

Abstract
Introduction
Polycystic ovary syndrome (PCOS) is a common condition characterized by reproductive, hyperandrogenic and dysmetabolic features, and often becomes clinically manifest during adolescence, particularly with weight-gain.
Sources of data
Pubmed search.
Areas of agreement
PCOS is heritable and closely associates with obesity (based on data from both epidemiological and genetic studies). Furthermore, insulin resistance forms a central cornerstone of the pathogenesis of PCOS and mediates a close association between obesity and the severity of the phenotypic features of PCOS.
Areas of controversy
Our understanding of the pathogenesis of PCOS remains incomplete, especially regarding its missing heritability (with only a small fraction having been identified from the genome-wide association studies reported to date), and its developmental origins.
Growing points
A challenge for the future is to explore a role for epigenetic modifications in the development of PCOS, and implications for the in utero environment and novel therapeutic opportunities.
Keywords: polycystic ovary syndrome, obesity, genetics, environment
Introduction
Polycystic ovary syndrome (PCOS) affects between 6 and 10% of women of reproductive age1 and typically manifests during adolescence, especially in the context of weight-gain and obesity.2 The internationally agreed ‘Rotterdam’ diagnostic criteria for PCOS require at least two of three criteria: (i) polycystic ovarian morphology; (ii) hyperandrogenic features (characterized clinically as hirsutism, androgenic alopecia and acne and biochemically as raised serum androgens [testosterone, androstenedione and free androgen index]) and (iii) reproductive features (oligo-amenorrhoea and sub-fertility).2,3 PCOS also associates with dysmetabolic features, mediated primarily through insulin resistance. Indeed, amongst women with PCOS, 10% have Type 2 Diabetes Mellitus (T2D), ~70% have dyslipidaemia, there is a 5–10-fold increased risk for the development of obstructive sleep apnoea, and the prevalence of metabolic syndrome varies between 34 and 46%.4–7
The purpose of this review is to explore why women with PCOS are obese, requiring an exploration of the pathogenesis of PCOS (outlined in Fig. 1).
Fig. 1.
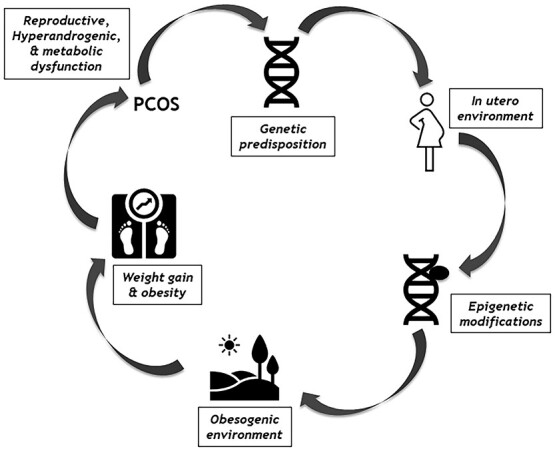
The Pathogenic mechanisms that underlie the development of PCOS.
Sources of data
Pubmed was used solely to access the relevant medical literature. Only articles written in the English language were used. The keywords ‘polycystic ovary syndrome’ and ‘obesity’ were used as search terms. There was a preference for more recently published articles where applicable.
Areas of agreement
Some aspects of the pathophysiology of PCOS are clearly defined, with widespread agreement amongst colleagues, including factors pertinent to its association with obesity: epidemiology/genetics and the role of insulin resistance.
Epidemiological and genetic interlinks between PCOS and obesity
One area of broad agreement is that PCOS and obesity are closely linked both from an epidemiological and genetic perspective. Regarding the former, obesity or overweight affects between 38 and 88% of women with PCOS.2 Based on data from the Northern Finland Birth Cohort 1966, weight-gain (particularly in early adulthood) is important for the subsequent development of PCOS.8 Furthermore, even modest (5%) weight-loss in obese women with PCOS associates with an improved phenotypic profile of PCOS (including reproductive, hyperandrogenic and dysmetabolic features).9
PCOS is a heritable condition.10,11 Based on data from a large Dutch twin study, the concordance of PCOS symptoms in monozygotic twins (70%) was double that in dizygotic twins.12 Furthermore, genetic evidence further entwines PCOS and obesity. One example is from my own UK-based case-control study conducted in Oxford, in which a variant (rs9939609) within the FTO gene (that associates with increased fat mass), was shown to associate with an increased risk for the development of PCOS (OR 1.30 per minor allele copy).13 In a further study from central Europe, a different variant (rs1421085) within the FTO gene was also shown to associate with the development of PCOS amongst a cohort of women with obesity or metabolic syndrome.14 Finally, variants within the FTO and Melanocortin Receptor 4 (MC4R) genes associated with obesity in women with PCOS.15 These studies demonstrate that at least some of the heritability of PCOS is likely mediated through effects of gene variants (such as those within FTO) that associate with increased fat mass and propensity for weight-gain and obesity. This is perhaps not surprising given the close epidemiological links between PCOS and obesity. These insights also help us to re-define how we conceptualise the role of genetics in health and disease. Rather than considering a gene ‘for’ PCOS, or a gene ‘for’ obesity, we should embrace the sheer scale and complexity of genetic effects within the broader scheme of the entire genome, and the almost unimaginable complexity of the web of proteins that it encodes. The example of variants within FTO enables a broader perspective on genetic effects, beyond merely influencing fat mass in isolation but also obesity-related conditions such as PCOS.
An extensive overview of the genetics of PCOS has been covered in detail elsewhere.10,11 Within the context of the recently reported genome-wide association studies (GWAS) on PCOS across cohorts from both China and Europe, there are some areas of agreement, including a total of 16 replicated loci that have been identified for association with PCOS.16–21 These genetic data provide pathogenic insights. This includes variants within FSHB that influence gonadotrophin (LH) production and secretion, variants within LHCGR and FSHR that influence gonadotrophin action through the functionality of the LH and FSH receptors, respectively and a possible role for variants within GATA4 in the control of steroidogenesis.19
In summary, the association between PCOS and obesity is incontrovertible. Widespread agreement from epidemiological studies2,8,9 provides compelling evidence that is further strengthened by evidence from genetics studies outlined here,13–15 based on variants in genes such as FTO that play an important role in the determination of body fat mass. The association between PCOS and obesity is mediated through effects of the latter on insulin resistance (explored in the section ‘The central role of insulin resistance in the development of PCOS’). This hypothesis is strengthened by a recently published study using bidirectional Mendelian randomization that revealed increasing body mass index (BMI) to be causal for PCOS but having PCOS did not appear to affect BMI.22
The central role of insulin resistance in the development of PCOS
Insulin resistance, affecting 50–90% of women with PCOS, forms a central component of its pathogenesis.23 There are two main mechanisms. The first relates to the metabolic implications of insulin resistance per se, such as the development of T2D, hypertension and dyslipidaemia. The second relates to the steroidogenic effects of secondary hyperinsulinaemia, acting through unaffected components of the post-receptor insulin cellular pathway.
Although the term ‘insulin resistance’ implies total resistance to the effects of insulin, in women with PCOS (as in T2D) this is selective, only affecting the ‘PI3-Kinase’ (PI3-K) post-receptor insulin pathway that confers the metabolic effects of insulin. Importantly, the severity of dysfunction of the PI3-K post-receptor insulin pathway is commensurate with the degree of weight-gain and obesity, possibly mediated through inflammatory pathways.2,5,24,25 Through this mechanism, the dysmetabolic features of PCOS (such as T2D, dyslipidaemia and hypertension) become manifest. The degree of weight-gain required for the clinical manifestation of obesity-related conditions such as PCOS and T2D varies between individuals and is influenced by the multiple other factors that constitute their underlying complex pathogeneses.
Selective resistance of the PI3-K post-receptor insulin pathway to the effects of insulin results in secondary and compensatory hyperinsulinaemia.24 Physiologically, the effects of insulin are pleiotropic, and extend well beyond merely coordinating metabolic processes. In addition to the PI3-K post-receptor insulin pathway, the ‘mitogen-activated protein kinase’ (MAP-K) post-receptor insulin pathway represents the other major pathway that mediates the cellular effects of insulin. The MAP-K post-receptor insulin pathway confers the atherogenic, steroidogenic and mitogenic effects of insulin.26 Importantly, in women with PCOS (and other dysmetabolic conditions such as T2D), the MAP-K post-receptor insulin pathway remains intact.2 Therefore, in the context of compensatory hyperinsulinaemia, the MAP-K post-receptor insulin pathway becomes hyper-stimulated, and underlies much of the reproductive dysfunction and hyperandrogenic features of PCOS.2,5,27 The former stems from the effects of hyperinsulinaemia on the impairment and arrest of ovarian preantral follicle development,2 with the clinical sequelae of oligo-amenorrhoea and sub-fertility. The latter stems from the synergistic effects of hyperinsulinaemia on the action of luteinising hormone (LH) within the ovarian theca cells, thereby stimulating excessive production of testosterone with the clinical sequelae of hirsutism, acne and androgenic alopecia.28,29 In addition to effects within the ovary, hyperinsulinaemia also stimulates adrenal androgen production (through the activation of CYP17 [P450c17α], a key enzyme in androgen biosynthesis).30 Hyperinsulinaemia also suppresses the production of SHBG within the liver (resulting in elevated free androgens),2 and may augment the pulse amplitude of LH secretion (further enhancing ovarian hyperandrogenaemia).31
To summarize this sub-section, insulin resistance in PCOS stems from aberrations in the PI3-K post-receptor insulin pathway. This underlies metabolic dysfunction and results in compensatory hyperinsulinaemia, that in turn drives the steroidogenic effects of insulin (including the reproductive and hyperandrogenic features of PCOS), mediated by the intact MAP-K post-receptor insulin pathway. Given the close association between body fat and dysfunction of the PI3-K post-receptor insulin pathway,2,5,24,25 this explains the importance of body weight in the phenotypic expression of PCOS, including why weight-gain often precedes the onset of the clinical and biochemical features of PCOS, and why effective and sustained weight-loss in patients with PCOS, in the context of obesity and overweight, remains central as a therapeutic strategy.
Areas of controversy
As outlined above, research published over many decades has enabled substantial agreement between clinicians and researchers regarding the pathophysiology of PCOS, including its heritability, epidemiological and genetic interlinks with obesity and the central role of insulin resistance. However, despite this progress and widespread agreement, there are important aspects of the pathophysiology of PCOS that remain incompletely understood and controversial. Most notably, these contentious areas include the role of epigenetic factors and the developmental origins of PCOS.
Epigenetic factors
As outlined above, the heritability of PCOS is incontrovertible. Furthermore, recently published data from GWAS studies in PCOS have provided novel insights into its genetic architecture.19,32,33 However, despite these trans-continental GWAS efforts involving many thousands of probands and controls, we have only identified a tiny minority (<5%) of the overall heritability of PCOS.12 An obvious question is what accounts for the vast majority of PCOS heritability, which hitherto has remained elusive and undetected. To answer this question, it is necessary to take a journey into the inner workings of the genetic code.
Our traditional notion of heritability implicates changes in the nucleotide sequences within our DNA that in turn modifies in some way the encoded protein expressed. The DNA nucleotide sequence can be viewed as the genetic hardware that is also targeted during GWAS studies. However, in recent years, a novel perspective on heritability has emerged, that does not implicate the DNA nucleotide sequence itself, but rather additional proteins that attach to the DNA molecule and alter its expression profile. These additional proteins can be viewed as the genetic software, and importantly do not alter in any way the underlying nucleotide sequence within the DNA molecule itself and are therefore not detectable by traditional GWAS techniques. Proteins that attach onto the DNA molecule without changing the nucleotide sequence, but which do influence gene expression profiles (through transcription and/or translation) are termed ‘epigenetic’ modifications.
There are two main theories regarding the hidden heritability of PCOS. The first is simply that the GWAS studies to date,19,32,33 despite the inclusion of many thousands of probands and controls, are simply under-powered to detect many (in fact, the majority) of the gene variants that contribute towards the development of PCOS. For this hypothesis to be true, it would require a scenario in which many (perhaps hundreds or even thousands) gene variants, each with relatively small effect sizes (below the detection of the GWAS performed to date) account for the overall heritability of PCOS. To detect such variants would likely require global collaboration with future GWAS including probands and controls in numbers that are perhaps an order of magnitude greater than those published on to date.5 The other main theory is that much of the heritability of PCOS stems from epigenetic modifications, which by their nature would not be detected through standard GWAS approaches. The reality is that the hidden heritability of PCOS probably stems from a combination of these two factors: multiple gene variants with relatively small effect sizes and epigenetic modifications.
The notion of epigenetics in the context of the pathogenesis of PCOS is compelling. There is much potential for altered gene expression to play an important role in the pathogenesis of PCOS, including for example genetic pathways implicated in steroid synthesis, insulin signalling, cell communication, reproductive function and carbohydrate metabolism.34 Indeed, in the case of other conditions with a complex pathogenesis (such as T2D, depression and certain malignancies) in which GWAS data also reveal only a small fraction of overall heritability, epigenetic modifications also likely contribute to at least some of the hidden heritability.34
Epigenetic modifications originate from exposure to environmental factors (including the in utero environment), and implicate protein-based additions and subtractions to the DNA molecule itself (including histone modifications and/or DNA hypo- or hypermethylation).34 Such protein-based modifications are mediated either directly or indirectly through micro-RNA (miRNA) -induced changes, the expression of miRNA itself being influenced by direct epigenetic modifications.34,35 MiRNAs are small, noncoding single-strands of RNA (length: 20–24 nucleotides)34 that can influence gene expression through multiple mechanisms. This includes modulation of the translation of mRNA, the degradation of mRNA transcripts and targeting of the epigenetic machinery itself (the latter termed ‘epi-miRNA’, with examples of DNA methyltransferases, histone deacetylases and ‘polycomb repressive complex’ genes).34 The pathogenesis of some well-characterized human diseases has aberrant miRNA as a central pathogenic factor.34,36 It is plausible that mi-RNAs also play a role in the pathogenesis of PCOS, although conclusive evidence remains elusive. Within the literature to date, miRNA-222 appears to show a consistent association with PCOS and should be a focus for future mechanistic research.34
Some studies have explored methylation of DNA in the context of PCOS. This includes the most comprehensive study of PCOS epigenetics reported on to date, which identified 106 differentially methylated CpG sites (associated with 88 genes) from genome-wide DNA methylation profiling of ovarian granulosa lutein cells derived from women with PCOS versus healthy controls. These methylated CpG sites implicated some genes involved in ovarian function and disruption to some gene networks with diverse functions.37 Other studies have revealed hypomethylation within the promoter region of the LH/choriogonadotrophin receptor (LHCGR) gene in women with PCOS, which may enhance its transcription and thereby activate the gonadal axis.34
To summarize this sub-section, although the heritability of PCOS is incontrovertible, GWAS studies have revealed only a tiny fraction of it. Epigenetic modifications (hidden from GWAS data) may account for at least some of the hidden heritability of PCOS. Given the dynamic nature of epigenetic modifications, their temporal transmutability, their sheer complexity and their elusiveness, a challenge for the future is to develop techniques that accurately and reliably identify these epigenetic modifications, and to develop adequately powered studies that can discern the genetic and epigenetic contributors to the development of PCOS. Such data should enable novel preventive and therapeutic approaches to PCOS, based on epigenetic targets (either through indirect approaches to environmental change and/or direct modifications to the epigenome itself).
The developmental origins of PCOS
Aside from epigenetic effects, the other main area of controversy regarding the pathogenesis of PCOS relates to its developmental origins. Our traditional perspective of PCOS development implicates weight-gain with worsening insulin resistance in the context of a genetic predisposition, with little emphasis on factors that originate in utero. However, in recent years, evidence primarily from animal-based studies support a broader perspective on the developmental origins of PCOS incorporating the intra-uterine environment, known as the ‘Fetal Programming Hypothesis’ (FPH). Within the FPH, there are two proposed mechanisms that implicate fetal exposure to either excessive levels of androgens (with ‘programming’ effects on the developing fetal tissues that are androgen-sensitive) or restricted nutrients (with effects on the levels of insulin and insulin resistance within the fetus). To conflate the two main areas of controversy in PCOS pathogenesis, some of the clinical manifestations of the FPH may be mediated through epigenetic effects in utero.38
Regarding the excessive androgen exposure component of the FPH, most of our evidence stems from animal-based studies such as rodents, in which the development of polycystic ovaries and anovulatory sterility occurs in female offspring following early intra-uterine exposure to raised levels of testosterone.38 Furthermore, in rhesus monkeys the later development of irregular ovulatory menstrual cycles, ovarian hyperandrogenism, hypersecretion of LH and enlarged poly-follicular ovaries (features of PCOS) in addition to metabolic dysfunction (visceral adiposity, insulin resistance and increased risk for the development of T2D) in female offspring was demonstrated following androgenization during early in utero development.38,39 In support of a potential role for epigenetic factors, the female offspring of rhesus monkeys that had experienced early in utero androgenization also displayed changes in gene expression profiles in later life, including enhanced 5α-reductase activity, reduced aromatase activity and enhanced transcription of genes expressed within the granulosa cells (such as FSHR and IGF-1).38,40 Consistent with a role for tissue programming effects, it appears that the timing of in utero androgenization is important, with exposure to excessive androgens during early gestation having a greater impact on the subsequent manifestation of the phenotypic features of PCOS. There is also evidence from studies on sheep and rodents that in utero exposure to excessive maternal androgens associates with low birth weight offspring.38
Regarding the potential mechanisms by which early intra-uterine exposure to excessive levels of maternal androgens may contribute towards the future development of insulin resistance and PCOS in female offspring, this remains contentious although various hypotheses have been suggested. These include possible effects on reducing fetal growth and birth weight through impaired placental function, induction of relative insulin hypersecretion within the fetus that may then program adrenal hyperandrogenism and induced changes in the hypothalamic regulation of gonadotrophin secretion.38
The excessive androgen exposure component of the FPH is also supported by human-based observational studies, although rather than conclusive proof as such, these data only provide circumstantial evidence with observations consistent with the FPH. In fact, human in utero exposure to excessive maternal androgens is relatively rare given the protective effect of placental aromatase (that converts testosterone into oestradiol).38 Very occasionally, in the context of extremely elevated levels of androgens (either from maternal or fetal origins such as congenital adrenal hyperplasia [due to 21-hydroxylase deficiency] and congenital virilizing tumours respectively), the protective effect of placental aromatase is exceeded resulting in fetal exposure to excessive androgens, and female offspring may then develop PCOS later in life.38 Another rare clinical scenario in which this may occur is in the context of maternal aromatase enzyme dysfunction (stemming from mutations within the CYP19 gene that encodes the aromatase enzyme). Female offspring of mothers with such rare mutations can also develop PCOS later in life.38
Unlike human-based observations, the animal-based studies outlined occurred in the context of artificial exposure to excessive androgens in utero (including the maternal administration of exogenous androgens). It should be noted that in the context of PCOS at least, maternal serum androgens are usually only mildly or moderately elevated, and female offspring are therefore usually protected from exposure to hyperandrogenaemia in utero through effective placental aromatase activity.38 However, there is a possible mechanism implicating anti-Mullerian Hormone (AMH), whereby the female offspring of mothers with PCOS may have greater exposure to androgens in utero. In one study, there was demonstration of significantly elevated maternal serum levels of AMH during pregnancy in women with PCOS compared with controls.41 Maternally elevated AMH may influence female fetal development, as evidenced by murine studies in which there was diminished placental conversion of testosterone to estradiol, elevated maternal levels of testosterone and fetal masculinization with a PCOS-like phenotype in adulthood.41 Furthermore, the notion of the excessive androgen exposure FPH is strengthened by a fetal origin of androgen excess,42 and its mediation via epigenetic effects through the transgenerational heredity of PCOS.43 Therefore, based on the current literature there are compelling data to suggest that, at least in principle, fetal exposure to excessive androgens in utero may predispose to the development of phenotypic features of PCOS later in life in female offspring, through tissue programming and epigenetic effects. However, the extent to which these proposed mechanisms occur naturally and underlie PCOS pathogenesis remains contentious and should be a focus for future research.
As with excessive androgen exposure, generalized restricted nutrients in utero (as may occur during famine) may also predispose to the development of PCOS in female offspring, and forms an important component of the FPH.44 Our best theory is that in this context, there is a reduced need for fetal insulin secretion, and insulin resistance then develops within fetal target tissues as a protective compensatory response (with a metabolic dysfunctional legacy in later life).38 This is compounded by the association of restricted fetal nutrients with low birth weight, which in turn associates with the development of metabolic dysfunction (including insulin resistance and T2D) in both male and female offspring and PCOS in adolescent girls.38 As with excessive androgen exposure in utero, the legacy effects of fetal nutritional restriction may be mediated through epigenetic effects.38
Although fetal nutritional restriction may appear relatively unusual in our modern-day well-fed western society, it does remain an important concern on a global level. In addition to its associated socio-economic, political and healthcare problems, fetal nutritional restriction may also predispose to future metabolic dysfunction in offspring (including PCOS in female offspring). We should also recognize that maternal nutritional deficiencies do not just occur in the context of famine, but also not uncommonly in the context of obesity. Therefore, the importance of the establishment and maintenance of a healthy and nutritionally replete diet in pregnant mothers (including in the pre-conception period and in western societies) cannot be over-stated, not just for maternal and fetal health and wellbeing, but for the longer-term metabolic and reproductive legacy effects within offspring.
Areas timely for developing research
Future GWAS are required with larger numbers of probands and controls, and broader (possibly even global) collaborative efforts. Furthermore, we need to develop techniques for accurate and reliable identification of epigenetic changes, and to explore how the epigenome influences gene expression and the development (and heritability) of conditions like PCOS. Regarding the FPH for the development of PCOS, one area of research interest relates to adipose tissue development and distribution. As outlined, PCOS associates with obesity and insulin resistance, and it is known that visceral adiposity in particular associates with insulin resistance and features of the metabolic syndrome.45 This raises an intriguing question regarding possible effects of the in utero environment (through programming or other mechanisms), on the development and ultimate distribution of adipose tissue depots, thereby contributing towards visceral fat-related insulin resistance and metabolic dysfunction, and the development of PCOS later in life in female offspring. To counter enthusiasm for such a mechanism, in one of my own studies, use of an MRI-based technique showed no discernible differences in visceral adipose depots amongst women with PCOS versus BMI and fat mass matched controls.46 However, it remains possible that the in utero environment may still influence adipose tissue development in some ways that are relevant for PCOS pathogenesis, such as insulin sensitivity within adipocytes, worthy of further investigation.
Concluding remarks
Exploring the pathogenesis of PCOS (summarized in Table 1) can sometimes seem like peering into a fractal in ever greater detail. Although frustrating from one perspective in that a complete understanding of PCOS pathogenesis seems continuously out of reach and to elude us, this nevertheless bodes well for the future research potential of PCOS and its endlessly fascinating pathogenesis. We now reach the titular question of why women with PCOS are obese. To answer this requires insights into the outlined epidemiology, genetic and epigenetic aspects of PCOS, including the underlying role of insulin resistance. Obesity, acting through enhanced insulin resistance, promotes the clinical manifestation of PCOS in those girls and women who are genetically predisposed. Therefore, obesity increases the propensity for PCOS, and this is the true explanation for why women with PCOS are obese.
Table 1.
Summary of the main studies explored
| Main findings | Study design | Ref. |
|---|---|---|
| Obesity and overweight affects between 38 and 88% of women with PCOS | Epidemiological | 2 |
| Weight-gain results in the clinical manifestation of PCOS | NFBC 1966 | 8 |
| Modest weight-loss (5%) in obese women with PCOS results in improved phenotype | Epidemiological | 9 |
| Concordance of PCOS in 70% of monozygotic twins | Dutch twin study | 12 |
| Variant within the FTO gene associates with the development of PCOS with an odds ratio of 1.3 per minor allele copy | UK-based case–control study | 14 |
| 16 replicated loci identified for association with PCOS | GWAS | 16–21 |
| Insulin resistance affects between 50 and 90% of women with PCOS | Case–control studies | 23 |
| PI3-K post-receptor insulin pathway dysfunction is commensurate with the degree of weight-gain | Lab-based study | 2 , 5 , 24 , 25 |
| miRNA-222 associates with PCOS | Epigenetic case–control study | 34 |
| 106 differentially methylated CpG sites in PCOS (associated with 88 genes) based on genome-wide DNA methylation profiling of ovarian granulosa lutein cells | Lab-based study (PCOS vs controls) | 37 |
| Development of polycystic ovaries and anovulatory sterility in female offspring following maternal early intra-uterine exposure to raised androgens | Rodent-based studies | 38 |
| Development of irregular ovulatory menstrual cycles, ovarian hyperandrogenism, hypersecretion of LH, enlarged poly-follicular ovaries and metabolic dysfunction in female offspring following maternal early intra-uterine exposure to raised androgens | Rhesus monkey-based studies | 38 , 39 |
| Female offspring of mothers with aromatase enzyme dysfunction (mutations within the CYP19 gene) can develop PCOS later in life | Case reports | 38 |
Abbreviations: GWAS: genome-wide association study; miRNA: micro-RNA; NFBC 1966: Northern Finland Birth Cohort of 1966; PCOS: polycystic ovary syndrome; PI3-K: PI3-kinase.
Finally, as healthcare professionals, we need to manage our patients with PCOS and obesity with kindness and compassion. Although management of the reproductive, hyperandrogenic and metabolic sequelae is important, we also need to adopt a person-centred approach to management, including consideration of the personal and social implications of PCOS and obesity.
Conflicts of interest statement
The author has no potential conflict of interest.
Financial disclosure
None declared.
Data availability
No new data were generated or analysed in support of this review.
Acknowledgements
I acknowledge the many patients, relatives, nurses and physicians who contributed to the ascertainment of the various clinical samples reported on in this review.
References
- 1. Asuncion M, Calvo RM, San Millan JL, et al. A prospective study of the prevalence of the polycystic ovary syndrome in unselected Caucasian women from Spain. J Clin Endocrinol Metab 2000;85:2434–8. [DOI] [PubMed] [Google Scholar]
- 2. Barber TM, McCarthy MI, Wass JA, et al. Obesity and polycystic ovary syndrome. Clin Endocrinol (Oxf) 2006;65:137–45. [DOI] [PubMed] [Google Scholar]
- 3. Rotterdam EA-SPcwg . Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 2004;19:41–7. [DOI] [PubMed] [Google Scholar]
- 4. Barber TM, McCarthy MI, Franks S, et al. Metabolic syndrome in polycystic ovary syndrome. Endokrynol Pol 2007;58:34–41. [PubMed] [Google Scholar]
- 5. Barber TM, Franks S. Obesity and polycystic ovary syndrome. Clin Endocrinol (Oxf) 2021;95:531–41. [DOI] [PubMed] [Google Scholar]
- 6. Barber TM, Dimitriadis GK, Andreou A, et al. Polycystic ovary syndrome: insight into pathogenesis and a common association with insulin resistance. Clin Med (Lond) 2015;15:s72–6. [DOI] [PubMed] [Google Scholar]
- 7. Joharatnam J, Barber TM, Webber L, et al. Determinants of dyslipidaemia in probands with polycystic ovary syndrome and their sisters. Clin Endocrinol (Oxf) 2011;74:714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ollila MM, Piltonen T, Puukka K, et al. Weight gain and dyslipidemia in early adulthood associate with polycystic ovary syndrome: prospective cohort study. J Clin Endocrinol Metab 2016;101:739–47. [DOI] [PubMed] [Google Scholar]
- 9. Holte J, Bergh T, Berne C, et al. Restored insulin sensitivity but persistently increased early insulin secretion after weight loss in obese women with polycystic ovary syndrome. J Clin Endocrinol Metab 1995;80:2586–93. [DOI] [PubMed] [Google Scholar]
- 10. Barber TM, Franks S. Genetic basis of polycystic ovary syndrome. Expert Rev Endocrinol Metab 2010;5:549–61. [DOI] [PubMed] [Google Scholar]
- 11. Barber TM, Franks S. Genetics of polycystic ovary syndrome. Front Horm Res 2013;40:28–39. [DOI] [PubMed] [Google Scholar]
- 12. Vink JM, Sadrzadeh S, Lambalk CB, et al. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab 2006;91:2100–4. [DOI] [PubMed] [Google Scholar]
- 13. Barber TM, Bennett AJ, Groves CJ, et al. Association of variants in the fat mass and obesity associated (FTO) gene with polycystic ovary syndrome. Diabetologia 2008;51:1153–8. [DOI] [PubMed] [Google Scholar]
- 14. Attaoua R, Ait El Mkadem S, Radian S, et al. FTO gene associates to metabolic syndrome in women with polycystic ovary syndrome. Biochem Biophys Res Commun 2008;373:230–4. [DOI] [PubMed] [Google Scholar]
- 15. Ewens KG, Jones MR, Ankener W, et al. FTO and MC4R gene variants are associated with obesity in polycystic ovary syndrome. PLoS One 2011;6:e16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Welt CK, Styrkarsdottir U, Ehrmann DA, et al. Variants in DENND1A are associated with polycystic ovary syndrome in women of European ancestry. J Clin Endocrinol Metab 2012;97:E1342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mutharasan P, Galdones E, Penalver Bernabe B, et al. Evidence for chromosome 2p16.3 polycystic ovary syndrome susceptibility locus in affected women of European ancestry. J Clin Endocrinol Metab 2013;98:E185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goodarzi MO, Jones MR, Li X, et al. Replication of association of DENND1A and THADA variants with polycystic ovary syndrome in European cohorts. J Med Genet 2012;49:90–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayes MG, Urbanek M, Ehrmann DA, et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun 2015;6:7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen ZJ, Zhao H, He L, et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat Genet 2011;43:55–9. [DOI] [PubMed] [Google Scholar]
- 21. Shi Y, Zhao H, Cao Y, et al. Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat Genet 2012;44:1020–5. [DOI] [PubMed] [Google Scholar]
- 22. Brower MA, Hai Y, Jones MR, et al. Bidirectional Mendelian randomization to explore the causal relationships between body mass index and polycystic ovary syndrome. Hum Reprod 2019;34:127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Venkatesan AM, Dunaif A, Corbould A. Insulin resistance in polycystic ovary syndrome: progress and paradoxes. Recent Prog Horm Res 2001;56:295–308. [DOI] [PubMed] [Google Scholar]
- 24. Muntoni S, Muntoni S. Insulin resistance: pathophysiology and rationale for treatment. Ann Nutr Metab 2011;58:25–36. [DOI] [PubMed] [Google Scholar]
- 25. Reaven GM. The metabolic syndrome: requiescat in pace. Clin Chem 2005;51:931–8. [DOI] [PubMed] [Google Scholar]
- 26. Rice S, Christoforidis N, Gadd C, et al. Impaired insulin-dependent glucose metabolism in granulosa-lutein cells from anovulatory women with polycystic ovaries. Hum Reprod 2005;20:373–81. [DOI] [PubMed] [Google Scholar]
- 27. Barber TM, Franks S. Adipocyte biology in polycystic ovary syndrome. Mol Cell Endocrinol 2013;373:68–76. [DOI] [PubMed] [Google Scholar]
- 28. Franks SM, White D, Willis D. In: Filicori MFC (ed.). Mechanisms of Anovulation in Polycystic Ovary Syndrome. Amsterdam: Elsevier, 1996,183–6 [Google Scholar]
- 29. White D, Leigh A, Wilson C, et al. Gonadotrophin and gonadal steroid response to a single dose of a long-acting agonist of gonadotrophin-releasing hormone in ovulatory and anovulatory women with polycystic ovary syndrome. Clin Endocrinol (Oxf) 1995;42:475–81. [DOI] [PubMed] [Google Scholar]
- 30. Morin-Papunen LC, Vauhkonen I, Koivunen RM, et al. Insulin sensitivity, insulin secretion, and metabolic and hormonal parameters in healthy women and women with polycystic ovarian syndrome. Hum Reprod 2000;15:1266–74. [DOI] [PubMed] [Google Scholar]
- 31. Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev 1997;18:774–800. [DOI] [PubMed] [Google Scholar]
- 32. Day F, Karaderi T, Jones MR, et al. Large-scale genome-wide meta-analysis of polycystic ovary syndrome suggests shared genetic architecture for different diagnosis criteria. PLoS Genet 2018;14:e1007813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tian Y, Li J, Su S, et al. PCOS-GWAS susceptibility variants in THADA, INSR, TOX3, and DENND1A are associated with metabolic syndrome or insulin resistance in women with PCOS. Front Endocrinol (Lausanne) 2020;11:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ilie IR, Georgescu CE. Polycystic ovary syndrome-epigenetic mechanisms and aberrant MicroRNA. Adv Clin Chem 2015;71:25–45. [DOI] [PubMed] [Google Scholar]
- 35. Iorio MV, Piovan C, Croce CM. Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim Biophys Acta 2010;1799:694–701. [DOI] [PubMed] [Google Scholar]
- 36. Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res 2012;110:483–95. [DOI] [PubMed] [Google Scholar]
- 37. Makrinou E, Drong AW, Christopoulos G, et al. Genome-wide methylation profiling in granulosa lutein cells of women with polycystic ovary syndrome (PCOS). Mol Cell Endocrinol 2020;500:110611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gur EB, Karadeniz M, Turan GA. Fetal programming of polycystic ovary syndrome. World J Diabetes 2015;6:936–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abbott DH, Barnett DK, Bruns CM, et al. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update 2005;11:357–74. [DOI] [PubMed] [Google Scholar]
- 40. Dumesic DA, Schramm RD, Bird IM, et al. Reduced intrafollicular androstenedione and estradiol levels in early-treated prenatally androgenized female rhesus monkeys receiving follicle-stimulating hormone therapy for in vitro fertilization. Biol Reprod 2003;69:1213–9. [DOI] [PubMed] [Google Scholar]
- 41. Tata B, Mimouni NEH, Barbotin AL, et al. Elevated prenatal anti-Mullerian hormone reprograms the fetus and induces polycystic ovary syndrome in adulthood. Nat Med 2018;24:834–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abbott DH, Dumesic DA, Franks S. Developmental origin of polycystic ovary syndrome - a hypothesis. J Endocrinol 2002;174:1–5. [DOI] [PubMed] [Google Scholar]
- 43. Risal S, Pei Y, Lu H, et al. Prenatal androgen exposure and transgenerational susceptibility to polycystic ovary syndrome. Nat Med 2019;25:1894–904. [DOI] [PubMed] [Google Scholar]
- 44. Dumesic DA, Schramm RD, Abbott DH. Early origins of polycystic ovary syndrome. Reprod Fertil Dev 2005;17:349–60. [DOI] [PubMed] [Google Scholar]
- 45. Mongraw-Chaffin M, Hairston KG, Hanley AJG, et al. Association of Visceral Adipose Tissue and Insulin Resistance with incident metabolic syndrome independent of obesity status: the IRAS family study. Obesity (Silver Spring) 2021;29:1195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barber TM, Golding SJ, Alvey C, et al. Global adiposity rather than abnormal regional fat distribution characterizes women with polycystic ovary syndrome. J Clin Endocrinol Metab 2008;93:999–1004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analysed in support of this review.