

Abstract
Background
Pneumococcal carriage has often been studied from a serotype perspective; however, little is known about the strain-specific carriage and inter-strain interactions. Here, we examined the strain-level carriage and co-colonization dynamics of Streptococcus pneumoniae in a Swiss birth cohort by PacBio single-molecule real-time (SMRT) sequencing of the plyNCR marker.
Methods
A total of 872 nasal swab (NS) samples were included from 47 healthy infants during the first year of life. Pneumococcal carriage was determined based on the quantitative real-time polymerase chain reaction (qPCR) targeting the lytA gene. The plyNCR marker was amplified from 214 samples having lytA-based carriage for pneumococcal strain resolution. Amplicons were sequenced using SMRT technology, and sequences were analyzed with the DADA2 pipeline. In addition, pneumococcal serotypes were determined using conventional, multiplex PCR (cPCR).
Results
PCR-based plyNCR amplification demonstrated a 94.2% sensitivity and 100% specificity for Streptococcus pneumoniae if compared to lytA qPCR. The overall carriage prevalence was 63.8%, and pneumococcal co-colonization (≥ 2 plyNCR amplicon sequence variants (ASVs)) was detected in 38/213 (17.8%) sequenced samples with the relative proportion of the least abundant strain(s) ranging from 1.1 to 48.8% (median, 17.2%; IQR, 5.8–33.4%). The median age to first acquisition was 147 days, and having ≥ 2 siblings increased the risk of acquisition.
Conclusion
The plyNCR amplicon sequencing is species-specific and enables pneumococcal strain resolution. We therefore recommend its application for longitudinal strain-level carriage studies of Streptococcus pneumoniae.
Video Abstract
Supplementary Information
The online version contains supplementary material available at 10.1186/s40168-022-01344-6.
Keywords: Respiratory microbiome, PacBio single-molecule real-time (SMRT) sequencing, DADA2, Co-colonization, Streptococcus pneumoniae, Carriage duration, First year of life
Introduction
Carriage studies are essential for monitoring the sero- and molecular type changes of circulating pneumococcal strains, especially in the current era of pneumococcal conjugate vaccines (PCVs). However, reliance on conventional, culture-based methods with a known bias towards dominant strains [1] may limit the perception of the true and full dynamics (multiple carriage, new acquisition versus unmasking, etc.) of pneumococcal strains in the nasopharynx. Although the development of more sensitive measures like the microarray has improved serotype detection [2], strain-level insights are still necessary for pneumococcal carriage studies, as they can aid in identifying and tracking strains across samples (independent of the serotype) and easily distinguish new strain acquisitions from old strain re-acquisitions, identifying potential sources of transmission.
Previous studies have attempted to ascertain pneumococcal profiles from nasopharyngeal samples via shotgun metagenomics [3, 4]; however, resolution of “unique” pneumococcal strains remained limited, as shotgun metagenomics requires a very high read coverage to detect minor variants, which is even more challenging to achieve with samples that have a high quantity of host DNA. Alternative approaches are therefore needed for a thorough exploration of S. pneumoniae strains and their dynamics during carriage.
In recent years, amplicon-based high-throughput sequencing (HTS) via Illumina, PacBio, etc. has revolutionized a plethora of culture-independent gut-microbiome investigations, offering insights on strain-level diversity and resolving structures of complex, microbial communities [5, 6]. This increased resolution (beyond the 16S rRNA level) is assured by using a species-specific marker gene as a measure of inter-strain genomic variability. Strains are then subsequently identified based on the presence or absence of at least one single nucleotide variant (SNV) via computational methods such as the Divisive Amplicon Denoising Algorithm (DADA2) [7, 8], oligotyping [9, 10], and UNOISE [11]. Although proven successful in microbiome studies, this strategy is yet to be applied to longitudinal carriage studies seeking to explore finer-grained dynamics of S. pneumoniae in the nasopharynx.
The ~ 1300-bp pneumolysin non-coding region (plyNCR) has been previously reported as a highly specific and conserved marker for S. pneumoniae [12–14]. The resolution of the plyNCR region for strain typing was found to be comparable with multi-locus sequence typing (MLST) [12]. In addition, the plyNCR region has also been investigated and validated for the detection of co-colonization using terminal restriction length polymorphism (T-RFLP) [13]. However, the usefulness of the plyNCR has so far only been tested by restriction enzymes, which may limit the strain resolution, as polymorphisms of specific enzyme cuts are usually low [13, 15].
In this study, we aimed to assess the application of PacBio single-molecule real-time (SMRT) sequencing of the plyNCR marker for pneumococcal strain-level resolution. Using nasal swab (NS) samples of infants in a longitudinal cohort, we reveal an accurate and in-depth visualization of strain-resolved pneumococcal profiles and associated serotypes during carriage over a 12-month period, including estimations for acquisition and carriage duration since the introduction of the 13-valent pneumococcal conjugate vaccine (PCV13) in Switzerland.
Materials and methods
In silico screening of the plyNCR marker in pneumococcal and streptococcal genomes
Using a perl script (https://github.com/egonozer/in_silico_pcr), an in-silico search for the plyNCR marker was performed on 8325 reference sequences (RefSeq) of S. pneumoniae genomes available on the public NCBI database (https://www.ncbi.nlm.nih.gov/genome/?term=Streptococcus+pneumoniae, accessed on March 25, 2021). For a specificity assessment, the plyNCR marker was also screened against the Genbank assembly database (https://www.ncbi.nlm.nih.gov/assembly) of the viridans group streptococci (VGS): S. mitis (138), S. pseudopneumoniae (87), S. infantis (12) and S. oralis (122). Extracted sequences with indel regions were verified by mapping published NCBI assemblies to raw reads available on the sequence read archive (SRA) using minimap2 v2.17 and visualizing the mapped regions corresponding to the plyNCR on the Integrative Genomics Viewer (IGV). Sequences with unverifiable indel regions due to an absence of data on the sequence read archive (SRA) or low read coverage mapping (< 10 reads per site) were excluded. Final sequences were aligned using muscle v3.8.1551 [16], and a maximum likelihood (ML) phylogenetic tree was constructed under the generalized time-reversible (GTR) + gamma model using fasttree v2.1.10 [17]. Tree annotation and visualization were performed using the interactive tree of life (iTOL) [18].
Study participants, swab collection, and pneumococci screening
A total of 48 infants, who participated in our previous study [19] and were enrolled in the Basel Bern Infant Lung Development (BILD) Cohort (www.bild-cohort.ch) were followed during the first year of life [20]. Starting with the fifth week after birth [initiation time point; (T0)], nasal swabs (Verridial E. Mueller, Blonay, Switzerland) were collected biweekly from the study infants between February 2010 and April 2014 as previously described [19]. Ethical approval was obtained from the Ethics Committee of the Canton of Bern.
DNA was extracted directly from swabs (QIAamp DNA Minikit, Qiagen, Hilden, Germany) in 200 μl of transport medium and screened for pneumococci via quantitative real-time PCR (qPCR) targeting the lytA [21, 22]. A negative control was included for each qPCR run. The lower limit of detection (LLD) was defined as 10 lytA copies, and samples having < 10 lytA copies in 2/3 triplicate measurements were presumptively ruled as being negative for S. pneumoniae.
Mock control design
Four non-encapsulated S. pneumoniae strains of known plyNCR genomic signatures were chosen from our previous study [23] (Additional file 1: Table S1). Genomic DNA of strains were diluted to equal masses (1 ng) and mixed in varying proportions to ascertain the resolution limit and sensitivity of the plyNCR as an amplicon marker during SMRT sequencing (Fig. 1). Mock communities (MC) were prepared as follows: (group 1) MC1–5—MC1 and 2; single strains and MC3, 4, and 5; mixtures of MC1 and 2 in ratios 1:9, 1:49, and 1:89, respectively; (group 2) MC6–10—MC6 and 7; single strains and MC8, 9, and 10; mixtures of MC6 and MC7 in ratios 1:99, 1:119, and 1:139, respectively.
Fig. 1.

Experimental design of mock controls for plyNCR sensitivity. Ten mock communities were created using in-house pneumococcal strains of known composition and genomic signatures. The samples were prepared for plyNCR amplicon sequencing and sequenced in quadruplicates as controls together with nasal swab amplicons on the PacBio Sequel
PCR for plyNCR marker amplification
PCR amplification of the plyNCR marker was performed using previously described primers [12], modified with tailed PacBio Universal sequences for downstream amplicon sequencing (Table 1). The plyNCR PCR reaction and cycling conditions were followed for in vitro samples (mock communities) and NS swabs as previously described [13], with the only modification being that 10 μl of swab DNA was used per reaction and 40 amplification cycles were used for NS samples with low yields from the first amplification.
Table 1.
Primers used in this study
| Gene | Primer | Sequence (5′-3′) | Reference |
|---|---|---|---|
| lytA | Forward | ACGCAATCTAGCAGATGAAGCA | [22] |
| Reverse | TCGTGCGTTTTAATTCCAGCT | ||
| Probe | FAM-GCCGAAAACGCTTGATACAGGGAG-BHQ1 | ||
| plyNCRa | Forward | [GCAGTCGAACATGTAGCTGACTCAGGTCAC]AAAGGCTGCACGGACATTG | [12] |
| Reverse | [TGGATCACTTGTGCAAGCATCACATCGTAG]CCGATTTGCCACTAGTGCGTAAGC | ||
| cpsA | Forward | GCAGTACAGCAGTTTGTTGGACTGACC | [24] |
| Reverse | GAATATTTTCATTATCAGTCCCAGTC |
aPacbio Universal Sequence overhang in []. Primers were HPLC purified and blocked at the 5′ site with an amino group
PCR products were visualized on a 1% agarose gel and samples having the expected plyNCR amplicon product size of ~ 1.3 kb were purified using the QIAcube (Qiagen, Germany) or AMPure PB Beads (Beckman Coulter Inc., USA) and quantified on the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA).
PacBio SMRT amplicon sequencing of the plyNCR marker
Equimolar pooling of amplified DNA was performed using the Barcoded Universal F/R Primers Plate 96 v2 (Pacific Biosciences, USA) to allow multiplexing in pools of 25–26 amplicons per SMRTcell (Additional file 1: Table S2). In total, 11 SMRTcells were used. Four technical replicates of each mock control group (MC1–5 and MC6–10) were included in four respective SMRTcells per group (Fig. 1; Additional file 1: Table S2).
SMRTbell libraries from the pooled amplicons were prepared using SMRTbell Express Template Prep Kit 2.0 according to the manufacturer’s instructions (Pacific Biosciences, USA). Sequencing on the Sequel I platform was performed using the Sequel Binding kit 3.0, Sequel Sequencing Kit 3.0, and Sequencing Chemistry S/P3-C3/5.0. Each pool of purified SMRTbell libraries was sequenced on single 10-h movie SMRT cells (SMRT cell 1M v3) with a pre-extension time of 1.3 h.
Bioinformatics and sequence analysis
Subreads from each SMRTcell were first demultiplexed with Lima v1.10.0 (https://github.com/PacificBiosciences/barcoding) and then aligned to an extracted plyNCR sequence region from the reference strain 106.66 (NCBI BioProject Accession: PRJNA554545) using Pbalign v0.4.1 (https://github.com/PacificBiosciences/pbalign/) with the parameters hitPolicy=allbest and minAnchorSize=19. Circular consensus sequence (CCS) reads were generated from the aligned subreads with minPredictedAccuracy = 0.999 using the CCS v4.0.0 (https://github.com/PacificBiosciences/ccs). All bioinformatic analyses were conducted on UBELIX (http://www.id.unibe.ch/hpc), the high-performance computing (HPC) cluster at the University of Bern. All raw PacBio sequencing reads were deposited in the European Nucleotide Archive (ENA) under study accession number PRJEB45241.
The R package, DADA2, was used to denoise the CCS reads and identify the relative abundance of each amplicon sequence variant (ASV) following the online DADA2 + PacBio protocol by Callahan (https://benjjneb.github.io/LRASManuscript/LRASms_Zymo.html). Data was analyzed per SMRTcell. ASVs were further processed by length exclusion (≤ 1150 bp) and false-positive removal (Additional file 1: Fig. S1). For the finalized ASV output, ASVs sharing 100% identity were defined as “unique.”
Molecular serotype/serogroup identification
Serotypes/serogroups were determined via conventional PCR (cPCR) either in singleplex or multiplex for all samples having ≥ 10 lytA copies, based on established protocols by the Streptococcus Laboratory of the Centers for Disease Control and Prevention (CDC; https://www.cdc.gov/streplab/pneumococcus/resources.html). Samples with ≥ 10 lytA copies but no serotype and no cpsA amplification bands were considered non-typeable pneumococci (NT). We found no samples with missing serotype but presenting cpsA bands. In samples, where co-colonization was present (≥ 2 plyNCR ASVs) but only one serotype was identified by cPCR, the identified serotype was assumed to belong to the most abundant ASV and the presumed unidentified serotype(s) were classified as “unknown.” The term “unknown” was used due to the possibility of the presumed unidentified serotype(s) being (i) the same serotype as the most abundant ASV, (ii) a NT, or (iii) a serotype not included in the 40-serotype multiplex primer panel.
Data analyses
Pneumococcal carriage prevalence was defined as the proportion of infants with ≥ 10 lytA copies in a swab sample. Time to acquisition, carriage duration, and clearance were calculated based on previous definitions [25, 26]. In brief, the time of acquisition was estimated to be the midpoint from the last of two negative swabs to the first positive swab while clearance was inferred as the midpoint between the last positive swab and the first of two negative swabs. Time of acquisition for infants with detectable pneumococci at T0 (first swab taken) was inferred to be the midpoint from birth to T0. The period between acquisition and clearance was considered the duration of carriage, and this was calculated for both pneumococcal strains (unique plyNCR ASV) and serotypes.
Statistical analyses were conducted in the R studio environment v3.6.1 (https://www.r-project.org/). Time to first acquisition and carriage duration were analyzed using the Kaplan-Meier survival curve. The Cox regression model was used to assess the association between time to first acquisition and epidemiologic/socio-economic factors such as respiratory symptoms, smoking exposure, season of birth, parental education, having two or more siblings, and pneumococcal vaccination. Carriage duration was right censored at the last swab time point. New acquisitions at the final swab time point were excluded from the duration analyses. Co-colonization dynamics were categorized and adapted from previous definitions [27]: (i) strain displacement (a primary colonizer is outcompeted by the secondary strain and subsequently, cleared), (ii) strain dominance (carriage of a primary colonizer is maintained and newly acquired secondary strains are transient), (iii) stable co-colonization (two strains are carried across ≥ 2 time points), and (iv) short-term co-colonization (multiple strains are observed at a single time point). The chi-squared test was applied for categorical testing and P < 0.05 was considered significant. The results were reported with 95% confidence intervals (CI).
Results
In silico evaluation of the plyNCR marker reveals pneumococcal specificity and plyNCR homolog presence among viridans group streptococci (VGS)
In order to investigate the presence and exclusivity of the plyNCR marker to pneumococcal genomes, an in silico screening of the marker was performed on all S. pneumoniae RefSeqs as well as Genbank assemblies of the closely related VGS species. Of the 8325 pneumococcal genome RefSeqs on the NCBI database (accessed, March 25, 2021), the plyNCR marker was present in all but one genome (0.01%; accession: GCF_001113845.1), which only gave a partial hit (~ 500 bp), due to the presence of multiple large deletions, which is likely indicative of inadequate sequencing coverage prior to assembly. Among non-pneumococcal streptococci, no hits for the marker were found against S. oralis and S. infantis genomes. However, homologous of the marker, which were identical in size (~ 1100 bp), were present in 6/138 (4.3%) S. mitis and 12/87 (13.8%) S. pseudopneumoniae genomes. All homolog sequences clustered separately due to similar deletions and were within the same clade as five pneumococcal plyNCR sequences, which also exhibited deletions (Fig. 2; Additional file 1: Table S3). The deletions in these five pneumococcal plyNCR sequences were not identical to the homologs but were confirmed by mapping the raw reads to the assemblies. Therefore, the in silico specificity for the “right size” plyNCR is 100% for S. pneumoniae.
Fig. 2.
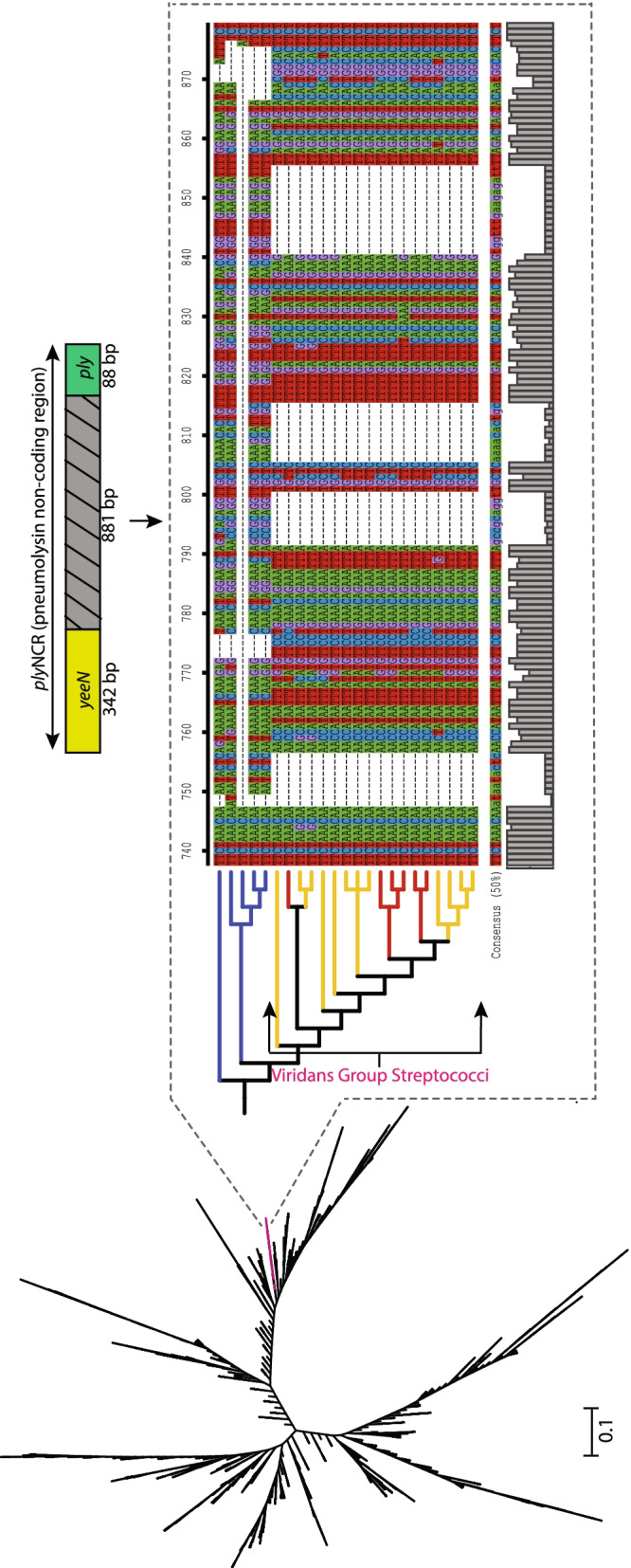
Maximum likelihood phylogeny of the plyNCR marker among strains of the Streptococcus mitis group. The ML tree was inferred using the generalized time reversible (GTR) + gamma model with FastTree and is unrooted with unscaled branches. A zoomed-in subtree (dashed rectangle) shows a cladistic clustering and a portion of the sequence alignment from five pneumococci (in blue) and 18 viridans group streptococci (VGS; S. mitis and S. pseudopneumoniae in red and yellow, respectively) with the latter having characteristic deletions. Above the subtree, the plyNCR is illustrated as a non-coding region flanked by partial sections of the conserved genes, yeeN (hypothetical regulatory protein) and ply (pneumolysin)
Comparison of plyNCR PCR to the lytA qPCR for pneumococcal detection in NS samples
Given a demonstrated in silico specificity of the plyNCR among S. pneumoniae genomes, we next compared the PCR detection of the plyNCR to the qPCR-based assay targeting the highly conserved lytA gene using NS samples from 47 BILD cohort infants (samples from one infant were excluded due to low quality as shown below). Between February 2010 and April 2014, a total of 1068 NS were collected. After exclusion of low-quality samples and those taken during antibiotic therapy or respiratory infection (n = 196), a total of 872 high-quality NS samples were included from 47 infants with a median of 19 samples per infant (range, 11–25). The full characteristics of the 47 BILD cohort infants are presented in Additional file 1: Table S4. Overall, 631/871 (72.4%) high-quality NS samples had < 10 lytA copies and were considered negative for pneumococci. This was corroborated by negative plyNCR (617/631 samples) or “wrong size” (~ 1100 bp; Additional file 1: Fig. S2) (14/631 samples) amplification in all samples, indicating a 100% specificity of the marker PCR. Of the remaining 240 (27.6%) samples with ≥ 10 lytA copies, only 14 (5.8%) failed to amplify the plyNCR marker by PCR, resulting in a sensitivity of 94.2% (Additional file 1: Table S5).
PacBio amplicon sequencing for strain (plyNCR) resolution
After exclusion of low-yield amplicons (i.e., those that produced faint bands on the gel) and low-quality amplicons (i.e., did not pass the PacBio quality control), a total of 214 (88.4%) plyNCR amplicon products from 242 NS samples were sequenced (Fig. 3). This included PCR products from two of fourteen NS samples suspected of being VGS, due to having < 10 lytA copies and amplifying a short fragment with the wrong size (~ 1100 bp) during plyNCR PCR (Fig. 3). These two products were included to confirm the genomic signature differences between true plyNCR sequences and homolog sequences. Sequencing failed for only one of the 214 (0.5%) amplicons, despite using three replicates. Additional file 1: Table S6 summarizes the sequencing statistics and DADA2 denoising process for each SMRTcell.
Fig. 3.

Flowchart of nasal swab (NS) sample processing for PacBio SMRT sequencing. Molecular processing of NS samples for plyNCR sequencing (strain identification) highlighted in peach. 1PCR performed using primers with PacBio universal sequence overhang. *QC: quality control excludes amplicons with no observable peak or multiple peaks on the fragment analyzer. VGS, viridans group streptococci
Sensitivity and resolution limit of PacBio sequenced plyNCR in mock control mixtures
To assess the sensitivity and resolution limit of the plyNCR for strain discrimination in a 25-sample-multiplex library, 10 mock community mixtures were sequenced in quadruplicates with NS amplicons, and strains from each community were resolved as amplicon sequence variants (ASVs) using DADA2. Figure 4 illustrates the results of the DADA2 error model. All strains were correctly identified down to the 2% variant level (1:49 mixture). The least abundant strain at the 1.1% variant level (1:89 mixture) was resolved in only one of four replicates, though with two additional, incorrect SNPs.
Fig. 4.

Sensitivity of the plyNCR for pneumococcal detection in sequenced mock samples. The plyNCR was amplified from 10 pneumococcal mock communities (MC) and sequenced in technical quadruplicates by SMRT sequencing. The top and bottom panels show the abundance of all strains recovered as amplicon sequence variants (ASVs) by DADA2 in MC1-5 (group 1) and MC6-10 (group 2), respectively. MC1, 2, 6, and 7 represent single strain communities. MC3, 4, and 5 contain mixtures of MC1 and 2 in ratios 1:9, 1:49, and 1:89, respectively, while MC8, 9, and 10 contain mixtures of MC6 and MC7 in ratios 1:99, 1:119, and 1:139, respectively. “Two SNPs off” indicates that the least abundant strain in MC5 (1:89) was recovered with all 17 expected SNPs and two additional, erroneous SNPs
The plyNCR marker enables pneumococcal strain-level identification in NS samples
Given a demonstrated discriminatory power of the plyNCR in mock samples, sequenced plyNCR amplicons from NS samples were analyzed. In total, DADA2 identified 250 plyNCR ASVs from 209/213 (98%) sequenced samples. Thirty of these 250 (12%) ASVs were unique and represented distinct strain profiles in each infant during the 12-month period (Fig. 5). Co-colonization was detected in 38/213 (17.8%) sequenced samples with the relative proportion of the least abundant strain(s) ranging from 1.1 to 48.8% (median, 17.2%; IQR, 5.8–33.4%). No ASV output was observed in four samples; two of which (having < 10 lytA copies) had amplified a plyNCR homolog. On relaxation of the DADA2 read length stringency settings, all four samples outputted homolog sequences with an average read length of 1055 bp (range, 953–1072 bp), sharing high BLAST identities (range, 92.8–97%) to VGS species. This confirmed the two suspicious samples that amplified a plyNCR homolog to be indeed VGS species.
Fig. 5.

Non-culture-based Amplicon sequencing of the plyNCR marker for pneumococcal strain resolution. Nasal swabs (NS) are illustrated as crosses (“x”; lytA < 10 copies) or circles (lytA ≥ 10 copies) for pneumococcal absence and presence during the first year of life, respectively. Infants with only negative pneumococcal swabs were omitted. Each unique amplicon sequence variant (ASV) recovered by DADA2 after plyNCR sequencing represents a pneumococcal strain and is denoted as a single count (1–30) within the dark brown circles. The major (most abundant) plyNCR ASV is shown first, followed by minor (lower abundant) plyNCR ASVs. Swabs where pneumococci were expected (lytA ≥ 10 copies), but having no plyNCR ASV (due to failed/sub-optimal amplification, low QC quality, failed sequencing, or no DADA2 output) are illustrated as empty tan circles. Serotypes/serogroups identified via conventional multiplex PCR (cPCR) are shown at the top of each circle. Question mark (?) indicates an expected, but indeterminable serotype or non-typeable (NT) S. pneumoniae in a co-colonizing sample, due to the limitations of the multiplex panel
Serotype identification of plyNCR ASVs using conventional PCR (cPCR)
Of the 240 NS samples having ≥ 10 lytA copies, a total of 24 serotypes/serogroups were identified via multiplex/singleplex cPCR. Overall, 59.3% (166/280) of serotypes were non-PCV13, 29.6% (83/280) were PCV13, 6.8% (19/280) were unknown, and 4.3% (12/280) were NT (Fig. 5; Additional file 1: Table S7). A distinct serotype or NT was linked to each unique plyNCR ASV strain profile in an infant, except for infants #15 and #29, where two distinct serotypes/serogroups were identified at different time points for their respective plyNCR ASV (#16 and #8) strain profiles, indicating new strain acquisitions (Fig. 5). New strain acquisitions were also inferred for infant #3, where the same serotype/serogroup was identified for two unique plyNCR ASVs (#28 and #20) (Fig. 5). Multiple serotype identification by cPCR was limited for 44.7% (17/38) of samples with identified co-colonization, preventing serotype attribution for 12 plyNCR ASVs in nine infants (Fig. 5; Additional file 1: Table S7). In addition, it is not possible to link the amplicon sequencing variants and serotypes in samples containing co-colonizing serotypes unless the major types are clearly more abundant as compared to the minor types.
Pneumococcal carriage prevalence, acquisition, and dynamics
Overall, 30/47 (63.8%) infants carried pneumococci (lytA-based carriage) at least once during the 12-month study period. Carriage increased from 10.3% (4/39; 95% CI 3.3–25.2%) during the first month of enrollment (before 2 months of age), reaching a peak of 50% (22/44; 35.8–64.2%) during the 8-month of life (Fig. 6A).
Fig. 6.

Pneumococcal carriage prevalence, acquisition, and dynamics. A Carriage prevalence by age. B Age at first acquisition for infants (dotted line shows median). C Serotype distribution
The median age at first acquisition was 147 days (IQR 63–303 days) (Fig. 6B). Of the assessed cohort characteristics, having two or more siblings was found to be significantly associated with an increased rate of first acquisition (p = 0.02; Additional file 1: Table S8). Among those with carriage episodes, the number of observed pneumococcal acquisitions ranged from one to six per child, with a total of 86 pneumococcal acquisitions for all 47 infants.
The median duration of carriage for the first acquired pneumococcal episode was 98 days (IQR 28–140 days), compared to 41 days (25–75 days) for successive acquisitions (p = 0.054, log-rank test). Among serotypes, the median duration of carriage was longer for non-PCV13 serotypes at 71 days (IQR 41–101 days), compared to 63 days (31–148 days) for PCV13 serotypes (Additional file 1: Table S9). Serotype 15B/C was the most prevalent serotype among all infants (Fig. 6C). Co-colonization was observed at least once among 50% of infants carrying pneumococci. Majority (52.6%; 20/38) of strains exhibited a stable co-colonization dynamic, followed by strain dominance (28.9%), short-term co-colonization (13.2%), and displacement (5.3%) (Additional file 1: Fig. S3).
Discussion
For the first time, we report an amplicon-based high-throughput sequencing (HTS) approach that incorporates the DADA2 algorithm, enabling pneumococcal strain resolution and longitudinal characterization of carriage dynamics based on SNPs in the plyNCR marker. Our method is less labor-intensive and more accurate than conventional methods, as it allows for direct detection of major and minor (low abundance) pneumococcal strain variants and, therefore, circumvents the need of picking and analyzing individual pneumococcal colonies to fully grasp the extent of diversity. Also, our method does not require prior culture enrichment which may disrupt the original bacterial composition as certain variants may outcompete others.
We first performed qPCR of lytA which is a gold standard method for the detection of S. pneumoniae. While it is known that the specificity is not 100%, it is very well accepted to use lytA for the detection of S. pneumoniae [22]. Our results further demonstrate the plyNCR’s potential as a top-performing pneumococcal marker with an in silico specificity among only S. pneumoniae strains as well as a high-molecular (PCR) performance (100% PPV and 100% specificity) matching that of the gold standard (lytA qPCR). The modest rates achieved in sensitivity (94.2%) and NPV (97.8%) appear to stem from the use of a conserved amount of template DNA (10 μl), which in light of the assay’s previously described detection limit (1 pg) at a higher DNA amount (31.4 μl) [13], may explain the reduced sensitivity and NPV. Nevertheless, compared to previous marker discoveries like the SPN9802 [28, 29] and SP2020 [30], the plyNCR shows more promise, given an absence of misidentified strains (false positives) during the molecular assessment, as well as its ability to easily discriminate pneumococci from closely related VGS, which may harbor plyNCR homologs at a size difference of ~ 200 bp. To our knowledge, this is the first carriage study that provides adequate resolution of “unique” nasopharyngeal pneumococcal strains in a longitudinal infant sampling without a prior culture step. Only two other studies have attempted to characterize S. pneumoniae at the strain level using infant longitudinal samples [3, 4]. However, both studies employed a metagenomic approach, which requires (i) a general enrichment process for Streptococcus sp., (ii) a high coverage that is mostly outbalanced by host DNA, (iii) a short-read sequencing library requiring assembly, and (iv) a more computationally intensive (and mostly database-dependent) approach [31], all of which may be cost-limiting for large-scale studies. On the other hand, our amplicon sequencing strategy utilizes a variable, species-specific marker that can be (i) amplified directly from clinical samples, (ii) sequenced as long reads on a 3rd generation platform (PacBio) with an accuracy of ≥ 99.8%, and (iii) easily resolved as true genetic strain variants down to a single nucleotide difference via the DADA2 pipeline. However, and as shown very recently, deep shotgun sequencing has of course unsurpassed sensitivity for detecting multiple colonization presupposed prior culturing maintains the original “in vivo” co-colonization ratio [32].
The parallel use of cPCR enabled serotype identification directly from NS samples and allowed serotypes to be primarily linked to most strain (plyNCR ASV) profiles. However, co-colonizing serotypes were unidentified in 45% of NS samples, demonstrating a significant limitation of this method for multiple serotype detection, as previously shown [2, 33]. In addition, it is not possible to unambiguously link the amplicon sequencing variants and serotypes in samples containing multiple samples. The microarray, which is currently considered the best method for detecting multiple serotypes [2], notably overcomes these limitations, as it is able to detect all known serotypes, including NTs and other bacterial species as well as their relative abundances, though at a higher cost and with a culture-amplification step, which is not required for cPCR. The same is true for the abovementioned very recent study using shotgun sequencing [32].
By itself, the plyNCR does not offer any serotype information for resolved strains in carriage, and likewise, serotyping does not provide any genomic background details for pneumococci in carriage. The advantage of the plyNCR for carriage studies is therefore complemented by serotyping, as it confirms the identity of a particular strain (plyNCR ASV) and allows instances of new acquisitions (with an identical serotype, but a different strain) and re-acquisitions to be easily identified. Thus, plyNCR combined with serotyping offers a more detailed insight into the carriage dynamics including duration.
Based on our findings, the median age to first acquisition (147 days) was much higher in our study compared to reports from higher carriage countries such as South Africa, Malawi, Indonesia, and the Gambia [25, 27, 34, 35]. Given higher rates of PCV uptake in Switzerland [36], the longer time to acquisition in our birth cohort may therefore reflect reduced chances for pneumococcal transmission. Also, the very first swab was taken once the infants were 6 weeks of age in our study. We may have missed earlier pneumococcal carriage, though pneumococcal colonization might be quite rare in the first days/weeks of life.
The presence of two or more siblings in the home was the only risk factor associated with the first pneumococcal acquisition, supporting previous reports of older siblings being the transmission sources for infants [37, 38]. We additionally observed a trend of longer carriage durations for the first acquired pneumococcal episode compared to subsequent episodes, which is similar to a recent study in Indonesia [27]. Longer duration of carriage has been shown to be positively correlated with more prevalent serotypes and negatively associated with age [39], which may partially explain our findings, as 66% of first acquisitions were the more prevalent non-PCV13 serotypes.
Contrary to a previous study, which demonstrated stabilized co-colonization rates during the pre-vaccine (2004–2005) and PCV7 era (2006–2009) in Switzerland [14], our current findings indicate that the rate of co-colonization is ~ 2× higher since PCV13 implementation (2010–2014) in the country. Interestingly, our estimated rate of 17.8% is not far off when compared to studies conducted during similar periods in the Netherlands (22%) [40], Portugal (25%) [41], and Nepal (20.2%) [42]. Although it remains unclear whether overall co-colonization rates are affected by PCV selection pressure or simply unmasking following the use of more sensitive detection measures, the lower rates observed in the previous Swiss study may imply an underestimation bias resulting from sampling acute otitis media (AOM) and pneumonia patients, who represent populations with compromised nasopharyngeal flora [43], compared to healthy (asymptomatic) young children, who are considered the major reservoir and drivers of pneumococcal transmission [38, 44]. At the same time, methodological differences may have also contributed to differing estimations.
The most common pneumococcal co-colonization dynamic observed in this birth cohort was a stable co-colonization, with varied relative strain abundances, indicating continued inter-strain competition. This is contrary to a recent study in Indonesian infants [27], where displacement of the primary colonizer by the secondary serotype was more common. As there is no national PCV program in Indonesia, a disparity in the serotype distributions of both countries may explain the observed differences in co-colonization dynamics. This is mostly evidenced by reported high carriage episodes of serotype 6B in Indonesia [27], which has all but disappeared from Swiss carriage isolates [45]. As the Indonesian study did not include any information on the serotypes involved in co-colonization, no further comparisons can be made between both studies. However, the identification of serotype 15B/C as a frequent initial/primary co-colonizer [41, 42] is consistent with the observations in this study.
Our study has a number of limitations. First, the NS samples used here were collected by the parents up to a decade ago, and as a result, we cannot rule out the possibility of a substandard collection as well as potential sample degradation from previous frequent use of the samples in earlier studies. We have chosen “deep nasal” over nasopharyngeal swabs due to logistical reasons as we told the parents rather than study nurses to weekly swab their children during the first year of life. However, we received a high pneumococcal carriage rate (~ 64%). In comparison, the pneumococcal carriage rate found by PCR was 51.6% in Swiss children in 2009, and we are therefore confident that our data does not underestimate the carriage rate [13]. Second, it is possible that the unavoidable addition of the PacBio Universal sequencing overhangs to the plyNCR primers may have reduced the amplification sensitivity, especially in the presence of low template DNA. This is mostly supported by successful PCR amplifications (without overhangs) in 50% of the 14 NS samples that failed to amplify with the use of overhang primers (results not shown). This trade-off in specificity due to the overhangs suggests that additional PCR optimization may be required for studies seeking to apply PacBio SMRT amplicon sequencing to their research. Third, there was no plyNCR ASV output for two samples, despite successful plyNCR amplification, suggesting a preferential sequencing bias for homologs, which were observed when the DADA2 length filter was lifted (results not shown). We are yet to estimate the frequency of this event in future studies. Fourth, sequencing had to be repeated for one amplicon, but no technical reason was found for its failure during troubleshooting. Therefore, the cost of PacBio amplicon sequencing could be exacerbated by random repetitions as well as the required sequencing depth for variant detection during multiplexing. However, this limitation is applicable to all HTS approaches. Lastly, a major limitation of this study was the absence of a culture-based serotyping method like the Quellung or microarray for cPCR verification. This was not feasible as the nasal swabs used in this study were not intended for the investigation of viable bacterial cultures. Thus, our use of the cPCR was therefore unavoidable, though we argue that despite its limitations, cPCR still provided reliable serotyping results due to our study’s close sampling time frame (biweekly swabs), where previous and/or ensuing time points with identical serotypes and plyNCR ASVs provided additional support and justification for serotype attribution, even in 84% of co-colonizing samples.
Despite these limitations, our amplicon-based sequencing approach was successful in resolving strain profiles of S. pneumoniae in the nasopharynx and distinguishing them from those of the viridans group streptococci. In addition, our infant cohort represents an intensively sampled study population, preventing underestimation bias in carriage estimates and allowing us to closely track changing carriage and co-colonization dynamics over a 12-month period.
Conclusions
We therefore conclude that the use of the plyNCR as an amplicon marker can further our understanding of carriage and co-colonization dynamics on a strain level. In so doing, we demonstrated pneumococcal strain acquisitions as unique plyNCR sequences with associated serotypes in an infant birth cohort and presented accurate estimations for carriage acquisition and durations on a strain level. As PCV13 implementation had and still has an impact on pneumococcal carriage (and therefore invasive diseases), consistent monitoring and increased follow-up remain a priority for understanding the carriage strain dynamics in a future era of next-generation vaccines.
Supplementary Information
Additional file 1: Table S1. Details of the in-house pneumococcal strains used for mock community preparation. Table S2. Pacbio SMRTcell Sample pooling. Table S3. Assembly details of S. mitis, S. pseudopneumoniae and S. pneumoniae strains showing close plyNCR relatedness during in-silico analysis. Table S4. Characteristics of the BILD infant cohort (n=47). Table S5. Comparison of discordant qPCR and PCR amplification results of nasal swab samples (n=25) from BILD cohort infants. Table S6. Mean read output statistics from PacBio SMRT sequencing and DADA2 pipeline. Table S7. Conventional serotyping of nasal swab (NS) samples (n=240) from BILD cohort infants. Table S8. Multivariable Cox regression analysis of potential risk factors associated with time to first pneumococcal acquisition. A Hazard Ratio (HR) < 1 indicates a reduced hazard (rate) of pneumococcal acquisition while an HR > 1 indicates an increased rate of pneumococcal acquisition. Table S9. Duration of pneumococcal carriage by acquisition and serotype. Fig. S1. False positive removal during DADA2 ASV calling. Fig. S2. Detection of S. pneumoniae by plyNCR PCR. Fig. S3. Patterns of co-colonization dynamics in four infants.
Acknowledgements
We would like to thank Dr. Insa Korten and the study nurses of the BILD cohort for their involvement in the sample and data collection process as well as the participants. We also appreciate the efforts of the Next Generation Sequencing Platform of the University of Bern, especially Catia Gomes for performing the high-throughput PacBio sequencing. We would also like to thank Dr. Simone Oberhänsli for her help in the data transfer and UBELIX (http://www.id.unibe.ch/hpc), the HPC cluster at the University of Bern for providing a platform for the bioinformatic analyses conducted in this study.
Authors’ contributions
O.R-A.O, S.D.B, and M.H. designed the study. P.L. has been in charge of the clinical data and metadata. O.R-A.O prepared the samples for amplicon sequencing and performed the validation experiments. O.R-A.O performed the bioinformatic analyses. M. H. supervised the study. O.R-A.O wrote the manuscript with input from the co-authors. All authors read and approved the final manuscript.
Funding
The samples used in this study are part of the Basel Bern Infant Lung Development (BILD) cohort study, which is funded by the Swiss National Science Foundation (grants 1209932473B_124654 and 324730_144280). A part of this work has also been funded by the Swiss National Science Foundation (grant No. 310030_197083) to M. H.
Availability of data and materials
All raw PacBio sequencing reads were deposited in the European Nucleotide Archive (ENA) under study accession number PRJEB45241.
Declarations
Ethics approval and consent to participate
Ethical approval was obtained from the Ethics Committee of the Canton of Bern.
Consent for publication
Not applicable.
Competing interests
MH has received an investigator-initiated (research) grant from Pfizer for partial support for the submitted work. However, Pfizer had no role in the data analysis and content of the manuscript of this study.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Huebner RE, Dagan R, Porath N, Wasas AD, Klugman KP. Lack of utility of serotyping multiple colonies for detection of simultaneous nasopharyngeal carriage of different pneumococcal serotypes. Pediatr Infect Dis J. 2000;19(10):1017–1020. doi: 10.1097/00006454-200010000-00019. [DOI] [PubMed] [Google Scholar]
- 2.Satzke C, Dunne EM, Porter BD, Klugman KP, Mulholland EK, PneuCarriage project g The PneuCarriage Project: a multi-centre comparative study to identify the best serotyping methods for examining pneumococcal carriage in vaccine evaluation studies. PLoS Med. 2015;12(11):e1001903. doi: 10.1371/journal.pmed.1001903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manenzhe RI, Dube FS, Wright M, Lennard K, Mounaud S, Lo SW, et al. Characterization of pneumococcal colonization dynamics and antimicrobial resistance using shotgun metagenomic sequencing in intensively sampled South African infants. Front Public Health. 2020;8:543898. doi: 10.3389/fpubh.2020.543898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wright MS, McCorrison J, Gomez AM, Beck E, Harkins D, Shankar J, et al. Strain level Streptococcus colonization patterns during the first year of life. Front Microbiol. 2017;8:1661. doi: 10.3389/fmicb.2017.01661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobay LM, Wissel EF, Raymann K. Strain structure and dynamics revealed by targeted deep sequencing of the honey bee gut microbiome. mSphere. 2020;5(4). 10.1128/mSphere.00694-20. [DOI] [PMC free article] [PubMed]
- 6.Yan Y, Nguyen LH, Franzosa EA, Huttenhower C. Strain-level epidemiology of microbial communities and the human microbiome. Genome Med. 2020;12(1):71. doi: 10.1186/s13073-020-00765-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Callahan BJ, Wong J, Heiner C, Oh S, Theriot CM, Gulati AS, et al. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019;47(18):e103. doi: 10.1093/nar/gkz569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eren AM, Borisy GG, Huse SM, Mark Welch JL. Oligotyping analysis of the human oral microbiome. Proc Nat Acad Sci USA. 2014;111(28):E2875–E2884. doi: 10.1073/pnas.1409644111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eren AM, Maignien L, Sul WJ, Murphy LG, Grim SL, Morrison HG, et al. Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol Evol. 2013;4(12):1111-9. [DOI] [PMC free article] [PubMed]
- 11.Edgar RC, Flyvbjerg H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics. 2015;31(21):3476–3482. doi: 10.1093/bioinformatics/btv401. [DOI] [PubMed] [Google Scholar]
- 12.Hathaway LJ, Brugger S, Martynova A, Aebi S, Muhlemann K. Use of the Agilent 2100 bioanalyzer for rapid and reproducible molecular typing of Streptococcus pneumoniae. J Clin Microbiol. 2007;45(3):803–809. doi: 10.1128/JCM.02169-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brugger SD, Hathaway LJ, Muhlemann K. Detection of Streptococcus pneumoniae strain cocolonization in the nasopharynx. J Clin Microbiol. 2009;47(6):1750–1756. doi: 10.1128/JCM.01877-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brugger SD, Frey P, Aebi S, Hinds J, Muhlemann K. Multiple colonization with S. pneumoniae before and after introduction of the seven-valent conjugated pneumococcal polysaccharide vaccine. PLoS One. 2010;5(7):e11638. doi: 10.1371/journal.pone.0011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang W, Kang X, Yang Q, Lin Y, Fang M. Review on the development of genotyping methods for assessing farm animal diversity. J Anim Sci Biotechnol. 2013;4(1):2. doi: 10.1186/2049-1891-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mika M, Mack I, Korten I, Qi W, Aebi S, Frey U, et al. Dynamics of the nasal microbiota in infancy: a prospective cohort study. J Allergy Clin Immunol. 2015;135(4):905–912 e911. doi: 10.1016/j.jaci.2014.12.1909. [DOI] [PubMed] [Google Scholar]
- 20.Fuchs O, Latzin P, Kuehni CE, Frey U. Cohort profile: the Bern infant lung development cohort. Int J Epidemiol. 2012;41(2):366–376. doi: 10.1093/ije/dyq239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mika M, Maurer J, Korten I, Allemann A, Aebi S, Brugger SD, et al. Influence of the pneumococcal conjugate vaccines on the temporal variation of pneumococcal carriage and the nasal microbiota in healthy infants: a longitudinal analysis of a case-control study. Microbiome. 2017;5(1):85. doi: 10.1186/s40168-017-0302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carvalho Mda G, Tondella ML, McCaustland K, Weidlich L, McGee L, Mayer LW, et al. Evaluation and improvement of real-time PCR assays targeting lytA, ply, and psaA genes for detection of pneumococcal DNA. J Clin Microbiol. 2007;45(8):2460–2466. doi: 10.1128/JCM.02498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hilty M, Wuthrich D, Salter SJ, Engel H, Campbell S, Sa-Leao R, et al. Global phylogenomic analysis of nonencapsulated Streptococcus pneumoniae reveals a deep-branching classic lineage that is distinct from multiple sporadic lineages. Genome Biol Evol. 2014;6(12):3281–3294. doi: 10.1093/gbe/evu263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pai R, Gertz RE, Beall B. Sequential multiplex PCR approach for determining capsular serotypes of Streptococcus pneumoniae isolates. J Clin Microbiol. 2006;44(1):124–131. doi: 10.1128/JCM.44.1.124-131.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dube FS, Ramjith J, Gardner-Lubbe S, Nduru P, Robberts FJL, Wolter N, et al. Longitudinal characterization of nasopharyngeal colonization with Streptococcus pneumoniae in a South African birth cohort post 13-valent pneumococcal conjugate vaccine implementation. Sci Rep. 2018;8(1):12497. doi: 10.1038/s41598-018-30345-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill PC, Cheung YB, Akisanya A, Sankareh K, Lahai G, Greenwood BM, et al. Nasopharyngeal carriage of Streptococcus pneumoniae in Gambian infants: a longitudinal study. Clin Infect Dis. 2008;46(6):807–814. doi: 10.1086/528688. [DOI] [PubMed] [Google Scholar]
- 27.Murad C, Dunne EM, Sudigdoadi S, Fadlyana E, Tarigan R, Pell CL, et al. Pneumococcal carriage, density, and co-colonization dynamics: a longitudinal study in Indonesian infants. Int J Infect Dis. 2019;86:73–81. doi: 10.1016/j.ijid.2019.06.024. [DOI] [PubMed] [Google Scholar]
- 28.Abdeldaim GM, Stralin K, Olcen P, Blomberg J, Herrmann B. Toward a quantitative DNA-based definition of pneumococcal pneumonia: a comparison of Streptococcus pneumoniae target genes, with special reference to the Spn9802 fragment. Diagn Microbiol Infect Dis. 2008;60(2):143–150. doi: 10.1016/j.diagmicrobio.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki N, Yuyama M, Maeda S, Ogawa H, Mashiko K, Kiyoura Y. Genotypic identification of presumptive Streptococcus pneumoniae by PCR using four genes highly specific for S. pneumoniae. J Med Microbiol. 2006;55(Pt 6):709–714. doi: 10.1099/jmm.0.46296-0. [DOI] [PubMed] [Google Scholar]
- 30.Tavares DA, Handem S, Carvalho RJ, Paulo AC, de Lencastre H, Hinds J, et al. Identification of Streptococcus pneumoniae by a real-time PCR assay targeting SP2020. Sci Rep. 2019;9(1):3285. doi: 10.1038/s41598-019-39791-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anyansi C, Straub TJ, Manson AL, Earl AM, Abeel T. Computational methods for strain-level microbial detection in colony and metagenome sequencing data. Front Microbiol. 2020;11:1925. doi: 10.3389/fmicb.2020.01925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tonkin-Hill G, Ling C, Chaguza C, Salter SJ, Hinfonthong P, Nikolaou E, et al. Pneumococcal within-host diversity during colonisation, transmission and treatment. bioRxiv. 2022:2022.2002.2020.480002. [DOI] [PMC free article] [PubMed]
- 33.Mauffrey F, Fournier E, Demczuk W, Martin I, Mulvey M, Martineau C, et al. Comparison of sequential multiplex PCR, sequetyping and whole genome sequencing for serotyping of Streptococcus pneumoniae. PLoS One. 2017;12(12):e0189163. doi: 10.1371/journal.pone.0189163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinsbroek E, Tafatatha T, Phiri A, Swarthout TD, Alaerts M, Crampin AC, et al. Pneumococcal carriage in households in Karonga District, Malawi, before and after introduction of 13-valent pneumococcal conjugate vaccination. Vaccine. 2018;36(48):7369–7376. doi: 10.1016/j.vaccine.2018.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaguza C, Senghore M, Bojang E, Lo SW, Ebruke C, Gladstone RA, et al. Carriage dynamics of pneumococcal serotypes in naturally colonized infants in a rural African setting during the first year of life. Front Pediatr. 2020;8:587730. doi: 10.3389/fped.2020.587730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oyewole OR, Lang P, Albrich WC, Wissel K, Leib SL, Casanova C, et al. The impact of pneumococcal conjugate vaccine (PCV) coverage heterogeneities on the changing epidemiology of invasive pneumococcal disease in Switzerland, 2005-2019. Microorganisms. 2021;9(5). 10.3390/microorganisms9051078. [DOI] [PMC free article] [PubMed]
- 37.Althouse BM, Hammitt LL, Grant L, Wagner BG, Reid R, Larzelere-Hinton F, et al. Identifying transmission routes of Streptococcus pneumoniae and sources of acquisitions in high transmission communities. Epidemiol Infect. 2017;145(13):2750–2758. doi: 10.1017/S095026881700125X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flasche S, Lipsitch M, Ojal J, Pinsent A. Estimating the contribution of different age strata to vaccine serotype pneumococcal transmission in the pre vaccine era: a modelling study. BMC Med. 2020;18(1):129. doi: 10.1186/s12916-020-01601-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lipsitch M, Abdullahi O, D’Amour A, Xie W, Weinberger DM, Tchetgen Tchetgen E, et al. Estimating rates of carriage acquisition and clearance and competitive ability for pneumococcal serotypes in Kenya with a Markov transition model. Epidemiology. 2012;23(4):510–519. doi: 10.1097/EDE.0b013e31824f2f32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wyllie AL, Wijmenga-Monsuur AJ, van Houten MA, Bosch A, Groot JA, van Engelsdorp GJ, et al. Molecular surveillance of nasopharyngeal carriage of Streptococcus pneumoniae in children vaccinated with conjugated polysaccharide pneumococcal vaccines. Sci Rep. 2016;6:23809. doi: 10.1038/srep23809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valente C, Hinds J, Gould KA, Pinto FR, de Lencastre H, Sa-Leao R. Impact of the 13-valent pneumococcal conjugate vaccine on Streptococcus pneumoniae multiple serotype carriage. Vaccine. 2016;34(34):4072–4078. doi: 10.1016/j.vaccine.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 42.Kandasamy R, Gurung M, Thapa A, Ndimah S, Adhikari N, Murdoch DR, et al. Multi-serotype pneumococcal nasopharyngeal carriage prevalence in vaccine naive Nepalese children, assessed using molecular serotyping. PLoS One. 2015;10(2):e0114286. doi: 10.1371/journal.pone.0114286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan KC, Subramanian R, Chong P, Nelson EA, Lam HS, Li AM, et al. Pneumococcal carriage in young children after introduction of PCV13 in Hong Kong. Vaccine. 2016;34(33):3867–3874. doi: 10.1016/j.vaccine.2016.05.047. [DOI] [PubMed] [Google Scholar]
- 44.Simell B, Auranen K, Kayhty H, Goldblatt D, Dagan R, O’Brien KL, et al. The fundamental link between pneumococcal carriage and disease. Expert Rev Vaccines. 2012;11(7):841–855. doi: 10.1586/erv.12.53. [DOI] [PubMed] [Google Scholar]
- 45.Allemann A, Frey PM, Brugger SD, Hilty M. Pneumococcal carriage and serotype variation before and after introduction of pneumococcal conjugate vaccines in patients with acute otitis media in Switzerland. Vaccine. 2017;35(15):1946–1953. doi: 10.1016/j.vaccine.2017.02.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Details of the in-house pneumococcal strains used for mock community preparation. Table S2. Pacbio SMRTcell Sample pooling. Table S3. Assembly details of S. mitis, S. pseudopneumoniae and S. pneumoniae strains showing close plyNCR relatedness during in-silico analysis. Table S4. Characteristics of the BILD infant cohort (n=47). Table S5. Comparison of discordant qPCR and PCR amplification results of nasal swab samples (n=25) from BILD cohort infants. Table S6. Mean read output statistics from PacBio SMRT sequencing and DADA2 pipeline. Table S7. Conventional serotyping of nasal swab (NS) samples (n=240) from BILD cohort infants. Table S8. Multivariable Cox regression analysis of potential risk factors associated with time to first pneumococcal acquisition. A Hazard Ratio (HR) < 1 indicates a reduced hazard (rate) of pneumococcal acquisition while an HR > 1 indicates an increased rate of pneumococcal acquisition. Table S9. Duration of pneumococcal carriage by acquisition and serotype. Fig. S1. False positive removal during DADA2 ASV calling. Fig. S2. Detection of S. pneumoniae by plyNCR PCR. Fig. S3. Patterns of co-colonization dynamics in four infants.
Data Availability Statement
All raw PacBio sequencing reads were deposited in the European Nucleotide Archive (ENA) under study accession number PRJEB45241.