
Figure 7.
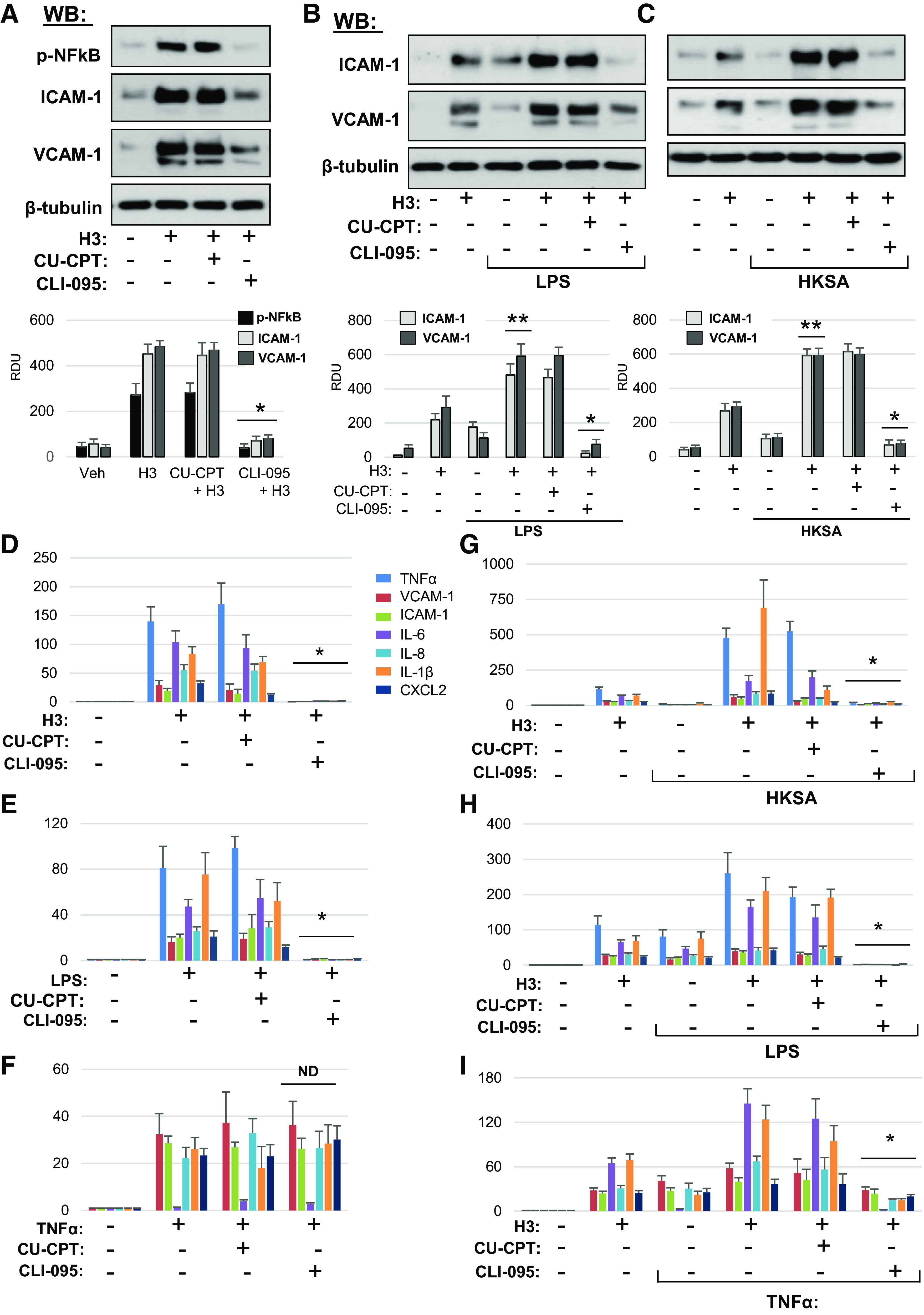
Inhibition of TLR4 signaling rescues histone H3-induced inflammation in lung EC. A: HPAEC pretreated with CU-CPT22 (5 µM) or CLI-095 (1 µM) for 30 min were stimulated with histone H3 (30 µg/mL, 5 h). NF-κB phosphorylation, ICAM-1, and VCAM1 protein levels in cell lysates were determined by Western blotting. Probing for β-tubulin was used as normalization control. Bar graphs depict quantitative densitometry analysis of Western blot data; *P < 0.01 vs. H3 alone; n = 4. Cells preincubated with CLI-095 or CU-CPT22 were treated with LPS (25 ng/mL) (B) or HKSA (5 × 108 particles/mL) (C) for 1 h followed by addition of 15 µg/mL H3 for 5 h. ICAM-1 and VCAM1 protein expression was measured by Western blotting. Probing for β-tubulin was used as normalization control. Bar graphs depict quantitative densitometry analysis of Western blot data; *P < 0.01 vs. HKSA+H3 or LPS+H3; **P < 0.01 vs. H3, LPS, or HKSA alone; n = 4. Effects of EC pretreatment with CU-CPT22 and CLI-095 on mRNA expression of inflammatory markers caused by H3 (D), LPS (E), and TNFα (F). HPAEC preincubated with vehicle, CLI-095, or CU-CPT22 were stimulated with H3 in combination with HKSA (G), LPS (H), or TNFα (I). Expression of markers of inflammation was analyzed by qRT-PCR. Data represent means ± SD, n = 4–6, *P < 0.01 vs. treatment without inhibitors. HPAEC, human pulmonary artery endothelial cell; HKSA, heat-killed S. aureus; LPS, lipopolysaccharide; ND, no difference; TNF-α, tumor necrosis factor-α; LPS, lipopolysaccharide.