


Abstract
DNA lesions in S phase threaten genome stability. The DNA damage tolerance (DDT) pathways overcome these obstacles and allow completion of DNA synthesis by the use of specialised translesion (TLS) DNA polymerases or through recombination-related processes. However, how these mechanisms coordinate with each other and with bulk replication remains elusive. To address these issues, we monitored the variation of replication intermediate architecture in response to ultraviolet irradiation using transmission electron microscopy. We show that the TLS polymerase η, able to accurately bypass the major UV lesion and mutated in the skin cancer-prone xeroderma pigmentosum variant (XPV) syndrome, acts at the replication fork to resolve uncoupling and prevent post-replicative gap accumulation. Repriming occurs as a compensatory mechanism when this on-the-fly mechanism cannot operate, and is therefore predominant in XPV cells. Interestingly, our data support a recombination-independent function of RAD51 at the replication fork to sustain repriming. Finally, we provide evidence for the post-replicative commitment of recombination in gap repair and for pioneering observations of in vivo recombination intermediates. Altogether, we propose a chronology of UV damage tolerance in human cells that highlights the key role of polη in shaping this response and ensuring the continuity of DNA synthesis.
INTRODUCTION
Accurate DNA replication is necessary to maintain genome stability. However, various endogenous or exogenous DNA damage constantly threaten the progression of the high-fidelity replicative DNA polymerases. The DNA damage response (DDR) organizes the multiple intricate molecular mechanisms controlling cell cycle progression, DNA repair and DNA damage tolerance (DDT). DDT comprises two pathways, conserved throughout evolution, that allow completion of DNA replication in spite of the presence of DNA lesions in the parental strands. Translesion synthesis (TLS) relies on the replacement of stalled replicative DNA polymerases by specific error-prone specialised DNA polymerases able to directly insert nucleotides opposite the lesion, in an error-free or an error-prone manner (1). Alternatively, homologous recombination (HR)-like mechanisms, globally referred as damage avoidance (DA) or homology-dependent repair (HDR), use the sister chromatid to circumvent the damage by strand exchange or template switch (2).
Whether the DDT pathways compete, compensate or collaborate is still unclear (3). Data from bacteria and yeast suggest a chronological occurrence, with TLS acting before HDR (4,5). In human, a certain specialisation of the TLS polymerases is illustrated by the xeroderma pigmentosum variant (XPV) syndrome, a hereditary skin cancer-prone disease caused by inactivating mutations in the gene coding for the Y-family TLS polymerase η (polη). Polη has the unique capacity to accurately overcome cyclobutane thymine dimers (TT-CPDs), which are the most abundant lesions induced by UV irradiation (6–8). Error-prone bypass by alternative TLS polymerases accounts for the hypermutability by UV of XPV cells (9–12). XPV cells also show increased recombination, as evidenced by a higher number of sister chromatid exchanges (SCEs) (13), but it is currently unclear whether this contributes to the disease. Hence, polη plays a unique role in the tolerance of UV damage and cannot be efficiently and accurately replaced by other DDT mechanisms.
How DDT coordinates with replication fork (RF) progression is also a matter of debate and DDT timing was proposed to impact on its accuracy and on the correct maintenance of the epigenetic marks in the surrounding chromatin (14,15). Various studies showed that TLS and HR are post-replicative mechanisms in yeast, proceeding uncoupled from the bulk replication to fill in gaps left behind RFs through stalling of the synthesis of an Okazaki fragment on the lagging strand or repriming of a stalled leading strand (5,16–18). A similar post-replicative model was proposed for TLS in human cells (14,19) and the recently identified primase-TLS polymerase PrimPol has been proposed to mediate repriming at stalled RFs (20–23). However, numerous studies showed that TLS contributes to RF progression (12,20,24–27) and it is likely that TLS can take place both at and behind the RFs in vertebrates (25,28,29). Single-strand DNA (ssDNA) gaps are excellent substrates for HR in yeast (30,31). Adar et al. showed, in a plasmid model, that ssDNA gaps located opposite to lesions can be filled in by RAD51-dependent HR mechanism in mammalian cells (32). Yet, several recombination-like RF restart pathways were also described. Moreover, in the last few years, fork reversal has become a widespread structural model to describe the response of RFs to any kind of replication stress, from nucleotide pool imbalance to bulky DNA lesions (33,34). RAD51 plays a critical role in this process, although its precise functions in RF remodelling, protection from nucleases and restart is not entirely clear.
In this work, we investigated the coordination of genome replication with DDT pathways in human fibroblasts following exposure to UVC. We used transmission electron microscopy (TEM) to assess the variations of replication intermediate (RI) architecture at early and late time points after irradiation in polη-proficient and polη-deficient backgrounds, coupled to analysis of RF progression by DNA fiber assay and monitoring of the recruitment of specific proteins to replicating chromatin by iPOND. We showed that polη activity configures the immediate response of RFs to UV-induced lesions and promotes their progression and the continuity of DNA synthesis. In the absence of polη, lesion tolerance mechanisms are diverted from the RF to post-replicative mechanisms. We present evidences that PrimPol contributes to but is not the main actor of RF repriming in XPV cells. Finally, our observations clearly indicate that recombination intermediates appear later after UV radiation exposure and their accumulation correlates with the detection of RAD51 behind RFs where it participates in gap repair. Interestingly, polη deficiency also led to the recruitment of RAD51 at the RFs without a parallel increase in the detection of recombination-like DNA intermediates, suggesting a strand invasion-independent role to overcome RF uncoupling, presumably by contributing to repriming events.
MATERIALS AND METHODS
Cell culture and treatments
SV40-transformed normal (MRC5-V1) and XPV (XP30RO) human fibroblasts were grown in minimum Eagle's medium (MEM, Gibco). XP30RO cells stably complemented by wild-type polη (XPVpolη cells) or polη mutants were described elsewhere (35,36) and were grown in the presence of 100 μg/ml zeocin (Invivogen). Mouse embryonic fibroblasts (kind gift from R. Wood) were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco). Media were supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin and cells were cultured under a 5% CO2 atmosphere. For UVC irradiation (254 nm), cells were washed with pre-heated Phosphate Buffered Saline (PBS) and irradiated without any medium at a fluency of 0.65 J m–2 s–1. Stock solutions of B02 and mirin (Sigma) were prepared in DMSO.
siRNA transfection
siRNAs purchased from Dharmacon were used to transiently down-regulate the expression of POLH (5′-GAAGUUAUGUCCAGAUCUU-3′), POLK (On-TARGET plus SMART pool), PRIMPOL (On-TARGET plus SMART pool), RAD51 (On-TARGET plus SMART pool, siGENOME duplex 2) and REV3L (On-TARGET plus SMART pool). Unspecific siRNAs (siNT) were used as control. Cells were transfected with 30 nM of siRNAs using Interferin (PolyPlus) according to the manufacturer's instructions, and were treated 48–72 h after transfection. For RAD51 depletion, 15 nM of siRNAs were used for 36 h.
Proliferation assay
Cells were plated at 1.5 × 105 per well of a six-well plate 24 h before UVC exposure. Cells were treated as indicated, incubated for 72 h and then counted with trypan blue staining using a Neubauer haemocytometer.
Cellular fractionation
Cells were harvested by trypsinization, washed in PBS and then subjected to cell fractionation. Cells (0.5–1 × 106) were resuspended in 1 ml of CytoSKeleton (CSK) 100 buffer (100 mM NaCl, 300 mM Sucrose, 3 mM MgCl2, 10 mM PIPES pH 6.8, 1 mM ethylene glycol-tetra-acetic acid (EGTA), 0.2% Triton X-100 and protease inhibitor cocktail (Roche)) and incubated for 15 min on ice. Samples were centrifuged at 7000 rpm for 5 min at 4°C, and washed once in CSK buffer. The pellet, corresponding to the insoluble protein fraction, was solubilized in 100 μl lysis buffer (10 mM Tris–HCl pH 7.5, 20 mM NaCl, 0.4% SDS, protease inhibitor cocktail) supplemented with 150 U/ml benzonase (Millipore) for 30 min at 37°C. For whole-cell extracts (WCE), cell pellets were directly lysed in lysis buffer with benzonase for 10 min at room temperature. Protein amounts were determined using Nanodrop (Thermo) or Bradford assay. Proteins were denatured for 10 min at 90°C in the presence of Laemmli buffer.
iPOND
Cells were irradiated with 25 J m–2 UVC and pulse-labelled with 10 μM 5-ethynyl-2′-deoxyuridine (EdU, Invitrogen) for 10 min. In Supplementary Figure 5D, the UVC dose was reduced to 5 J m–2 and the labelling period was extended to 20 min. iPOND was performed immediately after the pulse or after a chase in fresh medium supplemented with 10 μM thymidine (Sigma) as already described (37). Briefly, cells were crosslinked with 1% formaldehyde (Sigma), harvested by scrapping and permeabilized in PBS with 0.5% Triton X100. Biotin-azide (Molecular Probes) was conjugated to EdU by click chemistry for 2 h in click reaction buffer (10 mM sodium-l-ascorbate, 10 mM biotin-azide, 2 mM CuSO4 in PBS). Cells were lysed in iPOND lysis buffer (10 mM HEPES–NaOH pH 7.9, 100 mM NaCl, 2 mM EDTA, 1 mM EGTA, 1 mM PMSF, 0.2% SDS, 0.1% sarkozyl, antiproteases) and sonicated on a Bioruptor device. Solubilized chromatin was retrieved by centrifugation and subjected to streptavidin pull-down overnight (Dynabeabs MyOne Streptavidine C1, Invitrogen). After extensive bead washing, pull-downed proteins were eluted by boiling in 1× Laemmli buffer at 95°C for 30 min.
Western Blot
Samples were subjected to SDS-PAGE followed by PVDF membrane transfer. Membranes were immunoblotted with the following antibodies: mouse anti-β-actin (AC-15, Sigma #A5441), mouse anti-β-catenin (cl14, BD Biosciences, #610153), rabbit anti-histone H2B (V119, Cell Signaling #8135), rabbit anti-histone H3 (Abcam #ab1791), mouse anti-histone H4 (Abcam #ab31830), mouse anti-LaminA/C (Santa Cruz #sc-7292), mouse anti-PCNA (PC10, Santa Cruz #sc-56), goat anti-POLA1 (G-16, Santa Cruz #sc-5921), goat anti-POLD1 (C-20, Santa Cruz #sc-8797), rabbit anti-polκ (Bethyl #A301-975A), rabbit anti-polη (Abcam #ab17725, Bethyl #A301-231A), mouse anti-polη (B-7, Santa Cruz #sc-17770), rabbit anti-PrimPol (kind gift from L. Blanco), rabbit anti-RAD51 (Calbiochem #PC130-100UL), mouse anti-RPA32 (Calbiochem #NA19L) and mouse anti-RPA70 (Calbiochem #NA13).
Genomic DNA preparation and RIs enrichment
Replication intermediates were isolated as follows. Cells subjected to the indicated treatments were washed with PBS and DNA was crosslinked by two rounds of incubation on ice in a dark room for 5 min with 10 μg/ml trioxalen (Sigma) followed by irradiation with 45 kJ UVA, in order to generate interstrand crosslinks in the genomic DNA. Cells were then washed and harvested by scraping, then subjected to nuclei extraction (Nuclei EZ prep kit, Sigma). Genomic DNA was isolated using DNAzoL reagent (Invitrogen) following the manufacturer's instructions and resuspended in water. Thorough deproteinization, which is necessary to minimize intra- and inter- molecular crossovers (38), was performed by two rounds of hydrolysis by 1 mg/ml proteinase K overnight at 55°C in the presence of 1% SDS, followed by a gentle phenol/chloroform extraction. After precipitation, genomic DNA was treated at 37°C for 2 h with RNaseA (DNase and protease-free, Thermo Scientific) and BamHI (New England Biolabs). Solution was adjusted to 300 mM NaCl and loaded onto a benzoylated naphtoylated DEAE-cellulose (BND, Sigma) chromatography column. Fractions corresponding to dsDNA molecules were collected with DS Elution buffer (10 mM Tris–HCl pH 7.5, 5 mM EDTA and 0.8 M NaCl). Genomic DNA fragments containing ssDNA regions were eluted by the addition of SS Elution buffer (10 mM Tris–HCl pH 7.5, 5 mM EDTA and 1 M NaCl, 0.9% caffeine (Sigma)). SS fractions were pooled, precipitated with isopropanol, resuspended in water, quality-controlled by agarose gel electrophoresis and dosed with Nanodrop. In order to ensure that RIs structures are not lost during the whole procedure, the UVA/trioxalen crosslinking efficiency was systematically checked by TEM observation after heat/formamide denaturation of the samples.
RI analysis by transmission electron microscopy
We specifically developed a method to optimize recruitment, deployment of long ds-ssDNA molecules and their visualization in darkfield imaging mode. We have combined BAC (Benzyldimethyl-alkyl-ammonium chloride) hyperphase method with positive staining direct adsorption method. The hyperphase method initially developed by Vollenweider et al. (39) allows to stretch DNA intermediates and to avoid secondary structures and random intra- or inter-molecular crossovers. The direct adsorption method consists in the treatment of the carbon film with a glow discharge in presence of amylamine (positive charges) as initially demonstrated by Dubochet et al. (40). It favors the recruitment of DNA intermediates onto the surface, allows a positive staining of DNA by uranyl acetate and its observation in dark-field imaging mode. We obtain high-resolution images with an optimal contrast (38).
Briefly, ssDNA-enriched samples were added to a mix of formamide, BAC and glyoxal, to generate a hyperphase film onto the surface of a water bath. The DNA was picked up on positive functionalized carbon-coated grids by a brief contact with the hyperphase film surface. Grids were then washed in a large volume of purified milliQ water followed by 100% ethanol and dried with filter paper. Finally, molecules adsorbed onto the grids were positively stained with 2% aqueous uranyl acetate and dried. Samples were visualized in annular dark field mode, filtered in zero loss, with a Zeiss EM902 transmission electron microscope equipped with Electron Energy Loss Spectrometer (EELS). Electron micrographs were obtained at 50 000 or 85 000 magnifications (140 000 for the inlays) acquired using a Veletta high-resolution CCD camera associated to a dedicated iTEM software (Olympus, Soft Imaging Solutions). Series of at least 50 replication forks were analyzed for each condition, where single-stranded versus double-stranded areas were mapped. Configurations containing four (X forms) or more branches were analyzed and counted in reference with 50 canonical forks series. Common criteria to validate reversed fork structures (symmetry of daughter strands and morphological traits) were considered (41).
We have compared our approach with classical spreading method used to analyse DNA intermediates as previously described (41). Briefly, carbon-coated grids were incubated for 45 min with 33 μg/ml aqueous EtBr solution and dried. DNA intermediates were subjected to BAC spreading, incubated for 15 s in 0.02% UrAc solution, and finally washed with pure water and 100% ethanol. Carbon-coated grids supporting the DNA molecules after their spreading were subjected to Pt/C rotary shadowing using the MED20 BALTEC coating system. The angle between the specimen and the Pt/C gun was adjusted to 5°. The vacuum was adjusted to 2.5 × 10–5 mbar and the evaporation rate was maintained at 0.03 nm/s to reach a thickness of 11 nm measured by the QSG 100 thin film monitor.
Strand exchange assay
Rad51 and RPA proteins from yeast Saccharomyces cerevisiae were purified as previously described (42). 13 μM Rad51 were incubated with 40 μM of circular viral ssDNA (+) PhiX174 (previously purified in ion exchange MiniQ column) for 3 min, in 10 mM Tris–HCl pH 7.5, 100 mM NaCl, 3 mM MgCl2, 2 mM ATP, 1 mM DTT and 2 mM Spermidine, and 1.3 μM RPA was then added into reaction for 10 minutes. 20 μM of linear dsDNA PhiX174 replicative form I (Phix174 RFI homologous plasmid purchased from New England Biolabs and linearized by digestion with the restriction enzyme XhoI or PstI followed by phenol-chloroform extraction) was added into the reaction for 30 min, to a 10 μL final volume. The reaction was performed at 37°C. Reactions were crosslinked with psoralen (final concentration 10 μg/mL) during 4 min under UVA (crosslink density of 1 per 200–300 nt) and stopped using 0.5 mg/ml Proteinase K, 1% SDS, 12.5 mM EDTA and incubated overnight at room temperature. For TEM analysis, four strand exchange reactions were pooled, DNA was purified using phenol-chloroform and ethanol precipitation then spread using BAC hyperphase method (as for in vivo RIs TEM samples preparation).
RAD51 filament TEM analysis
RAD51 filaments were formed by incubating 9 μM in nucleotides of a 3′ overhang DNA substrate (designed as previously described (42)) with 3 μM RAD51 (1 protein per 3 nt) in a buffer containing 10 mM Tris–HCl pH7.5, 50 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 1.5 mM ATP and 1 mM DTT for 5 minutes at 37°C, then adding 0.04 μM RPA during 15 min. B02 was introduced in varying concentrations (0, 5, 10, 20 and 50 μM) at the same time as RAD51. For filaments observation, the reaction was quickly diluted 40 times in a buffer containing 10 mM Tris–HCl pH 7.5, 50 mM NaCl, 2 mM MgCl2, 2 mM Cacl2 and observed by TEM (DNA-protein samples). During one minute, a 5 μl drop of the dilution was deposited on a 600-mesh copper grid previously covered with a thin carbon film and pre-activated by glow-discharge in the presence of amylamine (Sigma-Aldrich, France) (40,43). Grids were rinsed and positively stained with aqueous 2% (w/v) uranyl acetate, dried carefully with a filter paper and observed in the annular dark-field mode in zero loss filtered imaging, using a Zeiss 902 transmission electron microscope. Images were captured as described above. Filament lengths were measured on 50 molecules for each B02 concentration and statistical analysis was performed with Kruskal–Wallis test followed by Dunn's multiple comparisons test.
DNA fiber assay
Cells were incubated for 20 min in medium supplemented with 25 μM iododeoxyuridine (IdU, Sigma). Cells were washed, irradiated with UVC and further incubated in medium supplemented with 50 μM chlorodeoxyuridine (CldU, Sigma) for 20–60 min as indicated. Alternatively, cells were first labelled with 25 μM CldU, then with 25 μM IdU. 1000 cells, resuspended in 2 μl of PBS, were spotted on microscope glass slides and lysed in 7 μl of spreading buffer (200 mM Tris–HCl pH 7.5, 50 mM EDTA pH 8.0, 0.5% SDS). DNA was spread by tilting the slides. DNA was fixed in methanol/acetic acid 3:1 and denatured in 2.5 M HCl for 1 h at room temperature. Samples were blocked in blocking buffer (1% BSA, 0.1% Tween20 in PBS) for 1 h. IdU, CldU and total DNA were detected using the following protocol (44): (i) mouse anti-BrdU (Becton Dickinson, 1/20, IdU) + rat anti-BrdU (Abcys, 1/100, CldU) for 1 h at room temperature, (ii) goat anti-mouse AF594 + goat anti-rat AF488 for 30 min at 37°C, (iii) mouse anti-single stranded DNA (Millipore, 1/100) for 45 min at 37°C, (iv) rabbit anti-mouse AF350 for 20 min at 37°C and (v) goat anti-rabbit AF350 for 20 min at 37°C. All fluorescent antibodies were from Molecular Probes and used at 1/100 dilution. Antibodies were diluted in blocking buffer. Samples were washed four times in PBS between each antibody incubation. Samples were mounted in fluorescent mounting medium (DAKO). Images were acquired and analysed on an Axio Imager Z1 microscope using the Axio Vision software (Zeiss). Broken replication tracks were excluded from the analysis.
S1 nuclease DNA fiber assay
Detection of ssDNA gaps on nascent DNA was performed as already described (45) with minor modifications. Analogue pulses and treatments were performed as described above and in the figure legends. Cells were then extracted in 100 mM NaCl, 300 mM Sucrose, 3 mM MgCl2, 10 mM PIPES pH 6.8, 0.5% Triton X-100 for 7 min at room temperature and incubated for 20 min at 37°C in S1 nuclease buffer (50 mM NaCl, 30 mM sodium acetate pH 4.6, 2 mM zinc acetate and 5% glycerol) supplemented or not with 20 U ml–1 of S1 nuclease (Invitrogen). Nuclei were collected by scrapping in PBS 0.1% BSA followed by centrifugation at 1500 × g for 5 min at 4°C. Nuclei were resuspended in PBS 10 mM EDTA. DNA was spread and immune-stained as described above. Images were acquired on an Axio Observer Z1 microscope using Axio Vision software (Zeiss) and analysed on ImageJ software. All bicolor replication signals were included in the analysis.
Immunofluorescence
Cells were pre-extracted in CSK100 buffer for 5 min on ice under gentle agitation. Cells were fixed in 4% paraformaldehyde for 20 min and permeabilized in methanol at –20°C for 10 s. Cells were incubated for 1 h at room temperature with primary antibodies (rabbit anti-RAD51 Calbiochem PC130 1/200 and mouse anti-PCNA Santa Cruz PC10 1/500) diluted in IF buffer (3% BSA, 0.5% Tween 20 in PBS). Cells were washed three times in PBS and stained for 30 min with secondary antibodies from Molecular Probes (goat anti-rabbit AF488 1/1000, goat anti-mouse AF594 1/1000). Coverslips were mounted in fluorescent mounting medium (DAKO) supplemented with DAPI (1.5 μg/ml). Images were acquired on an AxioImager Z1 microscope using the Axio Vision software (Zeiss). Intensity was quantified with the ImageJ software.
In situ proximity ligation assay
PLA to nascent DNA was performed as already described (37). Briefly, cells were irradiated with UVC (25 J m–2) and incubated for 30 min. 10 μM EdU was added for the last 7 min of the incubation. Cells were pre-extracted in CSK100 buffer and fixed prior to biotin-azide conjugation to EdU. Cells were incubated with primary antibodies against RAD51 (rabbit anti-RAD51 1/200, Calbiochem PC130) and biotin (mouse anti-biotin 1/6000, Jackson ImmunoResearch #200-002-211). PLA and EdU counterstaining were performed according to the manufacturer's instructions using the Duolink In Situ Red kit (Sigma) and goat anti-mouse Alexa Fluor 488 antibody (Molecular Probes).
RT-qPCR
Total RNA extraction was performed using Maxwell RSC simply RNA cell kit (Promega) according to the manufacturer's instructions. 1 μg RNA was reverse transcribed to cDNA with Maxima H minus Reverse Transcriptase (Thermo Scientific) using the following amplification program: 10 min at 25°C, 60 min at 50°C, 5 min at 85°C. Samples were treated with RNaseH (Thermo Scientific) for 20 min at 37°C and 10 min at 65°C. qPCR was performed in duplicates with SYBR Green ROX qPCR Master Mix on CFX96 RealTime Systen (Biorad) according to the manufacturer's instructions using the following primers: hREV3L-F: ACCCCCGGAGTACCACTTATCCAGC, hREV3L-R: CCGGAGATATGGTGCCTTGGACA, hGAPDH-F: ACCACAGTCCATGCCATCAC, hGAPDH-R: TCCACCACCCTGTTGCTGTA. ‘No template’ and ‘no reverse transcriptase’ controls were performed in parallel to exclude any contamination. Fold changes were calculated using the ΔΔCq method.
Statistical analysis
Statistical analyses were performed with Prism 9 (GraphPad software). The statistical tests used are indicated in the legends of the figures. ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
RESULTS
Polη is recruited in the vicinity of replication forks to promote their progression after UVC irradiation
It is well documented that polη accumulates at replication foci after UV in human cells (46,47). However, it remains unclear if this recruitment occurs at stalled RFs or during post-replicative gap filling. To address this question, we performed an iPOND analysis, a procedure that allows discriminating between proteins enriched at and behind RFs (48). Cells were pulse-labelled with 5-ethynyl-2′-deoxyuridine (EdU) after UVC irradiation and cross-linked immediately or after a thymidine chase. Proteins bound to EdU-labelled DNA were pulled-down and analysed by western blot (Figure 1A, Supplementary Figure S1A). Polη could readily be detected at RFs in the mock-irradiated sample, as we have previously shown (37). After UV, Polη amounts increased and then, like PCNA amounts, decreased with chase, showing that polη is recruited preferentially in the vicinity of RFs after UV. Conversely, RAD51 levels increased in post-replicative chromatin, suggesting a spatio-temporal separation between polη-dependent TLS and recombination events.
Figure 1.

Polη promotes replication fork progression after UV. (A) MRC5-V1 cells stably expressing polη were irradiated or not with UVC (25 J m–2) and pulse-labelled with 10 μM EdU for 10 min. Cells were cross-linked immediately after the pulse or after a 30- or 60-min thymidine chase. Biotin was conjugated to EdU by click chemistry (click rxn) prior to cell lysis and chromatin shearing. Labelled DNA was retrieved on streptavidin beads and input and bound proteins (iPOND) were analysed by western blot using the indicated antibodies. iPOND band intensity was corrected by the corresponding input band intensity and expressed as a ratio of the mock-treated condition. An independent experiment is shown in Supplementary Figure S1A. (B) XPV cells and their corrected counterpart (XPVpolη) were irradiated or not at 25 J m–2 between 20-min pulses of IdU and CldU. DNA was spread on glass slides and IdU, CldU and total DNA were detected by immunofluorescence. Replicated tracks were measured on unbroken bicolor signals (1 of 3 independent experiments, n = 300, box-plot with interquartile range and 5–95 percentile whiskers, +: mean of the distribution, dots: outliers, Kruskal-Wallis followed by Dunn's multiple comparisons test, ns: not significant, ***P < 0.001). (C) MRC5-V1 cells were depleted for polη using siRNAs and treated like in B (1 of 2 independent experiments, n = 150, Mann–Whitney test, siNT: non specific siRNA, siPOLH: siRNA against POLH mRNAs). siPOLH efficiency was confirmed by western blot. (D) XPV and XPVpolη cells were treated as in B but with a CldU pulse of 20, 40 or 60 min. Left panel shows the distributions of IdU and CldU tracks (n > 90, Kruskal-Wallis followed by Dunn's multiple comparisons test, ns: not significant, **P < 0.01, ***P < 0.001). Right panel shows the linear regression of the mean CldU calculated from these distributions. Slopes and r2 coefficients are indicated.
To determine if polη deficiency impacts RF progression in the presence of UV lesions, we performed DNA fiber assays in XPV cells and XPV cells stably complemented with wild-type polη (herein referred as XPVpolη cells). Replication tracks were shorter after UV irradiation and this defect was further aggravated by polη deficiency (Figure 1B, Supplementary Table S1). Similar results were obtained by inverting the pulses with thymine analogues (Supplementary Figure S1B, Supplementary Table S1) or after depletion of endogenous polη in SV40-immortalized human MRC5-V1 fibroblasts (Figure 1C, Supplementary Table S2). These results indicate that polη promotes RF progression in the presence of UV lesions, in agreement with previous findings (24–26). This required polη catalytic activity, as well as its PCNA- and ubiquitin-binding motifs, two regulatory domains that cooperate for polη accumulation in replication foci (47) (Supplementary Figure S1C, Supplementary Table S1). Increasing the duration of the post-UV pulse led to longer tracks in both cell lines (Figure 1D, Supplementary Table S3). Linear regression analyses showed that, on the average, replication tracks extend at the same rate in the first 60 min following UV irradiation, this rate being lower in polη-deficient cells (Figure 1D, right panel). Forks presenting a severe slow down (defined as post-UV length lower than 30% of the pre-UV length) were more abundant in XPV cells than in XPVpolη cells 20 min after UV, but they did not persist at later time points (Supplementary Figure S1D). Altogether, these results show that RFs are not permanently stalled in irradiated XPV cells and that alternative restart mechanisms can take place, although with a decreased efficiency and/or with a delay.
In polη-deficient cells, residual error-prone TLS was shown to involve two pathways depending on polζ and polκ, respectively (9–11). However, depletion of polκ and/or of REV3L, the catalytic subunit of polζ, did not further aggravate the RF progression defect observed in XPV cells (Supplementary Figure S1E, Supplementary Table S4), suggesting that, although required for XPV cell survival and for UV-induced mutagenesis, these alternative polymerases act on a different substrate than stalled RFs, presumably to fill-in post-replicative gaps (25,49).
Polη limits ssDNA accumulation at and behind replication forks after UVC
To determine which mechanisms sustain RF progression in the absence of polη, we analysed the molecular architecture of RFs by transmission electron microscopy (TEM) at high resolution. Cells were irradiated or not with UVC and subjected to UVA/psoralen in vivo crosslinking to stabilize DNA replication intermediates (RIs) by avoiding branch migration. Purified DNA was enriched in RI-containing material as already described (5,41). To characterize the structure of the different DNA RIs, we implemented a protocol that combines the BAC method and positive staining (BPS) to optimize the adsorption and the spreading of the DNA onto the carbon film and to provide an optimal contrast and resolution using a darkfield imaging mode (38).
As expected, in mock-irradiated samples, the majority of RFs displayed no or short ssDNA stretches at the elongation point (Figure 2A), presumably on the lagging strand, as previously shown (5,50). Thirty min after UVC irradiation (25 J m–2), two types of events were observed. First, some RFs displayed an extended ssDNA stretch at one side of the elongation point (Figures 2B, D, E, G), corresponding to a blocked leading strand and its subsequent uncoupling from the helicase activity, as previously observed by TEM on plasmid models (50) and later confirmed by in vitro and in vivo studies in yeast and human models (5,18,33,51). Secondly, some RFs showed single-stranded regions behind the elongation point, on one or both daughter strands (Figures 2C–I). RI analyses revealed DNA molecules with successive gaps on the same strand (Figure 2H), gaps in opposite strands (Figure 2F) or both uncoupling and post-replicative gap(s) (Figures 2D, E, G), presumably because UV lesions are formed at a higher frequency in specific genomic regions (52). Very long internal gaps were rarely observed (Figure 2I).
Figure 2.

UVC irradiation leads to long ssDNA stretches at the elongation point and to post-replicative gaps as analysed by TEM in annular dark field mode. (A) Usual replication fork arising from the hydrolysis of a replication bubble by enzymatic restriction. The fork does not display ssDNA regions and is symmetric as regards to the identical daughter strand lengths. (B–I) Replication intermediates observed 30 min after exposure to UVC (25 J m–2). Inlays represent enlargements of the regions framed with dotted lines (scale bar = 50 nm). Panels B, D, E and G show uncoupled replication forks with ssDNA starting from the elongation point because of DNA synthesis block on the leading strand. Panel C shows a fork with an internal ssDNA gap on one daughter strand. The gap can appear on the leading strand (D), the lagging strand (E) or both (F, G). Gaps can accumulate successively on the same daughter strand (H). Forks displaying ssDNA at and behind the elongation point can also be observed (D, E, G). Unusually long gaps are rarely observed (I, internal gap length = 545 nm). ep: elongation point; ss: ssDNA. All images are from irradiated XPV cell samples.
To allow quantitative comparison of polη-proficient and polη-deficient cells, ssDNA regions were measured on >50 RFs in three independent experiments. This quantitative analysis highlighted that UVC irradiation led to a significant increase of the median length of ssDNA stretches both at and behind RFs in XPV cells compared to XPVpolη cells (Figures 3A, B, Supplementary Table S5). Lengths of ssDNA at the elongation point rarely exceed 450 nucleotides, even in polη-deficient cells, supporting the notion that uncoupling between the replicative helicase and the stalled leading strand is spatially limited. Given that short ssDNA regions were also observed in untreated conditions, we chose the 95th percentiles of the distributions of untreated XPVpolη cells as the thresholds to identify abnormally long ssDNA likely resulting from impaired DNA synthesis. Interestingly, these thresholds were similar at and behind the forks (82 and 81 nm, respectively), and correspond to a distance of approximately 160 nucleotides, close to the size of an Okazaki fragment (53). Consequently, we consider that RFs bearing ssDNA at or behind the elongation point above these thresholds are the result of lesion-induced RF uncoupling and post-replicative gaps, respectively. In irradiated XPV cells, 47% of forks were uncoupled and 61.6% displayed above-threshold internal gaps (herein referred as gapped forks), with structures bearing both particularities representing 28.7% (versus 3.1% in XPVpolη cells) (Supplementary Figure S2A). Taking these numbers together, we conclude that most of the forks encountered at least one lesion in our experimental conditions.
Figure 3.
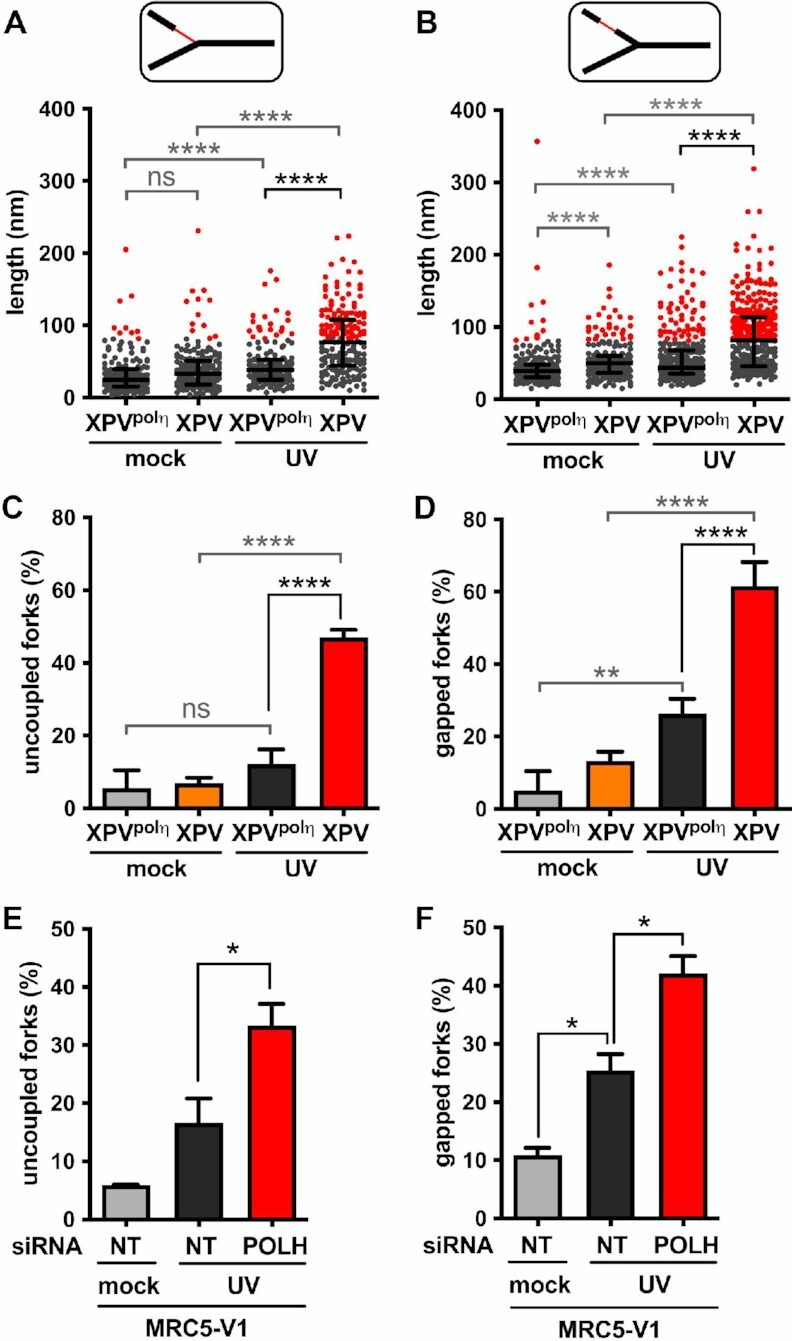
Polη prevents accumulation of ssDNA at and behind the elongation point after UV. TEM analysis was performed in XPV and XPVpolη cells 30 min after UVC irradiation at 25 J m–2 in three independent experiments. For each condition, at least 50 forks were analysed per experiment. (A) Distribution of the lengths of the ssDNA regions located at RFs (pool from the three experiments, n > 160). (B) Distribution of the lengths of the ssDNA regions located behind RFs. Data are shown as dot plots with median and interquartile range. Statistical significance was tested by Kruskal-Wallis followed by Dunn's multiple comparisons test (ns: not significant, ****P < 0.0001). The 95th percentiles of the distributions of untreated XPVpolη cells (81.55 and 81.25 nm at and behind RFs, respectively) were used as thresholds to identify abnormally long ssDNA stretches at and behind the elongation point (red dots in the graphes). (C) The number of uncoupled RFs (i.e. bearing a ssDNA stretch above the threshold at the elongation point) was expressed as a percentage of total RFs analysed. (D) The number of gapped RFs (i.e. bearing at least a ssDNA gap above the threshold behind the elongation point) was expressed as a percentage of total RFs analysed. Values are the mean ± SD of the three independent experiments. Statistical significance was tested by ordinary one-way ANOVA followed by Tukey's multiple comparisons test (ns: not significant *P < 0.05, **P < 0.01, ****P < 0.0001). (E) Proportion of uncoupled forks and (F) proportion of gapped forks were determined as described above in MRC5-V1 cells depleted or not for polη (N = 2).
After UVC irradiation, XPV cells showed a 3.8-fold increase of RF uncoupling compared to XPVpolη cells (Figure 3C), in agreement with the stronger slow-down observed with the DNA fiber assays. Gapped forks were 2.3 times more frequent and displayed more long gaps in polη-deficient cells (Figure 3D, Supplementary Figure S2B). In support of these observations, daughter strands were more sensitive to digestion by the ssDNA-specific nuclease S1 in XPV cells than in XPVpolη cells (Supplementary Figures S2C, D, Supplementary Table S6). Considering that long ssDNA at the elongation point is indicative of an impairment of the leading strand synthesis (see above), it became possible to discriminate the leading and the lagging strands of uncoupled RFs. By restricting our analysis to these structures in irradiated XPV samples (n = 30, 29 and 23 in three independent experiments), we observed that long post-replicative gaps were preferentially located in the lagging strand and were found in 43.9% ± 2.7 of the molecules. Nonetheless, these events were not rare in the leading strand and were observed in 28% ± 5.8 of the RIs. The distributions of UV-induced post-replicative gap lengths were similar in XPV and XPVpolη cells (Supplementary Figure S2E), indicative of a common mechanism for gap formation in the presence or absence of polη. Moreover, this suggests that the gaps formed in XPVpolη cells are unlikely to be filled-in in a polη-dependent manner in the first 30 min following UV exposure, since gap lengths should then have been statistically reduced compared to XPV cells. A similar conclusion was made in yeast studies (5). Dynamic accumulation of ssDNA at and behind RFs in XPV cells was associated with an enhanced recruitment of the ssDNA-binding complex RPA on chromatin (Supplementary Figure S2F), as previously described (24). Interestingly, while the RPA signal remained elevated up to 24 h in XPV cells after 25 J m–2, in XPVpolη cells it peaked at 2–5 h, then slowly decreased over 20 h. We consider that this decrease corresponds to gap-filling repair events, which will be further characterized below. Importantly, we recapitulated these findings in MRC5-V1 fibroblasts depleted of polη by siRNAs (Figures 3E, F and Supplementary Figures S2 G-I, Supplementary Table S7), confirming that our observations are entirely dependent on the loss of polη.
While we were able to observe 4-branch DNA figures, also called X shaped, on our TEM grids, a careful analysis implementing the common criteria used to discriminate bona fide fork reversal from accidental crossing of long linear DNA molecules (41) indicated that while some features were compatible with the occurrence of reversed forks (symmetry), other features were not (absence of open-centring, not aligned arms). These structures accounted for less than 4% of the RIs in our conditions and their abundance was not altered after UVC irradiation or polη invalidation (Supplementary Figure S3A). These numbers are in perfect agreement with the yields detected in a recent work (54). We made similar observations by using the standard TEM procedure in bright field mode after Pt/C shadowing commonly used to study RIs (41) (Supplementary Figure S3B).
Altogether, our data show that polη-dependent TLS at RFs is a critical mechanism to sustain a continuous DNA synthesis, preventing accumulation of uncoupled forks and post-replicative ssDNA gaps in response to UV. Altered forks were also observed in polη-proficient cells to a lesser extent, suggesting that a subset of UV lesions cannot be bypassed at RFs even when polη is functional. These lesions are presumably the 6–4 photoproducts (6–4 PPs), which represent 20–25% of UV-induced DNA lesions and cannot be bypassed by polη alone (25). Moreover, our data support a higher occurrence of repriming events when polη is not functional.
PrimPol sustains RF progression in XPV cells but is not the only actor of repriming downstream of the UV lesions
Actors allowing the repriming of DNA replication downstream DNA damage remain elusive in eukaryotes. Recent studies revealed that the primase activity of PrimPol, an archaic primase and TLS polymerase, mediates RF progression after UV and hydroxyurea (HU) in vertebrates (20–22). However, to date, no structural data fully support this model. Moreover, PrimPol impairment does not lead to a dramatic sensitization to UV light, even when polη is not functional (55).
First, we showed that PrimPol, but not the catalytic polymerase sub-unit of polα (POLA1), was recruited on chromatin as early as 30 min after UVC and to a higher extent in XPV cells compared to XPVpolη cells (Figure 4A, Supplementary Figure S4A), suggesting an increased usage of PrimPol in absence of polη. DNA fiber assays showed that PrimPol depletion impeded RF progression after UV to a higher extent in XPV cells than in their complemented counterpart (Figure 4B, Supplementary Figure S4B, Supplementary Table S8), and suppressed the accumulation of ssDNA post-replicative gaps detected by the S1 nuclease DNA fiber assay (Supplementary Figure S4C, Supplementary Table S8), in agreement with previous findings (20,55,56). However, even in XPV cells, the impact of PrimPol deficiency was moderate in the first 20 min following UVC irradiation and became prominent in the 20–40 min time window further analysed, with a significant 2-fold decrease in the mean fork rate and the persistence of forks presenting a severe slow down (Figure 4B), suggesting that PrimPol is involved in a delayed backup mechanism for fork progression. The lack of effect of PrimPol depletion on RPA recruitment to chromatin early after UV irradiation also suggested that PrimPol activity was not required for most ssDNA accumulation (Figure 4C).
Figure 4.

PrimPol depletion further reduces replication track length and impacts on RI architecture in irradiated XPV cells. (A) Cellular fractionation was performed at the indicated time points following UVC irradiation at 25 J m–2 in XPV and XPVpolη cells (N = 3). Whole cell extracts (WCE) of unirradiated cells and chromatin-bound proteins were analysed by western blot using the indicated antibodies. (B) XPV cells were depleted for PrimPol (PP) and DNA fiber assay was performed as described in Figure 1D. Upper panel show the distributions of replication track length (n > 300, Kruskal-Wallis followed by Dunn's multiple comparisons test, ns: not significant, ***P < 0.001, ns: not significant; **P < 0.01, ****P < 0.0001). siPRIMPOL efficiency was confirmed by western blot. An independent experiment was performed with CldU and IdU as pre- and post-UVC labels, respectively, and gave the same results. The middle and lower panels show the average post-UV length (mean ± SD, t-test, *: P < 0.05) and the proportion of replication signals with post-UV length shorter than 30% of the corresponding pre-UV length (mean ± SD, ordinary two-way ANOVA followed by Sidak's multiple comparisons test, **P < 0.01, ***P < 0.001) for these two experiments, respectively. (C) Upper panel: kinetics of recruitment of RPA70 on chromatin after UVC (25 J m–2) in XPV and XPVpolη cells upon PrimPol depletion (N = 3). Lower panel: WCEs were used to confirm the efficiency of PrimPol depletion. (D) Relative uncoupled forks and distribution of ssDNA lengths at the elongation point in irradiated XPV cells following PrimPol depletion. (E) Relative gapped forks and distribution of gap lengths in irradiated XPV cells following PrimPol depletion. At least 50 forks were characterized in two independent experiments. Histograms are the mean ± SD of the two experiments (one sample t-test, ns: not significant, *P < 0.05). ssDNA length distributions are pools of the two experiments (dot plot with median and interquartile range).
TEM analysis did not reveal any significant impact of PrimPol depletion on RI molecular architecture in polη-expressing cells 30 min after UV (Supplementary Figure S4D, Supplementary Table S9). In XPV cells, the median ssDNA length at the elongation point unexpectedly decreased when PrimPol was depleted (Figure 4D, Supplementary Table S9). Although this difference was not statistically significant, it was observed in two independent experiments (data not shown) and was not observed in polη-expressing cells (Supplementary Figure S4D). One simple explanation could be that decreased RF rate observed upon PrimPol depletion reduces the chance to encounter a UV lesion, mitigating RF uncoupling. Alternatively, remodelling of some RIs might occur in absence of both polη and PrimPol. In support with this latter hypothesis, we observed a transient decrease of the amounts of PCNA and other replisome components on chromatin in PrimPol-depleted XPV cells (Supplementary Figure S4E).
Although the overall number of long internal gaps is lower in the absence of PrimPol (65 gaps versus 113 gaps on the 102 RFs analysed in siPRIMPOL- and siNT-treated XPV cells, respectively), numerous long post-replicative gaps still formed in PrimPol-depleted XPV cells (Figure 4E and Supplementary Figure S4F). PrimPol depletion led to a 25% decrease in the proportion of gapped RFs (Figure 4E). Interestingly, this reduction seemed specifically related to the RIs bearing both uncoupling and internal gaps (Supplementary Figure S4G), suggesting that PrimPol primase activity may deal with regions bearing a high density of DNA damage. On this type of RIs, the extended ssDNA at the elongation point allows identifying the leading strand. We observed that PrimPol depletion preferentially impaired gap formation on the leading strand (Supplementary Figure S4H), thus suggesting that PrimPol can indeed contribute to reprime some stalled leading strands in XPV cells. However, although siRNAs against PrimPol were highly efficient (Figure 4C), the impairment in gap formation was only partial, indicating that PrimPol-independent mechanisms also occur. These mechanisms, nonetheless, did not involve a compensatory increase in recruitment of POLA1 on chromatin (Supplementary Figure S4D).
Hence, our data suggest that PrimPol primase activity is required in specific conditions, i.e high density of UV-induced lesions and unavailability of polη-dependent bypass at the RF. The global effect of PrimPol depletion on RF progression observed in DNA spreading assay may therefore reflect the involvement of its TLS polymerase or lesion skipping activities. Moreover, we cannot rule out the possibility that impaired mitochondrial DNA replication induced by PrimPol deficiency indirectly contributes to the impaired genome replication in presence of DNA damage (21,55,57).
RAD51 promotes the repriming of uncoupled RFs
In addition to its role in DSB repair, HR was proposed to restart stalled RFs and to contribute to fill in post-replicative gaps both in yeast and mammalian cells (5,32,58–60). Moreover, HR was proposed to act earlier and to a greater extent in irradiated XPV cells than in polη-proficient cells to compensate for polη deficiency (13). However, it is not clear whether this earlier commitment of HR drives RF progression or results from the precocious and larger accumulation of post-replicative gaps in absence of polη.
As stated before, we detected by iPOND an accumulation of RAD51 behind RFs in irradiated polη-expressing cells (Figure 1A, Supplementary Figure S1A). This was also observed in wild-type immortalised human and mouse fibroblasts (Supplementary Figures S5A, B) and is indicative of a post-replicative commitment of RAD51 in gap repair. The accumulation of RAD51 behind RFs could be observed after UV irradiation of both XPVpolη and XPV cells, and was thus independent of the polη status (Figure 5A). However, a specific increase of RAD51 in the vicinity of RFs upon UV irradiation was observed in XPV cells compared to polη-complemented cells (Figure 5A). This was confirmed by proximity ligation assay (PLA) between RAD51 and nascent DNA (Supplementary Figure S5C, Supplementary Table S10). Accumulation of RAD51 at RFs was also observed in XPV cells at a lower UVC dose (Supplementary Figure S5D).
Figure 5.

RAD51 limits RF uncoupling in UV-irradiated XPV cells. (A) iPOND was performed in XPV and XPVpolη cells as described in Figure 1A with a 1-hour thymidine chase when indicated. Immunoblotting was done with the indicated antibodies. (B) DNA fiber assay was performed in XPV cells pre-treated or not with 20 μM B02 for 8 h. The distributions of replication track lengths are shown (n > 200, Kruskal–Wallis followed by Dunn's multiple comparisons test, ns: not significant; ****P < 0.0001). (C) TEM analysis was performed 30 min after UVC exposure (25 J m–2) in XPV cells pre-treated or not with 20 μM B02 for 8 h in two independent experiments. At least 50 RIs were analysed per condition in each experiment. Upper and lower left panels: distributions of all the ssDNA stretches measured at and behind the elongation point, respectively (dot plots with median and interquartile range, pool of two experiments). Upper and lower right panels: number of uncoupled and gapped forks, respectively, expressed as a percentage of all the forks analysed (mean ± SD of two independent experiments, t-test, **P < 0.01, ***P < 0.001). (D) XPV cells were depleted or not for PrimPol and pre-treated with 20 μM B02 for 8 h prior to UVC exposure (25 J m–2). TEM analysis was performed 30 min after irradiation. Values are the mean ± SD of two independent experiments with 50 RFs analysed per condition and per experiment. siPRIMPOL (siPP) efficiency was assessed by western blot.
To further dissect the functions of RAD51 at and behind RFs, we decided to use the RAD51 inhibitor B02, which was proposed to inhibit the binding of RAD51 to the DNA, subsequently inhibiting its strand invasion activity (61,62). First, we better delineated the effects of B02 on the RAD51 filament assembly and morphology in in vitro reactions coupled to TEM imaging (Supplementary Figure S6A, Supplementary Table S11). While RAD51 filaments had a regular and homogeneous helical structure in the absence of B02, addition of increasing concentrations of B02 led to the formation of heterogeneous filaments, interrupted by the presence of clusters (‘bright spots’) (Supplementary Figure S6A). This was associated with a significant decrease in the filament length. The increase of free RAD51 in the background, as well as the appearance of DNA molecules completely devoid of RAD51 (about 5% at 20 μM of B02), suggested a dissociation of RAD51, rarely total and mainly local, which could explain the formation of these disorganized filaments and the reported loss of strand invasion enzymatic activity (62). Although the in vitro effects of B02 on RAD51 filaments occur relatively quickly, the kinetic of its cellular uptake and in vivo activity is still unclear (61,63) and we observed a decrease of the chromatin-bound RAD51 after 6 h of B02 treatment in unirradiated XPV cells (Supplementary Figure S6B).
We next addressed the consequences of B02-mediated RAD51 inhibition in cells early after UV irradiation. We pre-treated XPV cells with B02 for 7–8 h prior to UV exposure and confirmed by immunofluorescence that this treatment impaired the UV-induced recruitment of RAD51 on chromatin in S phase cells (Supplementary Figure S6C, Supplementary Table S10). In these experimental conditions, the inhibition of RAD51 led to shorter post-UV replication tracks, while pre-UV tracks or tracks in unirradiated cells were not altered (Figure 5B, Supplementary Table S12). The post-UV track lengths were not restored by inhibition of the MRE11 3′-5′ exonuclease activity with Mirin (Supplementary Figure S6D, Supplementary Table S12), suggesting that post-UV track shortening upon RAD51 inhibition was not the consequence of extensive resection of neo-synthesised DNA. This hypothesis was further confirmed by TEM analysis of the RFs, since we observed similar ssDNA length distributions in DMSO- and B02-treated cells (Figure 5C, left panels). The TEM analysis though highlighted an increase of the proportion of uncoupled RFs upon B02-mediated RAD51 inhibition with the parallel observation of an overall reduction of the occurrence of gapped forks (Figure 5C right panels, Supplementary Table S13). These observations are in good agreement with the reduced susceptibility of nascent strands towards S1 nuclease cleavage in these conditions (Supplementary Figure S6E, Supplementary Table S12). Moreover, the proportion of gapped forks was not further reduced following PrimPol depletion in B02-treated cells (Figure 5D, Supplementary Table S14). Altogether, these data raise the possibility that RAD51 acts at RFs to promote repriming, an activity that, considering our in vitro analysis (Supplementary Figure S6A), would require the formation of a functional RAD51 filament.
To confirm the results obtained with the B02-mediated inhibition of RAD51, we decided to knock down RAD51 by RNA interference in XPV cells. Of note, since it drastically impaired cell growth and led to tetraploidization in our cellular model (data not shown), we lowered the amount of siRNAs and performed our experiments in the first 36 h following transfection. Similar to what we observed with a B02 treatment, the RAD51 depletion led to shorter post-UV replication tracks (Supplementary Figures S7A, Supplementary Table S15). This was associated with a progressive attrition of pre-UV replication tracks, as already described (26). Using TEM, we also observed a strong reduction in the proportion of RFs bearing post-replicative gaps in RAD51-depleted XPV cells (Supplementary Figure S7B, Supplementary Table S16), supporting the possibility of an impaired repriming. Under these conditions of acute depletion of RAD51, we observed a global decrease in the length of the ssDNA stretches at RFs (Supplementary Figure S7C) that could be explained by fork backtracking, an event that did not seem to occur under a milder inhibition of RAD51 with B02, where structures of the uncoupled RFs were maintained. Noteworthy, the impact of RAD51 depletion was less severe in polη-proficient cells, and it specifically affected RIs with both gaps and uncoupling (Supplementary Figures S7D, E, Supplementary Table S16). Once again, these results highlight the key role of polη at UV-stalled replication forks.
RAD51 induces recombination structures during post-replicative gap repair
After thorough characterization of RF architecture early after UV irradiation, we turned our attention to the DNA intermediates that could be detected by TEM in samples harvested 2 h after UV exposure (25 J m–2). This analysis revealed, in addition to RF figures, multi-branched (or H-shaped) structures, usually bearing ssDNA stretches, in both XPV and XPVpolη cells (Figure 6A). In XPV cells, these structures represented 21% of the observed intermediates while they were rarely observed in unirradiated samples (<2%) or 30 min after UVC. We determined these were RAD51-dependent structures, as their levels were reduced in the presence of B02 (Figure 6B). The global shape of these structures and the median length of the central bar of the H (65.2 nm with DMSO versus 55.9 nm with B02) were not altered by B02 treatment (Figure 6C, Supplementary Table S17). H-shaped structures could originate from converging forks at sites of replication termination, but the frequent presence of ssDNA at the central link of the H suggests another origin. Notably, TEM analysis of DNA intermediates resulting from in vitro RAD51-dependent strand-exchange assay revealed the formation of exactly the same type of H-shaped structures (Figure 6D). Interestingly, the link of these branched intermediates also corresponded to ssDNA resulting from the strand transfer between DNA molecules and displayed a length compatible with the in vivo H-shaped structures (97.7 ± 19 nm long, about 189 nucleotides). In addition, we also visualized in vivo rare X-shaped structures containing ssDNA at the junction, in an open-centred configuration (Figure 6A, panels v and vi), similar to what has been previously observed for natural (64,65) or synthetic (38) Holliday Junctions, but also for fork reversal figures (41). In our view, all these multi-branched structures correspond to post-replicative HR intermediates resulting from strand exchange. Accordingly, some of these intermediates were detected in the vicinity of the RF elongation point (Figure 6A, panels vii–ix).
Figure 6.

RAD51-dependent H-shaped structures accumulate in genomic DNA 2 h following UVC challenge. (A) Examples of in vivo five-branched structures (H-shaped) observed by TEM 2 h after UVC (25 J m–2) in XPV cells. Inlays represent enlargements of the regions indicated by dotted arrows. The central bar of the H-shaped structures can vary in length and be made of dsDNA (i, ii, vii), ssDNA (iii, viii) or both (iv). Very rarely, open-centred structures reminding Holliday Junctions were observed (v, vi). These structures can be located next to the elongation point of a fork (vii, viii, ix). (B) Relative levels of H-shaped structures (normalized to the count of 100 RFs). Values are the mean ± SD of two independent experiments (ordinary one-way ANOVA followed by Tukey's multiple comparisons test, **P < 0.01, ***P < 0.001). TEM analysis was performed in XPV cells 2 h after UVC (25 J m–2). When indicated, cells were pre-treated with 20 μM B02 for 8 h. (C) Lengths of the H-shaped structure central bars (pool from the two independent experiments, bar is the median of the distribution, Kruskal-Wallis with Dunn's multiple comparisons test, ns: not significant). (D) Examples of joint molecules formed in the in vitro RAD51-dependent strand-exchange assay observed by TEM. The circular ssDNA Phix174 virion was incubated with RAD51, followed by RPA addition, then linearized Phix174 RFI homologous plasmid was added to the reaction. RAD51 polymerizes onto the ssDNA substrate to form the presynaptic filament and then catalyses the strand invasion and strand transfer with the linear homologous dsDNA. Final products are linear ssDNA (lss) and nicked dsDNA plasmid (nc). For TEM observations, reactions were subjected to UVA/trioxalen in vitro crosslinking, deproteinization, and spread using the BAC hyperphase method. (i) circular ssDNA Phix174 virion (css) and linearized homolog plasmid (lds). (ii), (iii) Representative views of joint molecules. Formation of five-branched (H-shaped) structures is shown in (iii). (iv) Final products of the reaction. Scale bar = 200 nm.
To further investigate the post-replicative function of RAD51, we first assessed the long-term dynamics of RPA accumulation on chromatin of cells treated with B02 immediately after irradiation at a low UVC dose (2 J m–2). Whereas B02 did not alter RPA accumulation on chromatin 3 and 8 h after irradiation in XPV cells, it impaired RPA disappearance at later time points in a dose-dependent manner (Figure 7A and Supplementary Figure S8A). Of note, 2 J m–2 was too low to induce an increase of chromatin-bound RPA in polη-proficient cells (Figure 7A), but, to a lower extent, a similar effect was observed in XPVpolη cells at a higher UVC dose (Supplementary Figure S8B). These results indicate that functional RAD51 is required for efficient post-replicative gap repair. Indeed, this parallels the observation of gap repair defects upon B02 treatment in S1 nuclease DNA fiber assays in XPV cells (Figure 7B, Supplementary Table S18). Addition of B02 after UVC irradiation had no impact in RF progression or post-replicative gap formation in the initial 40-min labelling period, while showing a clear difference in completion of gap repair. Complete repair was achieved between 4 and 8 h in DMSO-treated cells, while gaps persisted in cells treated with B02 (Figure 7B, Supplementary Table S18). The lack of effect of B02 on the initial labelling period suggests that, unlike what was observed by pre-incubating cells with B02 before irradiation (Supplementary Figure S6D), the approach used here with addition of B02 after UVC irradiation allowed separating the replicative and post-replicative functions of RAD51. From the above data, we could infer that the accumulation of RAD51 on post-replicative chromatin correlates with the observation of multi-branched structures in vivo which illustrates post-replicative RAD51-dependent recombination events occurring to fill in the gaps accumulated behind the RFs.
Figure 7.

RAD51 is required for post-replicative gap filling and cell survival. (A) XPV and XPVpolη cells were irradiated with UVC at 2 J m–2. 10 μM B02 was added immediately after irradiation. RPA70 recruitment to chromatin was assessed at different times by cellular fractionation (N = 3). (B) XPV cells were pulsed with IdU for 20 min before UVC irradiation at 10 J m–2 followed by CldU pulse for 40 min. When indicated, B02 (20 μM) was added with CldU and maintained until permeabilization. S1 nuclease treatment and DNA spreading were performed immediately after the CldU pulse or after a chase in pre-heated medium for 4 or 8 h. CldU lengths were measured on bicolor signals and plotted as dot plots with median and interquartile range (n > 320, Kruskal–Wallis followed by Dunn's multiple comparisons test, N = 2). (C) XPV and XPVpolη cells were irradiated with increasing UVC doses. When indicated, 10 μM B02 was added post-irradiation. Cell survival was assessed 72 h after treatment (mean ± SD of three independent experiments. For each condition, 100% represents the number of cells in the non-irradiated conditions). (D) Cell survival following 2 J m–2 UVC upon increasing concentrations of B02 (mean ± SD of four independent experiments. For each cell line, 100% represents the number of cells in the DMSO condition).
Finally, we assessed the impact of RAD51 inhibition on cell survival after UVC irradiation. Whereas B02 treatment had similar effects on cell proliferation in unirradiated XPV and XPVpolη cells (data not shown), it increased UVC sensitivity to a higher extent in XPV cells than in XPVpolη cells (Figures 7C,D, Supplementary Table S19). In particular, at the sub-lethal dose of 2 J m–2, XPV cells were sensitized by lower concentrations of B02 than XPVpolη cells (Figure 7D). Importantly, these findings were fully recapitulated in polη-depleted MRC5-V1 cells (Supplementary Figures S8C,D).
Altogether, we thus propose that RAD51 acts in two different mechanisms during replication of UV-damaged DNA. First, it favours RF progression through repriming when bypass by polη is not possible. As we did not observe recombination-like structures early after UV, we think that this function does not involve RAD51 strand-invasion activity. Secondly, it accumulates behind RFs to allow recombination-based post-replicative gap-filling independently of the polη status. As XPV cells more strongly rely on repriming and gap filling for completion of DNA replication, this consequently results in an important dependence of XPV cells towards RAD51 activities.
DISCUSSION
The present time-course characterization of RIs architecture by TEM, combined with biochemical approaches, provides new insights for the understanding of the organisation of DDT after exposure of human cells to UV irradiation. From our data, we propose that polη-dependent TLS is the primary and main fork restart mechanism and that its efficiency configures the DDT response. When this pathway is not possible, either at UV lesions that cannot be bypassed by polη alone or because polη is not functional, DDT is diverted to a post-replicative mechanism through repriming. Interestingly, our results point to a participation of RAD51 in both the repriming and the fill in of the post-replicative gaps generated by repriming and that only this second function involves strand exchange transactions.
Impact of UV damage and polη deficiency on the architecture of replication forks
Our TEM analysis showed that the early response of RFs to UV damage is the generation of abnormally long ssDNA regions at and behind the elongation point, the amount of which is related to the capacity of polη to bypass lesions in the vicinity of RFs. Thanks to the unique structure of its catalytic site, polη ensures efficient and accurate bypass of TT-CPDs (8). Furthermore, we have previously shown that polη travels with RFs during unperturbed S phase (37) and here we show that it further rapidly accumulates at RFs after UV. Hence, replisomes of WT cells possess the enzyme allowing the on-the-fly bypass of the most abundant UV-induced lesion and, therefore, a limited impact of UV damage on RF architecture, whereas polη deficiency leads to RF uncoupling and accumulation of ssDNA gaps in both lagging and leading strands.
In a yeast reconstituted replisome, the stalling of the leading strand synthesis at a single lesion leads to the uncoupling of the replicative helicase and the slowdown of DNA unwinding (18,66). Our data support this model and the reduced replication track lengths measured after UV by DNA fiber experiments can be interpreted as the result of successive uncoupling/recoupling cycles at each leading strand lesion encountered by a single RF during the labelling period. As WT cells also show a significant decrease of post-UV replication track lengths, we infer that polη does not prevent uncoupling but rather allows rapid recoupling of the stalled leading strand in vivo, as recently shown in vitro (51).
On naked DNA templates, uncoupling can proceed to more than 1 kb past a leading strand CPD (18,66). However, our measurements indicate that uncoupling is limited in length in vivo, even when polη is not functional, and rarely exceeds 450 nucleotides. As we could observe longer ssDNA after treatment with camptothecin using the same experimental approach (38), this is unlikely to be due to DNA breaks during the DNA preparation, which could have led to the loss of longer ssDNA stretches. In the absence of polη, alternative tolerance pathways at the RF, including repriming and the recently described alternative bypass by DNA polymerase theta (polθ) (12) should limit the extent of uncoupling. Uncoupling may also be restricted in vivo by topological constraints, chromatin ahead of the RF or checkpoint activation. In support of the latter hypothesis, Rad53 has been shown to restrict CMG helicase activity in the yeast purified replisome system (67).
Numerous recombination-based mechanisms have been proposed to restart stalled RFs, such as HR or template switch. Moreover, fork reversal, which involved the key HR protein RAD51, was proposed as a universal response to replication stress in human cells (33). We, however, did not observe any structural evidence of recombination or fork reversal in the early response to UV damage, even when polη is not functional. This is in agreement with previous TEM studies, which showed that reversed forks are observed in specific conditions in response to bulky DNA lesions in yeast, i.e. in mutants inactivated for the checkpoint pathway (5) or unable to efficiently reprime replication (68). Accordingly, recent reports have unravelled a competition between TLS at RF, RF repriming and RF reversal (69,70) and distinct roles of fork remodelling and protection factors in response to HU or mitomycin C (71). It is therefore more than likely that the cell type and the origin of the replication stress dictate the choice between TLS, repriming and reversal. Here, 30 min after UVC, X-shaped structures were evidenced without open centred shape, contrary to those observed 2 h after UVC. Although we do not exclude that fork reversal may exist in our experimental conditions, it would appear as a minor event. We further demonstrate here that on-the-fly direct bypass by polη is the favoured early response to UV lesions in skin fibroblasts, outcompeting the two other mechanisms.
Repriming as a damage tolerance mechanism that compensates polη deficiency
Our structural data showed that repriming is the alternative mechanism used to promote RF progression when on-the-fly polη-dependent TLS is not possible. We therefore propose that repriming constitutes a tolerance mechanism per se. This blockage-skipping mechanism was characterized in bacteria and, although not yet fully understood, also described in yeast and human cells (19,25,33,72–74).
Recently, in silico prediction based on conserved motifs with archea/eukaryotes AEP superfamily of primase identified a novel primase-polymerase, named PrimPol, in most eukaryotes, including mammals (20–22,75). We confirmed that PrimPol depletion further impaired RF progression in irradiated XPV cells and provided structural evidence that PrimPol can indeed reprime leading strand synthesis in vivo. However, our TEM analysis also suggests that its primase activity has a limited role after UV, even in a polη-deficient background, which correlates with the modest impact of PrimPol deficiency on cell survival (55) and the observation that PrimPol is rather involved in an adaptive response to successive genotoxic exposures (69). PrimPol also possesses a TLS polymerase activity. However, its active site is too constrained to accommodate UV lesions (76), which it may rather skip through primer realignment (20,75,76). Therefore, PrimPol could contribute to RF progression in a primase-independent manner when polη is not functional. PrimPol has been recently shown to be controlled by the ATR checkpoint (69,78), a pathway that is over-activated in UV-exposed polη-deficient cells (24). It would therefore be interesting to assess the impact of PrimPol on UV-induced mutagenesis in XPV cells.
Beyond PrimPol, the polα primase-polymerase remains the only other currently identified candidate for repriming. The E. coli replisome possesses the inherent capacity to tolerate lesions in the leading strand by DnaG primase-dependent de novo priming (73), a mechanism that competes with TLS (79). Reconstitution of yeast replisome with purified proteins showed that repriming of a stalled leading strand by polα is rather inefficient (18). Yeast cells do not possess a PrimPol homologue but TEM analysis revealed that repriming occurs in vivo (5,68). It is therefore most likely that accessory factors, post-translational modifications and/or RF remodelling processes promote polα (or a currently unidentified other primase) access to the stalled leading strand in vivo. Their identification would provide further mechanistic insights into the repriming mechanism in eukaryotes.
Dual requirement for RAD51 at and behind RFs
Our data are in agreement with the existence of two spatiotemporally distinct functions of RAD51 in response to UV damage: a role at the RF to promote replication progression through repriming when bypass by polη is not possible, and a second role in DNA damage tolerance behind the fork, by recombination mediated post-replication gap-filling, in line with what has been recently shown at BPDE-induced DNA adducts (60).
Our data strongly suggest that RAD51 indeed works at stalled RFs in a strand-exchange independent manner to sustain their progression when encountering UV lesions. This is consistent with previous studies that shed light on non-canonical functions of RAD51 to escort and stabilize ongoing RFs, but also to promote the restart of stalled RFs through their remodelling (26,80–85), a role reportedly independent of RAD51 strand-exchange activity (86). RF restart activities are being proposed to constitute a separate RAD51-mediated pathway to that involved in canonical HR repair (81). However, contrary to previous reports (26,33,80), we showed that RAD51 inhibition or depletion led to a decreased number of UV-induced post-replicative gaps. Moreover, gap formation was not further reduced by PrimPol depletion in B02-treated cells. Hence, we hypothesize that RAD51 may contribute to repriming. This hypothesis contradicts the study from Vallerga et al that reported that RAD51 prevents PrimPol-mediated excessive fork elongation (26). This discrepancy might be due to different experimental conditions (e.g. U2OSsiPOLHvs XPV cells and RAD51 depletion vs RAD51 inhibition using B02 for Valerga's study and our study, respectively). However, our hypothesis is supported by a recent report showing that RAD51 prevents RF uncoupling at methylmethane sulfonate (MMS)- induced lesions in yeast (87).
How RAD51 promotes repriming will require further investigations. Yeast Polα activity on the leading strand is inhibited by RPA in vitro (18) and a direct interaction between RAD51 and polα has already been reported in Xenopus extracts (88). It is therefore tempting to speculate that RAD51 can promote access of polα to the stalled leading strand. Similarly, although RPA was shown to recruit PrimPol on DNA, short ssDNA templates saturated by RPA are not good substrates for PrimPol primase activity (77,89,90). Given that we showed that ssDNA generated at the RFs is limited in length, replacing RPA with RAD51 could also favour repriming by PrimPol. Alternatively, RAD51 could also indirectly promote repriming through its protection activity against nucleolytic degradation. Interestingly, in contrast to what was observed after other genotoxic insults (38,80,82,88,91), the exonuclease activity of MRE11 does not trigger fork degradation after UV. Further experiments are warranted to investigate the role of other nucleases in the processing of uncoupled and reprimed RFs.
The second role of RAD51 in response to UV-irradiation would entail the canonical functions of homology-search and strand-exchange in the context of ssDNA gaps. Here, our TEM analysis leads to the original observation of recombination-related intermediates occurring in vivo on genomic DNA in human cells in a RAD51-dependent manner. Of note, H-shaped recombination intermediates had already been evidenced by electron microscopy in E. coli infected with T4 phages, Xenopus oocytes extrachromosomal DNA, yeast mitochondrial DNA or in yeast cells treated with MMS (92–95). Previous studies conducted in budding yeast evidenced that UV-induced recombination does not rely on double-strand breaks but on ssDNA gap processing (59). Our in vitro studies showed that the five-branched structures observed in vivo are similar to intermediates formed during RAD51-dependent strand invasion processes, indicating that they can form independently of the presence of a fork structure and further supporting the idea that they form during post-replicative gap filling. Furthermore, they can be, although rarely, located behind a replication fork and their architectures are compatible with the models of template switch and recombinational gap repair described in the literature (2). Nonetheless, further work is needed to unravel the molecular mechanisms underlying their formation and resolution and determine if they are intermediates in the formation of UV-induced SCEs. Interestingly, a previous work conducted in yeast proposed that RAD51 loaded at RFs remains loaded on ssDNA gaps formed by repriming ahead of MMS lesions to allow their later repair by HR (96). This model may explain the increasing amounts of RAD51 behind RFs observed by iPOND in the present work, as multiple gaps accumulate in the wake of RFs.
Alternative TLS polymerases and mutagenesis in XPV syndrome
In WT cells, the bypass of 6–4 PPs is achieved through Polζ-dependent gap filling (25,97). Several studies demonstrated that polζ, polκ, and to a lesser extent polι, partially compensate for polη deficiency in XPV cells, leading to mutagenic CPD bypass (9,10,12). Here, we show that polζ and polκ do not contribute to the progression of RFs in the absence of polη, indicating that alternative TLS of CPDs proceeds post-replicatively. Importantly, in budding yeast, the mismatch repair (MMR) pathway operates coupled to DNA replication (98) and is therefore less efficient on mismatches created during polζ-mediated extension step of the gap filling process (99,100). MMR proteins were also shown to travel with the replication machinery in human cells (101). This suggests that the delayed bypass of TT-CPDs imposed by polη deficiency may further amplify the intrinsic error-proneness of the alternative TLS pathways taking place in XPV cells, by lowering mismatch correction efficiency, leading to untargeted mutagenesis beyond the lesions.
A recent study has identified polθ as the inserter TLS polymerase initiating the mutagenic bypass of CPDs before further extension by polζ or polκ (12). Intriguingly, polθ deficiency leads to impaired RF progression after UV, in a synergic manner with polη deficiency (12). This raises the possibility that the insertion made by polθ at the RF stimulates repriming, a hypothesis that could be tested in future TEM experiments. It would also be important to understand why alternative TLS polymerases do not bypass UV lesions at the RF, a situation that may favour MMR-mediated correction of their errors. We have previously shown that SUMOylation of polη promotes its association with the replication machinery (37). The SUMOylation site of polη is not conserved in other Y family DNA polymerases, suggesting a regulation specific to polη. Hence, TLS polymerases could be differentially regulated for access of the vicinity of RFs. Alternatively, when polη is not functional, the slower efficiency and/or kinetics of photoproduct bypass may also leave time for repriming to occur and RF to progress before TLS is completed.
Another open question concerns the respective contributions of TLS and HR to gap repair. Previous studies in budding yeast proposed a redundancy between TLS and HR to process UV-induced post-replicative gaps (59). In mammalian cells, depletion of polη and/or polθ leads to a higher number of RAD51-dependent SCEs after UV (12), suggesting that HR compensates for TLS deficiency. Elsewhere, a competition between TLS and HR pathways has been extensively suggested (58,102–105). The suggested role of TLS polymerases in HR itself further blurry the picture (106–108). Here we show that RAD51 is preferentially recruited behind RFs and that its inhibition sensitizes cells to UV, both in polη-deficient and polη-proficient cells. One possibility could be that the whole post-replicative tolerance is mediated through HR events but that they involved synthesis steps made by TLS polymerases. On the other hand, as the recombination intermediates we observed are rare compared to replication forks, RAD51 could also have a HR-independent function behind RFs, similar to its role at RFs, through protection of the gaps in order to allow their fill in by canonical TLS.
Altogether, our data allow drawing a chronology of DNA damage tolerance mechanisms in response to UV lesions with polη acting first at the RF, followed by repriming supported by a strand-exchange independent function of RAD51 if polη is not efficient and further post-replicative gap filling through TLS and/or HR. Hence, our study supports a collaboration between the different DDT pathways for replication completion of damaged DNA and the idea that the DDT pathway choice is dictated by the type of lesion and the most efficient mechanism available, giving a priority to the continuity of DNA synthesis.
DATA AVAILABILITY
All the data supporting the findings of this study are available with the online version of the article.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are indebted to L. Blanco for the kind gift of anti-PrimPol antibody. The authors thank L. Blanco, A. Doherty, and all members of the P.L.K. lab for helpful discussions and J.H. Guervilly for critical reading of the manuscript. The authors thank Barbara Ben-Yamin and Raphaël Corre for the RT-qPCR experiments.
Notes
Present address: Emmanuelle Despras, Sorbonne Université, INSERM, Unité Mixte de Recherche Scientifique 938 and SIRIC CURAMUS, Centre de Recherche Saint-Antoine, Equipe Instabilité des Microsatellites et Cancer, Equipe labellisée par la Ligue Nationale contre le Cancer, F-75012 Paris, France.
Contributor Information
Yann Benureau, UMR9019 CNRS, Genome Integrity and Cancers, Laboratory Genome Integrity, Immune Response and Cancers, Equipe Labellisée La Ligue Contre Le Cancer, Gustave Roussy 94805, Villejuif, France; UMR9019 CNRS, Genome Integrity and Cancers, Laboratory DSB Repair, Replication stress and Genome Integrity, Gustave Roussy 94805, Villejuif, France; Université Paris-Saclay, France.
Caroline Pouvelle, UMR9019 CNRS, Genome Integrity and Cancers, Laboratory Genome Integrity, Immune Response and Cancers, Equipe Labellisée La Ligue Contre Le Cancer, Gustave Roussy 94805, Villejuif, France; Université Paris-Saclay, France.
Pauline Dupaigne, UMR9019 CNRS, Genome Integrity and Cancers, Laboratory DSB Repair, Replication stress and Genome Integrity, Gustave Roussy 94805, Villejuif, France; Université Paris-Saclay, France.
Sonia Baconnais, UMR9019 CNRS, Genome Integrity and Cancers, Laboratory DSB Repair, Replication stress and Genome Integrity, Gustave Roussy 94805, Villejuif, France; Université Paris-Saclay, France.
Eliana Moreira Tavares, UMR9019 CNRS, Genome Integrity and Cancers, Laboratory DSB Repair, Replication stress and Genome Integrity, Gustave Roussy 94805, Villejuif, France; Université Paris-Saclay, France.
Gerard Mazón, UMR9019 CNRS, Genome Integrity and Cancers, Laboratory DSB Repair, Replication stress and Genome Integrity, Gustave Roussy 94805, Villejuif, France; Université Paris-Saclay, France.
Emmanuelle Despras, UMR9019 CNRS, Genome Integrity and Cancers, Laboratory Genome Integrity, Immune Response and Cancers, Equipe Labellisée La Ligue Contre Le Cancer, Gustave Roussy 94805, Villejuif, France; Université Paris-Saclay, France.
Eric Le Cam, UMR9019 CNRS, Genome Integrity and Cancers, Laboratory DSB Repair, Replication stress and Genome Integrity, Gustave Roussy 94805, Villejuif, France; Université Paris-Saclay, France.
Patricia L Kannouche, UMR9019 CNRS, Genome Integrity and Cancers, Laboratory Genome Integrity, Immune Response and Cancers, Equipe Labellisée La Ligue Contre Le Cancer, Gustave Roussy 94805, Villejuif, France; Université Paris-Saclay, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
La Ligue Nationale contre le Cancer ‘Equipe labellisée’; Institut National du Cancer [INCa-PLBio2016-159, INCa-DGOS-Inserm12551]; Agence Nationale de la Recherche [FIRE 17-CE12-0015]; Paris-Sud University (MRM); E.D. was supported by Institut National du Cancer [INCa-PLBio2016-144]; Y.B. was supported by the SIRIC SOCRATE, Gustave Roussy [INCa-DGOS-Inserm 12551]. Funding for open access charge: INCa-DGOS-Inserm [12551]
Conflict of interest statement. None declared.
REFERENCES
- 1. Sale J.E., Lehmann A.R., Woodgate R.. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012; 13:141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Branzei D., Szakal B.. DNA damage tolerance by recombination: molecular pathways and DNA structures. DNA Repair (Amst.). 2016; 44:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sale J.E. Competition, collaboration and coordination–determining how cells bypass DNA damage. J. Cell Sci. 2012; 125:1633–1643. [DOI] [PubMed] [Google Scholar]
- 4. Naiman K., Philippin G., Fuchs R.P., Pagès V.. Chronology in lesion tolerance gives priority to genetic variability. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:5526–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lopes M., Foiani M., Sogo J.M.. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell. 2006; 21:15–27. [DOI] [PubMed] [Google Scholar]
- 6. Johnson R.E., Kondratick C.M., Prakash S., Prakash L.. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999; 285:263–265. [DOI] [PubMed] [Google Scholar]
- 7. Masutani C., Kusumoto R., Yamada A., Dohmae N., Yokoi M., Yuasa M., Araki M., Iwai S., Takio K., Hanaoka F.. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999; 399:700–704. [DOI] [PubMed] [Google Scholar]
- 8. Biertümpfel C., Zhao Y., Kondo Y., Ramón-Maiques S., Gregory M., Lee J.Y., Masutani C., Lehmann A.R., Hanaoka F., Yang W.. Structure and mechanism of human DNA polymerase eta. Nature. 2010; 465:1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ziv O., Geacintov N., Nakajima S., Yasui A., Livneh Z.. DNA polymerase zeta cooperates with polymerases kappa and iota in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:11552–11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gueranger Q., Stary A., Aoufouchi S., Faili A., Sarasin A., Reynaud C.-A., Weill J.-C.. Role of DNA polymerases eta, iota and zeta in UV resistance and UV-induced mutagenesis in a human cell line. DNA Repair (Amst.). 2008; 7:1551–1562. [DOI] [PubMed] [Google Scholar]
- 11. Yoon J.-H., Prakash L., Prakash S.. Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:18219–18224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoon J.-H., McArthur M.J., Park J., Basu D., Wakamiya M., Prakash L., Prakash S.. Error-Prone replication through UV lesions by DNA polymerase θ protects against skin cancers. Cell. 2019; 176:1295–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Limoli C.L., Giedzinski E., Cleaver J.E.. Alternative recombination pathways in UV-irradiated XP variant cells. Oncogene. 2005; 24:3708–3714. [DOI] [PubMed] [Google Scholar]
- 14. Diamant N., Hendel A., Vered I., Carell T., Reissner T., de Wind N., Geacinov N., Livneh Z.. DNA damage bypass operates in the s and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res. 2012; 40:170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sarkies P., Reams C., Simpson L.J., Sale J.E.. Epigenetic instability due to defective replication of structured DNA. Mol. Cell. 2010; 40:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karras G.I., Jentsch S.. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond s phase. Cell. 2010; 141:255–267. [DOI] [PubMed] [Google Scholar]
- 17. Daigaku Y., Davies A.A., Ulrich H.D.. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010; 465:951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor M.R.G., Yeeles J.T.P.. The initial response of a eukaryotic replisome to DNA damage. Mol. Cell. 2018; 70:1067–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elvers I., Johansson F., Groth P., Erixon K., Helleday T.. UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res. 2011; 39:7049–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mourón S., Rodriguez-Acebes S., Martínez-Jiménez M.I., García-Gómez S., Chocrón S., Blanco L., Méndez J.. Repriming of DNA synthesis at stalled replication forks by human primpol. Nat. Struct. Mol. Biol. 2013; 20:1383–1389. [DOI] [PubMed] [Google Scholar]
- 21. García-Gómez S., Reyes A., Martínez-Jiménez M.I., Chocrón E.S., Mourón S., Terrados G., Powell C., Salido E., Méndez J., Holt I.J.et al.. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell. 2013; 52:541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bianchi J., Rudd S.G., Jozwiakowski S.K., Bailey L.J., Soura V., Taylor E., Stevanovic I., Green A.J., Stracker T.H., Lindsay H.D.et al.. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell. 2013; 52:566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wan L., Lou J., Xia Y., Su B., Liu T., Cui J., Sun Y., Lou H., Huang J.. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013; 14:1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Despras E., Daboussi F., Hyrien O., Marheineke K., Kannouche P.L.. ATR/Chk1 pathway is essential for resumption of DNA synthesis and cell survival in UV-irradiated XP variant cells. Hum. Mol. Genet. 2010; 19:1690–1701. [DOI] [PubMed] [Google Scholar]
- 25. Quinet A., Martins D.J., Vessoni A.T., Biard D., Sarasin A., Stary A., Menck C.F.M.. Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Res. 2016; 44:5717–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vallerga M.B., Mansilla S.F., Federico M.B., Bertolin A.P., Gottifredi V.. Rad51 recombinase prevents mre11 nuclease-dependent degradation and excessive primpol-mediated elongation of nascent DNA after UV irradiation. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E6624–E6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sokol A.M., Cruet-Hennequart S., Pasero P., Carty M.P.. DNA polymerase η modulates replication fork progression and DNA damage responses in platinum-treated human cells. Sci. Rep. 2013; 3:3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Edmunds C.E., Simpson L.J., Sale J.E.. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell. 2008; 30:519–529. [DOI] [PubMed] [Google Scholar]
- 29. Temviriyanukul P., van Hees-Stuivenberg S., Delbos F., Jacobs H., de Wind N., Jansen J.G.. Temporally distinct translesion synthesis pathways for ultraviolet light-induced photoproducts in the mammalian genome. DNA Repair (Amst.). 2012; 11:550–558. [DOI] [PubMed] [Google Scholar]
- 30. Lettier G., Feng Q., Mayolo A.A.de, Erdeniz N., Reid R.J.D., Lisby M., Mortensen U.H., Rothstein R. The role of DNA double-strand breaks in spontaneous homologous recombination in s. cerevisiae. PLoS Genet. 2006; 2:e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mozlin A.M., Fung C.W., Symington L.S.. Role of the saccharomyces cerevisiae rad51 paralogs in sister chromatid recombination. Genetics. 2008; 178:113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adar S., Izhar L., Hendel A., Geacintov N., Livneh Z.. Repair of gaps opposite lesions by homologous recombination in mammalian cells. Nucleic Acids Res. 2009; 37:5737–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J.A., Herrador R., Vindigni A., Lopes M.. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015; 208:563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cortez D. Replication-Coupled DNA repair. Mol. Cell. 2019; 74:866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stary A., Kannouche P., Lehmann A.R., Sarasin A.. Role of DNA polymerase eta in the UV mutation spectrum in human cells. J. Biol. Chem. 2003; 278:18767–18775. [DOI] [PubMed] [Google Scholar]
- 36. Zlatanou A., Despras E., Braz-Petta T., Boubakour-Azzouz I., Pouvelle C., Stewart G.S., Nakajima S., Yasui A., Ishchenko A.A., Kannouche P.L.. The hMsh2-hMsh6 complex acts in concert with monoubiquitinated PCNA and pol η in response to oxidative DNA damage in human cells. Mol. Cell. 2011; 43:649–662. [DOI] [PubMed] [Google Scholar]
- 37. Despras E., Sittewelle M., Pouvelle C., Delrieu N., Cordonnier A.M., Kannouche P.L.. Rad18-dependent SUMOylation of human specialized DNA polymerase eta is required to prevent under-replicated DNA. Nat. Commun. 2016; 7:13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Benureau Y., Moreira Tavares E., Muhammad A.-A., Baconnais S., Le Cam E., Dupaigne P.. Method combining BAC film and positive staining for the characterization of DNA intermediates by dark-field electron microscopy. Biol Methods Protoc. 2020; 5:bpaa012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vollenweider H.J., Sogo J.M., Koller T.. A routine method for protein-free spreading of double- and single-stranded nucleic acid molecules. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dubochet J., Ducommun M., Zollinger M., Kellenberger E.. A new preparation method for dark-field electron microscopy of biomacromolecules. J. Ultrastruct. Res. 1971; 35:147–167. [DOI] [PubMed] [Google Scholar]
- 41. Neelsen K.J., Chaudhuri A.R., Follonier C., Herrador R., Lopes M.. Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol. Biol. 2014; 1094:177–208. [DOI] [PubMed] [Google Scholar]
- 42. Tavares E.M., Wright W.D., Heyer W.-D., Le Cam E., Dupaigne P.. In vitro role of rad54 in Rad51-ssDNA filament-dependent homology search and synaptic complexes formation. Nat. Commun. 2019; 10:4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Veaute X., Jeusset J., Soustelle C., Kowalczykowski S.C., Le Cam E., Fabre F.. The srs2 helicase prevents recombination by disrupting rad51 nucleoprotein filaments. Nature. 2003; 423:309–312. [DOI] [PubMed] [Google Scholar]
- 44. Takedachi A., Despras E., Scaglione S., Guérois R., Guervilly J.H., Blin M., Audebert S., Camoin L., Hasanova Z., Schertzer M.et al.. SLX4 interacts with RTEL1 to prevent transcription-mediated DNA replication perturbations. Nat. Struct. Mol. Biol. 2020; 27:438–449. [DOI] [PubMed] [Google Scholar]
- 45. Quinet A., Carvajal-Maldonado D., Lemacon D., Vindigni A.. DNA fiber analysis: mind the gap!. Methods Enzymol. 2017; 591:55–82. [DOI] [PubMed] [Google Scholar]
- 46. Kannouche P., Broughton B.C., Volker M., Hanaoka F., Mullenders L.H., Lehmann A.R.. Domain structure, localization, and function of DNA polymerase eta, defective in xeroderma pigmentosum variant cells. Genes Dev. 2001; 15:158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Despras E., Delrieu N., Garandeau C., Ahmed-Seghir S., Kannouche P.L.. Regulation of the specialized DNA polymerase eta: revisiting the biological relevance of its PCNA- and ubiquitin-binding motifs. Environ. Mol. Mutagen. 2012; 53:752–765. [DOI] [PubMed] [Google Scholar]
- 48. Sirbu B.M., Couch F.B., Feigerle J.T., Bhaskara S., Hiebert S.W., Cortez D.. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011; 25:1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jansen J.G., Tsaalbi-Shtylik A., Hendriks G., Verspuy J., Gali H., Haracska L., de Wind N.. Mammalian polymerase zeta is essential for post-replication repair of UV-induced DNA lesions. DNA Repair (Amst.). 2009; 8:1444–1451. [DOI] [PubMed] [Google Scholar]
- 50. Cordeiro-Stone M., Makhov A.M., Zaritskaya L.S., Griffith J.D.. Analysis of DNA replication forks encountering a pyrimidine dimer in the template to the leading strand. J. Mol. Biol. 1999; 289:1207–1218. [DOI] [PubMed] [Google Scholar]
- 51. Guilliam T.A., Yeeles J.T.P.. Reconstitution of translesion synthesis reveals a mechanism of eukaryotic DNA replication restart. Nat. Struct. Mol. Biol. 2020; 27:450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. García-Nieto P.E., Schwartz E.K., King D.A., Paulsen J., Collas P., Herrera R.E., Morrison A.J.. Carcinogen susceptibility is regulated by genome architecture and predicts cancer mutagenesis. EMBO J. 2017; 36:2829–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ogawa T., Okazaki T.. Discontinuous DNA replication. Annu. Rev. Biochem. 1980; 49:421–457. [DOI] [PubMed] [Google Scholar]
- 54. Kosar M., Giannattasio M., Piccini D., Maya-Mendoza A., García-Benítez F., Bartkova J., Barroso S.I., Gaillard H., Martini E., Restuccia U.et al.. The human nucleoporin tpr protects cells from RNA-mediated replication stress. Nat. Commun. 2021; 12:3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bailey L.J., Bianchi J., Doherty A.J.. PrimPol is required for the maintenance of efficient nuclear and mitochondrial DNA replication in human cells. Nucleic Acids Res. 2019; 47:4026–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tirman S., Quinet A., Wood M., Meroni A., Cybulla E., Jackson J., Pegoraro S., Simoneau A., Zou L., Vindigni A.. Temporally distinct post-replicative repair mechanisms fill PRIMPOL-dependent ssDNA gaps in human cells. Mol. Cell. 2021; 81:4026–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Torregrosa-Muñumer R., Forslund J.M.E., Goffart S., Pfeiffer A., Stojkovič G., Carvalho G., Al-Furoukh N., Blanco L., Wanrooij S., Pohjoismäki J.L.O.. PrimPol is required for replication reinitiation after mtDNA damage. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:11398–11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pathania S., Nguyen J., Hill S.J., Scully R., Adelmant G.O., Marto J.A., Feunteun J., Livingston D.M.. BRCA1 is required for postreplication repair after UV-induced DNA damage. Mol. Cell. 2011; 44:235–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ma W., Westmoreland J.W., Resnick M.A.. Homologous recombination rescues ssDNA gaps generated by nucleotide excision repair and reduced translesion DNA synthesis in yeast G2 cells. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E2895–E2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Piberger A.L., Bowry A., Kelly R.D.W., Walker A.K., González-Acosta D., Bailey L.J., Doherty A.J., Méndez J., Morris J.R., Bryant H.E.et al.. PrimPol-dependent single-stranded gap formation mediates homologous recombination at bulky DNA adducts. Nat. Commun. 2020; 11:5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huang F., Mazina O.M., Zentner I.J., Cocklin S., Mazin A.V.. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J. Med. Chem. 2012; 55:3011–3020. [DOI] [PubMed] [Google Scholar]
- 62. Huang F., Motlekar N.A., Burgwin C.M., Napper A.D., Diamond S.L., Mazin A.V.. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 2011; 6:628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alagpulinsa D.A., Ayyadevara S., Shmookler Reis R.J.. A small-molecule inhibitor of RAD51 reduces homologous recombination and sensitizes multiple myeloma cells to doxorubicin. Front. Oncol. 2014; 4:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Potter H., Dressler D.. On the mechanism of genetic recombination: electron microscopic observation of recombination intermediates. Proc. Natl. Acad. Sci. U.S.A. 1976; 73:3000–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sogo J.M., Lopes M., Foiani M.. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002; 297:599–602. [DOI] [PubMed] [Google Scholar]
- 66. Taylor M.R.G., Yeeles J.T.P.. Dynamics of replication fork progression following helicase-polymerase uncoupling in eukaryotes. J. Mol. Biol. 2019; 431:2040–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Devbhandari S., Remus D.. Rad53 limits CMG helicase uncoupling from DNA synthesis at replication forks. Nat. Struct. Mol. Biol. 2020; 27:461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fumasoni M., Zwicky K., Vanoli F., Lopes M., Branzei D.. Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the polα/primase/ctf4 complex. Mol. Cell. 2015; 57:812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Quinet A., Tirman S., Jackson J., Šviković S., Lemaçon D., Carvajal-Maldonado D., González-Acosta D., Vessoni A.T., Cybulla E., Wood M.et al.. PRIMPOL-Mediated adaptive response suppresses replication fork reversal in BRCA-Deficient cells. Mol. Cell. 2020; 77:461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nayak S., Calvo J.A., Cong K., Peng M., Berthiaume E., Jackson J., Zaino A.M., Vindigni A., Hadden M.K., Cantor S.B.. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv. 2020; 6:eaaz7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rickman K.A., Noonan R.J., Lach F.P., Sridhar S., Wang A.T., Abhyankar A., Huang A., Kelly M., Auerbach A.D., Smogorzewska A.. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 2020; 34:832–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Heller R.C., Marians K.J.. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 2006; 439:557–562. [DOI] [PubMed] [Google Scholar]
- 73. Yeeles J.T.P., Marians K.J.. The escherichia coli replisome is inherently DNA damage tolerant. Science. 2011; 334:235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Callegari A.J., Clark E., Pneuman A., Kelly T.J.. Postreplication gaps at UV lesions are signals for checkpoint activation. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:8219–8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Iyer L.M., Koonin E.V., Leipe D.D., Aravind L.. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res. 2005; 33:3875–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rechkoblit O., Gupta Y.K., Malik R., Rajashankar K.R., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K.. Structure and mechanism of human primpol, a DNA polymerase with primase activity. Sci. Adv. 2016; 2:e1601317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guilliam T.A., Jozwiakowski S.K., Ehlinger A., Barnes R.P., Rudd S.G., Bailey L.J., Skehel J.M., Eckert K.A., Chazin W.J., Doherty A.J.. Human primpol is a highly error-prone polymerase regulated by single-stranded DNA binding proteins. Nucleic Acids Res. 2015; 43:1056–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mehta K.P.M., Thada V., Zhao R., Krishnamoorthy A., Leser M., Lindsey Rose K., Cortez D.. CHK1 phosphorylates PRIMPOL to promote replication stress tolerance. Sci. Adv. 2022; 8:eabm0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gabbai C.B., Yeeles J.T.P., Marians K.J.. Replisome-mediated translesion synthesis and leading strand template lesion skipping are competing bypass mechanisms. J. Biol. Chem. 2014; 289:32811–32823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hashimoto Y., Ray Chaudhuri A., Lopes M., Costanzo V.. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010; 17:1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Petermann E., Orta M.L., Issaeva N., Schultz N., Helleday T.. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010; 37:492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M.. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011; 145:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schlacher K., Wu H., Jasin M.. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012; 22:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cano-Linares M.I., Yáñez-Vilches A., García-Rodríguez N., Barrientos-Moreno M., González-Prieto R., San-Segundo P., Ulrich H.D., Prado F.. Non-recombinogenic roles for rad52 in translesion synthesis during DNA damage tolerance. EMBO Rep. 2021; 22:e50410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chen X., Bosques L., Sung P., Kupfer G.M.. A novel role for non-ubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene. 2016; 35:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mason J.M., Chan Y.-L., Weichselbaum R.W., Bishop D.K.. Non-enzymatic roles of human RAD51 at stalled replication forks. Nat. Commun. 2019; 10:4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Joseph C.R., Dusi S., Giannattasio M., Branzei D.. Rad51-mediated replication of damaged templates relies on monoSUMOylated DDK kinase. Nat. Commun. 2022; 13:2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kolinjivadi A.M., Sannino V., De Antoni A., Zadorozhny K., Kilkenny M., Técher H., Baldi G., Shen R., Ciccia A., Pellegrini L.et al.. Smarcal1-Mediated fork reversal triggers mre11-dependent degradation of nascent DNA in the absence of brca2 and stable rad51 nucleofilaments. Mol. Cell. 2017; 67:867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Guilliam T.A., Brissett N.C., Ehlinger A., Keen B.A., Kolesar P., Taylor E.M., Bailey L.J., Lindsay H.D., Chazin W.J., Doherty A.J.. Molecular basis for primpol recruitment to replication forks by RPA. Nat. Commun. 2017; 8:15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Martínez-Jiménez M.I., Lahera A., Blanco L.. Human primpol activity is enhanced by RPA. Sci. Rep. 2017; 7:783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lemaçon D., Jackson J., Quinet A., Brickner J.R., Li S., Yazinski S., You Z., Ira G., Zou L., Mosammaparast N.et al.. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017; 8:860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Broker T.R., Lehman I.R.. Branched DNA molecules: intermediates in T4 recombination. J. Mol. Biol. 1971; 60:131–149. [DOI] [PubMed] [Google Scholar]
- 93. Pont-Kingdon G., Dawson R.J., Carroll D.. Intermediates in extrachromosomal homologous recombination in xenopus laevis oocytes: characterization by electron microscopy. EMBO J. 1993; 12:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sena E.P., Revet B., Moustacchi E.. In vivo homologous recombination intermediates of yeast mitochondrial DNA analyzed by electron microscopy. Mol. Gen. Genet. 1986; 202:421–428. [DOI] [PubMed] [Google Scholar]
- 95. Giannattasio M., Zwicky K., Follonier C., Foiani M., Lopes M., Branzei D.. Visualization of recombination-mediated damage bypass by template switching. Nat. Struct. Mol. Biol. 2014; 21:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. González-Prieto R., Muñoz-Cabello A.M., Cabello-Lobato M.J., Prado F.. Rad51 replication fork recruitment is required for DNA damage tolerance. EMBO J. 2013; 32:1307–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Yoon J.-H., Prakash L., Prakash S.. Error-free replicative bypass of (6-4) photoproducts by DNA polymerase zeta in mouse and human cells. Genes Dev. 2010; 24:123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hombauer H., Srivatsan A., Putnam C.D., Kolodner R.D.. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science. 2011; 334:1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kochenova O.V., Daee D.L., Mertz T.M., Shcherbakova P.V.. DNA polymerase ζ-dependent lesion bypass in saccharomyces cerevisiae is accompanied by error-prone copying of long stretches of adjacent DNA. PLoS Genet. 2015; 11:e1005110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lehner K., Jinks-Robertson S.. The mismatch repair system promotes DNA polymerase zeta-dependent translesion synthesis in yeast. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:5749–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dungrawala H., Rose K.L., Bhat K.P., Mohni K.N., Glick G.G., Couch F.B., Cortez D.. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell. 2015; 59:998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Naiman K., Pagès V., Fuchs R.P.. A defect in homologous recombination leads to increased translesion synthesis in e. coli. Nucleic Acids Res. 2016; 44:7691–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Eppink B., Tafel A.A., Hanada K., van Drunen E., Hickson I.D., Essers J., Kanaar R.. The response of mammalian cells to UV-light reveals Rad54-dependent and independent pathways of homologous recombination. DNA Repair (Amst.). 2011; 10:1095–1105. [DOI] [PubMed] [Google Scholar]
- 104. Delmas S., Matic I.. Interplay between replication and recombination in escherichia coli: impact of the alternative DNA polymerases. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:4564–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kane D.P., Shusterman M., Rong Y., McVey M.. Competition between replicative and translesion polymerases during homologous recombination repair in drosophila. PLoS Genet. 2012; 8:e1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sharma S., Hicks J.K., Chute C.L., Brennan J.R., Ahn J.-Y., Glover T.W., Canman C.E.. REV1 and polymerase ζ facilitate homologous recombination repair. Nucleic Acids Res. 2012; 40:682–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. McIlwraith M.J., West S.C.. DNA repair synthesis facilitates RAD52-mediated second-end capture during DSB repair. Mol. Cell. 2008; 29:510–516. [DOI] [PubMed] [Google Scholar]
- 108. Kawamoto T., Araki K., Sonoda E., Yamashita Y.M., Harada K., Kikuchi K., Masutani C., Hanaoka F., Nozaki K., Hashimoto N.et al.. Dual roles for DNA polymerase eta in homologous DNA recombination and translesion DNA synthesis. Mol. Cell. 2005; 20:793–799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data supporting the findings of this study are available with the online version of the article.