

Abstract
The SARS-CoV-2 Omicron variant evades most neutralizing vaccine-induced antibodies and is associated with lower antibody titers upon breakthrough infections than previous variants. However, the mechanism remains unclear. Here, we find using a geometric deep-learning model that Omicron’s extensively mutated receptor binding site (RBS) features reduced antigenicity compared with previous variants. Mice immunization experiments with different recombinant receptor binding domain (RBD) variants confirm that the serological response to Omicron is drastically attenuated and less potent. Analyses of serum cross-reactivity and competitive ELISA reveal a reduction in antibody response across both variable and conserved RBD epitopes. Computational modeling confirms that the RBS has a potential for further antigenicity reduction while retaining efficient receptor binding. Finally, we find a similar trend of antigenicity reduction over decades for hCoV229E, a common cold coronavirus. Thus, our study explains the reduced antibody titers associated with Omicron infection and reveals a possible trajectory of future viral evolution.
Keywords: SARS-CoV-2, Omicron variant of concern, spike protein, antigenicity, computational structural biology, deep learning
Graphical abstract
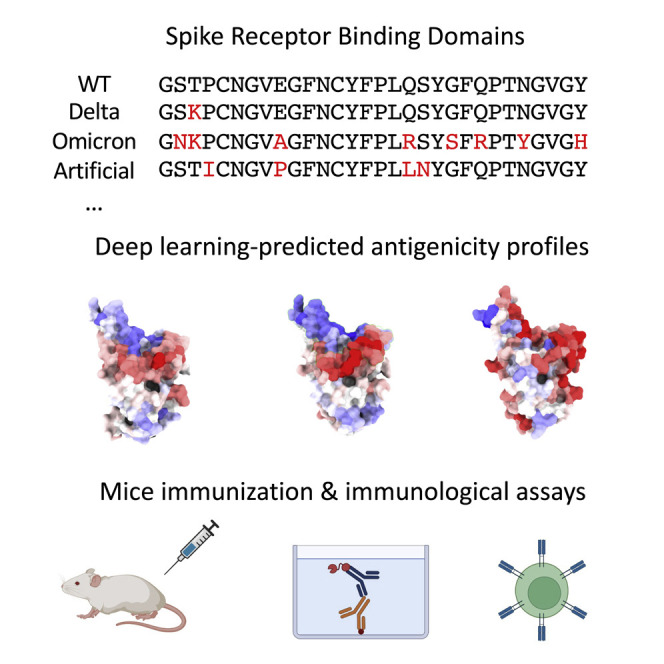
SARS-CoV-2 Omicron variant evades most neutralizing vaccine-induced antibodies and is associated with lower antibody titers upon breakthrough infections than previous variants. Tubiana et al. investigate the underlying mechanism using geometric deep learning, mice immunization experiments, and biochemical assays. Mutations reduce antigenicity of the receptor binding site, leading to lower antibody response.
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) continues to evolve, producing variants of concern (VOC) with improved transmissibility and abilities to evade host immunity. The newly identified VOC Omicron (B.1.1.529) contains many mutations including 11 that localize on the variable receptor binding site (RBS), which is the major target of serologic response (Piccoli et al., 2020). These mutations collectively facilitate the immune evasion of both vaccinated and convalescent sera while maintaining angiotensin converting enzyme 2 (ACE2) binding (Collie et al., 2021; Edara et al., 2022; Hoffmann et al., 2021; Liu et al., 2021; Rossler et al., 2022b; Schmidt et al., 2022; Servellita et al., 2022). Some of the Omicron mutations (S477N, E484K, N501Y, Q498R) previously emerged from an in vitro directed evolution experiment optimizing ACE2 binding (Zahradnik et al., 2021). Others, including K417N, E484A, and Q498R, induced immune escape from wild-type (WT)-elicited antibodies (Greaney et al., 2022a). However, it remains unclear if the extensive RBD mutations could affect the immunogenicity, antigenicity, and immunodominance hierarchy of the naive host antibody response (Greaney et al., 2022b).
Here, immunogenicity refers to the ability of an antigen to induce a humoral and/or cell-mediated immune response upon immunization or infection (Anfosso et al., 1979). B cell antigenicity refers to the magnitude of antigen binding by affinity-matured antibodies (Zhang and Tao, 2015). The immunodominance hierarchy corresponds to the spatial distribution of epitopes on the antigen structures (Angeletti and Yewdell, 2018). Despite their importance, however, high-throughput analysis of immunogenicity, antigenicity, or immunodominance hierarchy of a protein antigen remains very challenging (Angeletti and Yewdell, 2018). Empirical, data-driven approaches are appealing alternatives, as they can sidestep the slow and intractable affinity maturation process. However, while T cell epitope prediction is now well established, B cell epitope prediction has limited success. Indeed, antibodies frequently target conformational epitopes, sets of residues close in space but distal along the sequence. This hampers (1) comprehensive experimental mapping of antibody epitopes and (2) computational prediction from sequence only. Recently, we have developed ScanNet, a geometric deep-learning model for structure-based prediction of binding sites, including protein-protein binding sites and B cell epitopes (Tubiana et al., 2022). ScanNet is an end-to-end, interpretable deep-learning architecture that builds representations of atoms and amino acids based on the spatio-chemical arrangements of their neighbors. It exploits a large public dataset containing thousands of antigen structures with labeled epitopes to learn the three-dimensional structural patterns underlying antibody binding. Examples of structural patterns learned by the model include the prescribed absence of atoms, e.g., exposed side chain atoms or backbones nitrogens/oxygens available for hydrogen bond formation. ScanNet can make predictions using either an experimental structure or computational model as input and calculates a residue-wise epitope probability score, hereafter termed antigenicity profile. ScanNet predicts epitopes substantially more accurately than other established approaches, including those based on (1) comparative modeling, (2) physico-chemical and geometric features such as hydrophobicity scales and solvent accessibility, and (3) AlphaFold-Multimer. Importantly, the ScanNet-predicted antigenicity profile of the WT spike glycoprotein RBD correlated well with the empirical antibody hit rate estimated from available structures of spike protein-antibody complexes. Thus, its antigenic profile directly reflected the immunodominance hierarchy, i.e., the epitope distribution. This prompted us to systematically investigate the impact of VOC mutations on antigenicity and immunodominance hierarchy using ScanNet.
Results
Deep learning predicts a decrease in the Omicron RBS antigenicity
Here, we used a geometric deep-learning model (ScanNet [Tubiana et al., 2022]) together with available experimental RBD structures and structure modeling tools (Modeller, Rosetta) to systematically investigate the RBD antigenic profiles for WT (the Wuhan strain) and the VOCs (Figure 1 ). To validate our computational pipeline, we calculated the antigenicity profile for WT and found that it correlates well with the frequency of structurally determined RBD epitopes (Spearman rho = 0.77, STAR Methods, Figure S1A). The antigenicity profiles are overall similar for all the RBDs, as expected given the high sequential and structural similarity, and the RBS residues have high antibody binding propensity (Figure 1A). Alpha, Beta, and Delta VOCs have a moderate increase in the RBS antigenicity compared with WT (Figures 1B and 1E). However, the antigenicity of Omicron RBS (particularly, residues 470–500 and 445–455) was significantly reduced (Figures 1B–1E). Moderate increases in antigenicity were also detected for several residues (403–420, 501–505); however, these sites are less targeted than the dominant epitopes mapped experimentally (Figures S1A, S1C and S1D). A significantly negative correlation (Pearson correlation r = −0.43, p value = 2.4 × 10−10) was found between the residue-wise empirical antibody hit rate and the change in antigenicity upon Omicron mutations (Figure S1B). Together, our analysis indicates that the overall antigenicity of Omicron RBS is reduced, with the strongest reduction on the residues most frequently targeted by antibodies.
Figure 1.

Impact of Omicron mutations on antigenicity based on geometric deep learning
(A) Residue-wise antigenicity profile of WT and four VOCs computed with ScanNet. For each sequence, predictions are averaged over multiple structural conformations (STAR Methods).
(B) Difference between each VOC and WT, depicted as a scatterplot. The area of each point represents the statistical significance of the difference (larger is more significant): it is proportional to the absolute value of the associated Z score (clipped at |Z| = 10, the dots in the caption correspond to |Z| = 5).
(C) Omicron RBD colored by the difference of antigenicity (PDB: 7qnw) with respect to WT.
(D) Upper panel: Prevalence of mutations for each VOC based on GISAID. Bottom panel: Corresponding predicted change in overall antigenicity.
(E) Boxplots of RBS average antigenicity for WT and four VOCs calculated over multiple structures. p value annotated legend: ns: p > 5e-2, ∗∗∗p < 1e-4 (two-sided Wilcoxon-Mann-Whitney test).
(F) Distribution of changes in antigenicity across all single-point mutations and all stability-preserving single-point mutations previously identified by deep mutation scan (Starr et al., 2020), a cutoff of −0.5 in log-odds scale). The blue histogram denotes the distribution over structural models of the WT scores, and intuitively corresponds to the noise level induced by the structural modeling component of the prediction pipeline. The corresponding matrix is shown in Figure S2A.
To assess the significance of the change and dissect the individual contribution of the 15 Omicron mutations to the overall antigenicity, we modeled the structures of the corresponding 15 single-point mutants using Modeller (Webb and Sali, 2017) and calculated their antigenic profiles (STAR Methods). Eight mutations (53%) decreased the antigenicity, particularly Q493R, G496S, and Q498R (Figure 1D). Five mutations (33%) increased the antigenicity while the remaining had no obvious effect. Next, we modeled the structures of all the point mutants and calculated their antigenicity (Figure S2A). Only 26% decreased the antigenicity (Figure 1F). Therefore, the reduced antigenicity of Omicron is not random (p = 0.034, χ2 contingency test) and may result from evolutionary pressure. To evaluate the potential synergistic effects, we also investigated the combined effects of mutations, and found overall positive epistasis, meaning that mutations with similar effects tended to reinforce one another (STAR Methods).
Omicron mutations lead to a drastic and systemic reduction in RBD antigenicity in vivo
To substantiate the deep-learning analysis, we immunized mice via the mucosal delivery route with the recombinant RBDs from WT (n = 4) or VOCs (n = 5) and analyzed their adaptive immune responses (STAR Methods). All the animals showed robust and comparable T cell responses as indicated by the in vitro recall assays. Specifically, their splenocytes produced high levels of interferon gamma (IFNg) when re-stimulated with WT, Delta, or Omicron RBDs regardless of the immunogens that they originally received (Figure S3A), suggesting a successful initiation of Th1-mediated immune response. A strong Th17 response was also generated as expected for this type of mucosal immunization regimen (Clemente et al., 2017) (Figure S3B). Interleukin (IL)-17 levels appeared to be more consistent among all groups of mice suggesting they were mainly produced by antigen-specific CD4 T cells, whereas IFNg can come from natural killer or gamma-delta T cells without the need of antigen recognition. We also analyzed the local response in the lungs in the animals and observed comparable IFNg and IL-17 responses (Figures S3C and S3D).
Next, we performed ELISA to measure antibody titers of the immunized sera from 15 days (Figure S4) and 25 days (Figure 2 ) after the boost against the corresponding antigens. In contrast to the T cell response, we found that the antibody titers (half-maximal inhibitory reciprocal serum dilution or ID50) of the Omicron-immunized sera were consistently low in both bleeds and significantly reduced by over 15-fold (mean half-maximal inhibitory concentration [ID50] = 924) compared with that of WT (mean ID50 = 15,325) and other VOCs (mean ID50s = 11,564, 14,683, and 19,557 for Alpha, Beta, and Delta, respectively) (Figure 2A). Thus, our in vivo experiments were consistent with the deep-learning model, revealing that mutations can greatly reduce the antigenicity of Omicron RBD.
Figure 2.

Analysis of the RBD-immunized sera
(A) ELISA of RBD-immunized mouse sera (n = 4 mice for WT, n = 5 for VOCs) against the corresponding antigen. The binding titer was calculated as the ID50 (reciprocal serum dilution that inhibits the 50% maximal RBD binding).
(B) ELISA of RBD-immunized sera against five different RBDs (cross-reactivity analysis).
(C) The percentage change of binding titers against different RBDs.
(D) Pseudovirus neutralization assay evaluating the potencies of WT and Omicron RBD-immunized sera against either SARS-CoV-2 WT (Wuhan-Hu-1, D614G) strain or Omicron. The neutralization titer was calculated as the ID50 (reciprocal serum dilution that inhibits 50% of the maximal pseudovirus infection). Two connected dots referred to the pseudovirus neutralization results of the same animal serum. The dashed line indicates the highest serum concentration (i.e., dilution of 22, which is the lowest reciprocal serum dilution) used in the study.
Previous structural analysis revealed that most antibodies target the variable RBS (Yuan et al., 2021). The remaining antibodies bind conserved epitopes that are cross-reactive among VOCs (Barnes et al., 2020; Cameroni et al., 2021; Gaebler et al., 2021; Xiang et al., 2021). To better understand the antigenicity and immunodominance hierarchy of RBD variants, we evaluated the cross-reactivity of immunized sera by ELISA (Figures 2B and 2C). WT-immunized sera had comparably high titers against Alpha (ID50 = 15,176) and Delta (ID50 = 13,985) RBDs but presented decreased activities against Beta (ID50 = 10,763; by 35%) and more substantially against Omicron (ID50 = 6,201; by 69%). The magnitudes of antibody evasion by VOCs were consistent with clinical data (Edara et al., 2022; Hoffmann et al., 2021; Liu et al., 2021; Rossler et al., 2022b; Schmidt et al., 2022; Servellita et al., 2022), indicating that the RBD immunodominance hierarchy is similar between mouse and human.
We found that Omicron-immunized sera had substantially lower antibody titers against all the VOCs (with the mean ID50s in the range of 388–626, Figure 2B). Despite the reductions, Omicron-immunized sera still bind most efficiently to its own antigen (Figure 2B), indicating that Omicron’s RBS remains to be highly antigenic while other conserved epitopes can also contribute to the overall antigenicity. Moreover, while the titer of Omicron-immunized sera against WT RBD was only a small fraction of that of WT-immunized sera against Omicron (388/6201 or 6%), the percentages of cross-reactive antibodies were highly comparable (∼31%, Figure 2C). Thus, the immunodominance hierarchy for Omicron remained largely unaltered and the reduction of response was rather systemic, contributed by both RBS and other conserved epitopes. This result was further supported by competitive ELISA using either the recombinant ACE2 or high-affinity nanobodies targeting distinct and highly conserved RBD epitopes (Figure S5, STAR Methods).
Since Beta RBD shares three mutation sites with Omicron (K417N, E484K/A, and N501Y) critical for antibody binding, we also evaluated the cross-reactivity of Beta-immunized sera and found that the titer only decreased by 51% against the Omicron RBD. These sera cross-reacted better with Omicron RBD than the WT-immunized sera, where a 69% titer reduction was observed against the Omicron RBD (Figure 2C). Since the antibody titers of the Beta-immunized mice are comparable to those of WT sera, we conclude that these three mutated residues do not significantly contribute to the antigenicity decrease (Figure 1D).
Next, we performed SARS-CoV-2 pseudovirus assay to evaluate the contribution of Omicron mutations to the neutralization potency of the immunized sera (Figure 2D). Despite some cross-reactivity of WT-immunized sera against Omicron (ID50 = 6,201), their neutralization activities were barely detectable. Strikingly, the potencies of the Omicron-immunized sera were generally inefficient against the Omicron virus (except for one serum) and their activities against WT (the Wuhan-Hu-1/D614G strain) were hardly detected.
Analysis of the evolution of hCoV229E reveals a decrease in antigenicity
hCoV229E is a common cold coronavirus that has been circulating in the human population for decades. As one of the first coronavirus strains being described, its sequences and structures have been well documented and can be used as a model system to study the evolution of antigenicity and host serologic response (Eguia et al., 2021; Li et al., 2019; Wong et al., 2017). Previous structural and immunological studies suggested that the hCoV229E alphacoronavirus has been undergoing extensive antibody escape (Eguia et al., 2021; Li et al., 2019; Wong et al., 2017), and that its evolution could reflect the future evolution of SARS-CoV-2 (Eguia et al., 2021). The hCoV229E proteome features a spike protein with a (structurally different) RBD that targets the human aminopeptidase N protein. Similar to SARS-CoV-2, the corresponding RBS, which consists of three loops, is also the major immunodominant region. We collected RBD sequences of all hCoV229E isolates with known collection dates and evaluated their antigenicity via structural modeling and ScanNet. Longitudinal analysis revealed an overall trend of decreasing antigenicity on RBS until the 2010s, with subsequent oscillation during the last decade (Figure 3A). Allegedly, these two phases might correspond to (1) a transitory adaptation period to the host humoral immunity and (2) an out-of-equilibrium, stationary phase where the virus continuously evolves to shield itself from antibodies elicited by past infections.
Figure 3.

Plausibility of further decrease of antigenicity in future variants
(A) Evolution of the antigenicity of hCoV229E RBS for isolates collected from the 1960s to date. Classes are assigned based on phylogeny and structural features of the RBS, following Li et al. (2019) and Wong et al. (2017). Black line denotes the isotonic regression fit (i.e., piecewise constant, monotonous least squares fit) using all points until 2010. A downward trend is observed for over 40 years (Spearman correlation coefficient: −0.82, p = 1e-18 ).
(B) ScanNet-predicted protein binding propensity (higher is better) versus antigenicity (lower is better) of the SARS-CoV-2 RBS for WT, four VOCs, all single-point mutants, and 1,000 artificial variants with 15 mutations from WT (same number as Omicron) generated using a sequence generative model (STAR Methods). Crosses indicate 95% confidence interval.
While we are unfamiliar with related works on the evolution of susceptibility to adaptive immunity, similar trends were reported for innate immunity (Di Gioacchino et al., 2021; Greenbaum et al., 2008, 2014). Notably, since the 1918 outbreak, the H1N1 strand has gradually evolved to hide from pattern recognition receptors by reducing its number of CpG dinucleotide motifs through synonymous mutations.
Computational exploration for the SARS-CoV-2 RBD sequence space opens up the possibility of a further decrease in antigenicity
Since SARS-CoV-2 was only recently introduced into the human population, there is insufficient information to witness a similar evolutionary trend. We can nonetheless evaluate the potential for additional reduction of antigenicity. Although the virtual deep mutational scan readily identifies multiple mutations that could lead to antigenicity reduction, particularly on sites 448, 449, and 506 (Figure S2A), it is unclear whether or not they are beneficial for the overall viral fitness. Viral fitness includes multiple factors, such as affinity and specificity of ACE2 binding, structural stability, equilibrium distribution of up and down conformations, and corresponding transition times. Therefore, we restricted the search space to variants that are likely to arise based on past evolutionary records. This was done in four steps (STAR Methods): (1) Construction of a multiple sequence alignment (MSA) of Beta coronaviruses RBDs; (2) selection, training, and validation of a sequence generative model, i.e., a probability distribution over RBD sequences P(S); (3) generation of a repertoire of de novo RBD variants by sampling from the sequence generative model in the vicinity of the original WT (a total of 1,000 variants, each contains 15 point mutations similar to those of Omicron); and (4) determination of the antigenicity and protein binding profiles using ScanNet for each de novo variant. The protein binding profile is similarly determined as the antigenicity profile using a ScanNet model trained for generic protein-protein binding site prediction. We monitored it to ensure that antigenicity reduction is not achieved by an overall loss of protein binding.
The sequences of the de novo variants obtained by sampling from the generative model preserved the conservation and coevolution patterns of the RBD protein family and the sarbecovirus subgenius (Figure S6). As expected, their sequences varied substantially on the RBS and exhibited diverse ranges of antigenicity and protein binding propensity (Figure 3B). We found that antigenicity and protein binding scores are correlated, indicating a possible evolutionary trade-off between ACE2 binding and the immune escape. A small fraction (7.4%) of the artificial variants showed potentially improved binding and reduced antigenicity than Omicron (Figure 3B, shaded square).
Based on Shannon entropy calculations, there are 0.074 × 2.715 = 200k such de novo 15-point variants, implying a trajectory of uncertainty with a possibility of further antigenic reduction of new variants. Enrichment analysis (STAR Methods) further revealed that multiple mutations, including Q493 I/V/L, P479I, L452Y, and K462Q, may contribute to the decrease while maintaining stability and ACE2 binding. However, we must stress that only a fraction of these RBD variants is likely viable for the virus. First, evolutionary-based generative modeling, despite being extensively validated (Hawkins-Hooker et al., 2021; Repecka et al., 2021; Russ et al., 2020; Wu et al., 2021), does not guarantee (100%) protein stability. Second, both the evolutionary model and the ScanNet binding propensity scores are host-agnostic. Hence, the designed sequences do not necessarily bind Human ACE2 but instead may bind other ACE2 orthologs, as observed, e.g., for ancestral sarbecovirus sequences (Starr et al., 2022). Last, the ScanNet antigenicity prediction is imperfect.
Discussion
In this study, we leveraged computational prediction facilitated by geometric deep-learning (ScanNet) and experimental approaches to systematically investigate the RBD antigenicity. ScanNet provides a rapid means to quantify the antigenicity of proteins from structure, at the individual residue level, for emerging viruses and their variants. The use of the mouse model for the investigation of RBD antigenicity enables comparison among variants, minimizing the potential bias and background complexity that are often associated with clinical samples. Competitive ELISA using pan-sarbecovirus-binding nanobodies allows experimental investigation of the immunodominance hierarchy.
We found that Omicron mutations resulted in substantial antigenicity reduction on the RBS site: the key target of neutralizing antibodies and, correspondingly, substantially lower antibody titers. Interestingly, we did not detect major changes in the immunodominance hierarchy, as Omicron-immunized sera also bound less efficiently the conserved, non-RBS epitopes than WT-immunized sera. This implies that the localized antigenicity reduction resulted in a lower overall immunogenicity. Hypothetically, proteins that are more antigenic are better associated by polyreactive immunoglobulin Gs which, in turn, could facilitate affinity maturation and trigger faster the humoral immune system.
We stress that such an immune concealing strategy differs from the immune escape one, where mutations prevent binding by matured antibodies elicited from past infections. Immune concealing could provide an absolute viral fitness improvement that might explain the rapid takeover of Omicron over Delta. Consistently, longitudinal study of the hCoV229E RBD using ScanNet showed a consistent decrease of antigenicity over decades (with short-term fluctuations), followed by an oscillatory phase possibly stirred by immune escape. It remains unclear which of the two strategies is preferable throughout the complete course of viral evolution. For SARS-CoV-2, computational analysis of artificial variants shows that a further decrease of antigenicity is plausible. However, only a fraction of these variants is likely viable, and future experimental validations will be necessary to better understand the properties of these possible future variants.
Our results corroborate findings from other studies. During our manuscript preparation, a preprint reported that an Omicron-specific mRNA vaccine boost appears to provide inferior protection against Omicron infection in non-human primates compared with boost using the WT mRNA vaccine (Gagne et al., 2022). Moreover, new clinical data suggested that the antibody titers after Omicron breakthrough cases were lower than those of after Delta infection (Collie et al., 2021; Edara et al., 2022; Hoffmann et al., 2021; Liu et al., 2021; Rossler et al., 2022b; Schmidt et al., 2022; Servellita et al., 2022). Finally, Omicron convalescent sera from unvaccinated individuals was found to only weakly neutralize Omicron virus while the serum neutralizing activities against other VOCs was below the detection limits (Khan et al., 2022; Rossler et al., 2022a). Thus, our study is consistent with both the preclinical vaccine trials and clinical convalescent data and provides critical insights into the underlying mechanism of the attenuated host serologic response against Omicron. Cumulatively, our investigations unravel a potential trajectory of future viral evolution and underlie the challenges to develop effective Omicron-specific vaccines.
Limitations of the study
Although ScanNet enables rapid and reliable assessment of antigenicity, we note that the correlation between our predicted antigenicity and the experimental results is most likely non-linear. In the current study, we only explored a single immunization protocol by the intraperitoneal route. Our results were also based on the recombinant RBD, which dominates host antibody response, instead of on the whole spike glycoprotein and/or viral infection. However, emerging evidence based on vaccine and infection support our central conclusion that Omicron is characterized by inferior antigenicity. In addition, the major focus of our study is to understand the antibody response against VOCs. Investigations on other components of cellular immunity (such as T cell immunity and Fc effectors functions) may yield more comprehensive insight into the disrupted host response by Omicron and the potential mechanism (He et al., 2022; Richardson et al., 2022). Finally, due to technical challenges, our current work did not experimentally explore the computationally designed variants. Future studies will be needed to carefully evaluate these variants to better understand the potential trajectory of viral evolution.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Invitrogen T7 Tag Polyclonal Antibody, HRP | Thermo Fisher | Cat#: PA1-31449; RRID: AB_1960906 |
| Pierce™ High Sensitivity NeutrAvidin™-HRP | Thermo Fisher | Cat#: 31030 |
| Invitrogen goat anti-mouse IgG (H + L) secondary antibody, HRP | Thermo Fisher | Cat#: G-21040; RRID: AB_2536527 |
| ELISA Mouse IL-17A | BioLegend | Cat#: 432504 |
| ELISA Mouse IFN-g | BioLegend | Cat#: 430804 |
| Bacterial and virus strains | ||
| SARS-CoV-2 (Wuhan-Hu-1, D614G) reporter virus particles (luciferase) | Integral Molecular | Cat#: RVP-702L |
| SARS-CoV-2 (Omicron) reporter virus particles (luciferase) | Integral Molecular | Cat#: VP-768L |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 (COVID-19) S protein RBD, MALS verified | Acro Biosystems | Cat#: SPD-C52H3 |
| SARS-CoV-2 (COVID-19) Spike RBD (N501Y/Alpha), MALS verified | Acro Biosystems | Cat#: SPD-C52Hn |
| SARS-CoV-2 (COVID-19) Spike RBD (K417N,E484K,N501Y/Beta), MALS verified | Acro Biosystems | Cat#: SPD-C52Hp |
| SARS-CoV-2 (COVID-19) Spike RBD (L452R,T478K/Delta), MALS verified | Acro Biosystems | Cat#: SPD-C52Hh |
| SARS-CoV-2 (2019-nCoV) Spike RBD (B.1.1.529/Omicron), MALS verified | Acro Biosystems | Cat#: SPD-C522e |
| ACE2 protein, Human, biotinylated | Sinobiologics | Cat#: 10108-H08H-B |
| Epitope 3 and 4 nanobodies | Xiang et al., 2022 | N\A |
| LPS-EB VacciGrade™ | InvivoGen | Cat#: vac-3pelps |
| Critical commercial assays | ||
| Renilla-Glo luciferase assay system | Promega | Cat#: E2720 |
| Experimental models: Cell lines | ||
| 293T-hsACE2 stable cell line | Integral Molecular | Cat# C-HA101; Lot#: TA060720MC |
| Experimental models: Organisms/strains | ||
| C57BL/6J, mus musculus | The Jackson Laboratory | IMSR_JAX:000664 |
| Software and algorithms | ||
| ScanNet | Tubiana et al., (2022) |
https://github.com/jertubiana/ScanNet http://bioinfo3d.cs.tau.ac.il/ScanNet/ |
| Restricted Boltzmann Machines | Tubiana et al. (2019) | https://github.com/jertubiana/PGM |
| Modeller | Webb and Sali (2017) | https://salilab.org/modeller/ |
| PyRosetta | Chaudhury et al., 2010 | https://www.pyrosetta.org |
| HHblits | Steinegger et al., (2019) | https://github.com/soedinglab/hh-suite |
| MAFFT | Nakamura et al., (2018) | https://mafft.cbrc.jp/alignment/software/ |
| ChimeraX | Pettersen et al., (2021) | https://www.cgl.ucsf.edu/chimerax/ |
Resource availability
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by lead contact Yi Shi. (wally.yis@gmail.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
8 weeks old female C57BL/6 mice were ordered from The Jackson Laboratory and housed in pathogen-free conditions at the core animal facility at the University of Pittsburgh Medical Center with the approval from the University of Pittsburgh Institutional Animal Care and Use Committee. 40μg recombinant RBD plus 5μg LPS-EB VacciGrade™ (InvivoGen) was given to isoflurane anesthetized mice in sterile PBS (50μL) intranasally on day 0 and day 10, a test bleed was draw on day 25 and mice were sacrificed on day 35 for bleed, spleen and lung harvesting.
Method details
ScanNet
Deep learning has been highly successful in protein structure prediction (AlQuraishi, 2019; Baek et al., 2021; Ingraham et al., 2018; Jumper et al., 2021; Senior et al., 2020; Wang et al., 2017). However, leveraging the structures for function prediction has remained a major challenge (Chruszcz et al., 2010). Recently, we have developed ScanNet, a geometric deep learning model for structure-based prediction of binding sites including protein-protein binding sites and B-cell epitopes (Tubiana et al., 2022). ScanNet is an end-to-end architecture learning representations of atoms and amino acids based on the spatio-chemical arrangements of their neighbors. Briefly, ScanNet first extracts an atomic neighborhood around each heavy atom (K = 16 neighbors, corresponding to about 4Å), and calculates their local coordinates in a frame centered around the atom and oriented using the covalent bonds. The neighborhood, formally a point cloud with attributes (atom group type) is then passed through a set of trainable spatio-chemical filters. Each filter detects a specific spatio-chemical pattern within the neighborhood, such as hydrogen bonds. Conversely, some filters also detect prescribed absences of atoms, e.g. exposed side chain atoms, or backbones nitrogens/oxygens available for hydrogen bond formation. The later filters are critical for epitope prediction, as reactive atom groups that are not engaged in intra-chain interactions are more prone to be targeted by antibodies. The resulting atom-wise embeddings are next pooled at the amino acid level, and the process is reiterated around each amino acid. Finally, the resulting amino acid-wise embeddings are converted to propensity scores via a neighborhood attention module, which projects the embeddings to scalar values and smoothes them (in a learnt fashion) across a neighborhood.
We previously trained ScanNet for detecting B-cell epitopes based on 3756 antibody-antigen complexes available from the PDB. ScanNet predicted known epitopes substantially more accurately than AlphaFold-multimer or previous works that relied on amino acid propensity scores and geometric features such as solvent accessibility. We previously found that for the Spike protein RBD of WT, the predicted antigenicity profile correlated well with the residue-wise antibody hit rate computed from 246 PDB structures of spike protein - antibody complexes, defined as the fraction of antibodies that bind to the residue (Tubiana et al., 2022). We successfully reproduced the analysis with the prediction pipeline described below (Figure S1).
VOCs antigenicity and protein binding profiles
Since the sequences considered are highly similar (92–99% sequence identity to WT) and the structures are virtually indistinguishable by human eye, the predicted epitope propensity profiles are overall similar. Additionally, ScanNet is sensitive to subtle structural features such as sidechain-backbone hydrogen bonds (especially for asparagines) that are not always consistent from one crystal structure to the other for a given variant. To maximize the signal-to-noise ratio, we proceeded as follows:
-
1.
We used the model version that only takes the sequence and structure as input and discards the position-weight matrix. For the antigenicity profile, this version achieves the same performance as the one using evolutionary information (Table S4 of Tubiana et al., 2022). For the protein binding profile, the performance is overall lower than the version using evolutionary information (Table 1 of Tubiana et al., 2022), but is nonetheless satisfactory for the Spike RBD.
-
2.
All predictions were averaged over 11 networks, each trained using a different random seed. All SARS-CoV-1/2 antibody-antigen complexes were excluded from the training set.
-
3.
We used multiple RBD structures per variant. For the WT, we selected 29 RBD structures. For the other VOCs, all the available RBD structures were taken (WT PDBs: 7eam:A, 7mzj:B, 7dhx:B, 7mfu:A, 7efr:B, 7kn3:A, 7mmo:C, 7kgj:A, 7n4j:A, 7mf1:A, 7mzm:A, 7jmo:A, 7vnb:B, 7s4s:A, 7lop:Z, 7r6w:R, 7kmg:C, 7deu:A, 7det:A, 7c8v:B, 7cjf:C, 7d2z:B, 7bnv:A, 7nx6:E, 6m0j:E, 7mzh:E, 7ean:A, 7n3i:C, 6yla:E. Alpha: 7fdg:E, 7neg:E, 7nx9:E, 7mji:B, 7mjl:A, 7mjn:B, 7ekf:B. Beta: 7ps4:E, 7ps6:E, 7ps0:E, 7ps7:E, 7ps2:G, 7ps0:A, 7ps5:E, 7q0h:E, 7prz:E, 7pry:E, 7ps1:E, 7q0g:E, 7nxa:E, 7e8m:E. Delta: 7w9f:E, 7w9i:E, 7wbq:B, 7wbq:D, 7v8b:A. Omicron: 7qnw:E, 7wbp:B, 7wbl:B, 7t9l:A. SARS-CoV-1: 3bgf:S, 7rks:R, 6waq:D, 2ajf:E, 3d0g:E, 3scl:E, 2ghv:E, 2ghw:A, 2dd8:S).
-
4.
Since some structures consistently missed many sidechain-backbone hydrogen bonds, we standardized them by applying to each structure the FastRelax protocol of PyRosetta (5 cycles) (Chaudhury et al., 2010; Nivón et al., 2013). To reduce the noise induced by Rosetta, we generated 20 relaxation runs per structure and averaged epitope profiles over them. This protocol reduced the intra-variant, inter-structure standard deviation by 10–25%.
Altogether, the antigenicity and protein binding propensity profiles of each single point mutant were averaged over 11 X 20 X N profiles where N was the number of available RBD structures. Based on the intra-variant, inter-structure variance, we estimated the average resolution of our differential antigenicity profiles as 0.008 (in probability units). RBS residues were defined based on available crystal structures as: 403, 417–421, 445–456, 473–505. The overall RBS antigenicity and the binding score were defined as the average across RBS residues of the corresponding profile.
Single-point mutants antigenicity profiles
Mutant structures were generated using comparative modeling. We first selected six representative templates for the WT RBD by clustering the aforementioned RBD structures (7jvb:A, 7eam:A, 7d2z:B, 7kgj:A, 7vnb:B, 7det:A). Next, we generated for each single point mutant and each template 20 structural models using Modeller (Webb and Sali, 2017). Homology modeling is sufficiently accurate here (and much faster than AlphaFold) because of the high sequence identity values (92–99%), and because VOCs have very small conformational variability (as evidenced by experimental crystal structures). We also verified on a few examples that Modeller models were almost identical to AlphaFold ones. As neutral controls, we also generated structural models for the original amino acid at each position (i.e. the WT sequence). Each model was scored using the 11 networks, obtaining 6x20x11 = 1320 profiles per mutant which were then averaged to yield a single antigenicity profile and a single binding propensity profile. The overall impact of a mutation to antigenicity was defined as the difference between the summed profiles across the entire protein. Despite the averaging, we found that conformational variability yielded changes in total propensity of the same order of magnitude as the one of changes upon single point mutants: 48% of the mutations had an insignificant impact on total antigenicity, i.e. within the [5%,95%] percentiles of the WT antigenicity distribution (Figure S7A). The predicted profiles notably featured small variations in regions far away from the mutation, arising solely because of modeling noise. To improve the signal to noise ratio, we instead computed a weighted sum of the difference of profiles, where the weight is a smoothing function of the distance to the mutated residue (Figure S7C). Since ScanNet predictions are based on local neighborhoods and the conformational noise away from the mutation is expected to average out anyway, the local estimator is unbiased and has lower variance. After smoothing, only 21% of mutations were insignificant (Figure S7B). Protein binding propensity profiles were calculated in the same manner using comparative models and ScanNet models trained for protein binding site prediction (Figures S7D and S7E). Finally, a positive correlation was found between changes in antigenicity and change in binding (Figure S7F). This reflects the trade-off between high receptor binding propensity and low antigenicity. The ∼470k structural models and ∼10 million profiles were generated in about ten days using a single computer with 64Gb RAM and an Intel Xeon Phi processor with 56 cores (52ms per profile).
Combined effect of mutations
To evaluate potential epistatic effects, we chose the seven RBS mutations that reduced antigenicity (N440K, G446S, S477N, T478K, Q493R, G496S, Q498R) and tested all 27 combinations of reverse mutations from Omicron background (i.e., the antigenicity should increase again). The same modeling protocol was used as for the single-point mutants, but with 15 templates instead of six (7jvb_A, 7eam_A, 7d2z_B, 7kgj_A, 7vnb_B, 7det_A, 7w9f_E, 7w9i)E, 7qnw_E, 7wbp_B, 7w7bl_B, 7t9k_A, 7t9c_B, 7u0d_B, 7wlc_E). Figure S8 shows the scatterplot of Hamming distance to Omicron sequence against the change in antigenicity, with full line and dashed line corresponding, respectively to the i) average over all mutants with k mutations and the ii) epistasis-free prediction based on the effect of the single-point mutations. Overall, the epistasis-free prediction underestimates the effect of combined mutations, meaning mutations tend to reinforce one another on average (positive epistasis).
Generation and screening of de novo variants
We investigated whether additional mutations of the SARS-CoV-2 RBD could further reduce its RBS antigenicity, without altering other components of the viral fitness such as structural stability and ACE2 binding. Using ScanNet, we tested sequences in the vicinity of the original WT sequence that are likely to arise based on past evolutionary records. These de novo RBD variant sequences were obtained by sampling from a sequence generative model trained on a multiple sequence alignment (MSA) of beta coronaviruses RBDs. Methodological details about the protocol and validation steps are described below.
-
1.
Construction of the MSA. Homologs of the WT RBD were first searched in the UniprotKB using BLAST. Top hits were manually aligned with MAFFT (Nakamura et al., 2018) (command: mafft —amino —localpair —maxiterate 1000 —op 5 —ep 0), and only the columns not gapped for the WT were kept. Next, additional homologs were searched in the UniRef30 (release 2020/06) using HH-blits (Steinegger et al., 2019). After filtering out hits with unknown residues and/or >25% of gaps, we obtained a (redundant) alignment of B = 521 sequences. The MSA covered all the five betacoronaviruses subgenii (sarbecovirus, embecovirus, merbecovirus, nobecovirus, hibecovirus). The effective number of sequences (defined as in (Morcos et al., 2011), approximately corresponding to the number of 90% sequence identity clusters) was Beff = 72.8, a relatively low value. The sequence profile of the MSA (Figure S6A) features conserved sites (most of which are buried), whereas the RBS region is highly variable.
-
2.
Sequence generative model. Herein, the objective is to learn a probability distribution over the sequence space P(S) by maximizing the average likelihood ˂logP(S)˃ of the previously observed viral sequences in the MSA. Intuitively, maximizing the likelihood amounts to assigning high probability values to seen (i.e. evolutionary selected) sequences and low elsewhere (i.e. sequences unexplored or washed away by selection), such that P is normalized to 1. The likelihood can therefore be interpreted as a proxy for viral fitness (Cocco et al., 2018). Importantly, a “smooth” parametric form P θ(S) must be chosen to ensure that the model also assigns high probability values to sequences that are close (and presumably evolutionary fit), but unobserved either due to limited sequencing or exploration of the sequence space throughout evolution. Possible choices for the parametric forms include the independent model (i.e. the position specific sequence model or equivalently, insertion-free HMM profiles), Potts model (i.e. the Boltzmann Machine, BM) (Morcos et al., 2011) or Restricted Boltzmann Machine (RBM) as well as various deep learning-based models (Riesselman et al., 2018; Wu et al., 2021). We used RBM here, which is an undirected graphical model that learns the conservation and coevolution patterns of the sequence distribution (Tubiana et al., 2019). In the context of RBD modeling, RBM enjoys two desirable properties over the more thoroughly validated BM model. First, its flexible number of parameters allows better optimization of the bias-variance trade-off. RBM has N × (M + 1)× q parameters, where N is the number of columns, M is the (tunable) number of hidden units and q = 21is the number of amino acids (+gap) compared to for the Potts model. Our selected model has 100Xfewer parameters than a regular Potts model. Second, it is able to model high-order epistasis arising from heterogeneous viral fitness landscapes. Indeed, since different subgenii target different receptors, they are expected to have related but distinct fitness landscapes.
RBM were trained using the PGM package (https://github.com/jertubiana/PGM) (Tubiana et al., 2019) using the Persistent Contrastive Divergence algorithm with the following parameters: number of hidden units: from 5 to 100; hidden unit potential: dReLU; batch size: 100; number of Markov chains: 100; number of Monte Carlo steps between each gradient evaluation: 100; number of gradient updates: 40000; optimizer: ADAM with initial learning rate: 5,10−4, exponentially decaying after 50% of the training to 5 × 10−6, β 1 = 0, β 2 = 0.99, ε = 10−3. For the regularization, we used a L1 2 penalty on the weights (of strength ranging from 0.0 to 5.0) and L2 penalty on the fields (of strength ). Samples were assigned a weight inversely proportional to their number of 90% sequence identity homologs in the MSA. Annealed importance sampling was used to evaluate the partition functions, using 3 × 104 intermediate temperatures and 10 repeats. The low depth of the alignment prompted us to thoroughly explore the hyperparameter space to best calibrate the model complexity (Figures S6B and S6C). We divided the MSA into five folds so that any pair of sequences belonging to different folds have at most 80% sequence identity. We then performed a grid search over the regularization strength and number of hidden units. We monitored i) the quality of convergence, ii) the cross-validation likelihood, iii) cross-validation pseudo-likelihood (not shown, correlated to the likelihood), and iv) the spearman correlation between the likelihood of all single-point variants of WT and their corresponding yeast-display expression levels - an experimental proxy for structural stability. The latter were measured in a deep mutational scan experiment performed by Starr et al. (Starr et al., 2020). We selected the model with M = 20 hidden units and regularization strength . It featured a per-site likelihood value of −1.31 (compared to −1.98 for the best independent model after grid search on pseudo-count values), Spearman correlation ρ = 0.53 (Figure S6D, compared to 0.42 for the best independent model and 0.54 for the Potts model as recently reported in (Rodriguez-Rivas et al., 2022)) and per-site entropy of 0.99 (corresponding to 2.7 amino acid choices per site). Its likelihood function also correlated with the changes in ACE2 binding affinity upon mutation as measured by deep mutational scan (Starr et al., Spearman correlation 0.41, p = 10−158). Finally, the generative properties of the model were deemed satisfactory: Monte Carlo samples obtained from P(S) reproduced the moments (Figures S6E and S6F) and the clustered topology of the distribution of natural sequences (Figure S6G).
-
3.
Artificial mutant generation. After model selection 1,000 artificial mutants were generated as follows. We sampled from the gap-less, focused distribution where 15 is the same number of mutations from WT as Omicron. Sampling from the conditional distribution was done by importance sampling Markov Chain Monte Carlo, i.e. by sampling from the modified distribution where λ = 1.9 was chosen such that We used the alternate Gibbs sampler, with 5000 burn-in steps, 100 steps between each sample and 100 independent chains. Generated samples with fewer or more mutations were discarded; approximately 5000 samples with exactly 15 mutations were kept. We extracted 1,000 representatives by agglomerative clustering (using as representative the cluster member with highest likelihood). The distribution of the mutations (Figure S6H) features high variability on the RBS in general and particularly at positions mutated in VOCs. In total, 1523 of the 19 × 15 = 3705 potential mutations are observed at least once. All VOCs mutations are observed at least once, except for Q498R and S375F, with the mutations Y505H and G446S being the most frequent (in 6.9% and 6.4% of the sequences).
-
4.
Scoring of artificial mutants. We used the same comparative modeling followed by ScanNet antigenicity and binding site prediction pipeline as for the single-point mutants.
-
5.
Mutation enrichment analysis. An artificial mutant “improves” over Omicron if (i) its ScanNet antigenicity score is lower or equal than the one of Omicron, and (ii) its ScanNet binding propensity score higher or equal than Omicron. Out of 1000 sampled artificial mutants, 74 improved mutants were found. For each of the 1523 mutations sampled, we tested its association with the improved phenotype using a χ2 contingency test, and used the Benjamini-Hochberg procedure to control the false discovery rate (0.05 cut-off). In total six statistically significant mutations were found: Q493 I/V/L, P479I, L452Y, K462Q. They are all viable for expression and ACE2 binding based the DMS data from (Starr et al., 2020).
Analysis of the human 229E alphacoronavirus
Previous structural and immunological studies suggested that the hCoV229E alphacoronavirus has been undergoing extensive antibody escape since its entry into human population (Eguia et al., 2021; Li et al., 2019; Wong et al., 2017), and that its evolution could reflect the future evolution of SARS-CoV-2 (Eguia et al., 2021). The hCoV229E proteome features a spike protein with a (structurally different) receptor binding domain which targets the human aminopeptidase N protein. Similarly to SARS-CoV-2, the corresponding receptor binding site, which consists of three loops, is also the major immunodominant region. We evaluated the evolution of antigenicity of the hCoV229E RBS using ScanNet as follows. We first collected six template structures for the 229E RBD (PDB 6u7h:A, 6atk:E, 6ixa:A, 6u7e:D, 6u7f:D, 6u7g:D) and constructed a structure-based multiple sequence alignment using ChimeraX (Pettersen et al., 2021), and an HMM profile model (using the hhalign utility (Steinegger et al., 2019)). Next, we retrieved all 203 available hCoV229E spike protein sequences from Uniprot, aligned them to the HMM profile (command hhalign -t one_strain_sequence.fasta -i template_sequences.fasta -oa3m output.fasta -all) and discarded sequences that did not cover at least 100 of the 134 columns of the alignment. The corresponding EMBL entry was used to retrieve the corresponding isolate/strain name and its collection date, if available. In total, we obtained 115 (redundant) sequences with known collection dates between 1967 and 2022. For each sequence, we generated 20 × 6 structural models with MODELLER (Webb and Sali, 2017) (20 per template). Since there was no antibody-hCoV229E spike protein complex in the training set of ScanNet, we used all the 55 networks trained for antibody binding site prediction (including the 11 used elsewhere that were not trained on SARS-CoV-1/2 data). To define the RBS residues, we first extracted all interface residues (6Å distance cut-off) of the template complexes (PDB 6atk, 6u7e, 6u7g, 6u7f) and labeled the corresponding MSA columns as RBS. Then, for a given strain, its RBS residues were identified as the ones mapped onto one of the RBS columns. The RBS antigenicity was defined for a given strain as the average over all networks, all structural models and all non-gapped RBS columns of the antigenicity profile. Note that due to the presence of deletions, the number of residues included in the RBS varied from one variant to the other and therefore summing rather than averaging yielded slightly different results. We tried both options and found a similar decreasing trend in both cases. Error bars (one standard deviation) were estimated based on the structural model variability (Figure 3A). The isotonic regression fit was performed using scikit-learn (sklearn.isotonic.IsotonicRegression, default parameters).
In vitro antigen restimulation assay
Individual lungs were collected, mechanically digested, and enzymatically digested with collagenase/DNase for 1 h at 37°C as described previously (Chen et al., 2011). Single cell suspensions were then passed through a 70-μm sterile filter. Red blood cells were lysed using a NH4Cl solution and the cells were enumerated then plated at 5 × 105 cells per well in 96-well, stimulated with 10μg/mL recombinant RBD proteins for 72 h. The supernatants were collected and analyzed by murine IFNg and IL-17A ELISA (BioLegend). Spleens were processed similar to the lungs without the need of enzymatic digestion.
ELISA (enzyme-linked immunosorbent assay)
Indirect ELISA was carried out to evaluate the serological responses of the total antibody in mice sera to an RBD. A 96-well ELISA plate (R&D system) was coated with the recombinant RBD protein (Acro Biosystems) at an amount of approximately 2–3 ng per well in a coating buffer (15 mM sodium carbonate, 35 mM sodium bicarbonate, pH 9.6) overnight at 4°C, with subsequent blockage with a blocking buffer (DPBS, v/v 0.05% Tween 20, 5% milk) at room temperature for 2 h. To test the immune response, the mice serum was serially 4 or 5-fold diluted starting from 1:27 (Omicron-immunized sera), 1:72 (WT) or 1:100 (other VOCs) in the blocking buffer and then incubated with the RBD-coated wells at room temperature for 2 h. HRP-conjugated secondary goat anti-mouse IgG (H + L) (Thermo Fisher, cat# G-21040) were diluted 1:1,500 in the blocking buffer and incubated with each well for an additional 1 h at room temperature. Three washes with 1x PBST (DPBS, v/v 0.05% Tween 20) were carried out to remove nonspecific absorbances between each incubation. After the final wash, the samples were further incubated in the dark with freshly prepared w3,3′,5,5′-Tetramethylbenzidine (TMB) substrate for 10 min at room temperature to develop the signals. After the STOP solution (R&D system), the plates were read at multiple wavelengths (450 nm and 550 nm) on a plate reader (Multiskan GO, Thermo Fisher). The raw data were processed by Prism 9 (GraphPad) to fit into a 4PL curve and to calculate IC50/logIC50.
Competitive ELISA with recombinant hACE2
A 96-well plate was pre-coated with either WT or Omicron recombinant RBD at 2–3 μg/mL at 4°C overnight. Mice serum was 3-fold diluted starting from 1:15 (Omicron) or 1:45 (WT) in the blocking buffer with a final amount of 50 ng biotinylated hACE2 (Sino Biological, cat# 10108-H08H-B)/8 ng epitope 3 nanobody/8 ng epitope 4 nanobody at each concentration and then incubated with the plate at room temperature for 2 h. The plate was washed by the washing buffer to remove the unbound hACE2. 1:5,000 diluted Pierce™ High Sensitivity NeutrAvidin-HRP (Thermo Fisher cat# 31,030) or 1:7,500 diluted T7-tag polyclonal antibody-HRP (Thermo Fisher, cat# PA1-31449) were incubated with the plate for 1 h at room temperature. TMB solution was added to react with the HRP conjugates for 10 min. The reaction was then stopped by the Stop Solution. The signal corresponding to the amount of the bound hACE2 or nanobodies was measured by a plate reader at 450 nm and 550 nm. The wells without sera were used as control to calculate the percentage of hACE2 or nanobody signal. The resulting data were analyzed by Prism 9 (GraphPad) and plotted.
Pseudotyped SARS-CoV-2 neutralization assay
The 293T-hsACE2 stable cell line (Integral Molecular, cat# C-HA101, Lot# TA060720MC) and pseudotyped SARS-CoV-2 (Wuhan-Hu-1 strain D614G and Omicron) particles with luciferase reporters were purchased from the Integral Molecular. The neutralization assay was carried out according to the manufacturers’ protocols. In brief, 2-fold serially diluted immunized mice serum starting from 1:22 dilution was incubated with the pseudotyped SARS-CoV-2-luciferase. For accurate measurements, seven concentrations were tested for each mice and at least two repeats were done. Pseudovirus in culture media without sera was used as a negative control. 100 μL of the mixtures were then incubated with 100 μL 293T-hsACE2 cells at 2.5 × 10e5 cells/mL in the 96-well plates. The infection took ∼72 h at 37°C with 5% CO2. The luciferase signal was measured using the Renilla-Glo luciferase assay system (Promega, cat# E2720) with the luminometer at 1 ms integration time. The obtained relative luminescence signals (RLU) from the negative control wells were normalized and used to calculate the neutralization percentage at each concentration. Data were processed by Prism 9 (GraphPad). Due to the poor neutralization of the serum at the highest concentration (lowest dilution), the IC50 was estimated as the maximal dilution that could inhibit ∼50% cell infections by the pseudovirus.
Quantification and statistical analysis
All statistical analysis was carried out using Python and the numpy scipy, and statannot packages. All technical details are provided throughout the manuscript, in the figure captions, or in the STAR Methods.
Acknowledgments
We thank Zhe Sang for the analysis of antibody binding. J.T. acknowledges helpful discussion with Andrea Di Gioacchino and Simona Cocco. Funding: This work was supported by NIH grants R35GM137905 (Y.S.), R01AI163011 (Y.S. and D.S.), R01HL137709 (K.C.), ISF 1466/18 and Israeli Ministry of Science and Technology (D.S.), the Edmond J. Safra Center for Bioinformatics at Tel Aviv University, the Human Frontier Science Program (cross-disciplinary postdoctoral fellowship LT001058/2019-C) (J.T.), and Len Blavatnik and the Blavatnik Family Foundation (H.J.W.).
Author contributions
D.S. and Y.S. conceived the study. J.T. performed all the computational analysis with the help of D.S. and H.J.W. Y.X., L.F., and K.C. performed the experiments. J.T.,Y.S., and D.S. drafted the manuscript with substantial input from Y.X. and K.C. All authors reviewed the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: October 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111512.
Supplemental information
Data and code availability
-
•The following datasets are made available from the Zenodo repository https://doi.org/10.5281/zenodo.7079268:
-
1.List of hCoV229E RBD sequences, with associated isolate and collection date identifiers, and ScanNet antigenicity score for reproducing Figure 3A.
- 2.
- 3.
-
4.Empirical epitope distribution for the RBD shown in Figures S1.
-
5.virtual Deep Mutational Scan performed with ScanNet and the sequence generative model shown in Figure S2.
-
1.
The remaining data analyzed (protein structure files) are publicly available from the Protein DataBank.
-
•
All computational analysis, statistical analysis and visualizations were carried out in Python 3.6.12 and 3.8.5 using publicly available software and standard packages (numpy, scipy, pandas, numba, scikit-learn, biopython, matplotlib, seaborn). Source code and trained models for ScanNet are available from https://github.com/jertubiana/ScanNet. ScanNet is also available as a public webserver from http://bioinfo3d.cs.tau.ac.il/ScanNet/. Source code for training, scoring and sampling Restricted Boltzmann Machines is available from (https://github.com/jertubiana/PGM). The following additional software were used: Modeller (https://salilab.org/modeller/), PyRosetta (https://www.pyrosetta.org), HHblits (https://github.com/soedinglab/hh-suite), MAFFT (https://mafft.cbrc.jp/alignment/software/), ChimeraX (https://www.cgl.ucsf.edu/chimerax/). BioRender was used for the graphical abstract.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- AlQuraishi M. End-to-end differentiable learning of protein structure. Cell Syst. 2019;8:292–301.e3. doi: 10.1016/j.cels.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfosso F., Soler M., Leyris R., Mallea M., Charpin J. Denaturization of allergen P: effect on allergenicity, antigenicity and immunogenicity. Ann. Allergy. 1979;42:384–389. [PubMed] [Google Scholar]
- Angeletti D., Yewdell J.W. Understanding and manipulating viral immunity: antibody immunodominance enters center stage. Trends Immunol. 2018;39:549–561. doi: 10.1016/j.it.2018.04.008. [DOI] [PubMed] [Google Scholar]
- Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G.R., Wang J., Cong Q., Kinch L.N., Schaeffer R.D., et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–876. doi: 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C.O., Jette C.A., Abernathy M.E., Dam K.M.A., Esswein S.R., Gristick H.B., Malyutin A.G., Sharaf N.G., Huey-Tubman K.E., Lee Y.E., et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588:682–687. doi: 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameroni E., Bowen J.E., Rosen L.E., Saliba C., Zepeda S.K., Culap K., Pinto D., VanBlargan L.A., De Marco A., di Iulio J., et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature. 2021;602:664–670. doi: 10.1038/s41586-021-04386-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S., Lyskov S., Gray J.J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26:689–691. doi: 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., McAleer J.P., Lin Y., Paterson D.L., Zheng M., Alcorn J.F., Weaver C.T., Kolls J.K. Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity. 2011;35:997–1009. doi: 10.1016/j.immuni.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chruszcz M., Domagalski M., Osinski T., Wlodawer A., Minor W. Unmet challenges of structural genomics. Curr. Opin. Struct. Biol. 2010;20:587–597. doi: 10.1016/j.sbi.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente A.M., Castronovo G., Antonelli A., D’Andrea M.M., Tanturli M., Perissi E., Paccosi S., Parenti A., Cozzolino F., Rossolini G.M., Torcia M.G. Differential Th17 response induced by the two clades of the pandemic ST258 Klebsiella pneumoniae clonal lineages producing KPC-type carbapenemase. PLoS One. 2017;12:e0178847. doi: 10.1371/journal.pone.0178847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco S., Feinauer C., Figliuzzi M., Monasson R., Weigt M. Inverse statistical physics of protein sequences: a key issues review. Rep. Prog. Phys. 2018;81:032601. doi: 10.1088/1361-6633/aa9965. [DOI] [PubMed] [Google Scholar]
- Collie S., Champion J., Moultrie H., Bekker L.-G., Gray G. Effectiveness of BNT162b2 vaccine against omicron variant in South Africa. N. Engl. J. Med. Overseas. Ed. 2021;386:494–496. doi: 10.1056/NEJMc2119270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Gioacchino A., Šulc P., Komarova A.V., Greenbaum B.D., Monasson R., Cocco S. The heterogeneous landscape and early evolution of pathogen-associated CpG dinucleotides in SARS-CoV-2. Mol. Biol. Evol. 2021;38:2428–2445. doi: 10.1093/molbev/msab036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edara V.-V., Manning K.E., Ellis M., Lai L., Moore K.M., Foster S.L., Floyd K., Davis-Gardner M.E., Mantus G., Nyhoff L.E., et al. mRNA-1273 and BNT162b2 mRNA vaccines have reduced neutralizing activity against the SARS-CoV-2 Omicron variant. Cell Rep. Med. 2022;3:100529. doi: 10.1016/j.xcrm.2022.100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguia R.T., Crawford K.H.D., Stevens-Ayers T., Kelnhofer-Millevolte L., Greninger A.L., Englund J.A., Boeckh M.J., Bloom J.D. A human coronavirus evolves antigenically to escape antibody immunity. PLoS Pathog. 2021;17:e1009453. doi: 10.1371/journal.ppat.1009453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaebler C., Wang Z., Lorenzi J.C.C., Muecksch F., Finkin S., Tokuyama M., Cho A., Jankovic M., Schaefer-Babajew D., Oliveira T.Y., et al. Evolution of antibody immunity to SARS-CoV-2. Nature. 2021;591:639–644. doi: 10.1038/s41586-021-03207-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagne M., Moliva J.I., Foulds K.E., Andrew S.F., Flynn B.J., Werner A.P., Wagner D.A., Teng I.-T., Lin B.C., Moore C., et al. mRNA-1273 or mRNA-Omicron boost in vaccinated macaques elicits comparable B cell expansion, neutralizing antibodies and protection against Omicron. bioRxiv. 2022 doi: 10.1101/2022.02.03.479037. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney A.J., Starr T.N., Bloom J.D. An antibody-escape estimator for mutations to the SARS-CoV-2 receptor-binding domain. Virus Evol. 2022;8:veac021. doi: 10.1093/ve/veac021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney A.J., Starr T.N., Eguia R.T., Loes A.N., Khan K., Karim F., Cele S., Bowen J.E., Logue J.K., Corti D., et al. A SARS-CoV-2 variant elicits an antibody response with a shifted immunodominance hierarchy. PLoS Pathog. 2022;18:e1010248. doi: 10.1371/journal.ppat.1010248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum B.D., Cocco S., Levine A.J., Monasson R. Quantitative theory of entropic forces acting on constrained nucleotide sequences applied to viruses. Proc. Natl. Acad. Sci. USA. 2014;111:5054–5059. doi: 10.1073/pnas.1402285111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum B.D., Levine A.J., Bhanot G., Rabadan R. Patterns of evolution and host gene mimicry in influenza and other RNA viruses. PLoS Pathog. 2008;4:e1000079. doi: 10.1371/journal.ppat.1000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins-Hooker A., Depardieu F., Baur S., Couairon G., Chen A., Bikard D. Generating functional protein variants with variational autoencoders. PLoS Comput. Biol. 2021;17:e1008736. doi: 10.1371/journal.pcbi.1008736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C., He X., Yang J., Lei H., Hong W., Song X., Yang L., Li J., Wang W., Shen G., Wei X. Spike protein of SARS-CoV-2 Omicron (B. 1.1. 529) variant has a reduced ability to induce the immune response. Signal Transduct. Target. Ther. 2022;7:119. doi: 10.1038/s41392-022-00980-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Krüger N., Schulz S., Cossmann A., Rocha C., Kempf A., Nehlmeier I., Graichen L., Moldenhauer A.-S., Winkler M.S. The Omicron variant is highly resistant against antibody-mediated neutralization–implications for control of the COVID-19 pandemic. Cell. 2021;185:447–456.e11. doi: 10.1016/j.cell.2021.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingraham J., Riesselman A., Sander C., Marks D. 2018. Learning Protein Structure with a Differentiable Simulator. [Google Scholar]
- Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan K., Karim F., Cele S., Reedoy K., San J.E., Lustig G., Tegally H., Rosenberg Y., Bernstein M., Jule Z., et al. Omicron infection enhances Delta antibody immunity in vaccinated persons. Nature. 2022;607:356–359. doi: 10.1038/s41586-022-04830-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Tomlinson A.C., Wong A.H., Zhou D., Desforges M., Talbot P.J., Benlekbir S., Rubinstein J.L., Rini J.M. The human coronavirus HCoV-229E S-protein structure and receptor binding. Elife. 2019;8:e51230. doi: 10.7554/eLife.51230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Iketani S., Guo Y., Chan J.F.W., Wang M., Liu L., Luo Y., Chu H., Huang Y., Nair M.S., et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature. 2021;602:676–681. doi: 10.1038/s41586-021-04388-0. [DOI] [PubMed] [Google Scholar]
- Morcos F., Pagnani A., Lunt B., Bertolino A., Marks D.S., Sander C., Zecchina R., Onuchic J.N., Hwa T., Weigt M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA. 2011;108:E1293–E1301. doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T., Yamada K.D., Tomii K., Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics. 2018;34:2490–2492. doi: 10.1093/bioinformatics/bty121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivón L.G., Moretti R., Baker D. A Pareto-optimal refinement method for protein design scaffolds. PLoS One. 2013;8:e59004. doi: 10.1371/journal.pone.0059004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.H., Ferrin T.E. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli L., Park Y.-J., Tortorici M.A., Czudnochowski N., Walls A.C., Beltramello M., Silacci-Fregni C., Pinto D., Rosen L.E., Bowen J.E., et al. Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell. 2020;183:1024–1042.e21. doi: 10.1016/j.cell.2020.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repecka D., Jauniskis V., Karpus L., Rembeza E., Rokaitis I., Zrimec J., Poviloniene S., Laurynenas A., Viknander S., Abuajwa W., et al. Expanding functional protein sequence spaces using generative adversarial networks. Nat. Mach. Intell. 2021;3:324–333. [Google Scholar]
- Richardson S.I., Madzorera V.S., Spencer H., Manamela N.P., van der Mescht M.A., Lambson B.E., Moore P.L., et al. SARS-CoV-2 Omicron triggers cross-reactive neutralization and Fc effector functions in previously vaccinated, but not unvaccinated, individuals. Cell Host Microbe. 2022;30:880–886.e4. doi: 10.1016/j.chom.2022.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesselman A.J., Ingraham J.B., Marks D.S. Deep generative models of genetic variation capture the effects of mutations. Nat. Methods. 2018;15:816–822. doi: 10.1038/s41592-018-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rivas J., Croce G., Muscat M., Weigt M. Epistatic models predict mutable sites in SARS-CoV-2 proteins and epitopes. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2113118119. e2113118119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rössler A., Knabl L., von Laer D., Kimpel J. Neutralization profile after recovery from SARS-CoV-2 omicron infection. N. Engl. J. Med. 2022;386:1764–1766. doi: 10.1056/NEJMc2201607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossler A., Riepler L., Bante D., von Laer D., Kimpel J. SARS-CoV-2 omicron variant neutralization in serum from vaccinated and convalescent persons. N. Engl. J. Med. 2022 doi: 10.1056/NEJMc2119236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ W.P., Figliuzzi M., Stocker C., Barrat-Charlaix P., Socolich M., Kast P., Hilvert D., Monasson R., Cocco S., Weigt M., Ranganathan R. An evolution-based model for designing chorismate mutase enzymes. Science. 2020;369:440–445. doi: 10.1126/science.aba3304. [DOI] [PubMed] [Google Scholar]
- Schmidt F., Muecksch F., Weisblum Y., Da Silva J., Bednarski E., Cho A., Wang Z., Gaebler C., Caskey M., Nussenzweig M.C., et al. Plasma neutralization of the SARS-CoV-2 omicron variant. N. Engl. J. Med. 2022;386:599–601. doi: 10.1056/NEJMc2119641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior A.W., Evans R., Jumper J., Kirkpatrick J., Sifre L., Green T., Qin C., Žídek A., Nelson A.W.R., Bridgland A., et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706–710. doi: 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Servellita V., Syed A.M., Brazer N., Saldhi P., Garcia-Knight M., Sreekumar B., Khalid M.M., Ciling A., Chen P.-Y., Kumar G.R. Neutralizing immunity in vaccine breakthrough infections from the SARS-CoV-2 Omicron and Delta variants. medRxiv. 2022 doi: 10.1101/2022.01.25.22269794. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.N., Greaney A.J., Hilton S.K., Ellis D., Crawford K.H.D., Dingens A.S., Navarro M.J., Bowen J.E., Tortorici M.A., Walls A.C., et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295–1310.e20. doi: 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.N., Zepeda S.K., Walls A.C., Greaney A.J., Alkhovsky S., Veesler D., Bloom J.D. ACE2 binding is an ancestral and evolvable trait of sarbecoviruses. Nature. 2022;603:913–918. doi: 10.1038/s41586-022-04464-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M., Meier M., Mirdita M., Vöhringer H., Haunsberger S.J., Söding J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20:473. doi: 10.1186/s12859-019-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubiana J., Cocco S., Monasson R. Learning protein constitutive motifs from sequence data. Elife. 2019;8:e39397. doi: 10.7554/eLife.39397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubiana J., Schneidman-Duhovny D., Wolfson H.J. ScanNet: an interpretable geometric deep learning model for structure-based protein binding site prediction. Nat. Methods. 2022;19:730–739. doi: 10.1038/s41592-022-01490-7. [DOI] [PubMed] [Google Scholar]
- Wang S., Sun S., Li Z., Zhang R., Xu J. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput. Biol. 2017;13:e1005324. doi: 10.1371/journal.pcbi.1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B., Sali A. Functional Genomics. Springer; 2017. Protein structure modeling with MODELLER; pp. 39–54. [Google Scholar]
- Wong A.H.M., Tomlinson A.C.A., Zhou D., Satkunarajah M., Chen K., Sharon C., Desforges M., Talbot P.J., Rini J.M. Receptor-binding loops in alphacoronavirus adaptation and evolution. Nat. Commun. 2017;8:1735. doi: 10.1038/s41467-017-01706-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Johnston K.E., Arnold F.H., Yang K.K. Protein sequence design with deep generative models. Curr. Opin. Chem. Biol. 2021;65:18–27. doi: 10.1016/j.cbpa.2021.04.004. [DOI] [PubMed] [Google Scholar]
- Xiang Y., Huang W., Liu H., Sang Z., Nambulli S., Tubiana J., Shi Y. Superimmunity by pan-sarbecovirus nanobodies. Cell Reports. 2022;39:111004. doi: 10.1016/j.celrep.2022.111004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Huang W., Liu H., Sang Z., Nambulli S., Tubiana J., Williams K.L., Duprex P., Schneidman-Duhovny D., Wilson I.A. Super-immunity by broadly protective nanobodies to sarbecoviruses. bioRxiv. 2021 doi: 10.1101/2021.12.26.474192. Preprint at. [DOI] [Google Scholar]
- Yuan M., Huang D., Lee C.C.D., Wu N.C., Jackson A.M., Zhu X., Liu H., Peng L., van Gils M.J., Sanders R.W., et al. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science. 2021;373:818–823. doi: 10.1126/science.abh1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradník J., Marciano S., Shemesh M., Zoler E., Harari D., Chiaravalli J., Meyer B., Rudich Y., Li C., Marton I., et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 2021;6:1188–1198. doi: 10.1038/s41564-021-00954-4. [DOI] [PubMed] [Google Scholar]
- Zhang J., Tao A. Allergy Bioinformatics. Springer; 2015. Antigenicity, immunogenicity, allergenicity; pp. 175–186. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•The following datasets are made available from the Zenodo repository https://doi.org/10.5281/zenodo.7079268:
-
1.List of hCoV229E RBD sequences, with associated isolate and collection date identifiers, and ScanNet antigenicity score for reproducing Figure 3A.
- 2.
- 3.
-
4.Empirical epitope distribution for the RBD shown in Figures S1.
-
5.virtual Deep Mutational Scan performed with ScanNet and the sequence generative model shown in Figure S2.
-
1.
The remaining data analyzed (protein structure files) are publicly available from the Protein DataBank.
-
•
All computational analysis, statistical analysis and visualizations were carried out in Python 3.6.12 and 3.8.5 using publicly available software and standard packages (numpy, scipy, pandas, numba, scikit-learn, biopython, matplotlib, seaborn). Source code and trained models for ScanNet are available from https://github.com/jertubiana/ScanNet. ScanNet is also available as a public webserver from http://bioinfo3d.cs.tau.ac.il/ScanNet/. Source code for training, scoring and sampling Restricted Boltzmann Machines is available from (https://github.com/jertubiana/PGM). The following additional software were used: Modeller (https://salilab.org/modeller/), PyRosetta (https://www.pyrosetta.org), HHblits (https://github.com/soedinglab/hh-suite), MAFFT (https://mafft.cbrc.jp/alignment/software/), ChimeraX (https://www.cgl.ucsf.edu/chimerax/). BioRender was used for the graphical abstract.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.