

Abstract
Prime editing is a new CRISPR-based, genome-editing technology that relies on the prime editor (PE), a fusion protein of Cas9-nickase and M-MLV reverse transcriptase (RT), and a prime editing guide RNA (pegRNA) that serves both to target PE to the desired genomic locus and to carry the edit to be introduced. Here, we make advancements to the RT moiety to improve prime editing efficiencies and truncations to mitigate issues with adeno-associated virus (AAV) viral vector size limitations, which currently do not support efficient delivery of the large prime editing components. These efforts include RT variant screening, codon optimization, and PE truncation by removal of the RNase H domain and further trimming. This led to a codon-optimized and size-minimized PE that has an expression advantage (1.4-fold) and size advantage (621 bp shorter). In addition, we optimize the split intein PE system and identify Rma-based Cas9 split sites (573-574 and 673-674) that combined with the truncated PE delivered by dual AAVs result in superior AAV titer and prime editing efficiency. We also show that this minimized PE gives rise to superior lentiviral vector titers (46-fold) over the regular PE in an all-in-one PE lentiviral vector. We finally deliver the minimized PE to mouse liver by dual AAV8 vectors and show up to 6% precise editing of the PCSK9 gene, thereby demonstrating the value of this truncated split PE system for in vivo applications.
Keywords: CRISPR-Cas9, prime editing, reverse transcriptase, AAV vectors, in vivo delivery, gene editing, gene therapy, PASTE
Graphical abstract
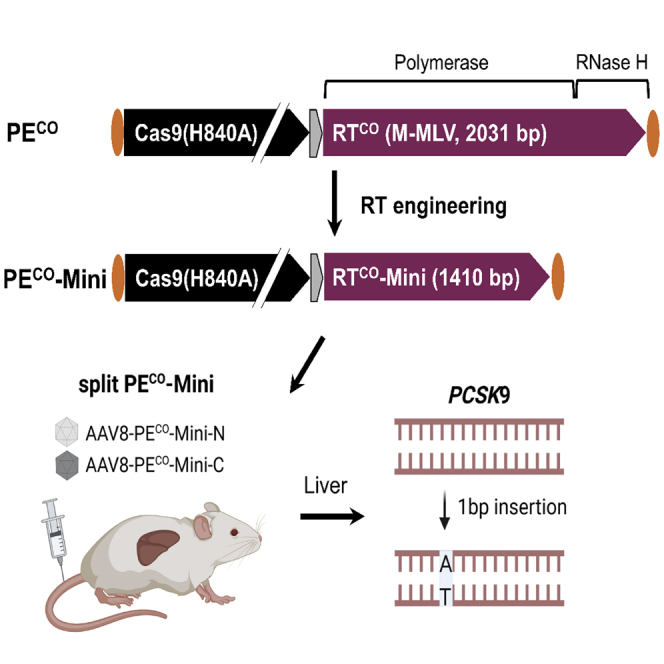
Gao et al. developed an optimized, truncated prime editor by engineering the reverse transcriptase domain. Combined with an optimized split-intein system, they showed that this truncated prime editor can be split between two AAVs, resulting in superior AAV titers and prime editing in vitro and in mouse liver.
Introduction
Prime editing enables versatile and precise genome editing independent of double-strand DNA breaks and exogenous donor template DNA and thus exhibits great potential as a tool for biomedical research and gene therapy.1 The prime editing system consists of (1) a prime editor (PE), an SpCas9 nickase (nCas9) fused to a mutant Moloney murine leukemia virus (M-MLV) reverse transcriptase (RT) (Figure 1A); (2) a prime editing guide RNA (pegRNA) that carries the desired edit; and (3) a nicking guide RNA (ngRNA). Despite its versatile genome editing capacity, prime editing is challenged by the large PE gene (∼6.4 kb), which makes delivery of the prime editing system (PE, pegRNA, and ngRNA) challenging. For instance, it is impossible to package the prime editing system into a single adeno-associated virus (AAV) vector, which is limited to ∼5 kb with the inclusion of the inverted terminal repeats (ITRs). Although two recent studies reported that the PE gene can be divided in two and delivered by dual AAV vectors relying on trans-splicing intein,2,3 the large size of PE severely restricts the position of the split site, which leads to suboptimal prime editing efficiency and leaves limited space for additional functional elements.
Figure 1.

RT variant screening and codon optimization
(A) Schematic of the PE gene cassette employed in this study. PE consists of Cas9-nickase (Cas9 [H840A]) and M-MLV RT. The bipartite SV40 nuclear localization signals (bpNLSs) are marked in orange and a linker in gray. (B) Screening of prime editing activity of 11 RT variants codon optimized by GenScript showed activity only for XMRV RT is shown (all variant data shown in Figure S1). PE constructs were transfected with plasmids encoding pegRNA and ngRNA into HEK293T cells, and prime editing results were analyzed after 3 days. (C) The effect of RT codon optimization on PE expression using algorithms from different companies is shown. Same amounts of PE plasmids were transfected into HEK293T cells, and western blotting was conducted after 3 days. PE protein expression levels were normalized to β-actin protein levels, and wild-type (WT) PE protein expression level was arbitrarily set to 1.0 (indicated below the PE blot). The image shows a representative blot from two independent experiments with similar results. (D) Comparison of prime editing frequencies of the original PE and the codon-optimized PECO by plasmid delivery is shown. HEK293T cells were transfected with varying amounts of PE or PECO (1,500, 300, and 60 ng) together with fixed amounts of plasmid encoding pegRNA and ngRNA. After 3 days, PCR products were subjected to Sanger sequencing and ICE analysis to evaluate prime editing frequencies. (E) Comparison of prime editing frequencies of PE and PECO by all-RNA delivery is shown. PE mRNA and synthetic pegRNA and ngRNA were electroporated into HEK293T cells. Cells were subjected to ICE analysis 3 days post-transfection. Bars represent mean values ± SD, and all data points for individual replicates are shown. ns, not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
In addition, the efficiency of prime editing can vary greatly across edit types, target loci, and cell types. Hence, there is a need for an improved prime editing system. Very recently, prime editing activity improvements were achieved by pegRNA engineering, nuclear localization signal (NLS) optimization to the PE, and manipulation of cellular DNA repair pathways.4, 5, 6
In this study, by engineering the RT of the PE, we developed a codon-optimized and size-minimized PE. Combining the optimized intein split PE system, a dual AAV split PE system was generated that has vastly improved AAV titer and prime editing activity.
Results
M-MLV RT performs the best among RT variants
With an attempt to improve prime editing activity and obtain a shorter PE with an RT of reduced size, we tested whether RTs from other species could replace M-MLV RT to constitute a functional PE. Using PE2 developed by Anzalone et al.,1 we screened 11 human codon-optimized RT variants, including nine retroviral RTs and two bacterial group II intron RTs (Data S1). The results showed that only xenotropic-murine-leukemia-virus-related virus (XMRV) RT was functional in inducing prime editing (HEK3 +3 CTT insertion) in the HEK3 gene (Figures 1B and S1). However, this XMRV-RT-containing PE induced much lower prime editing than that of M-MLV RT (17% versus 58%).
RT codon optimization improves prime editing activity
We next attempted to improve prime editing activity by increasing PE expression level. Since the SpCas9 has been widely developed, we focused on the RT moiety. We generated M-MLV RT variants using six different codon optimization algorithms from different companies: IDT, GenScript, GeneArt, GeneWIZ, Twist Bioscience, and Benchling. Four out of six were observed to increase PE protein expression level compared with the previously published variant with GenScript (hereafter termed PECO) expressing highest protein levels (1.4-fold increase) (Figure 1C). We next investigated the performance of PECO by plasmid delivery of varying PE and PECO amounts to HEK293T cells. Results consistently showed that PECO exhibited higher prime editing frequencies at all three tested targets (HBB, HEK3, and RNF2) (Figure 1D). We also performed this comparison with all-RNA delivery (PE mRNA with chemically modified pegRNAs and ngRNA), which is more therapeutically relevant in primary cells, and we observed a similar increase in editing by PECO (31.3% versus 23.6%) (Figure 1E).
Engineering a minimal M-MLV RT with uncompromised prime editing activity
Next, we sought to shorten the size of M-MLV RT. RT consists of an RNA-dependent polymerase domain and an RNase H domain (Figure 2A). The polymerase domain uses single-strand RNA as template to synthesize single-strand DNA, and the RNase H domain selectively degrades the RNA template of RNA/DNA hybrids.7 Considering that the RNase H domain is only for RNA template degradation, we hypothesized that the RNase H domain would be dispensable. We also hypothesized that the polymerase region might be trimmed at both ends while still retaining full activity. To test this, we deleted the RNase H domain of M-MLV RT (471 bp) to make a truncated PECO (PECO-ΔR), and based on PECO-ΔR, we made further truncations by 30 bp serial N- or C-terminal truncations to RT, creating a total of 11 different truncated PECO variants (Figure 2A). Prime editing experiments in HEK293T cells for two targets (HEK3 and HBB) showed that PECO-ΔR variants with a maximally 60-bp N-terminal deletion and 120-bp C-terminal deletion did not affect prime editing activity compared with the PECO-ΔR variant (Figure 2B). We then tested four combinatorial N- and C-terminal truncations and showed that a combined 150-bp deletion (ΔN60 + C90) to PECO-ΔR did not affect the prime editing efficiency compared with the PECO-ΔR variant, leading to a minimal PE (PECO-Mini) (Figure 2C). To finally assess the performance of PECO-Mini, we performed a parallel comparison to the full-length PECO and PECO-ΔR. These data showed that the PECO-Mini displayed very similar prime editing frequencies at three different target genes (Figure 2D). Furthermore, we observed no difference in the editing outcome (specific prime edits and unspecific indels) of the three PE variants (Figure S2). These results show that the RNase H domain is dispensable to PE, which was further supported by the observed functionality of an RNase-H-deleted XMRV RT PE variant (Figure S1). Intriguingly, PE with XMRV RT-ΔR outperformed XMRV RT (functional RNase H domain) in prime editing in the HBB gene (17% versus 28%), indicating a negative effect from the RNase H domain. However, we still did not observe any prime editing from other RNase-H-deleted RT PE variants (Figure S1). In conclusion, we engineered a codon-optimized and size-minimized PECO-Mini with an overall size reduction of 621 bp that retains prime editing activity compared with the full-length PE.
Figure 2.

Generation of a minimal PE by truncating M-MLV RT
(A) Schematic of PECO and truncated PECO variants used in this study. The RT is composed of a polymerase and an RNase H domain. Initially, a truncated PE was made with the RNase H domain deleted (471 bp) to form PECO-ΔR. Subsequently, serial 30-bp truncations at both ends were tested with the aim of generating a minimal PECO (PECO-Mini). (B) Minimizing M-MLV RT by deleting the RNase H domain (PECO-ΔR) and by further N- and C-terminal trimming is shown. The same molar amounts of PECO-ΔR and trimmed PECO-ΔR plasmids were transfected into HEK293T cells together with fixed amounts of plasmids encoding pegRNA and ngRNA. Cells were subjected to ICE analysis for evaluating prime editing activity 3 days post-transfection. (C) Combinatorial N- and C-terminal truncations were analyzed for prime editing efficiencies as in (B). Based on the activity, we selected RT-ΔR with ΔN60 + C90 as the minimal RT, which is 1,410 bp long. (D) Comparison of PECO and the two truncated variants PECO-ΔR and PECO-Mini performed as in (B) is shown. Bars represent mean values ± SD with all data points from independent experiments shown. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Optimization of an intein split PE system
To pave the way for effective dual AAV delivery of PE, it is important to establish an efficient split PE system. To do this, we constructed various intein-based split PECO systems (Figure 3A). Previous studies have shown that Cas9 activity is highly dependent on the position of the intein split site. We therefore chose four commonly used Cas9 split sites (573-574, 637-638, 674-675, and 713-714) and two different inteins (Npu and Rma).8,9 The split and non-split PECO systems were tested at the HBB and HEK3 targets in HEK293T cells. The efficiency of the eight two-plasmid split PECO systems varied markedly, and as expected, all were lower than the single PECO plasmid (no splitting) (Figure 3B). Rma 573-74 was the most efficient (85% activity of that of single-plasmid full-length PECO) followed by Rma 674-675 (75% activity) (Figure 3B). A direct comparison of Rma 573-574 with three previously reported split sites (Rma 1005-1006, Rma 1024-1025,3 and SD/SA 1035-103610) showed that Rma 573-574 mediated the most potent prime editing (Figure S3). An AAV genome size calculation showed that, if using the two most efficient intein sites (Rma 573-574 and 674-675) to split non-truncated PECO, the PECO C-terminal part would amount to >5 kb, thereby exceeding the ∼5-kb AAV packaging limit (including ITRs),11 even with the use of a small promoter (EFS) and poly(A) signal (Figure 3A). However, the two intein split sites could be used with the PECO-Mini with vector genome sizes below 4.7 kb. Despite the slightly higher activity of the Rma 573-574 split site, we chose the Rma 674-675 split site, as it leaves 300 bp of space in the vector encoding the C-terminal part, which would allow a more flexible promoter choice (total AAV vector size of 4.4 kb) (Figure 3C). Analysis of plasmid-based delivery of split PECO, PECO-ΔR, and PECO-Mini systems with the Rma 674-675 split site showed similar editing activity for all three targets tested, which is around 75% of the single all-in-one PECO plasmid system (Figure 3D).
Figure 3.

The engineered minimal PE variant enhances prime editing when delivered by dual AAV vectors
(A) Schematic of the split PE system where the full-length PE is reconstituted by intein protein-mediated trans-splicing. (B) Screening of split PE systems with different inteins and Cas9 split positions by plasmid transfection of HEK293T cells is shown. (C) Schematic of AAV vector genomes encoding the C-terminal part of the split PEs (PECO-C or PECO-Mini-C) is shown. ITRs, inverted terminal repeats; rbGlob, rabbit beta globin poly(A) signal. (D) Comparable activity of the split full-length and the two split truncated PE systems (PECO-ΔR split and PECO-Mini split) by plasmid transfection in HEK293T cells is shown. The single plasmid non-truncated PECO was included for comparison. (E) AAV6 vectors encoding the C-terminal part of the split PECO-Mini (PECO-Mini-C) were produced in parallel with vectors encoding the C-terminal part of the non-truncated PECO (PECO-C). AAV titers were determined by ddPCR with the titers of PECO-C set to 1. (F) HEK293T cells were transduced with equal amounts of AAV vectors (5 × 105 vg/cell) of the generated split AAV vectors, and prime editing frequencies were evaluated 5 days post-transduction by Sanger sequencing and ICE analysis. Bars represent mean values ± SD. For (B), (D), and (E), data points are from independent experiments. For (F), each data point is an independent transduction with one AAV preparation. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
The truncated PE enhances PE delivery by AAV and lentiviral vectors
To investigate the performance of the PECO-Mini compared with full-length PECO for AAV delivery, we packaged dual AAV6 vectors based on the Rma 674-675 split site (Figures 3C and S4) and measured the vector titers and performed prime editing assays. RNA polymerase III (RNA Pol III) cassettes for the pegRNA (U6 promoter) and ngRNA (7SK promoter) were placed in the vector encoding the PE N-terminal part, amounting to a vector genome size of 3.5 kb (Figure S4). AAV vectors were produced in parallel, and titer measurements by droplet digital PCR (ddPCR) showed a >5-fold higher titer of the vector carrying the C-terminal part of PECO-Mini (PECO-Mini-C; 4.4 kb) compared with the vector carrying the C-terminal part of the full-length PECO (PECO-C; 5.1 kb) (Figure 3E). Transduction of HEK293T cells using the same number of vector genomes per cell (vg/cell) of split PECO and split PECO-Mini systems showed 10%–40% editing with the split PECO-Mini system compared with 0%–10% editing by the split PECO system (Figure 3F). Since cells were transduced with equal dosages (vg/cell), this indicated that the PECO-C vector genomes were too large to produce functional vector particles, leading to inefficient PECO expression in target cells. Thus, PECO-Mini is highly advantageous for dual AAV split PE delivery.
Previous reports have demonstrated that an increasing size of lentiviral vectors (LVs) also correlates with decreased transductional titer, and the large size of CRISPR-Cas gene-editing platforms have been reported to yield low LV titers.11,12 To evaluate whether the minimized PE could benefit LV titer, we constructed two all-in-one LVs (LV_PECO and LV_PECO-Mini) encoding the pegRNA, ngRNA, and PE expression cassettes along an antibiotic selection gene (blasticidin) (Figure S5A). We used the same molecular amount of transfer plasmid DNA for LV production and found that the LV_PECO-Mini produced a 46.4-fold higher titer than LV_PECO, although the titer was still low (around 1 × 103 colony-forming units [CFUs]/mL) compared with titers for standard small- or medium-sized LVs (Figure S5B). We next tested these two LVs by transduction of HEK293T cells with equal volumes of crude LV followed by antibiotic selection of transduced cells. Prime editing analysis at different time points following selection showed that LV_PECO-Mini gave rise to efficient prime editing in a time-dependent manner, reaching 74% editing on day 14 after selection. In contrast, full-length LV_PECO showed no editing above background at all time points, indicating that packaging or transfer of functional full-length vector genomes was hampered by the large size of the LV_PECO cassette (10,413 bp) (Figure S5C).
Delivery of split-PECO-mini by dual AAVs for in vivo prime editing
Finally, we explored whether the PECO-Mini could be delivered by dual AAVs for in vivo prime editing. For this, we designed pegRNAs to insert a single bp (+1 A insertion) in the first exon of mouse proprotein convertase subtilisin/kexin type 9 (PCSK9) to disrupt PCSK9 function, which has a therapeutic benefit for familial hypercholesterolemia. Screening in the Hepa 1–6 murine liver cell line of 15 pegRNAs with varying lengths of the primer binding site (PBS) and the reverse transcription template (RTT) combined with one of two different ngRNAs showed that +1 A insertion could be achieved at up to 70% editing frequencies with ngRNA 1 and PBS and RTT lengths of 12 and 12 nt (Figure 4A).
Figure 4.

Delivery of PECO-Mini by dual AAVs for in vivo editing
(A) Screening of pegRNAs and ngRNAs by plasmid transfection of the Hepa 1–6 cell line. Cells were co-transfected with a plasmid encoding a puromycin resistance gene (PAC), and cells were transiently selected for transfection-positive cells by applying puromycin for 2 days, starting at 24 h post-transfection. Prime editing frequencies were quantified by Sanger sequencing and ICE analysis. (B) Schematic figure of the two AAV genomes carrying the split PECO-Mini system is shown. (C) Schematic overview of animal studies is shown. (D) Quantification of editing efficiency in mouse livers is shown. Four weeks after AAV vector injection into six mice (three mice for each dose), mice livers were isolated for genomic DNA extraction. Editing in the PCSK9 target gene was quantified by NGS. Bars represent means ± SD with individual data points from biological replicates shown.
We next constructed two AAV vectors with pegRNA and ngRNA of this configuration (Figure 4B). The PECO-Mini-N harbors cassettes expressing the pegRNA, ngRNA, and the N-terminal part of PECO-Mini. PECO-Mini-C expresses the C-terminal part of PECO-Mini and the pegRNA. Placing pegRNA expression cassettes on both vectors rather than on a single vector was found to enhance prime editing (Figure S6). AAV8 vectors were produced from these two constructs and injected intravenously into 10-week-old C57BL/6J mice at an equal vector amount of either 1 × 1011 or 1 × 1012 vg per vector (Figure 4C). Livers were isolated 4 weeks after injection, and next-generation sequencing (NGS) showed an average of 1.4% and 5.4% precise prime editing with the low and high AAV doses, respectively (Figure 4D). This demonstrates that PECO-Mini can be delivered by dual AAVs for in vivo prime editing.
Discussion
The versatile gene editing ability of prime editing not only makes it a very useful biomedical research tool but also holds great promise as a therapeutic approach to correct genetic mutations. However, the prime editing efficiency greatly varies across target loci and cell types, impeding its broad application.1 Importantly, the large size of PE makes delivery very challenging, especially for in vivo application, where AAV vectors constitute a popular vector choice but suffer from limited packaging capacity. Although PE system delivery has been achieved by intein-mediated splicing between dual AAV vectors carrying a split PE, the large size of the PE and the AAV packaging limitation severely restrict the position where the intein can be placed in the PE gene. Consequently, this leads to suboptimal prime editing efficiency and leaves limited space for additional functional elements in the vectors.
In this study, we engineered the M-MLV RT to develop a codon-optimized and minimal PE: PECO-Mini. Screening of several RT variants from other species revealed only XMRV RT to be functional but displaying much less activity than the M-MLV. It is interesting that most RTs were not able to give rise to prime editing. One can speculate that the fusion protein configuration for these variants simply do not support functional reverse transcription. Several enhancing mutations to the M-MLV RT were already engineered in the original prime editing study,1 and further optimization of RT variants has the potential to develop alternative and more potent PE platforms. Codon optimization to RT increased PE expression level by 1.4-fold and consequently improved prime editing activity as demonstrated by both plasmid and all-RNA delivery. Removal of the RNase H domain and subsequent trimming of RT resulted in a minimal PE that is 621 bp shorter than the original PE but maintains editing efficiency. Intriguingly, we found that the XMRV RT was less active than XMRV RT-ΔR in mediating prime editing (17% versus 28%), indicating a negative effect of this RNase H domain. One could speculate that this domain might degrade the pegRNA region, where it anneals to the RT PBS of the DNA target. However, this was not observed by M-MLV. By testing different Cas9 split sites and inteins, we identified two split sites (573-574 and 673-674) together with the Rma intein to be the most potent split PE systems. We demonstrated that, when using 673-674/Rma, PECO-Mini enables a split PE system for AAV packaging in which the two vector genomes are well below the AAV packaging limit in contrast to non-truncated PECO (Figure 3D). As a result, PECO-Mini leads to superior AAV titers and prime editing activity following PE delivery by AAV transduction. We also demonstrated that the PECO-Mini (673-674/Rma) could be delivered by dual AAVs into the liver of mice, giving rise to prime editing. Similar to the beneficial effect on AAV vector packaging, we hypothesized that PECO-Mini would also benefit other viral vector systems. Indeed, we showed that PECO-Mini provided a superior LV titer (46.4-fold) over the regular PECO in the all-in-one PE system. Furthermore, the all-in-one LV vector harboring PECO-Mini induced potent prime editing, whereas the full-length PECO was non-functional, highlighting the value of PECO-Mini in LV applications. Similarly, PECO-Mini may benefit other vector systems and enable more flexible vector design.
Intein-mediated split PE system delivery by dual AAV vectors enables versatile gene editing for in vivo gene therapy. We show here that choice of intein and the position of the Cas9 split site is critical to prime editing efficiency, which matches findings for base editing.8,9 For instance, a previous publication employed the Npu intein and Cas9 713-714 split site for PE delivery by dual AAV vectors.2 However, in our hands, this system displayed suboptimal prime editing efficiency (Figure 3B). Although we have identified two efficient split positions for PE delivery, a larger screening of Cas9 split sites and inteins might identify a more robust split PE system. Such investigations can be reinforced by the extra 621-bp space provided by the PECO-Mini. This extra capacity allows the accommodation of a more flexible promoter choice, e.g., the use of tissue-specific promoters or other functional elements.
Materials and methods
Plasmid construction
The plasmids pCMV-PE2 (Addgene no. 132775, a gift from David Liu) expressing prime editor 2 and pU6-pegRNA-GG-acceptor (Addgene no. 132777, a gift from David Liu) expressing pegRNA were gifts from David Liu.1 To test the RT variants from other species, the 12 selected RT variants were human codon optimized by GenScript, and then the corresponding DNA fragments were ordered from Twist Bioscience, which were inserted into the PE2 plasmid to replace M-MLV RT by Gibson cloning using BamHI and AgeI restriction enzyme sites. To make PE constructs with RTs of different codon usage, M-MLV RT was codon optimized by different web sources and the DNA fragments were ordered from Twist Biosciences and inserted into PE2 by BamHI and AgeI sites. The DNA sequences of RT variants can be found in Data S1 and S2. The DNA sequences of two truncated M-MLV RTs are listed in Data S3. pegRNA plasmids were cloned following the protocol from David Liu.1 ngRNA expression plasmid was cloned using a homemade px330 plasmid (Addgene plasmid no. 42230, a kind gift from Feng Zhang) described in Jensen et al.,13 in which Cas9 was removed and the sgRNA expressed from the U6 promoter. Nucleotides encoding ngRNA and pegRNA spacers and 3ʹ extension sequences are listed in Table 1.
Table 1.
Spacer and 3ʹ extension sequences of pegRNAs and spacer sequences of ngRNAs
| Spacer sequence | 3ʹ extension sequence | PBS length (nt) | RTT length (nt) | ||
|---|---|---|---|---|---|
| pegRNA | HEK3 +1 CTT insertion | 5ʹ-GGCCCAGACTGAGCACGTGA-3ʹ | 5ʹ-AGACTTCTCCACA GGAGTCAGGTGCAC-3ʹ |
13 | 10 |
| HBB +3 A to T | 5ʹ-GCATGGTGCACCTGACTCCTG-3ʹ | 5ʹ-AGACTTCTCCACAG GAGTCAGGTGCAC-3ʹ |
13 | 14 | |
| RNF2 +5 G to T | 5ʹ-GTCATCTTAGTCATTACCTG-3ʹ | 5ʹ-AACGAACACATCA GGTAATGACTAAGATG-3ʹ |
15 | 14 | |
| PCSK9 +1 A insertion | 5ʹ-CCCATACCTTGGAGCAACGG-3ʹ | 5ʹ-ACCTTCCGCCGaT TGCTCCAAGGT-3ʹ |
12 | 12 | |
| ngRNA | HEK3 +1 CTT insertion | 5ʹ-GTCAACCAGTATCCCGGTGC-3ʹ | |||
| HBB +3 A to T | 5ʹ-CCTTGATACCAACCTGCCCA-3ʹ | ||||
| RNF2 +5 G to T | 5ʹ-TCAACCATTAAGCAAAACAT-3ʹ | ||||
| PCSK9 +1 A insertion (ngRNA1) | 5ʹ-ACCTTCCGCCGaTTGCTCCA-3ʹ | ||||
| PCSK9 +1 A insertion (ngRNA2) | 5ʹ-CCGCCGaTTGCTCCAAGGTA-3ʹ |
AAV vector plasmids were cloned in pAAV-MCS (Agilent Technologies) containing inverted terminal repeats from AAV2. The AAV plasmids were created by Gibson cloning, and the DNA fragments encoding the split PE system were PCR amplified or synthesized by Twist Bioscience. The schematic of AAV genomes is displayed in Figure S4.
Third-generation LVs were constructed based on pCCL-PGK-MCS-IRES-Puro, which originated from pCCL-WPS-PGK-EGFP-WHV.14 The DNA fragments were PCR amplified or synthesized by Twist Bioscience, and Gibson cloning was performed for DNA insertion using NheI and MluI enzyme sites. All constructs were verified by Sanger sequencing.
In vitro transcription (IVT)
IVT of PE mRNA was performed as described previously.15 Briefly, plasmids for IVT were made based on a backbone containing the T7 promoter, gene of interest (PE), a murine Hba-a1 3ʹ UTR, and a stretch of 50 adenines (poly(A)). The plasmid was then linearized with a BtgZI restriction enzyme cutting immediately after the poly(A). The linearized plasmid was concentrated and purified by ammonium acetate precipitation. IVT reactions were conducted using the MEGAscript Kit (Ambion, Thermo Fisher Scientific) according to the manufacturer’s instructions but with full substitution of uridine with pseudouridine (Trilink Biotechnologies or APExBio) and co-transcriptional capping with CleanCap AG (Trilink Biotechnologies) in a 1:4 ratio between GTP and CleanCap. The mRNA was purified and concentrated using the RNA Clean and Concentrator kit (Zymo Research). The quality of the IVT RNA was confirmed on a denaturing formaldehyde gel and quantified by ultraviolet visible (UV-vis) spectrophotometry.
sgRNAs
Synthetic pegRNA and ngRNA were purchased from Synthego as chemically modified sgRNAs containing 2ʹ-O-methyl groups at the three first and last bases and 3ʹ phosphorothioate bonds between the first three and the last two bases.
Cell culture
HEK293T cells were cultured in DMEM (Life Technologies) supplemented with 10% fetal calf serum (FCS), 2 mM L-glutamine, penicillin (100 U/mL), and streptomycin (100 mg/mL). Hepa 1–6 cells were cultured in DMEM (Life Technologies) supplemented with 5% FCS, 2 mM L-glutamine, penicillin (100 U/mL), and streptomycin (100 mg/mL).
Transfection and electroporation
Plasmid transfection was conducted by Lipofectamine 2000 (Invitrogen). Briefly, 0.5 mL culture medium with 1.3 × 105 cells was seeded per well in 24-well plates 1 day prior to transfection. Transfections were performed with Lipofectamine 2000 according to the manufacturer’s protocol. For Hepa 1–6 cells, 300 ng pLenti-CMV-GFP-SV-Puro plasmid (Addgene no. 73582) was co-transfected, and cells were selected by puromycin addition (2 μg/mL) 1 day post-transfection for 2 days. Unless specified, 1,500 ng PE plasmid, 500 ng pegRNA plasmid, and 245 ng ngRNA were used per well. For comparing the split and non-split PE systems, a same molar of split or non-split PE plasmids (equivalent to 1,500 ng for non-split PE plasmid) together with fixed amounts of pegRNA (500 ng) and ngRNA (245 ng) plasmids were transfected. Three days post-transfection, cells were harvested, and genomic DNA was extracted using QuickExtract DNA Extraction Solution (Lucigen) according to the manufacturer’s instructions. The genomic DNA was used for PCR amplification of the DNA flanking the prime editing target sites (see primers in Table 2). PCR fragments were gel purified and subjected to Sanger sequencing (Eurofins Genomics). The prime editing efficiency was analyzed by Inference of CRISPR Edits (ICE).
Table 2.
Primers used for PCR, Sanger sequencing, and NGS
| Locus and sequence change | Name | Primer sequence (5ʹ-3ʹ) | Sequencing type |
| HBB +3 A to T | HBB Fw | 5ʹ-CCAACTCCTAAGCCAGTGCCAGAAGAG-3ʹ | Sanger sequencing |
| HBB Rev | 5ʹ-AGTCAGTGCCTATCAGAAACCCAAGAG-3ʹ | ||
| HBB NGS Fw | 5ʹ-ACACTCTTTCCCTACACGACGCTCTTCCG ATCTCCAACTCCTAAGCCAGTGCCAGAAGAG-3ʹ |
NGS | |
| HBB NGS Rev | 5ʹ-GACTGGAGTTCAGACGTGTGCTCTTCCGAT CTAGTCAGTGCCTATCAGAAACCCAAGAG-3ʹ |
NGS | |
| HEK3 +1 CTT insertion |
HEK3 Fw | 5ʹ-ATGTGGGCTGCCTAGAAAGG-3ʹ | Sanger sequencing |
| HEK3 Rev | 5ʹ-CCCAGCCAAACTTGTCAACC-3ʹ | ||
| HEK3 NGS Fw | 5ʹ-ACACTCTTTCCCTACACGACGCTCTTCCGAT CTATGTGGGCTGCCTAGAAAGG-3ʹ |
NGS | |
| HEK3 NGS Rev | 5ʹ-GACTGGAGTTCAGACGTGTGCTCTTCCGA TCTCCCAGCCAAACTTGTCAACC-3ʹ |
NGS | |
| RNF2 +5 G to T | RNF2 Fw | 5ʹ-ACGTCTCATATGCCCCTTGG-3ʹ | Sanger sequencing |
| RNF2 Rev | 5ʹ-ACGTAGGAATTTTGGTGGGACA-3ʹ | ||
| RNF2 NGS Fw | 5ʹ-ACACTCTTTCCCTACACGACGCTCTTCCGATC TACGTCTCATATGCCCCTTGG-3ʹ |
NGS | |
| RNF2 NGS Rev | 5ʹ-GACTGGAGTTCAGACGTGTGCTCTTCCGATCT ACGTAGGAATTTTGGTGGGACA-3ʹ |
NGS | |
|
PCSK9 +1 A insertion (ngRNA2) |
PCSK9 Fw | 5ʹ-CTTGGCTCCCCAGAGACATC-3ʹ | Sanger sequencing |
| PCSK9 Rev | 5ʹ-CTAAGTCTTGCCCTCGCCTC-3ʹ | ||
| PCSK9 NGS Fw | 5ʹ-ACACTCTTTCCCTACACGACGCTCTTCCGATC TCTTGGCTCCCCAGAGACATC-3ʹ |
NGS | |
| PCSK9 NGS Rev | 5ʹ-GACTGGAGTTCAGACGTGTGCTCTTCCGATCTC TAAGTCTTGCCCTCGCCTC-3ʹ |
NGS |
RNA delivery was performed by electroporation using the 4D-Nucleofector device (Lonza) in 20 μL format Nucleocuvette Strips according to the manufacturer’s protocols. HEK293T cells were trypsinized just before electroporation, and 2 × 105 cells were used per 20-μL reaction. The RNA amounts used were 2 μg of PE/PECO mRNA, 1 μg of pegRNA, and 1 μg of ngRNA. The electroporation buffer and program used for HEK293T cells were Opti-MEM (Gibco) and P3-CM138, respectively. Prime editing analysis included genomic DNA extraction and Sanger sequencing performed as described above.
Western blot analysis
We seeded 2.5 × 105 HEK293T cells in 1 mL culture medium per well in 12-well plate 1 day prior to transfection. Five hundred nanograms PE plasmid was transfected using Lipofectamine 2000. Two days after transfection, the cells were lysed using the Blue Loading Buffer Pack (no. 7722, Cell Signaling Technology) according to the manufacturer’s protocol. The isolated total cellular protein was electrophoresed on a 4%–20% precast polyacrylamide gel (Bio-Rad) and transferred to a polyvinylidene fluoride (PVDF) membrane. PE protein was detected using an anti-Cas9 antibody (no. 14697, Cell Signal) with a 1:1,000 dilution according to the manufacturer’s instructions. The β-actin protein was detected by β-Actin (8H10D10) Mouse monoclonal antibody (mAb) (no. 3700, Cell Signal) with a 1:1,000 dilution. The signals were visualized by ChemiDoc MP Imaging System (Bio-Rad) and analyzed by ImageJ.
NGS and data analysis
NGS was performed by Eurofins using the INVIEW CRISPR Check second PCR service with MiSeq Sequencing, 60,000 read pairs (2 × 300 bp). Briefly, genomic sites of interest were PCR amplified using Phusion Hot Start II PCR Master Mix with primers containing forward and reverse adapters (see primers in Table 2). The PCR products were agarose gel purified and submitted to Eurofins for library construction and sequencing. Analysis of prime editing outcomes was performed by CRISPResso2 using the homology-directed repair (HDR) mode.16 Only sequencing reads with a minimum homology of 90% and an average quality score ≥30 were included in the analysis. The percentage of “HDR” in the report represented the percentage of correct prime edits, and the indel yields were calculated as (number of indel containing reads + imperfect HDR)/(total reads).
AAV vector production
AAV6 and AAV8 vectors were produced as described with a few modifications.17 In brief, HEK293T cells were seeded at 11 × 106 cells per 15-cm culture dish 1 day before transfection. For AAV6, one 15-cm dish was transfected using standard polyethylenimine (PEI) transfection with 6 μg ITR-containing plasmid and 22 μg pDGM6 (a kind gift from D. Russell), which contains the AAV6 cap genes, AAV2 rep genes, and adenovirus helper genes. For AAV8, one 15-cm dish was transfected using standard PEI transfection with 6 μg ITR-containing plasmid, 15.5 μg pAdDeltaF6 helper plasmid (kindly provided by James M. Wilson, AddGene no. 112867), and 7.4 μg pAAV2/8 packaging plasmid (kindly provided by James M. Wilson, AddGene no. 112864). AAV vectors were collected 48 h post-transfection from cells by three freeze-thaw cycles followed by a 45-min incubation with TurboNuclease at 250 U mL−1 (Sigma). AAV vectors were purified on an iodixanol density gradient by ultracentrifugation at 237,000g for 2 h at 18°C. AAV vectors were extracted at the 60%–40% iodixanol interface and concentrated using a 100K Amicon Ultra-15 centrifugal filter with 1× sorbitol containing 0.001% pluronic acid. Vectors were aliquoted and stored at −80°C until use. AAV6 vectors were titrated using ddPCR (QX-200 from Bio-Rad) to quantify number of vector genomes as described previously.18 The ITR primers and probe used for ddPCR were forward primer 5ʹ-GGAACCCCTAGTGATGGAGTT-3ʹ; reverse primer 5ʹ-CGGCCTCAGTGAGCGA-3ʹ; ITR probe: 5ʹ-FAM-CACTCCCTCTCTGCGCGCTCG-IBFQ-3ʹ (IDT).
Lentiviral vector production
LVs were produced by transient transfection of Lenti-X 293T cells (Takara Bio). Cells were seeded at a density of 3.75–4.00 × 106 in p10 dishes and incubated overnight. A calcium phosphate solution containing 3 μg pRSV-REV, 3.75 μg pMD2G (VSV-G), 13 μg pMDlg_p-RRE, and 13 μg transgene was dropwise added to p10 dishes and incubated overnight. The medium was replenished after 24 h, and the crude supernatant was harvested at 48 h. The supernatant was passed through a 0.45-μM filter and centrifuged at 1,200 rpm to precipitate residual debris. The CFU assay for titer determination was performed by limiting dilution transduction of the LVs on 1 × 105 HEK293T cells seeded in 6-well plates with polybrene (8 μg/mL). The medium was aspirated 24 h after transduction, and blasticidin selection was applied (5 μg/mL) for 12 days. Cells were fixed in 4% paraformaldehyde for 15 min at room temperature and stained with 0.1% crystal violet for 20 min, followed by PBS washing twice. The number of colonies were counted under a microscope. LV titers were calculated with the formula number of colonies/total volume in the well (mL) × dilution factor.
Dual AAV6 and LV transduction of HEK293T cells
For AAV transduction, 1 × 104 HEK293T cells in 100 μL culture medium were seeded in 96-well plates and transduced with two AAVs, each at 5 × 105 vg/cell. Five days post-transduction, cells were subjected to prime editing analysis as described above. For LV transduction, 1 × 105 HEK293T cells in 1 mL medium were seeded in 6-well plates and transduction was carried out in a total volume of 1 mL fresh medium containing polybrene (8 μg/mL) and 1 mL crude virus. The medium was aspirated after 24 h, and blasticidin selection (10 μg/mL) was initiated. Cells were harvested at the indicated time point after transduction and subjected to prime editing analysis by Sanger sequencing as described above.
Animal studies
Mice were housed in the Animal Facilities of the Department of Biomedicine, Aarhus University, Denmark with standard chow and water ad libitum. Animal experiments were approved by the Danish Animal Experiments Inspectorate. AAV particles were delivered intravenously (i.v.) in 200 μL saline to 10-week-old C57BL/6 male mice. Mice received two different doses of AAV particles, 1 × 1011 vg or 1 × 1012 vg of each of PECO-Mini-C and PECO-Mini-N AAV (total dose of 2 × 1011 or 2 × 1012 vg). Mice were sacrificed 4 weeks after AAV delivery, and the livers were harvested. The genomic DNA of liver samples was obtained by treatment with chorion villi buffer followed by isopropanol precipitation.
Statistical analysis
The data were analyzed by GraphPad Prism 9 and presented as mean ± SD. The significance was tested using a two-tailed unpaired Student’s t test, and p < 0.05 was considered significant.
Acknowledgments
Z.G. gratefully acknowledges support from an individual postdoctoral fellowship from the Lundbeck Foundation (R303-2018-3571). R.O.B. gratefully acknowledges the support from a Lundbeck Foundation Fellowship (R238-2016-3349), the Independent Research Fund Denmark (0134-00113B, 0242-00009B, and 9144-00001B), an AIAS-COFUND (Marie Curie) fellowship from Aarhus Institute of Advanced Studies (AIAS) co-funded by Aarhus University’s Research Foundation and the European Union’s Seventh Framework Program under grant agreement no. 609033, the Novo Nordisk Foundation (NNF19OC0058238 and NNF17OC0028894), Innovation Fund Denmark (8056-00010B), the Carlsberg Foundation (CF20-0424 and CF17-0129), Slagtermester Max Wørzner og Hustru Inger Wørzners Mindelegat, the A.P. Møller Foundation, the Riisfort Foundation, and a Genome Engineer Innovation Grant from Synthego.
Declaration of interests
The authors declare the following competing interests: R.O.B. holds equity in Graphite Bio and UNIKUM Tx. R.O.B. is a part-time employee in UNIKUM Tx. None of the companies were involved in the present study. The remaining authors declare no competing interests.
Author contributions
Z.G. and R.O.B. conceived the study and designed the experiments. Z.G. performed the bulk of the experiments and analyzed the data with assistance from S.R., N.S.M., J.H., H.C., X.D., and M.K.T. S.R.P., M.K.T., J.G.M., and R.O.B. supervised the study. Z.G. took lead in writing the manuscript with assistance from R.O.B. All authors reviewed, edited, and approved the final manuscript.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.07.001.
Supplemental information
Data availability
The raw sequencing data have been deposited to the NCBI Sequence Read Archive under accession NCBI: GSE205532. All raw data are available from the corresponding author upon request. The plasmids PECO, PECO-Mini-N (Rma 673-674), PECO-Mini-C (Rma 673-674), and LV_PECO-Mini will be deposited to Addgene.
References
- 1.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu P., Liang S.Q., Zheng C., Mintzer E., Zhao Y.G., Ponnienselvan K., Mir A., Sontheimer E.J., Gao G., Flotte T.R., et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 2021;12:2121. doi: 10.1038/s41467-021-22295-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhi S., Chen Y., Wu G., Wen J., Wu J., Liu Q., Li Y., Kang R., Hu S., Wang J. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 2021 doi: 10.1016/j.ymthe.2021.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y., Yang G., Huang S., Li X., Wang X., Li G., Chi T., Chen Y., Huang X., Wang X. Enhancing prime editing by Csy4-mediated processing of pegRNA. Cell Res. 2021;31:1134–1136. doi: 10.1038/s41422-021-00520-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson J.W., Randolph P.B., Shen S.P., Everette K.A., Chen P.J., Anzalone A.V., An M., Newby G.A., Chen J.C., Hsu A., Liu D.R. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2021 doi: 10.1038/s41587-021-01039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen P.J., Hussmann J.A., Yan J., Knipping F., Ravisankar P., Chen P.-F., Chen C., Nelson J.W., Newby G.A., Sahin M. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. 2021;184:5635–5652. doi: 10.1016/j.cell.2021.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schultz S.J., Champoux J.J. RNase H activity: structure, specificity, and function in reverse transcription. Virus Res. 2008;134:86–103. doi: 10.1016/j.virusres.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y., Zhi S., Liu W., Wen J., Hu S., Cao T., Sun H., Li Y., Huang L., Liu Y. Development of highly efficient dual-AAV split adenosine base editor for in vivo gene therapy. Small Methods. 2020;4:2000309. [Google Scholar]
- 9.Levy J.M., Yeh W.-H., Pendse N., Davis J.R., Hennessey E., Butcher R., Koblan L.W., Comander J., Liu Q., Liu D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jang H., Jo D.H., Cho C.S., Shin J.H., Seo J.H., Yu G., Gopalappa R., Kim D., Cho S.-R., Kim J.H. Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases. Nat. Biomed. Eng. 2022;6:181–194. doi: 10.1038/s41551-021-00788-9. [DOI] [PubMed] [Google Scholar]
- 11.Wu Z., Yang H., Colosi P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao Z., Herrera-Carrillo E., Berkhout B. A single H1 promoter can drive both guide RNA and endonuclease expression in the CRISPR-Cas9 system. Mol. Therapy-Nucleic Acids. 2019;14:32–40. doi: 10.1016/j.omtn.2018.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jensen T.I., Mikkelsen N.S., Gao Z., Fosselteder J., Pabst G., Axelgaard E., Laustsen A., Konig S., Reinisch A., Bak R.O. Targeted regulation of transcription in primary cells using CRISPRa and CRISPRi. Genome Res. 2021;31:2120–2130. doi: 10.1101/gr.275607.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raoul C., Abbas-Terki T., Bensadoun J.-C., Guillot S., Haase G., Szulc J., Henderson C.E., Aebischer P. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- 15.Jensen T.I., Mikkelsen N.S., Gao Z., Foßelteder J., Pabst G., Axelgaard E., Laustsen A., König S., Reinisch A., Bak R.O. Targeted regulation of transcription in primary cells using CRISPRa and CRISPRi. Genome Res. 2021:275121. doi: 10.1101/gr.275607.121. gr. 275607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furuta-Hanawa B., Yamaguchi T., Uchida E. Two-dimensional droplet digital pcr as a tool for titration and integrity evaluation of recombinant adeno-associated viral vectors. Hum. Gene Ther. Methods. 2019;30:127–136. doi: 10.1089/hgtb.2019.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing data have been deposited to the NCBI Sequence Read Archive under accession NCBI: GSE205532. All raw data are available from the corresponding author upon request. The plasmids PECO, PECO-Mini-N (Rma 673-674), PECO-Mini-C (Rma 673-674), and LV_PECO-Mini will be deposited to Addgene.