensitize to Venetoclax via a decrease in membrane potential because of OMA1 activation or other stress responses, but not because of depolarization per se.
Work by Bajpai, et al beautifully revealed the interconnectedness of ETC and apoptosis, using multiple myeloma cell models (Bajpai et al., 2020). They found that pharmacologic inhibition of complex I or complex II sensitized cells to Venetoclax treatment through induction of BIM and NOXA transcription. Furthermore, ATF4 was required for BIM/NOXA induction in response to ETC inhibition, perhaps through the newly described HRI-dependent mechanisms. This study “connects the dots” between new work on ETC dysfunction and how it signals through the ISR to promote apoptosis. To reiterate, stress-activated transcription of BIM and NOXA neutralize MCL1, which the cell relies on to prevent apoptosis in the context of BCL2 inhibition by Venetoclax.
How inhibiting MCL1 indirectly through the ISR could be clinically impactful
As described above, increased reliance on MCL1 is a widespread mechanism of Venetoclax resistance, particularly in leukemia. So, why not just directly target MCL1 with a small molecule? This has been shown to be a potent therapeutic combination in animal models (Bhatt et al., 2020), but the question remains: Is it too potent? Is there a therapeutic window that could enable this regimen in human patients?
Three clinical trials of direct antagonists of MCL1 have been initiated (Diepstraten et al., 2022). Two were halted, and one was explicitly terminated due to evidence of cardiac toxicity. Of note, this resembles the phenotype of MCL1 knockout mice, suggesting that on-target toxicity could explain adverse effects of MCL1 inhibitors (Thomas et al., 2013; Wang et al., 2013). That MCL1 inhibitors appear to be highly toxic in isolation does not breed confidence that these drugs will be efficacious in combination with Venetoclax, but this might still be worth pursuing. An issue that complicates many preclinical studies of MCL1 inhibitors is that these drugs have higher affinity for human MCL1 compared to mouse Mcl1 (Caenepeel et al., 2018). Therefore, a human tumor xenografted into a mouse underestimates the on-target (but off-tissue) toxicity of these drugs (Brennan et al., 2018).
The apparent toxicity of MCL1 inhibitors opens the door for alternate approaches to achieve pharmacologic synergy with Venetoclax. Given that the ISR antagonizes MCL1 directly and indirectly, it is not surprising that ISR activation downstream of ETC dysfunction synergizes with Venetoclax. In contrast to direct MCL1 antagonism, selective agonism of the ISR could provide an indirect means of blocking MCL1. Indeed, pharmacologic agonism of PERK, an eIF2α kinase teleologically similar to HRI, synergized with Venetoclax in a recent study (Grenier et al., 2022).
MCL1 is highly expressed in cardiac tissues, which could partly explain the cardiac toxicity seen in trials of MCL1 inhibitors (GTEx). In contrast, HRI is scarcely expressed in cardiac tissues but more abundantly expressed in hematopoietic lineages. In fact, when HRI was discovered nearly 30 years ago, HRI transcripts were found in peripheral blood and bone marrow of rabbits, but were undetectable in heart, liver, lung, spleen, brain, and pancreas (Crosby et al., 1994). Therefore, HRI agonism could be a way to preferentially antagonize MCL1 in leukemia cells while sparing cardiac tissue. Consistent with the feasibility of this approach, a recent paper by Opferman and colleagues demonstrated that pharmacologic HRI agonism synergized with the BH3 mimetic navitoclax in a murine xenograft model of leukemia (Smith et al., 2020).
Biasing the ISR towards apoptosis
To briefly review, ETC dysfunction activates the protease OMA1, which cleaves DELE1 to activate the eIF2α kinase HRI. Once HRI phosphorylates eIF2α, is the signaling output the same as that from ISR activation by other inputs, e.g. unfolded proteins and PERK? We contend that there is a key difference in ETC-activated ISR compared to activation of ISR by other means, at least as far as apoptosis is concerned. That key difference is the activation of OMA1, which cleaves OPA1 in addition to DELE1. Thus, the activation of the ISR downstream of ETC dysfunction (DELE1 cleavage) coincides with changes in mitochondrial morphology and cristae junctions (OPA1 cleavage); both are caused by OMA1 activation (Figure 3). We speculate that BIM and NOXA produced in response to the ISR could trigger accelerated or amplified apoptosis in mitochondria when those mitochondria have already lost cristae junctions. If this is true, then combining Venetoclax with ETC poisons—as opposed to downstream ISR agonists or MCL1 inhibitors—might lead to increased apoptotic potency.
Figure 3: Intersection of two stress responses secondary to mitochondrial dysfunction.
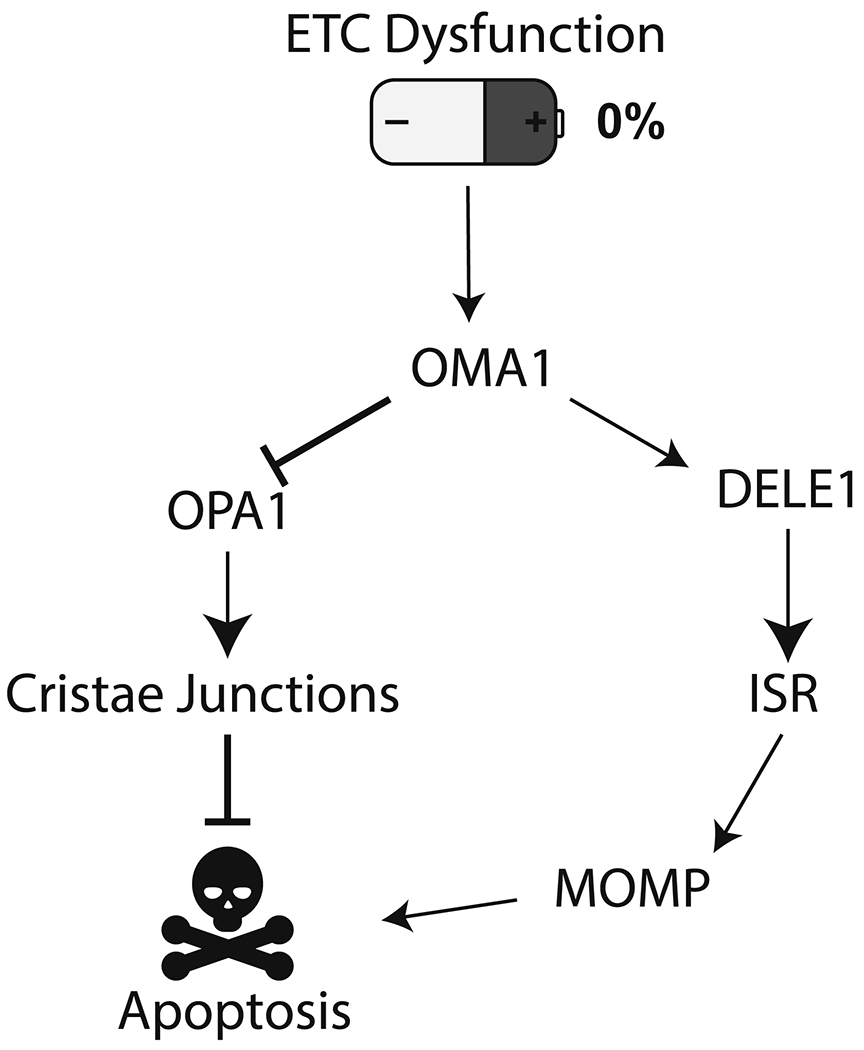
ETC dysfunction triggers OMA1 activation, which cleaves multiple substrates, including OPA1 and DELE1. Initially this activity serves a pro-homeostatic response. Prolonged stress activates the pro-apoptotic arm of the integrated stress response, which converges on BCL2 family proteins to trigger MOMP. Cleavage of OPA1 and dissolution of cristae junctions may act to internally prime mitochondria to commit to apoptosis in response to MOMP.
Why do respiration and apoptosis localize to the same intracellular structure?
The discovery that cytochrome C participates in both electron transport and caspase activation provided a profound illustration that mitochondria are central to both life and death of the cell (Liu et al., 1996). In the 26 years since that discovery, the question of why these two disparate functions co-localize to mitochondria has remained unanswered. The work described in this review could offer insights into this question. We speculate that the co-habitation of apoptosis and the ETC ensures that a single mitochondrial insult can trigger two responses simultaneously: One that is intrinsic to mitochondria (OPA1 cleavage, cardiolipin oxidation, increased fission) and another that acts as a mito-nuclear retrograde response (namely, activation of the integrated stress response through DELE1/HRI). Both responses initially act to restore homeostasis, such as by altering mitochondrial dynamics, increasing autophagy, and blocking translation to limit the proteostatic burden on mitochondria. In the event of continued dysfunction, the ISR pushes the cell toward apoptosis, in part through the production of BIM and NOXA and the inhibition of MCL1. These signals arrive at a mitochondrion that has been prepared for apoptosis by preceding activity of OMA1 and oxidation of IMM lipids, ensuring rapid and efficient cell death. It may be that these responses are optimally synchronized by co-localizing their underlying machinery to this fascinating organelle.
Conclusion
Our understanding of the functionally interaction of the ETC and apoptosis has greatly expanded. Respiration and “expiration” interface through biophysical means, such as sequestration of cytochrome C into cristae, and by retrograde signaling pathways, such as the ISR. The interface between these seemingly opposing processes holds much therapeutic promise and its investigation may uncover additional fundamental truths of mitochondria.
Funding:
1F30CA243440-01A1 to JMW; CA228346 and R35GM131854 to JR. J.R. is an investigator of the Howard Hughes Medical Institute. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (NIH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DI Statement:
The authors declare no competing interests.
REFERENCES:
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, and Langer T (2014). The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204, 919–929. 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archibald JM (2015). Endosymbiosis and Eukaryotic Cell Evolution. Current Biology 25, R911–R921. 10.1016/j.cub.2015.07.055. [DOI] [PubMed] [Google Scholar]
- Arnarez C, Marrink SJ, and Periole X (2016). Molecular mechanism of cardiolipin-mediated assembly of respiratory chain supercomplexes. Chem. Sci 7, 4435–4443. 10.1039/C5SC04664E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai R, Sharma A, Achreja A, Edgar CL, Wei C, Siddiqa AA, Gupta VA, Matulis SM, McBrayer SK, Mittal A, et al. (2020). Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat Commun 11, 1228. 10.1038/S41467-020-15051-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, and Langer T (2014). Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J 33, 578–593. 10.1002/embj.201386474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basch M, Wagner M, Rolland S, Carbonell A, Zeng R, Khosravi S, Schmidt A, Aftab W, Imhof A, Wagener J, et al. (2020). Msp1 cooperates with the proteasome for extraction of arrested mitochondrial import intermediates. MBoC 31, 753–767. 10.1091/mbe.E19-06-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belikova NA, Vladimirov YA, Osipov AN, Kapralov AA, Tyurin VA, Potapovich MV, Basova LV, Peterson J, Kurnikov IV, and Kagan VE (2006). Peroxidase Activity and Structural Transitions of Cytochrome c Bound to Cardiolipin-Containing Membranes. Biochemistry 45, 4998–5009. 10.1021/bi0525573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensard CL, Wisidagama DR, Olson KA, Berg JA, Krah NM, Schell JC, Nowinski SM, Fogarty S, Bott AJ, Wei P, et al. (2020). Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell Metabolism 31, 284–300.e7. 10.1016/j.cmet.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom CL, Beales PA, Lv Y, Vanderlick TK, and Groves JT (2013). Cytochrome c causes pore formation in cardiolipin-containing membranes. Proceedings of the National Academy of Sciences 110, 6269–6274. 10.1073/pnas.1303819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Pioso MS, Olesinski EA, Yilma B, Ryan JA, Mashaka T, Leutz B, Adamia S, Zhu H, Kuang Y, et al. (2020). Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell 38, 872–890.e6. 10.1016/j.ccell.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, and Sabatini DM (2015). An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551. 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blombery P, Anderson MA, Gong J, Thijssen R, Birkinshaw RW, Thompson ER, Teh CE, Nguyen T, Xu Z, Flensburg C, et al. (2019). Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov 9, 342–353. 10.1158/2159-8290.CD-18-1119. [DOI] [PubMed] [Google Scholar]
- Boos F, Krämer L, Groh C, Jung F, Haberkant P, Stein F, Wollweber F, Gackstatter A, Zöller E, van der Laan M, et al. (2019). Mitochondrial protein-induced stress triggers a global adaptive transcriptional programme. Nat Cell Biol 21, 442–451. 10.1038/S41556-019-0294-5. [DOI] [PubMed] [Google Scholar]
- Brennan MS, Chang C, Tai L, Lessene G, Strasser A, Dewson G, Kelly GL, and Herold MJ (2018). Humanized Mcl-1 mice enable accurate preclinical evaluation of MCL-1 inhibitors destined for clinical use. Blood 132, 1573–1583. 10.1182/blood-2018-06-859405. [DOI] [PubMed] [Google Scholar]
- Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, Whittington DA, Huang X, Poppe L, Cheng AC, et al. (2018). AMG 176, a Selective MCL1 Inhibitor, is Effective in Hematological Cancer Models Alone and in Combination with Established Therapies. Cancer Discovery CD-18-0387. 10.1158/2159-8290.CD-18-0387. [DOI] [PubMed] [Google Scholar]
- Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong W-J, Zhao F, Medeiros BC, Tyvoll DA, and Majeti R (2015). Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med 21, 178–184. 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, Sakellaropoulos T, Gong Y, Kloetgen A, Yap YS, et al. (2019). Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov 9, 890–909. 10.1158/2159-8290.CD-19-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon KJ, Orozco JM, Adelmann CH, Spinelli JB, Helm PW, van der, Roberts JM, Kunchok T, and Sabatini DM (2021). Genome-wide CRISPR screens reveal multitiered mechanisms through which mTORC1 senses mitochondrial dysfunction. PNAS 118. 10.1073/pnas.2022120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368, eaat5314. 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby JS, Lee K, London IM, and Chen JJ (1994). Erythroid expression of the heme-regulated eIF-2 alpha kinase. Molecular and Cellular Biology 10.1128/mcb.14.6.3906-3914.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupo RR, and Shorter J (2020). Skd3 (human ClpB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations. ELife 9, e55279. 10.7554/eLife.55279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daverey A, Levytskyy RM, Stanke KM, Viana MP, Swenson S, Hayward SL, Narasimhan M, Khalimonchuk O, and Kidambi S (2019). Depletion of mitochondrial protease OMA1 alters proliferative properties and promotes metastatic growth of breast cancer cells. Sci Rep 9, 14746. 10.1038/s41598-019-49327-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv 2, e1600200. 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2020). We need to talk about the Warburg effect. Nat Metab 2, 127–129. 10.1038/s42255-020-0172-2. [DOI] [PubMed] [Google Scholar]
- Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A, and Kelly GL (2022). The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer 22, 45–64. 10.1038/s41568-021-00407-4. [DOI] [PubMed] [Google Scholar]
- Eckl E-M, Ziegemann O, Krumwiede L, Fessler E, and Jae LT (2021). Sensing, signaling and surviving mitochondrial stress. Cell. Mol. Life Sci 78, 5925–5951. 10.1007/S00018-021-03887-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encarnación-Rosado J, and Kimmelman AC (2021). Harnessing metabolic dependencies in pancreatic cancers. Nat Rev Gastroenterol Hepatol 18, 482–492. 10.1038/S41575-021-00431-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessler E, Eckl E-M, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, and Jae LT (2020). A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579, 433–437. 10.1038/S41586-020-2076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessler E, Krumwiede L, and Jae LT (2022). DELE1 tracks perturbed protein import and processing in human mitochondria. Nat Commun 13, 1853. 10.1038/S41467-022-29479-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, et al. (2006). OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 126, 177–189. 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Friedman JR, and Nunnari J (2014). Mitochondrial form and function. Nature 505, 335. 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia D, and Shaw RJ (2017). AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Molecular Cell 66, 789–800. 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomello M, Pyakurel A, Glytsou C, and Scorrano L (2020). The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 21, 204–224. 10.1038/S41580-020-0210-7. [DOI] [PubMed] [Google Scholar]
- Greaves G, Milani M, Butterworth M, Carter RJ, Byrne DP, Eyers PA, Luo X, Cohen GM, and Varadarajan S (2018). BH3-only proteins are dispensable for apoptosis induced by pharmacological inhibition of both MCL-1 and BCL-X L. Cell Death & Differentiation 1. 10.1038/s41418-018-0183-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier A, Poulain L, Mondesir J, Jacquel A, Bosc C, Stuani L, Mouche S, Larrue C, Sahal A, Birsen R, et al. (2022). AMPK-PERK axis represses oxidative metabolism and enhances apoptotic priming of mitochondria in acute myeloid leukemia. Cell Reports 38, 110197. 10.1016/j.celrep.2021.110197. [DOI] [PubMed] [Google Scholar]
- Guièze R, Liu VM, Rosebrock D, Jourdain AA, Hernández-Sánchez M, Martinez Zurita A, Sun J, Ten Hacken E, Baranowski K, Thompson PA, et al. (2019). Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 36, 369–384.e13. 10.1016/j.ccell.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin Y-HT, Wiita AP, Xu K, Correia MA, and Kampmann M (2020). Mitochondrial stress is relayed to the cytosol by an OMA1–DELE1–HRI pathway. Nature 579, 427–432. 10.1038/S41586-020-2078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Molecular Cell 11, 619–633. 10.1016/S1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Harper JW, Ordureau A, and Heo J-M (2018). Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 19, 93–108. 10.1038/nrm.2017.129. [DOI] [PubMed] [Google Scholar]
- Herzig S, and Shaw RJ (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19, 121–135. 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG (2014). The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem 83, 779–812. 10.1146/annurev-biochem-060713-035802. [DOI] [PubMed] [Google Scholar]
- Jiang X, Jiang H, Shen Z, and Wang X (2014). Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. PNAS 111, 14782–14787. 10.1073/pnas.1417253111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, et al. (2005). Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1, 223–232. 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- Kale J, Osterlund EJ, and Andrews DW (2018). BCL-2 family proteins: changing partners in the dance towards death. Cell Death & Differentiation 25, 65–80. 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan K-L (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141. 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong J-N, Moujalled DM, et al. (2016). The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477. . [DOI] [PubMed] [Google Scholar]
- Letai A (2017). Apoptosis and Cancer. Annual Review of Cancer Biology 1, 275–294. 10.1146/annurev-cancerbio-050216-121933. [DOI] [Google Scholar]
- Lin KH, Xie A, Rutter JC, Ahn Y, Lloyd-Cowden JM, Nichols AG, Soderquist RS, Koves TR, Muoio DM, Maclver NJ, et al. (2019). Systematic Dissection of the Metabolic-Apoptotic Interface in AML Reveals Heme Biosynthesis to Be a Regulator of Drug Sensitivity. Cell Metabolism 29, 1217–1231.e7. 10.1016/j.cmet.2019.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, and Wang X (1996). Induction of Apoptotic Program in Cell-Free Extracts: Requirement for dATP and Cytochrome c. Cell 86, 147–157. 10.1016/S0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, Ferreira R, Naamati A, Ali A, Lewis CA, et al. (2021). Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Molecular Cell 81, 691–707.e6. 10.1016/j.molcel.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar T, and Langer T (2016). OPA1 processing in cell death and disease – the long and short of it. J Cell Sci 129, 2297–2306. 10.1242/jcs.159186. [DOI] [PubMed] [Google Scholar]
- MacVicar T, Ohba Y, Nolte H, Mayer FC, Tatsuta T, Sprenger H-G, Lindner B, Zhao Y, Li J, Bruns C, et al. (2019). Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature 575, 361–365. 10.1038/s41586-019-1738-6. [DOI] [PubMed] [Google Scholar]
- Mårtensson CU, Priesnitz C, Song J, Ellenrieder L, Doan KN, Boos F, Floerchinger A, Zufall N, Oeljeklaus S, Warscheid B, et al. (2019). Mitochondrial protein translocation-associated degradation. Nature 569, 679–683. 10.1038/S41586-019-1227-y. [DOI] [PubMed] [Google Scholar]
- Martínez-Reyes I, Cardona LR, Kong H, Vasan K, McElroy GS, Werner M, Kihshen H, Reczek CR, Weinberg SE, Gao P, et al. (2020). Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292. 10.1038/S41586-020-2475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick E, Titov DV, Skinner OS, Sharma R, Jourdain AA, and Mootha VK (2020). Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. ELife 9, e49178. 10.7554/eLife.49178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musatov A, and Robinson NC (2014). Bound cardiolipin is essential for cytochrome c oxidase proton translocation. Biochimie 105, 159–164. 10.1016/j.biochi.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, and Haynes CM (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okosun J, Wolfson RL, Wang J, Araf S, Wilkins L, Castellano BM, Escudero-Ibarz L, Al Seraihi AF, Richter J, Bernhart SH, et al. (2016). Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat Genet 48, 183–188. 10.1038/ng.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut M-N, Berthezène P, et al. (2017). Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun 8, 16031. 10.1038/ncomms16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, and Mihara K (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191, 1141–1158. 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Zhivotovsky B, and Orrenius S (2007). Role of cardiolipin in cytochrome c release from mitochondria. Cell Death & Differentiation 14, 1243–1247. 10.1038/sj.cdd.4402135. [DOI] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM (2016). The integrated stress response. EMBO Rep 17, 1374–1395. 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavitt GD, Ramaiah KV, Kimball SR, and Hinnebusch AG (1998). eIF2 independently binds two distinct eIF2B subcomplexes that catalyze and regulate guanine-nucleotide exchange. Genes Dev 12, 514–526. 10.1101/gad.12.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, and Youle RJ (2015). The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 85, 257–273. 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, and Auwerx J (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216, 2027–2045. 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raemy E, and Martinou J-C (2014). Involvement of cardiolipin in tBID-induced activation of BAX during apoptosis. Chemistry and Physics of Lipids 179, 70–74. 10.1016/j.chemphyslip.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Rainbolt TK, Lebeau J, Puchades C, and Wiseman RL (2016). Reciprocal Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress. Cell Reports 14, 2041–2049. 10.1016/j.celrep.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, et al. (2016). Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. New England Journal of Medicine 374, 311–322. 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca-Portoles A, Rodriguez-Blanco G, Sumpton D, Cloix C, Mullin M, Mackay GM, O’Neill K, Lemgruber L, Luo X, and Tait SWG (2020). Venetoclax causes metabolic reprogramming independent of BCL-2 inhibition. Cell Death Dis 11, 1–13. 10.1038/S41419-020-02867-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santucci R, Sinibaldi F, Cozza P, Polticelli F, and Fiorucci L (2019). Cytochrome c: An extreme multifunctional protein with a key role in cell fate. International Journal of Biological Macromolecules 136, 1237–1246. 10.1016/j.ijbiomac.2019.06.180. [DOI] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976. 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell JC, Olson KA, Jiang L, Hawkins AJ, Van Vranken JG, Xie J, Egnatchik RA, Earl EG, DeBerardinis RJ, and Rutter J (2014). A Role for the Mitochondrial Pyruvate Carrier as a Repressor of the Warburg Effect and Colon Cancer Cell Growth. Molecular Cell 56, 400–413. 10.1016/j.molcel.2014.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt O, Pfanner N, and Meisinger C (2010). Mitochondrial protein import: from proteomics to functional mechanisms. Nature Reviews Molecular Cell Biology 11, 655. 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- Sharon D, Cathelin S, Mirali S, Di Trani JM, Yanofsky DJ, Keon KA, Rubinstein JL, Schimmer AD, Ketela T, and Chan SM (2019). Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Science Translational Medicine 11, eaax2863. 10.1126/scitranslmed.aax2863. [DOI] [PubMed] [Google Scholar]
- Shi X, Reinstadler B, Shah H, To T-L, Byrne K, Summer L, Calvo SE, Goldberger O, Doench JG, Mootha VK, et al. (2022). Combinatorial GxGxE CRISPR screen identifies SLC25A39 in mitochondrial glutathione transport linking iron homeostasis to OXPHOS. Nat Commun 13, 2483. 10.1038/s41467-022-30126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KH, Budhraja A, Lynch J, Roberts K, Panetta JC, Connelly JP, Turnis ME, Pruett-Miller SM, Schuetz JD, Mullighan CG, et al. (2020). The Heme-Regulated Inhibitor Pathway Modulates Susceptibility of Poor Prognosis B-Lineage Acute Leukemia to BH3-Mimetics. Molecular Cancer Research 19, 636–650. 10.1158/1541-7786.MCR-20-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderquist RS, Crawford L, Liu E, Lu M, Agarwal A, Anderson GR, Lin KH, Winter PS, Cakir M, and Wood KC (2018). Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nat Commun 9. 10.1038/s41467-018-05815-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli JB, and Haigis MC (2018). The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 20, 745–754. 10.1038/s41556-018-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, and Chandel NS (2014). Mitochondrial reactive oxygen species and cancer. Cancer & Metabolism 2, 17. 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S, and Gustafsson ÅB (2013). Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 27, 1365–1377. 10.1101/gad.215871.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, et al. (2016). AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vranken JG, and Rutter J (2015). You Down With ETC? Yeah, You Know D! Cell 162, 471–473. 10.1016/j.cell.2015.07.027. [DOI] [PubMed] [Google Scholar]
- Vasan K, Werner M, and Chandel NS (2020). Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metabolism 32, 341–352. 10.1016/j.cmet.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, García-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Rupérez FJ, Barbas C, Ibañez B, and Langer T (2015). Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350, aad0116. 10.1126/science.aad0116. [DOI] [PubMed] [Google Scholar]
- Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, and Opferman JT (2013). Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 27, 1351–1364. 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yen FS, Zhu XG, Timson RC, Weber R, Xing C, Liu Y, Allwein B, Luo H, Yeh H-W, et al. (2021). SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 599, 136–140. 10.1038/s41586-021-04025-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1956). On the Origin of Cancer Cells. Science 123, 309–314. . [DOI] [PubMed] [Google Scholar]
- Weidberg H, and Amon A (2018). MitoCPR—A surveillance pathway that protects mitochondria in response to protein import stress. Science 360. 10.1126/science.aan4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, Kuwana T, Ellisman M, and Newmeyer DD (2008). Opa1-mediated cristae opening is Bax/Bak- and BH3-dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell 31, 557–569. 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]