

Abstract
Metagenomic classifiers are widely used for the taxonomic profiling of metagenomics data and estimation of taxa relative abundance. Small subunit rRNA genes are a gold standard for phylogenetic resolution of microbiota, although the power of this marker comes down to its use as full-length. We aimed at identifying the tools that can efficiently lead to taxonomic resolution down to the species level. To reach this goal, we benchmarked the performance and accuracy of rRNA-specialized versus general-purpose read mappers, reference-targeted assemblers and taxonomic classifiers. We then compiled the best tools (BBTools, FastQC, SortMeRNA, MetaRib, EMIRGE, VSEARCH, BBMap and QIIME 2’s Sklearn classifier) to build a pipeline called RiboTaxa. Using metagenomics datasets, RiboTaxa gave the best results compared to other tools (i.e. Kraken2, Centrifuge, METAXA2, phyloFlash, SPINGO, BLCA, MEGAN) with precise taxonomic identification and relative abundance description without false positive detection (F-measure of 100% and 83.7% at genus level and species level, respectively). Using real datasets from various environments (i.e. ocean, soil, human gut) and from different approaches (e.g. metagenomics and gene capture by hybridization), RiboTaxa revealed microbial novelties not discerned by current bioinformatics analysis opening new biological perspectives in human and environmental health.
INTRODUCTION
In recent decades, significant advancements in sequencing technologies have helped to better characterize microbiomes from human gut (1), soil (2) and oceans (3). Predicting the presence and relative abundance of taxa through analysis of phylogenetic markers like the 16S ribosomal RNA (rRNA) gene is a common approach adopted in microbial ecology (4). Widely used, PCR amplification and sequencing of the 16S rRNA gene through metabarcoding generally employs universal PCR primers to target highly variable regions of this gene. However, this approach can lead to PCR biases and sequence chimera (5), leading to incorrect microbial profiling. Furthermore, the amplicon length (about 400 bp corresponding to one to two variable regions) produced by the second-generation sequencing platform reduces the accuracy or reliability of phylogenetic resolution, limiting taxonomic affiliation to the family level, in general, or in the best cases, to genus level (6). Thus, using sequencing data, that allows access to all the 16S variable regions without primer and PCR biases, such as shotgun metagenomics or 16S-targeted gene capture by hybridization (7), appears to be the most suitable to describe precisely microbial communities based on a phylogenetic marker.
Commonly, microbial profiling of shotgun metagenomics data is performed through annotation of reads, de novo assembled genes or metagenome assembled genomes (8). However, deciphering taxonomic diversity is often limited by incomplete genome databases compared to large 16S rRNA gene repositories, despite relentless efforts to update them (9). Direct read annotation is largely favoured because of computational arduousness during assembly. Consequently, several approaches have been proposed and compared to estimate microbial diversity and relative abundance of species using whole genome or marker reference databases (10). Most methods such as MEGAN (11) rely on sequence alignment based on matched database sequences and use the lower common ancestor (LCA) algorithm to assign taxa to the query sequence. However, the LCA algorithm fails to consider the differing degrees of similarity between the query and the database hit sequences. To overcome this problem, BLCA (12) has adopted a Bayesian-based LCA method whereby the taxonomic assignment of the query sequence is weighted by a Bayesian probability based upon the sequence similarity of the database hit to the query. Nevertheless, alignment-based approaches are generally memory and time consuming (13) while the advantage of k-mer based approaches is its fast computational speed. As an example, Kraken (14)/Bracken (15) and SPINGO (16) employ alignment-free method by using exact k-mer matches between reference databases and reads to report the microbial diversity based on LCA of every taxon. Although k-mer approach is very efficient and fast on large metagenomics data, the choice of the length k highly influences the classification (17,18)
The hybrid pipeline METAXA2 (19) allows rRNA extracting reads from large sequencing data sets using Hidden Markov Models (HMM) (20) and directly subjects putative rRNA reads to a BLAST (21) search against a specialized rRNA database. METAXA2 can report read origin (archaeal, bacterial, nuclear eukaryote, mitochondrial or chloroplast) and hierarchical classification down to genus or species level. Here, the step of rRNA read extraction accelerates the algorithm execution while enhancing specificity of the analysis. Although HMM was used in METAXA2, other methods can be employed such as general-purpose mappers such as BBMap (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/) or specialized algorithm such as SortMeRNA (22) to align reads against a representative set of rRNA database.
Yet, the major concern remains that unassembled short-length sequences do not contain all the informative regions of the 16S rRNA genes, thus reducing the accuracy of taxonomic assignments to species level (23). Indeed, depending on the 16S rRNA region carried by each read, taxonomical level of affiliation may vary from phylum to species, impairing the true representation of the microbial community diversity. Converting short reads into full-length or nearly full-length rRNA gene sequences typically yields a more detailed taxonomic resolution, at the species or even strain level (24), providing a unique opportunity to thoroughly analyse microbial species that have never been identified before. Pipelines, including filtering, reconstruction of full-length SSU rRNA genes and their classification, may represent useful tools to facilitate data analysis for scientists. PhyloFlash (25) is a compilation of tools to rapidly filter and reconstruct the SSU rRNAs and explore phylogenetic composition from metagenomics or meta-transcriptomics datasets. To filter SSU reads from Illumina metagenomics dataset, phyloFlash proposes the general-purpose mapper BBMap and the rRNA-specialized tool SortMeRNA. To assemble full-length SSU rRNA sequences, the general-purpose genomic assembler SPAdes (26) and the reference-based assembler EMIRGE (27) are proposed. Taxonomic identification of assembled sequences is done using VSEARCH (28). Efficiency of 16S rRNA sequence reconstruction is the pivotal step of this approach. EMIRGE, that uses Bayesian approach to iteratively map short Illumina reads against a set of reference gene sequences with Bowtie (29), was selected for this pipeline but other reference-based targeted assemblers have been published since.
Released in 2020, MetaRib (30) has been developed to reconstruct rRNA gene sequences from total RNA meta-transcriptomic data. Using the same mapping in an iterative approach as EMIRGE, this tool provides several improvements such as integrating sub-assembly and dereplication before iterative mapping to reduce running time and memory usage. MATAM (31) also provides high-quality reconstructed full-length 16S rRNA gene sequences using the construction and exploitation of an overlap graph and is designed to minimize the error rate and the risk of chimera formation during rRNA gene assembly from metagenomics data.
Our aim was to propose an accurate and comprehensive tool based on SSU rRNA gene to exploit metagenomics data to the maximum, taking advantage of a widely studied phylogenetic marker that can overcome the lack of reference genomes and help to better characterize microbial communities and discover novelties.
In this article, we have evaluated the performance and accuracy of rRNA-specialized versus general-purpose read mappers, reference-targeted assemblers and taxonomic classifiers. Using a microbial mock, we aimed at identifying the tools that can efficiently lead to taxonomic resolution down to the species level from metagenomics data. Finally, we have compiled the selected tools to build a pipeline called RiboTaxa. This pipeline takes metagenomics Illumina sequences as input and rapidly filter and reconstruct SSU rRNA genes for taxonomic identification associated to relative abundance description. The efficiency of RiboTaxa was determined using a synthetic complex community of human gut microbiota and compared to existing 16S-based metagenomics classifiers (i.e. Centrifuge (32), Kraken2, METAXA2, phyloFlash, SPINGO, BLCA, MEGAN). Later, RiboTaxa was applied to various real metagenomics datasets (i.e. ocean, soil, human gut) but also to gene capture by hybridization datasets from previous works to reveal novelties not detected by current bioinformatics pipelines.
MATERIALS AND METHODS
Tool benchmarking on microbial MOCK community
Microbial MOCK community
The microbial MOCK community was composed of 21 bacterial and 7 archaeal species (Supplementary Figure S1), which was shotgun sequenced in paired-end (2 × 300 bp) MiSeq runs (Illumina) by Gasc and Peyret (33). Microbial strains were selected from the Leibniz Institute DSMZ collection provided the availability of their genome in GenBank database. The abundance of each species was then defined based on the 16S rRNA copy number per genome and the number of genomes in the mixture.
Raw Illumina reads from the shotgun sequencing (under accession number SRR5381736) were downloaded from NCBI open access Sequence Read Archive (SRA) using NCBI SRA Toolkit_v2.9.1 (https://www.ncbi.nlm.nih.gov/sra). Fastq-dump v2.8.2 (https://www.ncbi.nlm.nih.gov/sra/docs/sradownload/) was then used to extract Fastq files from SRR files using parameter -I -split-files to separate Fastq files into forward (R1) and reverse files (R2).
Sequence quality control and read trimming
Illumina shotgun reads were processed using bbduk.sh (k-mer = 21, https://jgi.doe.gov/data-and-tools/software-tools/bbtools/, BBTools v37.62) to remove Illumina adapters, known Illumina artifacts and to quality-trim both ends to Q20. Resulting reads containing more than one ‘N’, or with quality scores (before trimming) averaging <20 over the read, or length under 60 bp after trimming, were discarded. Quality control was performed using FastQC v0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) before and after trimming to ensure that high-quality Illumina reads are passed onto the next steps.
Database preparation and indexing
Unless specified otherwise, SILVA small subunit (SSU) ribosomal RNAs (16S/18S) 138.1 released on August 27, 2020 (http://www.arb-silva.de) was used for rRNA read extraction, full-length 16S rRNA gene reconstruction and taxonomic classification. SILVA 138.1 database (Fasta format) was clustered at 97% (NR97) with VSEARCH -cluster_fast v2.7.0, converted from RNA to DNA alphabet, and any ambiguous bases were replaced by random base characters using fix_nonstandard_chars.py. Specific database indexes were then built for each tool used in this study. The NR97 filtered and fixed database was indexed for BBMap with bbmap.sh (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/), for EMIRGE (27) and MetaRib (30) with Bowtie (29) and for SortMeRNA (22) and MATAM (31) with matam_db_preprocessing.py, for Kraken2/Bracken (34) with kraken2-build (kmer = 35) and for Centrifuge (32) with centrifuge-build.
SSU rRNA reads extraction
BBMap v38.87 (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/): The input reads were aligned (mapped) against the indexed SILVA 138.1 database, with minimum identity 70% by default, retaining all ambiguous alignments if there are multiple best scoring. Output was written in Fastq format, retaining all read pairs where at least one read could be aligned.
SortMeRNA v2.1b (22): Since SortMeRNA accepted only one file as input for the reads, forward and reverse paired-end reads were interleaved into a single Fastq file using merge-paired-reads.sh (SortMeRNA v2.1b). The reads were then aligned by SortMeRNA against the indexed SILVA 138.1, using min_lis of 2 (number of candidate alignments) and E-value cut-off of 1 by default. Aligned and unaligned reads were reported with options -aligned and -other, respectively and were written in Fastq format. To keep the order of paired-end reads –paired-in option was used and an overall statistics file (-log) was generated. After filtering, the aligned Fastq file containing the SSU rRNA reads was separated into forward and reverse paired-end reads using unmerge-paired-reads.sh (SortMeRNA v2.1b).
Assembly of nearly full-length SSU rRNA sequences
EMIRGE v0.61.1 (27): Two available scripts were tested, namely Emirge.py and Emirge_amplicon.py designed for metagenomics data and PCR amplicon data respectively. Both scripts were tested with the same following parameters. Average insert size of paired-end input was estimated from the initial mapping with Bowtie using mean_size.py (https://gist.github.com/timoast/af73c0e9fac00187ee49). EMIRGE was run with 120 iterations with -max_read_length = 300, -insert_mean = 500, -insert_stddev = 100 and -join_threshold = 1. Once all the iterations were completed, emirge_rename_fasta.py (EMIRGE) was run on the last iteration folder to convert Bam files into a single Fasta file containing all the reconstructed SSU rRNA genes and their relative abundances.
During sequence assembly, the join_threshold parameter influences sequence reconstruction as if two candidate sequences share ≥ join_threshold value over their bases with mapped reads, then both sequences are merged into one for the next iterations. Emirge_amplicon.py was run using the same above parameters with -join_threshold = 0.97 (as proposed in the original publication of EMIRGE) and the results were compared.
MetaRib (version from 13 November 2019) (30): For MetaRib, which uses the same iterative mapping algorithm as EMIRGE, the above parameters were used with -join_threshold = 1. The deduplication included in MetaRib was performed with the following mapping parameters: minid = 0.96, maxindel = 1, minhits = 2, idfilter = 0.98. Reconstructed SSU rRNA gene sequences were written in Fasta format and their relative abundances were output in a tsv file.
MATAM v1.6.0 (31): The forward and reverse paired-end reads were first reformatted to interleaved Fastq format with reformat.sh (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/). The resulting Fastq file was used as input and matam_assembly.py was run using a score threshold of 0.7, a minimum identity of 1 and an E-value cut-off of 1e−05. Reconstructed rRNA gene sequences were written in Fasta format and read counts were written in a tsv file.
Identification of 16S variants present in the microbial mock community
To build a 16S rRNA database of the microbial mock community, 16S rRNA sequences were downloaded from the NCBI (https://www.ncbi.nlm.nih.gov/gene/) for each microbial strain and concatenated into a single Fasta file (excluding low-quality variants containing Ns in sequence). The sequences were first deduplicated to get rid of strictly identical sequences and then clustered at 100% using VSEARCH v2.7.0. This 16S mock database was used in downstream affiliation of reconstructed sequences using blastn of NCBI BLAST + v2.11.0 (21) with an identity cut-off fixed at 99% to differentiate between microbial variants. Only the top hit alignment between the subject and the query sequence was targeted using HSP = 1 parameter.
Taxonomic affiliation
To classify nearly full-length reconstructed SSU sequences, Kraken2 v2.0.8-beta (14), Centrifuge v1.0.3-beta (32), mothur v1.33 (classify.seqs) (35), SPINGO v1.3 (16), BLCA v2.1 (12), QIIME 2’s Sklearn classifier (classify-sklearn plugin, QIIME2 v2020.8) (36) and RDP Classifier v11.5 (http://rdp.cme.msu.edu/) (37) were used. Kraken2 and Centrifuge had their own 16S databases (SILVA 138.1 SSU NR99 sequence set) from which indexes were created. Similarly, for SPINGO, the SILVA 138.1 SSU database was indexed using the provided makefile (https://github.com/tiendu/SPINGO_updated/tree/master/SPINGO_SILVA/database). Unlike other tools, BLCA does not require indexed database files. Instead, the SILVA SSU 138.1 database was provided to BLCA as a BLAST formatted library obtained from the Fasta file using the makeblastdb utility from NCBI BLAST + v2.11.0, together with a taxonomy file using write_taxonomy.py (https://github.com/FOI-Bioinformatics/flextaxd/tree/master/flextaxd/modules). Pre-trained full-length SSU SILVA 138.1 database was downloaded for mothur (https://mothur.org/wiki/silva_reference_files/) and QIIME 2’s Sklearn classifier (https://docs.qiime2.org/2020.11/data-resources/) and for RDP Classifier v11.5, pre-trained full-length 16S rRNA training set 18 released on 14 August 2020 (https://sourceforge.net/projects/rdp-classifier/files/), was used. Taxonomic classification using all the above tools was done with default parameters and a confidence cut-off set at 0.7.
Tool benchmarking on a synthetic complex human gut microbiota
Synthetic complex human gut microbiota
To mimic real dataset analysis, tool benchmarking was then done on a synthetic complex human gut community comprising of 100 microorganisms as described by Lu and Salzberg (38). To simulate this microbial community, 100 genomes (complete genomes, scaffolds, contigs) in Fasta format were downloaded from NCBI. Barrnap v0.9 (https://github.com/tseemann/barrnap) was used to extract all SSU rRNA sequences present in each genome using default parameters. They were clustered at 100% using VSEARCH to remove duplicates and used as references. ART simulator v2.5.8 (39) was then run on each individual genome file to generate 20X coverage of synthetic paired-end Illumina reads with length 250 bp using the following parameters: art-illumina -p -ss MSv3 -f 20 -l 250 -m 300 -s 100. To calculate theoretical abundance of each microorganism, the number of 16S rRNA reads was filtered from individual paired-end file using SortMeRNA and was divided by the sum of the 16S rRNA reads present in the community of 100 microorganisms. Finally, the synthetic metagenomics dataset was produced by concatenating all forward and reverse Fastq files into a single forward (HG_R1) and a single reverse (HG_R2) Fastq file respectively.
The paired-end human gut files were trimmed using bbduk.sh (k-mer = 21, https://jgi.doe.gov/data-and-tools/software-tools/bbtools/, BBTools v37.62) as described above. For SSU rRNA sequence reconstruction, MATAM, emirge.py and emirge_amplicon.py were provided with SSU reads filtered using SortMeRNA while MetaRib was provided with unfiltered high-quality reads. These choices were based on the results of the microbial MOCK community. After SSU rRNA gene assembly, the presence or absence of species was determined, instead of variants. For this, the SSU rRNA references, previously extracted by barrnap, were used in downstream affiliation of reconstructed sequences using blastn of NCBI BLAST + v2.11.0 (21) with an identity cut-off fixed at 97% to identify species. Only the top hit alignment between the subject and the query sequence was targeted using HSP = 1 parameter. Finally, microbial profiling was performed using all the above-mentioned tools applied to the microbial MOCK community.
RiboTaxa pipeline
RiboTaxa pipeline is written in bash and is available on GitHub (https://github.com/oschakoory/RiboTaxa). It can easily be installed using miniconda3 (https://docs.conda.io/en/latest/miniconda.html). Running conda_virt_env.sh will install all the necessary tools required by RiboTaxa to analyse raw Illumina metagenomics reads. To avoid conflicts between required dependencies and those in existing environment, RiboTaxa uses a virtual conda environment.
RiboTaxa pipeline includes SortMeRNA v2.1b and EMIRGE v0.61.1 tools, both of which need indexed databases of their own. For that, parameters for each tool are predefined in the configuration file, indexDB_arguments.conf, of RiboTaxa and indexDB_RiboTaxa.sh is run to create all necessary database indexes required by RiboTaxa to analyse raw metagenomics sequences. Any SSU database in Fasta format can be used, however, we recommend using SILVA SSU database as it is the most updated.
RiboTaxa pipeline is illustrated in Figure 1. The inputs for RiboTaxa are shotgun metagenomics singled-end or paired-end files, which have been generated by an Illumina sequencer. The sequence data can either be in uncompressed Fastq format or in compressed Fastq.gz format. Several samples can be handled in the same folder. RiboTaxa will extract the name of the sample from the name of the Fastq file. By running Pipeline_RiboTaxa.sh, RiboTaxa starts by removing Illumina adapters and trims Illumina reads using bbduk.sh (BBTools v37.62). Quality control is performed before and after trimming with FastQC v0.11.9 and summary statistics are reported in a standalone html file using MultiQC v1.11 (https://github.com/ewels/MultiQC/releases/tag/v1.11). For SSU rRNA gene reconstruction, RiboTaxa uses MetaRib (version from 13 November 2019) and EMIRGE v0.61.1 (Emirge_amplicon.py). MetaRib uses unfiltered high-quality reads for rRNA genes assembly while EMIRGE uses SSU reads that have been extracted using the rRNA-specialized tool SortMeRNA v2.1. The reconstructed sequences are then used as reference onto which unfiltered high-quality reads are mapped using BBmap v38.87 to calculate the relative abundance. Prior to taxonomic affiliation, the reconstructed rRNA gene sequences obtained from MetaRib and EMIRGE are clustered at 97% using VSEARCH v2.17.0 and abundances from BBmap are summed up for sequences sharing the same cluster. Finally, the clustered sequences are classified taxonomically using QIIME 2’s classify-sklearn plugin. Since RiboTaxa incorporates different tools, essential parameters for each tool (bbduk.sh, EMIRGE, MetaRib and skearn_classifier) need to be predefined in the RiboTaxa_arguments.conf file. This allows users to set parameters to fit to their own data and objectives. Threads and memory usage can also be set by the user.
Figure 1.

RiboTaxa pipeline. RiboTaxa takes raw metagenomics data as input and performs quality control using BBTools and FastQC. MetaRib reconstructs SSU rRNA sequences from high-quality reads. Also, SortMeRNA extracts SSU reads from the high-quality reads which are assembled into SSU rRNA sequences by EMIRGE. To estimate relative abundance, BBMap uses reconstructed sequences as reference to align high-quality reads. Prior to taxonomic identification, sequences from EMIRGE and MetaRib are clustered at 97% using VSEARCH and consensus sequences are classified using QIIME 2’s sklearn classifier.
Finally, RiboTaxa outputs a Fasta file containing all the reconstructed sequences clustered to 97% in SSU_sequences.fasta file, and taxonomic affiliation and relative abundances of each reconstructed sequences in SSU_taxonomy_abundance.tsv file. It is important to notice that RiboTaxa and the other metataxonomic tools could not assign certain sequences at species level and stopped at genus-level classification in case of nearly identical 16S rRNA gene sequences.
Parameters used for RiboTaxa analysis
The RiboTaxa_argument.conf file is available on GitHub and contains important parameters required for analysis. This config file needs to be properly filled to avoid errors. Compulsory parameters include directory paths while the remaining parameters can be left as default, except sequence reconstruction parameters –max_read_length, –insert_mean, –insert_stddev which exclusively depend on the sequencing length of the input data.
Comparison of RiboTaxa with other existing rRNA-based metagenomic classifiers on microbial MOCK and synthetic community data
Kraken2, Centrifuge, METAXA2, BLCA, SPINGO, MEGAN6 were evaluated for their ability to describe the microbial MOCK and synthetic community from short reads and labelled as ‘Without sequence reconstruction’. PhyloFlash and RiboTaxa, which included a step of sequence reconstruction from short reads, were labelled as ‘With sequence reconstruction’. All tools except RiboTaxa were run on high-quality metagenomics reads, trimmed and cleaned using bbduk.sh (from BBTools v37.62), while RiboTaxa performed its own quality control step using BBTools.
Kraken2 and centrifuge used default settings. To estimate the relative abundance, Bracken v2.6.1 was used to estimate microbial abundance from Kraken2’s output while Centrifuge outputs its own calculated abundances.
METAXA2 v2.1.2 (19): The function metaxa2 was run using default settings with option -t = bacteria, archaea to filter SSU reads of bacteria and archaea only and to discard any other SSU reads (e.g. mitochondrial, chloroplast). Classifying filtered sequences at different taxonomic levels was done using metaxa2_ttt with default parameters and sequence abundance was estimated using metaxa2_dc. Filtered SSU reads were written in Fasta format while taxonomic identification and read counts were written in txt format. Relative abundance was calculated by dividing each taxon count by the total number of reads classified at the species or genus level.
BLCA v2.1 (12): Taxonomic classification was performed by running 2.blca_main.py with the formatted database and taxonomy file on the interleaved paired-end HG files, in Fasta format (reformat.sh from https://jgi.doe.gov/data-and-tools/software-tools/bbtools/, BBTools v37.62).
SPINGO v1.3 (16): SPINGO was run on the interleaved paired-end HG files, in Fasta format, using default parameters (k = 8, bootstrap = 10). The output taxonomy file was then summarised using the provided python script, spingo_summary (https://github.com/GuyAllard/SPINGO)
MEGAN v6.21.0 (11): For taxonomic profiling, MEGAN takes as input DAA (DIAMOND alignment archive) formatted files. Thus, to generate DAA files, DIAMOND (40) v2.0.14 was used. First, the latest version of NCBI nr protein database was downloaded (on 19 July 2022) from https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz and indexed using diamond makedb with default parameters. The paired-end HG files were interleaved into a single fastq using reformat.sh (BBTools v37.62) and reads were aligned against the pre-built nr database using diamond blastx, specifying the parameter -f 100 to output alignments in DAA format. Finally, the meganizer tool, daa-meganizer, was applied to the DAA file for taxonomic binning based on the SILVA taxonomy mapping file obtained from https://software-ab.informatik.uni-tuebingen.de/download/megan6/welcome.html. Relative abundance was also calculated using the number of aligned reads output by DIAMOND.
phyloFlash v2.0 (25): Since phyloFlash offers two tools for rRNA reads extraction (BBMap or SortMeRNA) and full-length sequence reconstruction (SPAdes or EMIRGE), we tested several associations between these tools. Default tools (BBMap + SPAdes) were used to produce phyloFlash (BS) results. PhyloFlash was also run using SortMeRNA and EMIRGE (phyloFlash (SE)) or using BBMap and EMIRGE (phyloFlash (BE)). For all conditions, cluster identity was set at 100%, the maximum length of reads was 300 bp and the taxonomic classification was set at genus and species level. Output from SPAdes and EMIRGE were written in Fasta. Abundance estimates and summary statistics were reported in a standalone Html file. All reconstructed sequences were filtered using bbduk.sh (BBTools v37.62) to discard sequences below 800 bp. For taxonomic affiliation, phyloFlash classified the last common ancestor (LCA) consensus using the SILVA taxonomy and species-level classification was chosen.
RiboTaxa was run on raw metagenomics reads using default parameters except for the following: (MOCK: -max_read_length = 300, -insert_mean = 192, -insert_stddev = 98; synthetic: -max_read_length = 250, -insert_mean = 185, -insert_stddev = 90), which exclusively depend on the sequencing length of the input datasets.
Evaluation of RiboTaxa on real datasets
Metagenomics and 16S gene capture by hybridization are two well-adapted techniques to target the phylogenetic 16S marker gene. For RiboTaxa evaluation, metagenomics datasets (paired-end) as well as 16S gene capture by hybridization datasets (paired-end) from different environments including soil, ocean and human gut (Table 1) were downloaded from NCBI SRA using NCBI SRA Toolkit_v2.9.1 (https://www.ncbi.nlm.nih.gov/sra) as described earlier. The aim was to evaluate the versatility of RiboTaxa on different environments and on different techniques. All the parameters of RiboTaxa were kept as default except parameters associated with sequencing length of the input data (Ocean samples: -max_read_length = 101, -insert_mean = 121, -insert_stddev = 96; Human gut samples: -max_read_length = 151, -insert_mean = 203, -insert_stddev = 131; 16S gene capture by hybridization samples: -max_read_length = 151, -insert_mean = 233, -insert_stddev = 127).
Table 1.
Real datasets used to evaluate the versatility of RiboTaxa
| Method | Reference | Environment | Project (size) | # | Description |
|---|---|---|---|---|---|
| Metagenomics | (50) | Ocean | PRJEB13222 (21.07 Gb) | 20 | Healthy E. gazella coral (3 samples) Necrotic E. gazella coral (3) Healthy E. verrucosa coral (4) Healthy L. sarmentosa coral (3) Sediments (3) Seawater (4) |
| (51) | Human Gut | PRJNA553191 (151.16 Gb) | 62 | Young adults (mean age: 32.2 years) (11) Young elderly (mean age = 72.5 years) (13) Centenarians (mean age: 100.4 years) (15) Semi-supercentenarians (mean age: 106.3 years) (23) | |
| (52) | Soil | PRJNA647119 (23.04 Gb) | 12 | N10 (North-facing, 10 cm depth) (3) N160 (North-facing, 160 cm depth) (3) S10 (South-facing, 10 cm depth) (3) S160 (South-facing, 160 cm depth) (3) | |
| 16S gene capture by hybridization | (33) | Soil | SRR3648004 (1.2Gb) | 1 | Contaminated soil (1) |
RiboTaxa results (reconstructed sequences and taxonomies) for each dataset were analysed to measure any potential phylogenetic signals of interest present in the reconstructed SSU sequences that were probably missed by initial studies. For the sequence of interest, a similarity search was conducted using blastn of NCBI BLAST + v2.11.0 (21) with default parameters against the GenBank database. Phylogenetic analysis was performed using the pipeline phylogeny.fr (41). The 16S rRNA gene sequences in Fasta format were aligned with MUSCLE (42), followed by a curation step using Gblocks (43). A phylogenetic tree was reconstructed with PhyML by using the maximum-likelihood method (44). For each phylogenetic dataset, a percent identity matrix was also generated using Clustal Omega (45) and we used the identity thresholds defined by (46) to determine novel taxa, that is, 97 % for species, 94.5 % for genus, 86.5 % for family, 82.0 % for order.
SSU rRNA gene reconstruction versus metagenome-based approaches
To compare between SSU rRNA genes reconstruction by RiboTaxa versus metagenome-based approaches, taxonomic classification based on GTDB database (47) was also performed using Kraken2/Bracken. For this, 10 metagenomics samples from real datasets (3 from octocorals, 3 from semi-supercentenarians, 4 from N160 permafrost soil) were selected in which potentially new genus/species were identified. The aim was to see whether GTDB-based taxonomy could provide precise identification of novel species in different environments (i.e. ocean, human gut, soil) when there are few or no available genome references. The pipeline, struo (48), was used to build the index files of GTDB r202 for Kraken2 and Bracken. Kraken2 was then run on the metagenomics samples using default parameters to generate reports files which were then used by Bracken to calculate relative abundances.
Performance analyses
Precision, recall, and F-measure metrics were used to evaluate whether the presence or absence of taxa in a microbial community is correctly identified by a taxonomic classifier (36). At a given taxonomic level, L, a classification is:
True positive (TP), if that taxon is both observed and expected.
False positive (FP), if that taxon is observed but not expected.
False negative (FN), if a taxon is expected but not observed.
Precision is defined as the ratio of the true positives (TPs) to the sum of the TPs and the false positives (FPs)
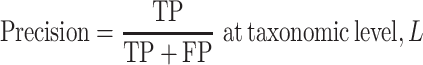 |
Recall is defined as the ratio of the TPs to the sum of the TPs and the false negatives (FNs).
 |
The F-measure is defined as the harmonic mean of precision and recall. This metric represents a synthesis of the performance of retrieval.
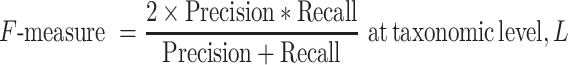 |
Precision and recall were multiplied by 100 to indicate results in percentages. Precision, recall and F-measure were calculated at each taxonomic level.
RESULTS
Tools benchmarking to build the best performing pipeline for 16S rRNA gene reconstruction from metagenomics data
The first part of this study aimed at identifying the best tools to achieve 16S rRNA reads filtering and 16S rRNA gene reconstruction, leading to species-level identification and relative abundancy description. For this, we used 1 246 376 pairs of Illumina sequences from a mock microbial community (designated MOCK) (Supplementary Figure S1). After filtering, 1 150 991 pairs of high-quality reads were kept for downstream processing analysis.
rRNA reads extraction and nearly full-length 16S rRNA gene reconstruction using the MOCK community
To extract SSU reads from metagenomics data of the MOCK community, we compared the widely used general-purpose mapper, BBMap and the rRNA-specialized tool SortMeRNA. Even though BBMap was faster than SortMeRNA, it filtered only 6339 pairs of SSU reads (0.55%) in contrast with SortMeRNA which filtered 10 675 pairs of SSU reads (0.93%) from the raw metagenomics dataset. Then we compared the efficiency of four reference-based targeted assemblers (Emirge amplicon.py, Emirge.py, MATAM and MetaRib) using three input datasets: unfiltered metagenomics reads, BBMap-filtered reads or SortMeRNA-filtered reads (Figure 2), resulting in 12 combinations.
Figure 2.

Sequence reconstruction by Emirge_amplicon.py, Emirge.py, MATAM and MetaRib using unfiltered metagenomics reads, BBMap-filtered reads and SortMeRNA-filtered reads. (A) Number of SSU sequences reconstructed by each tool. (B) Length of reconstructed sequences in base pairs (bp). Black dot inside violins represents the median length of the reconstructed sequences.
Emirge_amplicon.py reconstructed the highest number of sequences, followed by MetaRib, Emirge.py and finally MATAM. Emirge_amplicon.py output 54 sequences (375–1410 bp) from unfiltered reads, 50 sequences (497–1495 bp) from SortMeRNA-filtered reads and 42 sequences (437–1457 bp) from BBMap-filtered reads (Figure 2). In all cases, more 16S rRNA sequences were reconstructed from unfiltered reads followed by SortMeRNA-filtered reads and BBMap-filtered reads. The highest median length (1200 bp) resulted from sequence reconstruction by Emirge_amplicon.py using SortMeRNA-filtered reads with the longest sequence being 1495 bp. Similarly, MATAM reconstructed longer sequences having a median greater than 1125 bp using the three input datasets compared to Emirge.py (median < 1100 bp).
The rRNA sequences of the eight less abundant microorganisms of the MOCK (abundance lesser than 0.02%) were not reconstructed by any combination of tools and hence could not be detected. In fact, we confirmed the absence of reads related to these microorganisms by mapping raw reads on their 16S rRNA sequences (BBmap with default parameters). Here, the sequencing depth was too low to produce reads for these rare microorganisms. Henceforth, we focused our analysis on the 20 microorganisms of the MOCK that could be detected.
Reconstructed sequences obtained from the 12 tools combinations were compared to the 41 unique 16S rRNA variants originated from the 20 microbial species of the MOCK (Figure 3). In all cases, Emirge_amplicon.py reported the highest microbial (number of microorganisms detected) and sequence (number of 16S rRNA variants detected) diversities. Using SortMeRNA-filtered reads, 18 microorganisms (representing 38 variants) of the MOCK were identified followed by unfiltered reads (16 microorganisms) and BBMap-filtered reads (15 microorganisms). When the join threshold of Emirge_amplicon.py was reduced to 97% (as in the original publication), less variants (22 variants representing 18 microorganisms) were obtained from SortMeRNA-filtered reads. Hence, in this study, we kept the identity threshold at 100% to target the identification of 16S rRNA gene variants. Reconstructed sequences from MATAM least described the MOCK with at most 9 identified species from unfiltered reads. Although, MetaRib identified less variants (31 variants) than Emirge_amplicon.py from unfiltered reads, it was able to identify one variant of Halogeometricum borinquense DSM 11551 and two variants of Corynebacterium glutamicum DSM 20300 which were missed by all other combination of tools. Similarly, the sequence of Methanoculleus marisnigri DSM 1498 was identified only by Emirge_amplicon.py (from SortMeRNA-filtered reads).
Figure 3.

Reconstructed 16S rRNA genes. Reconstructed sequences are affiliated to the 16S MOCK database to identify the number of 16S gene variants of the MOCK with a cut-off at 99% identity. The number of variants per species is indicated in ‘Reference’. Only microorganisms showing 16S gene reads in the sequencing dataset are shown. The total number of detected variants is indicated in ‘Total’.
Thus, to maximise the number of identified microorganisms (or variants) in the microbial MOCK community, we decided to combine the two rRNA gene-targeted assemblers: Emirge_amplicon.py and MetaRib, to reconstruct the SSU rRNA genes. The reconstructed rRNA sequences obtained from Emirge_amplicon.py (using SortMeRNA-filtered reads) and MetaRib (using unfiltered reads) were clustered at 97% using VSEARCH. The final output resulted in 53 sequences and identified the 20 microorganisms of the mock (representing 41 variants). Most of the reconstructed sequences showed very high identity with the reference sequences, with an identity between 98–100% (34 sequences) or close to 97% (15 sequences). The remaining four sequences were relatively more distant from MOCK references (close to 94% identity) but did not impact taxonomic identification as they were well classified at genus level. This result indicates that minor artificial diversity was created during the reconstruction process of the nearly full-length 16S rRNA gene.
Taxonomic affiliation of reconstructed rRNA sequences from the MOCK community
Next, we evaluated the efficiency of seven taxonomic classifiers commonly used: Centrifuge, Kraken2, mothur, RDP classifier, SPINGO, BLCA and QIIME2’s Sklearn classifier. The first input data was the 53 clustered sequences obtained previously from Emirge_amplicon.py and MetaRib. Precision, recall and F-measure were then calculated by comparing the ‘expected’ classifications of the 20 detectable microorganisms of the mock community to the classifications predicted by each taxonomic classifier using the SILVA 138.1 database (Figure 4A).
Figure 4.

Performance of taxonomic classifiers used to classify nearly full-length 16S sequences based on the number of correctly assigned taxa, precision, recall and F-measure from class to species level. Error bars at 95% confidence intervals. (A) From the MOCK community. Left column: Using clustered sequences obtained from Emirge_amplicon.py (SortMeRNA-filtered reads) and MetaRib (unfiltered reads). Right column: Using MOCK reference 16S sequences. (B) Using clustered sequences obtained from Emirge_amplicon.py (SortMeRNA-filtered reads) and MetaRib (unfiltered reads).
Classification using Centrifuge, Kraken2 and mothur were limited to the genera level. Mothur and Kraken2 performed better than Centrifuge with a precision of 84.6% and 71.4%, respectively, compared to 69.2% with Centrifuge. Mothur also showed a higher sensitivity recall (55%) of the mock community compared to Kraken2 (50%) and Centrifuge (45%). On the other hand, RDP Classifier, SPINGO, BLCA and Sklearn classifier were more robust, achieving a level higher (species level) in their classifications. Alignment-based SPINGO and BLCA identified 11 and 5 species, respectively but were unable to infer taxa to 14 and 35 sequences, respectively which were left as ‘unclassified’. Overall, Sklearn classifier distinguished itself by identifying the 20 detectable microorganisms at genus level and 17 at species level while RDP classifier identified only 8 species. The precision score of both tools were outstanding (100% at all taxonomic levels up to genus) while at species level, the sensitivity recall of Sklearn classifier and RDP Classifier dropped to 85% and 40%, respectively.
To further confirm the efficiency of each taxonomic classifier, the second input data was the 16S rRNA reference sequences originated from the 20 detectable microbial strains (Figure 4A). Again, Sklearn classifier was the most efficient tool. Sklearn classifier identified 20 genera and 19 species using the 16S reference sequences of the mock community, with a precision at 100% at each level and a sensitivity recall of 95% at species level. The only species missed by Sklearn classifier using 16S reference sequences of the mock population was Pseudomonas putida, which was classified at genus level.
High performance of Sklearn classifier was further evidenced by F-measure which synthetizes the balance between recall and precision at different taxa levels (Figure 4A). Sklearn classifier had the highest score at species level, reaching 91.9% for the reconstructed sequences, which was not very far from the harmonic mean obtained for 16S reference sequences (97%).
Validation of benchmarked tools using a complex synthetic community
To validate the combined approach (i.e. Emirge_amplicon.py and MetaRib) during sequence reconstruction, we, next, evaluated the sequence reconstruction tools (i.e. Emirge_amplicon.py, Emirge.py, MATAM and MetaRib) on a synthetic sequencing data of 100 microorganisms from the human gut. This complex synthetic community was reproduced using 100 genomes (complete genomes, scaffolds and contigs), representing 45 genera and 100 species with 16S rRNA gene abundances varying from 0.273% to 2.789%. After filtering, 14 754 123 pairs of high-quality reads were kept for downstream processing analysis. Considering variants (<99% identity), a total of 347 sequences of the 16S rRNA gene from the 100 genomes were used as reference.
Nearly full-length 16S rRNA gene reconstruction using the synthetic community
Based on the results of the MOCK, Emirge.py, Emirge_amplicon.py and MATAM were provided with SSU reads extracted using SortMeRNA and MetaRib was run on unfiltered high-quality reads, trimmed and cleaned. Emirge_amplicon.py reconstructed the highest number of sequences (203 sequences) and Emirge.py reassembled the least (44 sequences). To determine the number of species represented by the reconstructed sequences, the latter were subjected to a blastn search against the 347 16S-reference sequences of the synthetic community and all sequence alignments with an identity of >97% was considered as a detected species. In this synthetic community, 44 microorganisms were successfully represented by the sequences reconstructed by the four tools. Emirge_amplicon.py identified 69 microorganisms, out of which, 8 species were exclusively identified by this tool. On the other hand, MetaRib and MATAM detected Bacteroides intestinalis, missed by Emirge_amplicon.py. In additional to this microorganism, MetaRib also detected two more bacteria: Oceanobacillus massiliensis and Roseburia intestinalis, both missed by MATAM and Emirge_amplicon.py.
Thus, these results confirmed that using a combined approach (i.e. Emirge_amplicon.py and MetaRib) during sequence reconstruction has the advantage of more accurately describing the microbial community. Hence, after clustering the reconstructed rRNA sequences obtained from Emirge_amplicon.py (using SortMeRNA-filtered reads) and MetaRib (using unfiltered reads) at 97%, it resulted in a total of 233 sequences, representing 72 species (>97% identity) of this synthetic community.
Taxonomic affiliation of reconstructed rRNA sequences from the synthetic community
To validate the best performance of QIIME2’s Sklearn classifier during microbial profiling, we evaluated the efficiency of previously used taxonomic classifiers (i.e. Centrifuge, Kraken2, mothur, RDP classifier, SPINGO, BLCA and QIIME2’s Sklearn classifier). The input data was the 233 clustered sequences obtained from Emirge_amplicon.py and MetaRib. Precision, recall and F-measure were then calculated at different taxa levels: from class to species (Figure 4B).
Taxonomic classification using Centrifuge, Kraken2 and mothur again stopped at genus level. This time, the performance of Kraken2 (precision = 86.8%) was slightly better than mothur (precision = 84.6%) and Centrifuge (precision = 79.5%), despite, both Kraken2 and mothur identified a total of 33 genera. On the other hand, BLCA, SPINGO, RDP and Sklearn classifier reached the species level classification. While BLCA and SPINGO displayed a precision of 91.6% and 76.6%, respectively, RDP and Sklearn classifier were 100% precise at inferring taxa. Moreover, the F-measure of Sklearn classifier was the highest (83.7%) compared to all taxonomic classifiers, confirming the best performance of Sklearn classifier for microbial profiling.
Compilation of RiboTaxa
After evaluating all the above tools, the best performing tools were compiled to build a pipeline enabling efficient and accurate taxonomic identification of any microbial community starting from raw shotgun metagenomics data. BBTools and FastQC were integrated to perform quality control. For SSU sequence reconstruction, Emirge_amplicon.py (giving the best results as described previously) was chosen. For optimal results, Emirge_amplicon.py was fed with SSU reads filtered by SortMeRNA. During tools comparison, MetaRib also performed outstandingly, identifying two microorganisms of the mock community, left out by Emirge_amplicon.py. Thus, to optimise SSU sequence reconstruction, MetaRib was also included. Complementarily, MetaRib was fed with unfiltered reads as this tool included a dereplication step during iteration and could handle large datasets in low computational time. To calculate the relative abundance, unfiltered high-quality reads were mapped onto the reconstructed sequences using BBMap (BBTools). Prior to taxonomic affiliation, the reconstructed rRNA gene sequences obtained from MetaRib and EMIRGE were clustered at 97% using VSEARCH and abundances from BBmap were summed up for sequences sharing the same cluster. Finally, Sklearn classifier (QIIME 2) was used to infer taxa. Throughout the study, the default database used for RiboTaxa was SILVA SSU 138.1.
RiboTaxa was then compared to existing taxonomic classifiers such as Centrifuge, Kraken2, METAXA2, phyloFlash, SPINGO, BLCA and MEGAN6 to evaluate their sensitivity, specificity and computation time during taxonomic classification using then MOCK and synthetic community.
Comparison of RiboTaxa with other existing tools on the MOCK dataset
Here, only the 20 detectable microorganisms were looked for in the resulting taxonomic classification. Centrifuge, Kraken2, MEGAN6, METAXA2, SPINGO and BLCA inferred taxa directly to short Illumina reads and were labelled as ‘Without sequence reconstruction’. RiboTaxa and phyloFlash included a step of SSU sequence assembly before microbial profiling and were labelled as ‘With sequence reconstruction’ (Table 2). All performance measures were computed at genus and species level (except for Centrifuge and Kraken2) to compare between all classifiers. Among classifiers ‘without sequence reconstruction’, METAXA2 performed the best at identifying the 20 genera correctly (recall 100%) while BLCA and Centrifuge, both, yielded the lowest recall (45%) of the microbial MOCK community with 11 and 9 correct taxa, respectively. On the other hand, RiboTaxa identified all the 20 genera with a recall of 100% and phyloFlash assigned 10 or 12 taxa at genus level with a recall of 60% (BS) and 50% (BE/SE). Direct short-length read classification led to unexpected taxa (false positives) (Figures 5A) while phyloFlash and RiboTaxa (‘with sequence reconstruction’) assigned taxa at genus and species level without false positive classification (Figure 5A), resulting in precision score of 100% (Table 2). Centrifuge and Kraken2 wrongly classified 2/3 of detected genera, reaching mean precision to 28% and 43% respectively, while METAXA2 misclassified 1/3 of detected taxa (precision 66.7%).
Table 2.
Statistics of the accuracy of different tools to describe the taxonomic composition of the MOCK community
| Tools | Precision (%) | Recall (%) | F-measure (%) | ||||
|---|---|---|---|---|---|---|---|
| Genus | Species | Genus | Species | Genus | Species | ||
| Without sequence reconstruction | BLCA | 40.9 | 42.9 | 45 | 30 | 42.8 | 32.3 |
| Centrifugea | 28.1 | - | 45 | - | 34.6 | - | |
| Kraken2a | 43.2 | - | 80 | - | 56.1 | - | |
| MEGAN6 | 55 | 64.3 | 55 | 45 | 55 | 52.9 | |
| METAXA2 | 66.7 | 100 | 100 | 50 | 80 | 66.7 | |
| SPINGO | 66.7 | 81.8 | 60 | 45 | 63.1 | 58 | |
| With sequence reconstruction | phyloFlash (BS) | 100 | 100 | 60 | 60 | 75 | 75 |
| phyloFlash (BE) | 66.7 | 100 | 50 | 50 | 57.1 | 66.7 | |
| phyloFlash (SE) | 50 | 100 | 50 | 50 | 50 | 66.7 | |
| RiboTaxa | 100 | 100 | 100 | 85 | 100 | 91.9 | |
aTaxonomic classification stopped at genus level.
Figure 5.

Tool performance to report genus and species diversity using high-quality metagenomics reads. (A) From the MOCK community (20 genera, 20 species). (B) From the synthetic human gut (HG) community (45 genera, 100 species). BLCA, Centrifuge, Kraken2, MEGAN6, METAXA2 and SPINGO infer taxa to short reads without sequence reconstruction. PhyloFlash and RiboTaxa reconstruct nearly full-length SSU sequences before assigning taxonomy.
Consequently, F-measure was 100% for RiboTaxa and between 67% and 75% for PhyloFlash compared to the ‘Without sequence reconstruction’ tools where F-measure was affected by the false positive classifications (Centrifuge = 35%, Kraken2 = 56%, METAXA2 = 80%, SPINGO = 63%, BLCA = 42%, MEGAN6 = 55%). Moreover, the difference between classifying short-length reads and longer sequences was more obvious at species level. RiboTaxa identified 17 species while phyloFlash assigned 10 or 12 species-level taxa using reconstructed sequences. Among the tools that inferred taxa to short-length reads, BLCA, MEGAN6, METAXA2 and SPINGO managed to reach the species level with 14, 14, 10 and 11 correctly assigned sequences (Figure 5A). The three microorganisms that RiboTaxa could not affiliate as species because of their highly similar 16S rRNA gene sequences to other species were Pseudomonas putida, Corynebacterium glutamicum and Geobacter lovleyi.
Relative abundance is another major criterion to consider in the analysis of microbial community diversity. Unfortunately, BLCA and SPINGO did not output read counts or provided relative abundance. RiboTaxa, similar to, phyloFlash and Centrifuge can estimate its own relative abundance. However, Bracken was used to estimate microbial abundance from Kraken2’s output. For METAXA2, relative abundance was calculated by dividing each taxon count by the total number of reads classified at the species or genus level. The estimated abundance of the correctly assigned taxa was relatively close to the theoretical ones with no under or over-estimation for 16S reconstruction-based tools (Figure 6). However, false positives (Figure 6, taxa in lighter colour) greatly impact the abundance profiles for Centrifuge, Kraken2 and METAXA2. RiboTaxa gave abundance profile close to the theoretical one due, in part, to excellent precision and recall results.
Figure 6.

Microbial mock community profiles obtained using metagenomic classifiers and RiboTaxa. (A) At genus level. Theoretical profile represents the relative genera abundance of the mock community. The genera identified by Centrifuge, Kraken2 and METAXA2 that are not part of the microbial mock community are indicated using lighter colour. (B) At species level. Theoretical profile represents the relative species abundance of the mock community. This profile is compared with the relative abundances output by METAXA2, phyloFlash and RiboTaxa.
Evaluation of RiboTaxa with other existing tools on the synthetic human gut community
Among the different taxonomic classifiers, Centrifuge and Kraken2 again stopped sequence affiliation at genus level with false positives classification, impacting negatively their respective F-measures (Kraken2 = 53.5%, Centrifuge = 47.3%) (Table 3). Moreover, despite METAXA2, SPINGO, BLCA and MEGAN6 reached species-level identification, they also produced many false positives due to short-reads classification (Figure 5B). PhyloFlash (SE) and RiboTaxa, which reconstructed nearly full-length SSU rRNA gene sequences prior to microbial classification, in turn, confirmed that short reads lack phylogenetic signals to be assigned to correct taxa. Both pipelines did not produce any false positive results and successfully classified sequences at genus and species level (Figure 5B) with a precision of 100% (Table 3). PhyloFlash (SE) reconstructed 198 SSU rRNA gene sequences with a mean length of 1011 bp. Taxonomic affiliation led to an identification of 42 genera (F-measure = 96.5%) and 48 species (F-measure = 64.9%) of the synthetic human gut community (Table 3).
Table 3.
Statistics of the accuracy of different tools to describe the taxonomic composition of the synthetic human gut community
| Tools | Precision (%) | Recall (%) | F-measure (%) | |||
|---|---|---|---|---|---|---|
| Genus | Species | Genus | Species | Genus | Species | |
| BLCA | 70.2 | 63.7 | 73.3 | 37 | 71.7 | 46.8 |
| Centrifugea | 32.2 | - | 88.9 | - | 47.3 | - |
| Kraken2a | 39.2 | - | 84.4 | - | 53.5 | - |
| MEGAN6 | 76 | 76.4 | 84.4 | 42 | 80 | 54.2 |
| METAXA2 | 72.4 | 64.5 | 93.3 | 31 | 81.6 | 41.9 |
| SPINGO | 59.1 | 59.3 | 64.4 | 35 | 61.7 | 44.0 |
| phyloFlash (SE) | 100 | 100 | 93.3 | 48 | 96.5 | 64.9 |
| RiboTaxa | 100 | 100 | 100 | 72 | 100 | 83.7 |
aTaxonomic classification stopped at genus level.
Yet, this synthetic community was best described by RiboTaxa. RiboTaxa reconstructed 233 SSU rRNA gene sequences with a mean length of 1512 bp. Microbial profiling led to 212 sequences assigned to genus level, identifying all the 45 genera of the synthetic human gut community (F-measure = 100%). Furthermore, 103 sequences out of 212 sequences classified at genus level (Figure 7A) were assigned to a lower taxonomic level and allowed the identification of 72 species with an F-measure of 83.7% (Table 3) and a species abundance relatively close to the theoretical profile (Figure 7B). Some of the species that could not be identified beyond genus level included closely related species of Escherichia and Clostridium which could not be differentiated by RiboTaxa (Supplementary Table S1) due to highly conserved 16S rRNA sequences limiting species discrimination. Nevertheless, taxonomic profiling of simulated human gut microbiota using RiboTaxa resulted in an F-measure of 100% and 83.7% at genus level and species level, respectively (Table 3). Few sequences stopped at family level and more specifically to two families, i.e. Enterobacteriaceae and Lachnospiraceae. These sequences were subjected to a blastn search against the 347 16S-reference sequences of the synthetic community to measure any artificial diversity that might have been produced during SSU sequence reconstruction. The parameter HSP was set to 1 to fetch the first best match only. The sequences majority shared an identity of >98% with the reference sequences, implying that the reconstructed sequences belonged to the synthetic community. It is known that, in certain cases, 16S rRNA gene may not be a reliable predictor of genus-level taxonomy (49).
Figure 7.

Tool performance to report correct taxonomic diversity of the synthetic human gut community from high-quality metagenomics reads. Theoretical abundance column represents the theoretical profile of the synthetic community. (A) At genus level, RiboTaxa, Kraken2/Bracken, Centrifuge, METAXA2, PhyloFlash(SE) and MEGAN6 were used for genus classification and relative abundance calculations. (B) At species level. RiboTaxa, METAXA2, PhyloFlash(SE) and MEGAN6 were used for species-level classification and relative abundance calculations. Detection of microorganisms are represented by dots. The colour and size of the dots vary according to the relative abundance calculated by the different tools. Undetected microorganisms by the different tools are denoted by the absence of dots.
All the tools (except BLCA and SPINGO) were also evaluated on their capacity to describe the relative abundances of this synthetic community. While RiboTaxa and phyloFlash (SE) have described the abundances relatively close to the theoretical ones, tools like Kraken2 and MEGAN6 have underestimated or overestimated the abundances of some microorganisms (Figure 7). For example, Kraken2 overestimated the abundances of Klebsiella, Streptococcus and Escherichia. Ruminococcus sp., Thermoanaerobacterium xylanolyticum, Corynebacterium halotolerans and Ruminococcus albus were all highly underestimated by MEGAN6, detecting them below 0.05% while their theoretical abundances were >1%.
Computational time comparison
16S-based metagenome classification using all the above tools was carried out on: i86linux32, 4.0GB RAM × 8 cores (32.8GB total).
Computational time for each tool is summarised in Figure 8. All the tools except RiboTaxa were run on high-quality metagenomic reads, trimmed and cleaned using BBTools while RiboTaxa was run on raw metagenomics reads as the latter performs its own quality control step using BBTools. Using the MOCK (Figure 8A) and the synthetic human gut communities (Figure 8B), RiboTaxa completed taxonomic assignment in 18 min 03 s and 26 min 08 s, respectively. Based on short k-mer approach, Kraken2 and Centrifuge were the fastest to infer taxa in both communities. On the other hand, alignment-based BLCA and DIAMOND-MEGAN took more than 1 hour to classify sequences of the synthetic community. Moreover, METAXA2, which uses HMM, was the most time-consuming and taxonomic classification lasted for 1hr45 min on the MOCK and 2 h 13 min on the synthetic community.
Figure 8.

Observed computational time of taxonomic profilers. (A) Required time for taxonomic profiling by Centrifuge, Kraken2, METAXA2, phyloFlash BBmap-SPAdes (BS), BBMap-EMIRGE (BE) and SortMeRNA-EMIRGE (SE), and RiboTaxa on the MOCK communtity. (B) Required time for taxonomic profiling by BLCA, Centrifuge, DIAMOND-MEGAN, Kraken2, METAXA2, phyloFlash SortMeRNA-EMIRGE (SE), RiboTaxa and, SPINGO on the synthetic human gut communtity.
Evaluation of RiboTaxa on real datasets
In addition to providing a fine taxonomic resolution down to the species level, RiboTaxa has proved to be a versatile tool with its ability to analyse metagenomics data from different environments including soil, ocean and human gut.
In a study focused on corals’ health involving 20 metagenomics samples (13 corals, 3 sediments and 4 seawater) (50), affiliation of prokaryotes was limited to the family level with Endozoicomonadaceae characterising healthy octocoral tissue. Using RiboTaxa, we identified that healthy octocoral harbors a total of 19 prokaryotic species, whereby E. gazella hosted 7, E. verrucosa 4 and L. sarmentosa 8 species. In comparison, 204, 67 and 22 species were detected in seawater, necrotic E. gazella tissue, and sediments respectively (Figure 9A; Supplementary Figure S2). Furthermore, RiboTaxa highlighted two species of uncultured Endozoicomonas which were dominant in the healthy tissue of all three octocorals species (average = 67.6%). The phylogenetic analysis coupled with percent identity matrix revealed that both species belonged to new genus (<95% similarity) despite being relatively close to the genus Endozoicomonas (Supplementary Figure S3; Table S2). Other very close unclassified sequences have been retrieved by similarity search in Genbank database confirming the robustness of our approach, but no genome seems to be available for these new species which opens new research perspectives on corals' health.
Figure 9.

Evaluation of RiboTaxa on metagenomics datasets from different environments. Number of species identified in: (A) octocorals samples (50). (B) Human gut samples (51). (C) Soil samples (52).
Applied to metagenomics data from a study on human gut and extreme longevity (51), RiboTaxa revealed a richer microbial diversity with 1135, 1495, 1756 and 2621 species in young adults, young elderly, centenarians and semi-supercentenarians, respectively (Figure 9B; Supplementary Figure S4). While the authors compared the relative abundance of different species between the four groups, we focused on the presence of specific microbial signatures in the different groups. Interestingly, among the different archaeal species identified, RiboTaxa detected the presence of an uncultured archaeon in 4 semi-supercentenarians. Phylogenetic analysis coupled with identity matrix revealed that the uncultured archaeon shared an identity <95% with Methanosphaera species highlighting a probable new archaeal genus not already described. By similarity search in Genbank database, we detected an uncultured clone, HM573433, initially isolated from faeces showing 99.68% identity but no available genome has been detected reinforcing the interest in exploring this new archaeal genus (Supplementary Figure S5; Table S2). Secondly, the genus Enorma, of the Coriobacteriaceae family, that has important functions such as the conversion of bile salts and steroids and the activation of dietary polyphenols, was only present in the centenarian and semi-supercentenarian groups. RiboTaxa allowed the identification of 3 new species belonging to this genus that could be species of interest participating in longevity process (Supplementary Figure S6; Table S2).
We also applied RiboTaxa on metagenomics data produced from underexplored permafrost mid-latitudinal alpine regions (52). Our results demonstrated that the richness was highest for the N160 (permafrost) soils (Supplementary Figure S7) with 97 detected species (Figure 9C) confirming the observed highest alpha diversity of the predicted protein-coding genes in N160 soils. We detected the presence of different species from Lysobacter and Arenimonas genera but also unknown species of Xanthomonadaceae family in permafrost N160 soils only. Phylogenetic analysis coupled with identity matrix revealed a new species belonging to the genus Thermomonas and 4 new species belonging to three new genera of the Xanthomonadaceae family (Supplementary Figure S8; Table S2).
The new 16S rRNA gene capture by hybridization (33) allows a significant enrichment of the 16S rRNA biomarker, providing a more accurate representation of microbial communities including the detection of rare microorganisms (<0.1%). RiboTaxa detected 495 species from the explored soil sample instead of 354 previously detected. Moreover, 23 archaeal species, belonging to the phylum Thaumarchaeota (initially known as mesophilic Crenarchaeota) were the dominant archaeal group found in the soil sample with a relative abundance of 0.25%. The two most abundant bacterial species detected by RiboTaxa were unclassified Luteimonas and unclassified Sphingomonas with a relative abundance of 13.3% and 8.9%, respectively. Phylogenetic analysis coupled with identity matrix revealed that the two new species belong to the genus Luteimonas (Supplementary Figure S9; Table S2) and to the genus Tardibacter (identity = 96.33%) instead of Sphingomonas (Supplementary Figure S10; Table S2), respectively. The initial misclassification into the genus Sphingomonas is due to the lack of Tardibacter sequences in SILVA 138.1 database.
SSU rRNA gene reconstruction versus metagenome-based approaches
Microbial profiling performed by Kraken2 using the GTDB database (47) revealed a high alpha diversity in each sample at all taxonomic levels. In healthy corals, an average of 12 949 species were detected compared to 20 321 species in necrotic corals. Due to this high microbial diversity, Bracken identified the most abundance family, Endozoicomonadaceae, at only 1.2%. Within this family, Kraken2 detected the genera Endozoicomonas (33 assigned reads), Parendozoicomonas (2 assigned reads) and Kistimonas (1 assigned read). However, unlike RiboTaxa, Kraken2 could not detect new genera within the Endozoicomonadaceae family consisting of two new species revealed by RiboTaxa. This is because Kraken only classifies the reads from genomes present in the reference database. Thus, lack of reference genomes in the GTDB database, particularly, for less explored microbial communities, hinders the identification of novel species (53). Similarly, in the semi-supercentenarian's samples, only bacterial diversity was detected with an average of 18 266 species compared to RiboTaxa, which identified the presence of an uncultured archaeon among other archaea detected. Finally, RiboTaxa revealed a microbial richness of 97 species in N160 permafrost soil samples which Kraken2 detected an average of 23 191 species in the same samples. This high alpha diversity provided by Kraken2 could be the result of short-reads misclassification conducting to diversity overestimation as demonstrated previously.
DISCUSSION
Shotgun metagenomics sequencing is a powerful method to characterise microbiota. Metagenomic classifiers have been developed to taxonomically classify metagenomics data and estimate taxa abundance profiles (54). Algorithmic approaches also ensure that classification speeds are fast enough to exploit very large numbers of sequencing reads. In the present study, we compared bioinformatics tools to exploit SSU reads for microbial community profiling from metagenomics data. A major advantage of rRNA gene analysis is that databases contain genes from hundreds of thousands species, making them far more comprehensive than current genome databases even if we observe continuous exponential genome accumulation. Furthermore, unknown microorganisms could be identified even for rare taxa using nearly full-length phylogenetic marker and positioned in phylogenic tree (33). Thus, phylogenetic approaches are designed to be able to detect distant homology, enabling the characterization of previously unidentified organisms. However, high conservation of 16S rRNA sequence cannot discriminate all species as it is the case for certain genera, or even in some cases, 16S rRNA gene may not be a reliable predictor of genus-level taxonomy (49). We observed this latter situation for a few microorganisms from Enterobacteriaceae and Lachnospiraceae families. To overcome these issues and reach strain-level identification, complete rDNA operon (which could still miss some proportion of bacterial species on account of unlinked rRNA genes) (55) or other marker databases for prokaryotic (56) and/or eukaryotes profiling (57) should be used as references. Low biomass microbiota, rare and unknown taxa characterizations are other challenges in metagenomics sample exploration. Hybridization capture targeting SSU rRNA genes overcomes such difficulties (33,58) and RiboTaxa has been designed and optimized for 16S/18S sequencing data analysis as well as other universal markers targeted by hybridization for microbial structure characterization (59).
Another difficulty of using shotgun DNA for rRNA gene analyses is the identification of SSU rRNA fragments in large sequence datasets (60). In this study, we extracted rRNA reads using SortMeRNA, an RNA-specialized tool and BBMap, a general-purpose read mapper. SortMeRNA performed better than BBMap in extracting reads that could be assembled using different tools such as EMIRGE (Emirge.py and Emirge_amplicon.py), MATAM and MetaRib. Nevertheless, tool selection was not based on the highest number of reconstructed sequences. Using SortMeRNA-filtered reads, Emirge_amplicon.py identified the highest number of microbial species and proved to be the most robust tool. In parallel, MetaRib which uses the same algorithm as EMIRGE but with a deduplication and subsampling step during sequence reconstruction, identified two additional microorganisms using unfiltered reads, which were missed by Emirge_amplicon.py. Thus, combining these two tools for SSU reconstruction led a greater population characterisation. The efficiency of this combined approach was first demonstrated using the MOCK where the sequences of the 20 detectable microorganisms were successfully reconstructed and secondly, using the synthetic human gut community, the reconstructed sequences allowed the identification of the 45 genera present in that community. Moreover, to target the identification of intragenomic variations between 16S gene copies (99% identity), SSU sequence reconstruction was optimised by using a joint threshold of 100%. We thus, provided evidence that it is possible to detect divergent copies of the 16S gene that exist within the same genome. According to Johnson et al (61), 99% sequence similarity can be an adequate threshold for clustering sequences originating from the same genome, reflecting polymorphisms in one or more 16S gene copies to differentiate between strains of the same species. Simultaneously, to ensure that the reconstructed sequence showed no or little artificial diversity from the real sequences, 16S reference sequences served as control. As we did not detect chimeric sequences during 16S rRNA gene reconstruction, we did not include chimera detection but such tool could be used (62). However, it is possible that chimeric database sequences could carry over into SSU reconstructions, if reads map across the full length of the chimera as indicated by EMIRGE developers (27).
This study emphasised on targeted-sequence reconstruction to maximise taxonomic assignment at species level. Comparing taxonomic profiling using short-length sequences and reconstructed SSU sequences proved that inferring taxa directly to short-length Illumina reads resulted in a high rate of false positives while reconstructed sequences were correctly assigned, though it required an assembly step prior to classification. As closely related species share very similar or identical genome segments, short reads often map to multiple species in the reference dataset restraining classification efficiency (18). This limitation was depicted by BLCA and DIAMOND-MEGAN6, which rely on sequence alignment based on matched database sequences. Despite classifying sequences at species level, they failed to consider the differing degrees of similarity between certain query and the database hit sequences (12) and either stopped at genus level or produced false-positives. Kraken2 and SPINGO, which used the exact k-mer approach and calculated the lowest common ancestor (LCA), were very rapid at classifying short reads. However, according to Tovo et al (63), the choice of the length k highly influences the classification and default value (k = 35) is frequently used because it is difficult or impossible to determine the optimal value for unknown environments. METAXA2 achieved the highest recall of the microbial community at genus level but also produced false sequence classifications, jeopardising the precision of the taxonomic profiling. Another study demonstrated that Centrifuge, Kraken2 and KrakenUniq yielded many more taxa than the number included in the test datasets (64). Thus, where reads are classified individually, multiple reference sequences can have identical levels of similarity, leading to a high number of false positives. On the contrary, using nearly full-length SSU sequences produced by phyloFlash and RiboTaxa resulted in correct taxa assignment of the mock community and yielded no false-positive results. We demonstrated that RiboTaxa has assigned the highest number of species-level taxa compared to other tools and identified even low abundant taxa.
The application of RiboTaxa to both the MOCK and synthetic human gut communities allowed the identification of all the microorganisms in each community, providing evidence that longer sequences contain stronger phylogenetic signals and yield higher precision for taxonomic profiling than short-read analysis (46). Moreover, nearly full-length 16S sequences contain a wealth of information that allows their accurate classification at appropriate taxonomic ranks, i.e. at a high rank when the sequences are divergent or highly novel and at a low rank when closely related organisms are present in the database (65). However, very closely related species evolve slowly and differ very little between their 16S rRNA genes (66). Consequently, due to their nearly identical 16S rRNA gene sequences, RiboTaxa could not assign certain sequences at species level and stopped at genus level classification. Although, 16S rRNA sequences provide real and significant advantages compared to targeted variable regions, it will never provide a perfect representation of bacterial species diversity (46). To improve this, the 16S ribosomal RNA gene was coupled with the 16S–23S rRNA internal transcribed spacer region sequences to characterize Escherichia species and to identify new strains (67). Likewise, combined with the 16S rRNA gene, the 16S–23S rDNA ITS region can improve the description of microbial diversity within and across group.
We used the SILVA SSU 138.1 NR99 database for taxonomic classification as it contains 510 508 SSU rRNA gene sequences from Bacteria, Archaea and Eukaryota domains (https://www.arb-silva.de/). However, RiboTaxa can also use other existing rRNA sequence databases such as the Ribosomal Database Project (RDP) (37) or Greengenes (68) or genome-based databases such as the GTDB (47). RiboTaxa could also classify eukaryotic organisms using 18S rRNA gene as phylogenetic marker. CCMetagen (64), which has recently been developed, uses the read mapping ConClave sorting scheme, implemented in the KMA software (69) to include microbial eukaryotes in metagenomics data exploration. In the same way, MetaPhlAn2 indexes several different gene families in its database to identify taxa from other microbial kingdoms (70).
Abundance estimation performance of different profilers varies considerably even on the same benchmark datasets (54). This apparently high-performance variation largely arises because the profilers report either one of two fundamentally different types of relative abundances: sequence abundance or taxonomic abundance (71). Furthermore, false positives and false negatives impact microbial structure characterization and by consequence true abundancies. As RiboTaxa shows high precision and recall, abundance evaluation appears close to true abundancies even if it is impossible to obtain exact profile. The presence of multiple copies of rRNA gene makes the community abundance data distorted and gene copy normalization should be necessary for correction (72). However, recent studies indicate that 16S rRNA gene copy number normalization does not provide more reliable conclusions in meta-taxonomic surveys (73,74), therefore, we choose not to use gene copy number correction for RiboTaxa. In all cases, cautions should be exercised when interpreting abundancies results. The use of single-copy marker genes like for MetaPhlan (75), mOTU (76) or PhyloSift (77) should, principally, make abundance estimation more precise, although it is impossible to know the copy number of a gene for a species with an incomplete genome and, of course, for unknown genomes. Many species of archaea and bacteria are polyploid and can contain more than ten copies of their chromosome that influence abundancies as gene copy number in haploid genome (78). Unknown microbial genomes/taxa, missing ploidy information, gene copy number and misclassification of reads from conserved regions across different species render the conversion very challenging, if not impossible (71). Expanded isolate genome availability is essential to improve detection capabilities as demonstrated recently by MetaPhlAn3 (79). However, it is still challenging to precisely analyze environmental samples using approaches based on genome databases, as most reference databases are mostly based on human-associated microorganisms (80).
The uniqueness of RiboTaxa is the benchmarking of tools and metrics optimization at each step, from quality control to taxonomic classification. The mock communities also proved to be integral for parameter optimization, and the accuracy of most taxonomic assignment was notably controlled for sequence artefacts. Thus, users can apply default optimized parameters on their datasets coming from any environment except for the sequence reconstruction parameters, –max_read_length, –insert_mean, –insert_stddev, which exclusively depend on the sequencing length of the input datasets. Run time is another recurring challenge for taxonomic classifiers. RiboTaxa is not as fast as short-read classifiers (i.e. Kraken2, Centrifuge). Indeed, the combined approach (EMIRGE-METARIB) during rRNA sequence reconstruction is responsible for this moderately high but reasonable computational time. However, it also allows to reach a higher precision of taxonomic assignments compared to fast short-read analysis which may assign sequences to wrong taxa (false positives). Thus, the user may need to choose between taxonomic accuracy and speed.
The efficiency of RiboTaxa was finally demonstrated using real datasets obtained from previous studies. In addition to providing a fine taxonomic resolution down to the species level, RiboTaxa has proved to be a versatile tool with its ability to analyse data from different environments including ocean, soil and human gut revealing novelties not detected by current approaches. Metagenome-based approaches were also surpassed by the dynamic approach of RiboTaxa. In fact, lack of reference genomes in the GTBD database, particularly, for less explored microbial communities, hinders the identification of novel species (53) while 16S rRNA genes have the advantage of more accurately identifying distinct microorganisms while reflecting real community richness (81). In octocoral samples, phylogenetic analysis of RiboTaxa derived sequences allowed the identification of a new genera within the Endozoicomonadaceae family consisting of two new species whose abundances are positively correlated with coral health, opening new perspectives to a better understanding of beneficial microbial interactions and coral preservation (82). It will be interesting to access genome information for these bacteria that are not currently available to decipher positive cross talks in coral holobiont. Similarly, we gave examples of specific signatures detected in human gut microbiota in a longevity study. We identified new species that could be of particular interest for health or well-being and potentially used as probiotics. However, precise characterization of these species through isolation and/or genome reconstruction is needed to validate such applications as recently described through new metabolic pathways discovery for the maintenance of intestinal homeostasis in centenarians (83). In the same way, specific microbial signatures in permafrost soils have been revealed using RiboTaxa. For instance, within the family Xanthomonadaceae, 4 novel species belonging to three new genera have been discovered. Fine characterization of these bacteria could also help in microbial adaptive process description in extreme environments and their evolution in the context of global change (84).
Throughout this study, we supported the identification of a potentially new genus or species with an alignment of > 97% with sequences from GenBank annotated as ‘uncultured’ bacteria from corresponding environment. We cannot exclude that a part of this diversity originates from artificial diversity created in silico during full-length sequence reconstruction even though intragenomic 16S gene sequence variation can be a valuable method to provide accurate representation of bacterial species (61). As indicated by EMIRGE developers, occasional presence of small indel errors in the reconstructed sequence could occur but in practice, these rare indels have little effect on taxonomic classification. Accuracy of 16S sequence reconstruction can be controlled as recently demonstrated by Marre et al (85). The authors designed specific primers targeting nearly full-length reconstructed 16S rRNA genes using the KASpOD algorithm (86) and tested them on biological samples through PCR experiments coupled with Sanger sequencing.
Using RiboTaxa, we detected rare microorganisms (<0.1%) in real datasets from different environments (soil, ocean, human gut) and different techniques (metagenomics, gene capture by hybridization), confirming the sensitivity of this approach. Most microbial studies focus on dominant taxa, bypassing the inexhaustible source of metabolic functions of rare microbial taxa, also called the ‘rare biosphere’ (87). Unfortunately, rare taxa detection is highly influenced by sequencing depth and deep sequencing renders downstream processes computationally exhaustive. In this study, we also demonstrated the ability of RiboTaxa to process huge datasets using hybridization capture, whereby SSU rRNA genes accounted for 55.12% of the reads in the soil compared with shotgun sequencing, in which usually <1% of sequences carry the biomarkers. We also revealed microbial novelties in such datasets.
In conclusion, RiboTaxa is a user-friendly metagenomic classifier which supports database preparation, read trimming and SSU read extraction, SSU rRNA gene targeted assembly and taxonomic profiling. This complete workflow demonstrates high specificity and sensitivity without false positive detection and output species relative abundance close to reality, enabling to manage large amount of metagenomics and gene capture by hybridization datasets from any environment. RiboTaxa could efficiently reveal microbial novelties not detected by current approaches as well as, nearly full-length 16S sequences could potentially be connected to metagenome-assembled genomes (MAGs) to improve linking between taxonomy and metabolic functions (88).
DATA AVAILABILITY
RiboTaxa is open source and free for all use. It is licensed under a GNU Affero General Public License 3.0. Source code is available at https://github.com/oschakoory/RiboTaxa and can be easily installed using miniconda3 (https://docs.conda.io/en/latest/miniconda.html).
All data generated or analysed during this study are included in this published article and its supplementary information files. All scripts used to generate graphs and figures are available at https://github.com/oschakoory/RiboTaxa_Supp.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the Mésocentre Clermont Auvergne University and AuBi platform for providing help and computing and storage resources. Computations have been performed on the supercomputer facilities of the Mésocentre Clermont Auvergne University. Figure 1 has been created with BioRender.com.
Contributor Information
Oshma Chakoory, Université Clermont Auvergne, INRAE, MEDIS, F-63000 Clermont-Ferrand, France.
Sophie Comtet-Marre, Université Clermont Auvergne, INRAE, MEDIS, F-63000 Clermont-Ferrand, France.
Pierre Peyret, Université Clermont Auvergne, INRAE, MEDIS, F-63000 Clermont-Ferrand, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Agence Nationale de la Recherche (project MICROPRONY) [ANR-19-CE02-0020] and ECOS-sud [ECOS n°C20B02]; O.C. and S.C.-M. were supported respectively by ANR Intelligence Artificielle (MIA: Artificial Intelligence for clerMont) co-financed by FEDER funds and MICROPRONY projects.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang W.-L., Xu S.-Y., Ren Z.-G., Tao L., Jiang J.-W., Zheng S.-S.. Application of metagenomics in the human gut microbiome. World J. Gastroenterol. 2015; 21:803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jansson J.K., Hofmockel K.S.. The soil microbiome-from metagenomics to metaphenomics. Curr. Opin. Microbiol. 2018; 43:162–168. [DOI] [PubMed] [Google Scholar]
- 3. Sunagawa S., Coelho L.P., Chaffron S., Kultima J.R., Labadie K., Salazar G., Djahanschiri B., Zeller G., Mende D.R., Alberti A.et al.. Ocean plankton. Structure and function of the global ocean microbiome. Science. 2015; 348:1261359. [DOI] [PubMed] [Google Scholar]
- 4. Edgar RC. Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. PeerJ. 2018; 6:e4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brooks J.P., Edwards D.J., Harwich M.D. Jr, Rivera M.C., Fettweis J.M., Serrano M.G., Reris R.A., Sheth N.U., Huang B., Girerd P.et al.. The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 2015; 15:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schloss P.D. The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene-based studies. PLoS Comput. Biol. 2010; 6:e1000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gasc C., Peyretaillade E., Peyret P.. Sequence capture by hybridization to explore modern and ancient genomic diversity in model and nonmodel organisms. Nucleic Acids Res. 2016; 44:4504–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen L.-X., Anantharaman K., Shaiber A., Eren A.M., Banfield J.F.. Accurate and complete genomes from metagenomes. Genome Res. 2020; 30:315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pasolli E., Asnicar F., Manara S., Zolfo M., Karcher N., Armanini F., Beghini F., Manghi P., Tett A., Ghensi P.et al.. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell. 2019; 176:649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parks D.H., Rigato F., Vera-Wolf P., Krause L., Hugenholtz P., Tyson G.W., Wood D.L.A.. Evaluation of the microba community profiler for taxonomic profiling of metagenomic datasets from the human gut microbiome. Front. Microbiol. 2021; 12:643682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huson D.H., Auch A.F., Qi J., Schuster S.C.. MEGAN analysis of metagenomic data. Genome Res. 2007; 17:377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao X., Lin H., Revanna K., Dong Q.. A bayesian taxonomic classification method for 16S rRNA gene sequences with improved species-level accuracy. BMC Bioinformatics. 2017; 18:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zielezinski A., Vinga S., Almeida J., Karlowski W.M.. Alignment-free sequence comparison: benefits, applications, and tools. Genome Biol. 2017; 18:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wood D.E., Salzberg S.L.. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014; 15:R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu J., Breitwieser F.P., Thielen P., Salzberg S.L.. Bracken: estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017; 3:e104. [Google Scholar]
- 16. Allard G., Ryan F.J., Jeffery I.B., Claesson M.J.. SPINGO: a rapid species-classifier for microbial amplicon sequences. BMC Bioinformatics. 2015; 16:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dubinkina V.B., Ischenko D.S., Ulyantsev V.I., Tyakht A.V., Alexeev D.G.. Assessment of k-mer spectrum applicability for metagenomic dissimilarity analysis. BMC Bioinformatics. 2016; 17:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nasko D.J., Koren S., Phillippy A.M., Treangen T.J.. RefSeq database growth influences the accuracy of k-mer-based lowest common ancestor species identification. Genome Biol. 2018; 19:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bengtsson-Palme J., Hartmann M., Eriksson K.M., Pal C., Thorell K., Larsson D.G.J., Nilsson R.H.. METAXA2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol. Ecol. Resour. 2015; 15:1403–1414. [DOI] [PubMed] [Google Scholar]
- 20. Bengtsson J., Eriksson K.M., Hartmann M., Wang Z., Shenoy B.D., Grelet G.-A., Abarenkov K., Petri A., Rosenblad M.A., Nilsson R.H.. Metaxa: a software tool for automated detection and discrimination among ribosomal small subunit (12S/16S/18S) sequences of archaea, bacteria, eukaryotes, mitochondria, and chloroplasts in metagenomes and environmental sequencing datasets. Antonie Van Leeuwenhoek. 2011; 100:471–475. [DOI] [PubMed] [Google Scholar]
- 21. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L.. BLAST+: architecture and applications. BMC Bioinformatics. 2009; 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kopylova E., Noé L., Touzet H.. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012; 28:3211–3217. [DOI] [PubMed] [Google Scholar]
- 23. Khachatryan L., de Leeuw R.H., Kraakman M.E.M., Pappas N., te Raa M., Mei H., de Knijff P., Laros J.F.J.. Taxonomic classification and abundance estimation using 16S and WGS—A comparison using controlled reference samples. Forensic Sci. Int. Genet. 2020; 46:102257. [DOI] [PubMed] [Google Scholar]
- 24. Johnson J.S., Spakowicz D.J., Hong B.Y., Petersen L.M., Demkowicz P., Chen L., Leopold S.R., Hanson B.M., Agresta H.O., Gerstein M.et al.. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019; 10:5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gruber-Vodicka H.R., Seah B.K.B., Pruesse E.. phyloFlash – rapid SSU rRNA profiling and targeted assembly from metagenomes. mSystems. 2020; 5: 10.1128/mSystems.00920-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D.et al.. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012; 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miller C.S., Baker B.J., Thomas B.C., Singer S.W., Banfield J.F.. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 2011; 12:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rognes T., Flouri T., Nichols B., Quince C., Mahé F.. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016; 4:e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Langmead B. Aligning short sequencing reads with bowtie. Curr. Protoc. Bioinforma. 2010; Chapter 11:Unit 11.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xue Y., Lanzén A., Jonassen I.. Reconstructing ribosomal genes from large scale total RNA meta-transcriptomic data. Bioinforma. Oxf. Engl. 2020; 36:3365–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pericard P., Dufresne Y., Couderc L., Blanquart S., Touzet H.. MATAM: reconstruction of phylogenetic marker genes from short sequencing reads in metagenomes. Bioinforma. Oxf. Engl. 2018; 34:585–591. [DOI] [PubMed] [Google Scholar]
- 32. Kim D., Song L., Breitwieser F.P., Salzberg S.L.. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016; 26:1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gasc C., Peyret P.. Hybridization capture reveals microbial diversity missed using current profiling methods. Microbiome. 2018; 6:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wood D.E., Lu J., Langmead B.. Improved metagenomic analysis with kraken 2. Genome Biol. 2019; 20:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schloss P.D. Reintroducing mothur: 10 years later. Appl. Environ. Microbiol. 2020; 86:e02343-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bokulich N.A., Kaehler B.D., Rideout J.R., Dillon M., Bolyen E., Knight R., Huttley G.A., Gregory Caporaso J.. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018; 6:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lan Y., Wang Q., Cole J.R., Rosen G.L.. Using the RDP classifier to predict taxonomic novelty and reduce the search space for finding novel organisms. PLoS One. 2012; 7:e32491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu J., Salzberg S.L.. Ultrafast and accurate 16S rRNA microbial community analysis using kraken 2. Microbiome. 2020; 8: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang W., Li L., Myers J.R., Marth G.T.. ART: a next-generation sequencing read simulator. Bioinformatics. 2012; 28:593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buchfink B., Reuter K., Drost H.-G.. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods. 2021; 18:366–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.-F., Guindon S., Lefort V., Lescot M.et al.. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008; 36:W465–W469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Edgar R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004; 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000; 17:540–552. [DOI] [PubMed] [Google Scholar]
- 44. Guindon S., Gascuel O.. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003; 52:696–704. [DOI] [PubMed] [Google Scholar]
- 45. Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J.et al.. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yarza P., Yilmaz P., Pruesse E., Glöckner F.O., Ludwig W., Schleifer K.-H., Whitman W.B., Euzéby J., Amann R., Rosselló-Móra R.. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014; 12:635–645. [DOI] [PubMed] [Google Scholar]
- 47. Parks D.H., Chuvochina M., Chaumeil P.-A., Rinke C., Mussig A.J., Hugenholtz P.. A complete domain-to-species taxonomy for bacteria and archaea. Nat. Biotechnol. 2020; 38:1079–1086. [DOI] [PubMed] [Google Scholar]
- 48. de la Cuesta-Zuluaga J., Ley R.E., Youngblut N.D.. Struo: a pipeline for building custom databases for common metagenome profilers. Bioinformatics. 2020; 36:2314–2315. [DOI] [PubMed] [Google Scholar]
- 49. Escobar-Zepeda A., Godoy-Lozano E.E., Raggi L., Segovia L., Merino E., Gutiérrez-Rios R.M., Juarez K., Licea-Navarro A.F., Pardo-Lopez L., Sanchez-Flores A.. Analysis of sequencing strategies and tools for taxonomic annotation: defining standards for progressive metagenomics. Sci. Rep. 2018; 8:12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Keller-Costa T., Lago-Lestón A., Saraiva J.P., Toscan R., Silva S.G., Gonçalves J., Cox C.J., Kyrpides N., Nunes da Rocha U., Costa R.. Metagenomic insights into the taxonomy, function, and dysbiosis of prokaryotic communities in octocorals. Microbiome. 2021; 9:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rampelli S., Soverini M., D’Amico F., Barone M., Tavella T., Monti D., Capri M., Astolfi A., Brigidi P., Biagi E.et al.. Shotgun metagenomics of gut microbiota in humans with up to extreme longevity and the increasing role of xenobiotic degradation. Msystems. (2020); 5:e00124-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Perez-Mon C., Qi W., Vikram S., Frossard A., Makhalanyane T., Cowan D., Frey B.. Shotgun metagenomics reveals distinct functional diversity and metabolic capabilities between 12 000-year-old permafrost and active layers on muot da barba peider (Swiss alps). Microb. Genomics. 2021; 7:000558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paoli L., Ruscheweyh H.-J., Forneris C.C., Kautsar S., Clayssen Q., Salazar G., Milanese A., Gehrig D., Larralde M., Carroll L.M.et al.. Uncharted biosynthetic potential of the ocean microbiome. Nature. 2022; 607:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ye S.H., Siddle K.J., Park D.J., Sabeti P.C.. Benchmarking metagenomics tools for taxonomic classification. Cell. 2019; 178:779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kinoshita Y., Niwa H., Uchida-Fujii E., Nukada T.. Establishment and assessment of an amplicon sequencing method targeting the 16S-ITS-23S rRNA operon for analysis of the equine gut microbiome. Sci. Rep. 2021; 11:11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mende D.R., Sunagawa S., Zeller G., Bork P.. Accurate and universal delineation of prokaryotic species. Nat. Methods. 2013; 10:881–884. [DOI] [PubMed] [Google Scholar]
- 57. Lind A.L., Pollard K.S.. Accurate and sensitive detection of microbial eukaryotes from whole metagenome shotgun sequencing. Microbiome. 2021; 9:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beaudry M.S., Wang J., Kieran T.J., Thomas J., Bayona-Vásquez N.J., Gao B., Devault A., Brunelle B., Lu K., Wang J.-S.et al.. Improved microbial community characterization of 16S rRNA via metagenome hybridization capture enrichment. Front. Microbiol. 2021; 12:644662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Links M.G., Dumonceaux T.J., McCarthy E.L., Hemmingsen S.M., Topp E., Town J.R.. CaptureSeq: hybridization-based enrichment of cpn60 gene fragments reveals the community structures of synthetic and natural microbial ecosystems. Microorganisms. 2021; 9:816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Guo J., Cole J.R., Zhang Q., Brown C.T., Tiedje J.M.. Microbial community analysis with ribosomal gene fragments from shotgun metagenomes. Appl. Environ. Microbiol. 2016; 82:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Johnson J.S., Spakowicz D.J., Hong B.-Y., Petersen L.M., Demkowicz P., Chen L., Leopold S.R., Hanson B.M., Agresta H.O., Gerstein M.et al.. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019; 10:5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Edgar R.C., Haas B.J., Clemente J.C., Quince C., Knight R.. UCHIME improves sensitivity and speed of chimera detection. Bioinforma. Oxf. Engl. 2011; 27:2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tovo A., Menzel P., Krogh A., Cosentino Lagomarsino M., Suweis S.. Taxonomic classification method for metagenomics based on core protein families with core-kaiju. Nucleic Acids Res. 2020; 48:e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Marcelino V.R., Clausen P.T.L.C., Buchmann J.P., Wille M., Iredell J.R., Meyer W., Lund O., Sorrell T.C., Holmes E.C.. CCMetagen: comprehensive and accurate identification of eukaryotes and prokaryotes in metagenomic data. Genome Biol. 2020; 21:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. von Meijenfeldt F.A.B., Arkhipova K., Cambuy D.D., Coutinho F.H., Dutilh B.E.. Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol. 2019; 20:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pei A.Y., Oberdorf W.E., Nossa C.W., Agarwal A., Chokshi P., Gerz E.A., Jin Z., Lee P., Yang L., Poles M.et al.. Diversity of 16S rRNA genes within individual prokaryotic genomes. Appl. Environ. Microbiol. 2010; 76:3886–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Magray M.S.U.D., Kumar A., Rawat A.K., Srivastava S.. Identification of Escherichia coli through analysis of 16S rRNA and 16S-23S rRNA internal transcribed spacer region sequences. Bioinformation. 2011; 6:370–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. DeSantis T.Z., Hugenholtz P., Larsen N., Rojas M., Brodie E.L., Keller K., Huber T., Dalevi D., Hu P., Andersen G.L.. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006; 72:5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Clausen P.T.L.C., Aarestrup F.M., Lund O.. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics. 2018; 19:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Truong D.T., Franzosa E.A., Tickle T.L., Scholz M., Weingart G., Pasolli E., Tett A., Huttenhower C., Segata N.. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods. 2015; 12:902–903. [DOI] [PubMed] [Google Scholar]
- 71. Sun Z., Huang S., Zhang M., Zhu Q., Haiminen N., Carrieri A.P., Vázquez-Baeza Y., Parida L., Kim H.-C., Knight R.et al.. Challenges in benchmarking metagenomic profilers. Nat. Methods. 2021; 18:618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Angly F.E., Dennis P.G., Skarshewski A., Vanwonterghem I., Hugenholtz P., Tyson G.W.. CopyRighter: a rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction. Microbiome. 2014; 2:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Louca S., Doebeli M., Parfrey L.W.. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome. 2018; 6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Starke R., Pylro V.S., Morais D.K.. 16S rRNA gene copy number normalization does not provide more reliable conclusions in metataxonomic surveys. Microb. Ecol. 2021; 81:535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Segata N., Waldron L., Ballarini A., Narasimhan V., Jousson O., Huttenhower C.. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods. 2012; 9:811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sunagawa S., Mende D.R., Zeller G., Izquierdo-Carrasco F., Berger S.A., Kultima J.R., Coelho L.P., Arumugam M., Tap J., Nielsen H.B.et al.. Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods. 2013; 10:1196–1199. [DOI] [PubMed] [Google Scholar]
- 77. Darling A.E., Jospin G., Lowe E., Matsen F.A., Bik H.M., Eisen J.A.. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ. 2014; 2:e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Soppa J. Polyploidy in archaea and bacteria: about desiccation resistance, giant cell size, long-term survival, enforcement by a eukaryotic host and additional aspects. J. Mol. Microbiol. Biotechnol. 2014; 24:409–419. [DOI] [PubMed] [Google Scholar]
- 79. Beghini F., McIver L.J., Blanco-Míguez A., Dubois L., Asnicar F., Maharjan S., Mailyan A., Manghi P., Scholz M., Thomas A.M.et al.. ntegrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. I. Elife. 2021; 10:e65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dueholm M.S., Andersen K.S., McIlroy S.J., Kristensen J.M., Yashiro E., Karst S.M., Albertsen M., Nielsen P.H.. Generation of comprehensive ecosystem-specific reference databases with species-level resolution by high-throughput full-length 16S rRNA gene sequencing and automated taxonomy assignment (AutoTax). Mbio. 2020; 11:e01557-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang Y., Liao S., Gai Y., Liu G., Jin T., Liu H., Gram L., Strube M.L., Fan G., Sahu S.K.et al.. Metagenomic analysis reveals microbial community structure and metabolic potential for nitrogen acquisition in the oligotrophic surface water of the indian ocean. Front. Microbiol. 2021; 12:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Thatcher C., Høj L., Bourne D.G.. Probiotics for coral aquaculture: challenges and considerations. Curr. Opin. Biotechnol. 2021; 73:380–386. [DOI] [PubMed] [Google Scholar]
- 83. Sato Y., Atarashi K., Plichta D.R., Arai Y., Sasajima S., Kearney S.M., Suda W., Takeshita K., Sasaki T., Okamoto S.et al.. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature. 2021; 599:458–464. [DOI] [PubMed] [Google Scholar]
- 84. Shen L., Liu Y., Allen M.A., Xu B., Wang N., Williams T.J., Wang F., Zhou Y., Liu Q., Cavicchioli R.. Linking genomic and physiological characteristics of psychrophilic arthrobacter to metagenomic data to explain global environmental distribution. Microbiome. 2021; 9:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Marre S., Gasc C., Forest C., Lebbaoui Y., Mosoni P., Peyret P.. Revealing microbial species diversity using sequence capture by hybridization. Microb. Genomics. 2021; 7:000714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Parisot N., Denonfoux J., Dugat-Bony E., Peyret P., Peyretaillade E.. KASpOD–a web service for highly specific and explorative oligonucleotide design. Bioinforma. Oxf. Engl. 2012; 28:3161–3162. [DOI] [PubMed] [Google Scholar]
- 87. Sogin M.L., Morrison H.G., Huber J.A., Mark Welch D., Huse S.M., Neal P.R., Arrieta J.M., Herndl G.J.. Microbial diversity in the deep sea and the underexplored ‘rare biosphere. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:12115–12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lesker T.R., Durairaj A.C., Gálvez E.J.C., Lagkouvardos I., Baines J.F., Clavel T., Sczyrba A., McHardy A.C., Strowig T.. An integrated metagenome catalog reveals new insights into the murine gut microbiome. Cell Rep. 2020; 30:2909–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RiboTaxa is open source and free for all use. It is licensed under a GNU Affero General Public License 3.0. Source code is available at https://github.com/oschakoory/RiboTaxa and can be easily installed using miniconda3 (https://docs.conda.io/en/latest/miniconda.html).
All data generated or analysed during this study are included in this published article and its supplementary information files. All scripts used to generate graphs and figures are available at https://github.com/oschakoory/RiboTaxa_Supp.