

Summary
Here, we found that heterozygous null of peroxisomal Nudt7 (Nudt7+/−) induced the typical NAFLD features, i.e. increased levels of hepatic triglyceride (TG) and fatty acid (FA), infiltration of inflammatory cells, impaired glucose tolerance and insulin sensitivity, and stimulation of lipolysis from adipose tissue. Particularly, in Nudt7+/− hepatocytes, de novo lipogenesis (DNL) was significantly increased. Ingenuity pathway analysis (IPA) and KEGG pathway analysis of RNA sequencing data suggested the activation of PPAR signaling in the liver of Nudt7+/− mice. Moreover, accumulation of palmitic acid in Nudt7+/− hepatocyte increased the level of H3K4me3 on the promoters of PPARγ resulting in the activation of PPARγ and induced the DNL in the hepatocytes of Nudt7+/− mice. Moreover, we found that liraglutide significantly reduced typical NAFLD features induced by NUDT7 deficiency. Our data suggest that dysregulation of peroxisomal NUDT7 is responsible for upregulation of hepatic DNL by accumulation of palmitic acid and PPARγ activation.
Subject areas: Biological sciences, Human metabolism, Cell biology, Transcriptomics
Graphical abstract
Highlights
-
•
Peroxisomal dysfunction is involved in the pathogenesis of NAFLD
-
•
Heterozygous peroxisomal Nudt7+/− mice develop NAFLD via activation of PPAR signaling
-
•
Palmitic acid accumulated by NUDT7 deficiency stimulates H3K4me3 on Pparg promoter
-
•
Liraglutide attenuates hepatic DNL induced by NUDT7 deficiency
Biological sciences; Human metabolism; Cell biology; Transcriptomics
Introduction
The earliest stage of nonalcoholic fatty liver disease is liver steatosis, which is characterized by the fat accumulation, inflammation, and fibrosis in hepatocytes, and potentially progresses to cirrhosis and hepatocellular carcinoma (Farrell and Larter, 2006; Benedict and Zhang, 2017). Since fatty liver has become an epidemic problem worldwide recently, understanding the underlying molecular and signaling mechanisms of hepatic fat accumulation will be a stepping stone for developing the potential therapeutic approaches and strategy to control the pathogenesis of fatty liver.
Dysregulation of hepatic lipid metabolism due to imbalance in fatty acid synthesis, storage, and catabolism closely linked to the development of fatty liver (Musso et al., 2009; Gong et al., 2017). The level of hepatic lipid could be governed by the balance between lipid acquisition through the uptake of circulating fatty acids (FA), lipid disposal through lipid oxidation or export, and/or de novo lipogenesis (DNL) (Fabbrini et al., 2010) by the complicated interactions among hormones, nuclear receptors, and transcription factors. Uptaking circulating FA in hepatocyte is affected by fatty acid transport proteins (FATPs), fatty acid translocase (CD36/FAT) (Chao et al., 2000), and fatty acid binding protein (FABP)1 (Wang et al., 2015) and exporting hepatic lipids are induced by microsomal triglyceride transfer protein (MTTP) and apolipoprotein B (apoB) to reduce hepatic lipid content (Hui et al., 2002). In addition, hepatic lipid accumulation could be resulted from imbalance between lipid acquisition and lipid disposal by an increased lipolysis or an increased fat intake, followed by the enhancement of free fatty acids (FFA). Plasma FFA, an important source for the TG synthesis in the liver, usually generated by white adipocytes via lipolysis can be taken up by FATP, caveolins, FAT/CD36, and FABP (Koo, 2013). FATP5 knockout (KO) mice showed the accumulation of hepatic triglycerides (TG) (Doege et al., 2006) whereas caveolin-1 KO mice exhibited lower hepatic TG accumulation (Fernandez et al., 2006). Moreover, in patients with NAFLD, increased level of hepatic FABP4 and FABP5 was observed and closely correlated with degree of hepatic fatty infiltration and percentage of liver fat (Westerbacka et al., 2007). Stimulation of DNL could contribute to fat accumulation in fatty liver. DNL starts with the conversion of acetyl-CoA to malonyl-CoA by acetyl-CoA carboxylase. Two regulatory transcription factors, sterol regulatory element-binding protein (SREBP)1c and carbohydrate regulatory element-binding protein (ChREBP) (Strable and Ntambi, 2010), are involved in the regulation of hepatic lipogenesis. SREBPs are responsible for activating almost 30 genes involved in lipid metabolism and resulted in the stimulation of DNL in hepatocytes (Shimano and Sato, 2017).
Fatty acid oxidation (FAO) significantly contributes to systemic lipid utilization and elevated level of FAO would increase oxidative capacity and lipid catabolism, reduce lipid load, and prevent the steatosis and lipotoxicity (Neuschwander-Tetri, 2010). FAs are oxidized primarily by mitochondrial and peroxisomal β-oxidation that catalyze the chain shortening of acyl-CoA (Hashimoto et al., 1999). As long-chain fatty acids cannot pass through the organelle membranes by simple diffusion, fatty acids have to be actively transported across both peroxisomal and mitochondrial membranes (Demarquoy and Le Borgne, 2015). Prior to transport, fatty acids are activated outside the organelles by conjugation to either coenzyme A (peroxisomes) or carnitine (mitochondria) (Schrader et al., 2015). Transport of acyl-CoA into the mitochondria is dependent on carnitine palmitoyltransferase-1 (CPT1) in the outer mitochondrial membranes and acyl-carnitines are transported across the inner mitochondrial membrane by CPT2 (Sharma and Black, 2009). Uncoupling of oxidation and phosphorylation in the mitochondria could induce the production of inflammatory cytokines and fibrogenic responses that are closely related to the development of fatty liver (Rolo et al., 2012). Unlike mitochondria that catalyze the β-oxidation of the short-, medium-, and long-chain fatty acids (LCFA), peroxisomes catalyze the β-oxidation of LCFAs and very long-chain fatty acids (VLCFAs) (Rolo et al., 2012). LCFAs and VLCFAs are oxidized in the peroxisomes to shortened fatty acids and transported into the mitochondria to be fully oxidized (Demarquoy and Le Borgne, 2015). Acyl-coenzyme A oxidase (ACOX1), a rate-limiting enzyme that is responsible for catabolism of VLCFAs in peroxisome, is related to the progression of fatty liver by regulating hepatic inflammation (Moreno-Fernandez et al., 2018). Acox1 deficiency in human fibroblasts significantly alters the inflammatory response, leading to the activation of the interleukin (IL)-1 pathway and the induction of IL-6 and IL-8 cytokines (El Hajj et al., 2012) suggesting that dysfunction of peroxisomal function might play an important role in the pathogenesis of fatty liver (Moreno-Fernandez et al., 2018). However, the functional role and underlying regulatory mechanism of peroxisome during the pathogenesis of fatty liver have not yet been well established.
In this study, we evaluated the important role of peroxisome in the pathogenesis of NAFLD and found that deficiency of peroxisomal nudix hydrolase 7 (NUDT7) is responsible for hepatic lipid accumulation and inflammatory responses through and the activation of hepatic DNL via activation of peroxisome proliferator-activated receptor (PPAR)γ. Moreover, we found that liraglutide, the most studied agent as a potential treatment option in NAFLD, reduced NAFLD induced by NUDT7 deficiency.
Results
Peroxisomal dysfunction may be responsible for the pathogenesis of fatty liver
To identify the responsible factor and regulatory mechanism in hepatic steatosis, we applied in silico analysis with GSE33814 (20 steatosis patient liver tissues vs. 13 normal patient liver tissues) and GSE39549 (high-fat diet (HFD) mouse liver vs. normal chow diet (NCD) mouse liver). Gene set enrichment analysis (GSEA) suggested that the impaired peroxisome homeostasis might be responsible for the development of fatty liver (Figure 1A).
Figure 1.

Peroxisome dysfunction may play an important role in the fatty liver
(A) Gene set analysis of GSE33814 (19 steatosis patient liver tissues vs. 13 normal patient liver tissues) and GSE39549 (3 HFD mouse liver vs. 3 NCD mouse liver).
(B) Analysis of peroxisomal genes in GSE39549 (liver of HFD mice vs. NCD mice) and the expression level of Nudt7 in GSE39549 (n = 3).
(C) Representative images of NUDT7 staining in HFD liver compared to NCD liver (n = 4). Scale bars, 100 μm.
(D) Expression level of peroxisomal genes of Cat+/+ and Cat−/− mouse liver using qRT-PCR (n = 3).
(E) Expression level of peroxisomal genes with shCon or shAcox1 lentivirus-injected mouse liver using qRT-PCR (n = 4) and analyzed of peroxisomal genes in GSE39549 and GSE49541. Values were expressed as means + SD An unpaired t-test was used for statistical analysis. ns = non-significant, ∗p ≤ 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To verify the significance of peroxisomal function in the development and progression of fatty liver, lentivirus containing shRNA specific to Acox1 (shAcox1), a peroxisomal β-oxidation initiation factor, was infected into C57BL/6 mice. Exposure of shACOX1 into NCD mouse significantly increased hepatic TG, FFA, lipid accumulation, and lipid reactive oxygen species (ROS) without altering body and liver weight (Figures S1A and S1B). The transcription levels of genes involved in lipid metabolism, such as Acaca, Cd36, Fasn, Fabp4, and Scd1, were dramatically increased in the liver of shAcox1-infected (shACOX1) NCD mouse (Figure S1C). The expression levels of FASN, PPARγ, and SCD1 were dramatically increased in shACOX1-infected liver compared to control (shCON) liver (Figure S1D). In addition, catalase knockout (Cat−/−) liver displayed the significantly increased levels of hepatic lipid accumulation (Figure S1E) with the increased the expression level of genes in lipid metabolism such as Acaca, Cd36, Fasn, Fabp4, and Scd1 compared to Cat+/+ liver (Figure S1F).
Peroxisomal NUDT7 deficiency stimulates lipid accumulation in liver
In silico analysis of the expression of peroxisome-specific 193 probes using HFD mouse liver and NCD mouse liver showed that NUDT7 was one of the most dramatically decreased in HFD mouse liver compared to NCD mouse liver (Figure 1B). Consistent with in silico analysis of GSE39549, we observed the increased expression level of Nudt7 in HFD mouse liver compared to NCD mouse liver (Figure 1C). We analyzed the expression levels of 42 peroxisome-specific genes using Cat−/− mouse liver and Nudt7 was one of the most dramatically decreased peroxisomal genes in Cat−/− mouse liver (Figure 1D). In addition, among 49 peroxisome-specific genes tested, Nudt7 was one of the most dramatically decreased genes in shACOX1-infected mouse liver compared to shCON mouse liver (Figure 1E).
We examined the expression level of NUDT7 in nonalcoholic fatty liver (NAFL) patient biopsy (Figure 2). Liver biopsy was collected from 27 patients who underwent bariatric and metabolic surgery. Among them, 5 patients without hepatic steatosis were excluded and 22 patients were enrolled. Histology of all of them showed NASH. Baseline characteristics are Table S1. Similar to in silico analysis, NUDT7 expression of liver tissue of patients with NASH was lower than that of normal subjects (Figures 2A and 2B). Patients with NASH were divided into two groups depending on the expression level of NUDT7 (Figures 2C and 2D). NAFLD activity-score (NAS) of patients with NASH with the low expression level of NUDT7 (n = 12) tends to be higher than those with a high expression level of NUDT7 (n = 10) (Figure 2C).
Figure 2.

NUDT7 expression is suppressed in patients with NASH
(A) Representative images of H&E, trichrome, and NUDT7 in normal and NASH patients liver biopsy. Scale bars, 200 μm.
(B) NUDT7 staining intensity (4 normal vs. 22 NASH patient liver).
(C) Individual staining intensity of NUDT7 and NAS score in NASH patient liver.
(D) Representative H&E staining images depend on the expression level of NUDT7. Scale bars, 100 μm. ∗∗∗p < 0.001 (Unpaired t-test).
To investigate the role of Nudt7 in the pathogenesis of hepatic steatosis, mouse primary hepatocytes were infected with shRNA specific for Nudt7 (shNudt7). In shNudt7-infected hepatocytes, the levels of LD, FFA,TG, free-, and ester-cholesterol were increased whereas activity of catalase was decreased (Figures S2A and S2B). The expression levels of genes in fatty acid accumulation such as Acaca, Cd36, Fasn, Pparg, and Scd1 were also increased by the knockdown of Nudt7 (Figure S2C). These increased levels of LD, FFA, TG, free-, and ester-cholesterol and increased levels of genes in fatty acid accumulation by the suppression of Nudt7 were recovered by the restoration of Nudt7. As well, decreased level of catalase activity was recovered by the restoration of Nudt7 (Figures S2A–S2C).
Heterozygous Nudt7+/− mice develop fatty liver along with adipose expansion
For further investigation of NUDT7 involvement of in the pathogenesis of fatty liver, we applied NUDT7 knockout (KO) mice previously reported from our laboratory (Song et al., 2018). The expression level of NUDT7 was confirmed with liver of wild, homozygous Nudt7 null (Nudt7−/−), and heterozygous Nudt7 null (Nudt7+/−) NCD mice (Figure S3A). Consistent with our report with chondrocyte, we also observed the dysregulation of mitochondrial genes in Nudt7+/− hepatocytes (Figure S3B). Significant increases in the weight of body and liver, the accumulation of lipid, and levels of serum AST, ALT, FFA, and hepatic TG were observed in Nudt7−/− NCD liver (Figure S3C). Interestingly, Nudt7+/− NCD mice become obese compared to wild-type (Nudt7+/+) NCD mice (Figures 3A and S4). Body weight and liver-to-body weight ratio (Figure 3B), epididymal white adipose tissue (eWAT), and total WAT-to-body weight ratio (Figure S4A) were significantly increased in Nudt7+/− NCD mice compared to Nudt7+/+ NCD mice. The accumulation of lipid droplets (LD) (Figure 3A), blood glucose, serum AST, ALT, hepatic FFA, and TG level was significantly increased in Nudt7+/− NCD mice compared to Nudt7+/+ NCD mice (Figure 3C) without alterations of food and water intake (Figure 3B). Furthermore, O2 consumption, CO2 production, and night heat production were significantly decreased in Nudt7+/− mice compared to Nudt7+/+ mice (Figure 3D). We observed the downregulation of Nudt7 expression in GSE39549 of HFD mouse eWAT compared to NCD mouse eWAT (Figure S4B). Moreover, adipocyte size and the number of peroxisome membrane protein 70 (PMP70)-positive cells were also significantly increased in Nudt7+/− adipose tissue (Figure S4C). Moreover, the expression levels of lipogenic genes such as Fasn, Scd1, Acaca, Cidea, Srebf2, and Fabp4, lipid metabolism such as Abca1, Prkaa1, Irs2, Crot, Sirt6, and Chrebf, adipokines such as Il1b, Il6, Tnfa, Ccl2, Ccl4, Cxcl2, Lep, and Lcn2, and lipolysis genes such as Atgl, Mgl, and Hsl (Figure S4D) were significantly increased in Nudt7+/− adipocyte compared to Nudt7+/+ adipocyte. The expression level of lipid transport genes such as Abca1, Abcg1, Apoa, and Apob was also significantly increased in in Nudt7+/− adipocyte (Figure S4E). Increased in the hypertrophy and expression level of adipokine genes, Lep and Lcn2 were significantly suppressed by the restoration of NUDT7 in Nudt7+/− adipocytes (Figure S4F). To confirm the possible involvement of adipose tissue in Nudt7+/−-induced NAFLD, primary hepatocyte of Nudt7+/+ and Nudt7+/− mice was co-cultured with either adipocyte of Nudt7+/+ or Nudt7+/− mice (Figure S5A). Lipid accumulation and expression levels of Fasn, MLL3, and Srebf1 were significantly upregulated in hepatocyte of Nudt7+/+ or Nudt7+/− mice co-cultured with adipocytes of Nudt7+/− mice (Figure S5B).
Figure 3.

Nudt7+/− mice display typical characteristics of fatty liver
(A) Representative images of liver, H&E, Oil Red O, and Filipin staining in Nudt7+/+ and Nudt7+/− mouse liver (n = 4).
(B) Food and water intake (n = 6), body and liver per body weight (n = 4).
(C) Analysis of blood glucose level (left panel) in Nudt7+/+ (n = 4) and Nudt7+/− (n = 4) mouse and serum AST and ALT, hepatic FFA and TG level (n = 4).
(D) Oxygen consumption (VO2), CO2 production (VCO2), and heat production were analyzed (n = 5).
(E) RNA sequencing data of 12-month-old Nudt7+/+ and Nudt7+/− liver were analyzed using KEGG pathway enrichment analysis (n = 3).
(F) Gene set analysis of Nudt7+/+ and Nudt7+/− mouse liver. ns = non-significant, ∗p ≤ 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (Unpaired t-test or two-way ANOVA).
Heterozygous Nudt7+/− mice develop fatty liver via activation of PPAR signaling
For further understanding of biological and signaling mechanism underlying NUDT7 deficiency-induced fatty liver, RNA sequencing was performed on RNA isolated from 12-month-old Nudt7+/+ and Nudt7+/− liver. Analysis of RNA sequencing showed 1,558 genes with fold change more than 1.5 (779 upregulated genes and 779 downregulated genes), whose expressions were significant differ between Nudt7+/+ and Nudt7+/− liver (Figure 3E). KEGG analysis suggested the PPAR signaling as one of the enriched signaling pathways in Nudt7+/− mouse liver (Figure 3E). Moreover, fatty acid metabolism and lipid metabolic process were significantly increased in Nudt7+/− liver (Figure 3F).
Histological analysis showed the increased level of PPARγ in Nudt7+/− mouse liver (Figures 4A and 4B). In addition, the expression levels of PPARγ target genes such as Cd36, Fabp4, and Il6 were significantly increased in Nudt7+/− liver (Figure 4C). To investigate the biological functional role of PPARγ in Nudt7+/− liver, genes known to interact with PPARγ were extracted from RNA sequencing and subjected to IPA (Figure 4D). IPA revealed that these PPARγ-interacting genes were involved in lipid metabolism and cholesterol uptake and absorption (Figure 4D). Exposure of rosiglitazone (Rosi), a known to PPARγ agonist, into primary hepatocytes of Nudt7+/+or Nudt7+/− mouse, significantly induced the LD accumulation and this increased LD accumulation was reduced by co-introduction of Nudt7 (Figures 4E and S6). In patients with NASH divided by the expression level of NUDT7 (high vs. low), we observed a significant increase of PPARγ in patients with NASH with low expression level of NUDT7 compared to patients with high expression of NUDT7 (Figure 4F).
Figure 4.

Activation of PPARγ in Nudt7+/− liver is responsible for hepatic lipid accumulation
(A) The expression level of Pparg in Nudt7+/+ and Nudt7+/− mouse liver was analyzed using qRT-PCR.
(B) Representative image of PPARγ in 12-month-old Nudt7+/− liver compared to Nudt7+/+ liver (n = 3; Scale bars, 20 μm) and positive staining ratio (n = 6 per group).
(C) The expression levels of PPARγ target genes (Cd36, Fabp4, and Il6) were analyzed by qRT-PCR (n = 3).
(D) Genes related to PPARγ signaling were extracted from the RNA sequencing data (n = 3) and subjected to IPA.
(E) Representative images of BODIPY493/508 staining and positive cell counting (n = 6 per group) in primary cultures of Nudt7+/− hepatocytes in the presence of 10 μM rosiglitazone (Rosi) with pcDNA-Nudt7 for 24 h. Scale bars, 200 μm.
(F) Representative images and intensity of PPARγ in NASH liver (n = 22) compared to normal liver (n = 4) biopsy. Scale bars, 100 μm. ∗p ≤ 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (Unpaired t-test or onw-way ANOVA).
Palmitic acid accumulated by NUDT7 deficiency stimulates H3K4me3 on Pparg promoter
To investigate the involvement of epigenetic regulation of NUDT7 deficiency in Pparg, we analyzed the methylation status of Pparg promoter. We observed a significant increase level of histone H3K4me3 in Nudt7+/− liver compared to Nudt7+/+ liver (Figure 5A). Moreover, the expression level of histone lysine methyltransferase (MLL) family member, particularly Mll3 and Mll4 known as H3K4 methyltransferase family member (Hu et al., 2013), was significantly increased in Nudt7+/− liver (Figure 5B). Nuclear staining of H3K4me3 was significantly increased in liver of Nudt7+/− mouse compared to Nudt7+/+ mouse (Figure 5C). Expression levels of DNA methyltransferases (DNMTs) known to inhibit H3K4me3 (Rose and Klose, 2014) such as Dnmt3b, Dnmt3l, and Mecp2 were significantly decreased in Nudt7+/− liver (Figure S7A). Exposure of DNA methylation inhibitors such as trichostatin A (TSA; 5 mM) or 5-Aza-2ʹ-deoxycytidine (5-aza; 10 mM) into primary mouse hepatocytes induced the expression of Mll3 (Figure S7B) as well as the expression level of DNL genes (Figure S7C). Chromatin immunoprecipitation (ChIP) qRT-PCR assay showed that H3K4me3 on Pparg promoter was significantly increased in Nudt7+/− liver compared with Nudt7+/+ liver (Figure 5D).
Figure 5.

Accumulated palmitic acid in Nudt7+/− liver is responsible for PPARγ activation
(A) The methylation levels of H3K4me2, H3K4me3, H3K27me2, and H3K27me3 were analyzed by immunoblotting in Nudt7+/+ and Nudt7+/− liver. Histone H3 was used for loading control (n = 3).
(B) The transcription levels of Mll3 and Mll4 were analyzed by qRT-PCR (n = 3).
(C) Representative images of H3K4me3 staining in the 12-month-old mouse liver (n = 3; Scale bars, 50 μm) and positive staining ratio (n = 6 per group).
(D) H3K4me3 level on PPARγ promoter was analyzed using qRT-PCR. DNA was isolated using chromatin immunoprecipitation (ChIP) in Nudt7+/+ and Nudt7+/− liver (n = 3).
(E) Palmitic acid level in Nudt7+/+ and Nudt7+/− liver (n = 5).
(F) Primary cultures of Nudt7+/+ hepatocytes were treated with BSA-conjugated with 50 μM palmitic acid (PA) or BSA alone (CON) and the expression level of H3K4me3, H3K27me3, H3K4me2, and H3K27me2 were analyzed by immunoblotting. Histone H3 was used for loading control.
(G) The expression levels of Mll3 and Mll4 were analyzed by qRT-PCR (n = 3).
(H) Primary cultures of Nudt7+/+ and Nudt7+/− hepatocytes were infected with lentiviruses containing NUDT7 (Nudt7-OE, +) or mock control (−) and the expression level of Mll3 was analyzed by qRT-PCR (n = 3).
(I) Representative images of BODIPY493/508 staining in primary cultures of Nudt7+/− hepatocytes transduced with shMll3 for 24 h. Scale bars, 100 μm. ∗p ≤ 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (Unpaired t-test or one-way ANOVA).
Lipid analysis showed that palmitic acid (C16:0, PA) was the most significantly increased FA in Nudt7+/− mouse (Figures 5E and S7D) and exposure of PA into primary hepatocytes induced the expression level of H3K4me3 (Figure 5F) as well as the expression levels of Mll3 and Mll4 (Figure 5G). Increased level of Mll3 in Nudt7+/− hepatocytes was reduced by Nudt7 restoration (Figure 5H). Increased level of lipid accumulation (Figure 5I) and inflammatory cytokines in Nudt7+/− hepatocytes was also significantly decreased by Mll3 knockdown (Figure S7E). Restoration of Nudt7 into Nudt7+/− mouse significantly decreased glucose and insulin tolerance (Figure S8) and the accumulation of LD in Nudt7+/− mouse liver (Figure 6A). The number of H3K4me3- and PPARγ-positive cells (Figure 6A), the levels of serum AST, ALT, FFA, and hepatic cholesterol (Figure 6B), expression level of PPARγ-downstream genes (Figure 6C), Mll3, Mll4, and chemokine genes (Figure 6D) were significantly decreased by the restoration of Nudt7. Moreover, restoration of Nudt7 reduced adipocyte size and the number of PMP70-positive cells in Nudt7+/− adipose tissue (Figure 6E).
Figure 6.

Restoration of Nudt7 reduces hepatic lipid accumulation, H3K4me3 of PPARγ, and inflammatory responses
(A) Nudt7+/− mice were tail-vein injected with lentiviruses containing Nudt7 or mock control (CON). Representative images of H&E and ORO staining and immunohistochemistry images of H3K4me3 and PPARγ staining (n = 3). Bar graph of H3K4me3 and PPARγ positive staining intensity was represented as the fold of CON (n = 6).
(B) The levels of serum AST, ALT, FFA, and hepatic cholesterol were analyzed (n = 4).
(C) The expression level of Pparg, Cd36, Fabp4, Il6, and Tnfa was analyzed using qRT-PCR (n = 3).
(D) The expression level of Nudt7, Mll3, Mll4, Ccl2, Ccl4, and Cxcl2 was analyzed using qRT-PCR (n = 3).
(E) Representative images of cell mask orange and PMP70 staining and graphs with adipocyte area per filed and percentage of PMP70-positive cell counting (n = 4; Scale bars, 100 μm). ∗p ≤ 0.05, ∗∗p < 0.01, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (Unpaired t-test).
Liraglutide attenuates hepatic lipid accumulation and hepatic DNL induced by NUDT7 deficiency
Liraglutide, the widely used glucagon-like peptide-1 receptor agonist (GLP-1 RA), known as an intrinsic PPARγ activator in endothelial cells (Onuma et al., 2014), is an agent considering as potent treatment option for NAFLD. The GLP-1 RA enhances intrinsic PPARγ activity. However, the mechanism of liraglutide underlying NAFLD is not well studied. In this study, to investigate whether liraglutide reduces the fatty liver induced by NUDT7 deficiency, we administrated 200 μg/kg body weight liraglutide into Nudt7+/− and Nudt7+/− mouse intraperitoneally. Interestingly, the expression level of NUDT7 was significantly increased in liraglutide-administrated Nudt7+/+ liver (Figure 7A). Furthermore, the expression level of PPARγ and H3K4me3 was significantly decreased in liraglutide-administrated Nudt7+/+ liver (Figure 7A). Administration of liraglutide significantly decreased the weight of body and liver (Figures S9A–S9C) as well as lipid accumulation (Figure 7B) in HFD Nudt7+/− mouse. Significantly reduced level of hepatic FFA, TG, and cholesterol accumulation (Figure 7C), FABP4, SCD1, PPARγ, and H3K4me3 (Figure 7D), and the expression level of gene in lipogenesis, lipid metabolism, and cytokine (Figure 7E) was observed in liraglutide-administrated Nudt7+/− liver.
Figure 7.

Liraglutide suppresses fatty liver induced by Nudt7 deficiency
(A) Representative images of H&E, NUDT7, PPARγ, and H3K4me3 in mouse liver of HFD-fed Nudt7+/+ mice with liraglutide or equal volume of sterile saline i.p. injected mouse.
(B) Representative image of H&E and BODIPY staining and the number of BODIPY positive cells in HFD-fed Nudt7+/− mice with liraglutide or equal volume of sterile saline i.p. injected mouse. (n = 4; Scale bar, 100 μm).
(C) Hepatic FFA, TG, and cholesterol were analyzed in HFD-fed Nudt7+/− mice liver (n = 6).
(D) Representative images of FABP4, SCD1, PPARγ, and H3K4me3 staining (n = 3; Scale bars, 100 μm).
(E) The expression level of lipogenic, lipid metabolism, and cytokine genes was analyzed using qRT-PCR (n = 3).
(F) Representative image of BODIPY staining and the number of BODIPY positive cells in primary hepatocyte with or without 100 nM liraglutide (n = 6; Scale bars, 100 μm).
(G) The expression level of Nudt7, Pparg, Mll3, Scd1, Fabp4, and Ldlr was analyzed using qRT-PCR (n = 3). ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (Unpaired t-test).
To rule out the possible involvement of adipose tissue in the action of liraglutide with Nudt7+/− mouse and verify the role of liraglutide in hepatocytes, we isolated hepatocyte from Nudt7+/− mouse and cultured with or without 100 nM liraglutide (Figure 7F). In primary cultured Nudt7+/− hepatocyte, lipid accumulation was significantly suppressed with treatment of liraglutide (Figure 7F) along with a significant increase in the expression level of Nudt7 and significant decrease in the expression level of Pparg, Mll3, Scd1, Fabp4, and Ldlr (Figure 7G).
Discussion
Even though increasing evidences suggested that peroxisomes that closely interact with mitochondria might be one of the key regulatory organelles in lipid metabolism (Demarquoy and Le Borgne, 2015) and peroxisomal function may play an important role in hepatic lipid metabolism (Lodhi and Semenkovich, 2014), only a few studies have been demonstrated the importance of peroxisomal function in the accumulation of hepatic lipid droplets (LD). Recently, it has been suggested that dysregulation of organelle interaction between peroxisome and mitochondria may be responsible for the development of fatty liver disease by abnormal regulation of fatty acid β-oxidation (Shai et al., 2016). Moreno-Fernandez and colleagues (2018) reported that the depletion of Acox1 caused hepatic lipid accumulation and inflammation (Moreno-Fernandez et al., 20182018). Consistent with this, our IPA using GEO dataset of advanced and mild nonalcoholic steatohepatitis (NASH) indicated that peroxisomal function, particularly fatty acid β-oxidation was significantly decreased in the advanced NASH compared to mild NASH. Among peroxisomal genes, we observed that peroxisomal nudix hydrolase 7 (Nudt7), one of family members of CoA diphosphatase mediating the cleavage of CoA, CoA esters, and oxidized CoA (Gasmi and McLennan, 2001), is significantly decreased in the primary hepatocytes isolated from Cat−/− mouse and HFD-induced mouse. Nudt7+/− mice displayed the typical characteristics of fatty liver i.e. increased levels of FFA, AST, ALT, and hepatic triglyceride (TG). Furthermore, decreased level of peroxisomal Nudt7 is responsible for the stimulation of inflammatory responses leading to the stimulation of inflammatory cytokines such as IL-1β, IL-6, and TNFα. These suggest that peroxisomal dysfunction via the suppression of Nudt7 may play an important role in the pathogenesis of fatty liver. Profound changes in adipose tissue are closely associated with the pathogenesis of fatty liver (Tamura and Shimomura, 2005). Adipocytes, the functional unit of adipose tissue performed the central role of lipid uptake and storage and releasing excess energy in the form of TG and FFA (Rector et al., 2008). Impairing the ability to absorb and store fatty acid in adipocytes or increasing lipolysis from adipose tissue led to the increased flux of fatty acid to the liver and inflammation and fibrosis in the liver (Rector et al., 2008). Recent study (Park et al., 2019) suggested that the disruption of peroxisomal biogenesis impaired adipose tissue thermogenesis. Adipose-specific KO of Pex16, the critical factor for assembly of the peroxisomal membrane and import of peroxisomal membrane proteins, decreased energy expenditure and increased the diet-induced obesity. On the other hand, adipose-specific KO of peroxisomal β-oxidation enzyme Acox1 did not affect adiposity and thermogenesis. Here, in our study, Nudt7+/− mice displayed the impaired WAT remodeling capacity, i.e. increased the lipolysis of adipose tissue suggesting that regulation of adipose tissue by NUDT7-induced secondary effects in the liver contributing to the development of NFALD. Interestingly, we observed the increased number of peroxisome in the adipose tissue of Nudt7+/− mice. We believe that this is due to a compensatory mechanism for peroxisomal dysfunction in Nudt7+/− mice, and research is currently ongoing to prove this. Moreover, since we found that co-culture of Nudt7+/+ hepatocyte with Nudt7+/− adipocyte induces lipid accumulation and activation of lipogenic genes suggesting the interaction between hepatocyte and adipocyte in the development of NFALD in Nudt7+/− mice, detailed studies on interaction between hepatocyte and adipocyte are also ongoing.
Peroxisome proliferator-activated receptors (PPARs), member of the nuclear receptor superfamily (Raney et al., 1997), are known to regulate lipid synthesis, glucose metabolism, and cholesterol efflux in various cell types including hepatocytes, adipocytes, and macrophages. PPARγ, highly expressed in adipose tissue and macrophage, plays important roles in lipid metabolism such as fatty acid uptake, β-oxidation and TG turnover, insulin resistance, and immune regulation (Cave et al., 2016; Lee et al., 1995). Hepatocyte PPARγ has been known as a steatogenic factors while others suggest the activation of PPARγ reduces hepatic steatosis. Previously, it has been reported that overexpression of PPARγ leads to ectopic fat deposition in the liver (Gross et al., 2017). Increase level of PPARγ has been shown in steatotic liver of animal models or patients with NAFLD (Matsusue et al., 2003). Hepatocyte-specific PPARγ knockout was associated with a reduction in the expression level of DNL genes. HFD-induced hepatic steatosis and lipogenesis genes such as Scd1, Srebf1c, and Acc were significantly reduced in hepatocyte-specific PPARγ knockout mice (Moran-Salvador et al., 2011). However, other groups (Wolf Greenstein et al., 2017) reported that liver (hepatocyte)-specific PPARγ knockdown in HFD mouse reduced the accumulation of hepatic TG but not the expression of DNL genes suggesting that PPARγ plays a minimal role in directly regulating hepatic DNL. In this study, we found that Nudt7 deficiency induced the over-activation of PPARγ and stimulated the accumulation of hepatic TG and DNL. Moreover, we found that the level of H3K4me3 in the Pparg promoter was significantly upregulated in the hepatocytes of Nudt7+/− liver due to increased level of an endogenous lipid that can activate PPARs, palmitic acid in Nudt7+/− liver. It has been reported that PPARs are activated by endogenous lipids such as free fatty acids (FFAs) and eicosanoids (Wahli and Michalik, 2012; Tailleux et al., 2012). Fatty acids such as palmitic acid, oleic acid, and prostaglandins have been identified as natural ligands for PPARs (Tailleux et al., 2012).
Since the controversial role of PPARγ on NALFD has been reported, the effect of rosiglitazone, a PPARγ agonist on improving or exacerbating NAFLD also contradictory [65, 66]. In HFD liver, treatment of rosiglitazone significantly increased in the accumulation of TG and lipid unlike to NCD liver (Gao et al., 2016). In A-ZIP/F-1 mouse, a model for the human disease lipoatrophic diabetes (Gavrilova et al., 2003), the treatment of rosiglitazone also significantly increased the accumulation of hepatic TG by the activation of PPARγ (Wahli and Michalik, 2012). In this study, we also found that exposure of rosiglitazone into Nudt7+/− hepatocytes accumulated LD and this lipid accumulation dramatically reduced the restoration of Nudt7. Recently, glucagon-like peptide-1 (GLP-1) receptors, known to express in hepatocytes, had been experimentally and clinically shown to ameliorate NAFLD (Lee et al., 2012). In liver biopsy specimens from patients with NASH, the number of GLP-1 receptors is reduced as well as in animal NAFLD models (Svegliati-Baroni et al., 2011). Liraglutide, the most widely used GLP-1 receptor agonists, is an attractive candidate for the treatment of NAFLD shown with multiple preclinical studies and clinical trials (Lv et al., 2020). Recent experimental study suggests that liraglutide might decrease hepatic inflammation and hepatocyte ballooning (Ipsen et al., 2018; Kojima et al., 2020). Moreover, liraglutide is known to reverse oxidative stress by activating PPARα to inhibit the expression of diacylglycerolacyl O-acetyltransferase (DGAT) in diabetes (Zhang et al., 2018) and reduce hepatic fatty acid flux by decreasing PPAR-γ in the liver of diet-induced obesity rats (Decara et al., 2016). However, the relationship between the effects of liraglutide on regulating hepatic inflammation and lipid metabolism remains unclear. Here, we observed that liraglutide reverses the NAFLD features induced by NUDT7 deficiency by regulating NUDT7-PPARγ axis. In addition, we also found the elevated level of PPARα in Nudt7+/− hepatocytes suggesting the possible role of PPARα in the pathogenesis of NAFLD induced by NUDT7 deficiency and the investigation on the role of other PPAR family including PPARα in NAFLD induced by NUDT7 deficiency is currently undergoing in our laboratory.
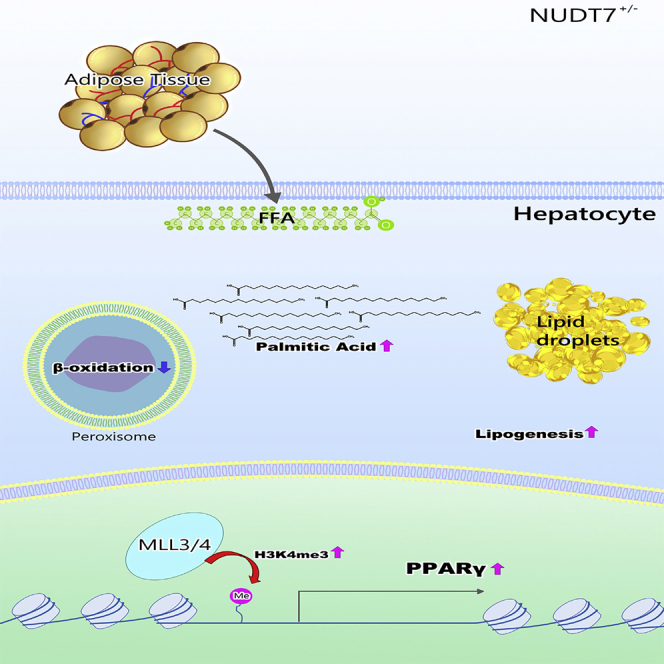
Taken together, here, peroxisomal Nudt7 deficiency displayed fatty liver due to an increased hepatic DNL via activation of PPARγ through upregulation of H3K4me3 and this could be reversed by liraglutide (Figure 8). Because of the interplay between peroxisomes and mitochondria in maintaining lipid homeostasis, we would expect that the mitochondrial dysfunction may be associated with NAFLD induced by NUDT7 deficiency and remains to be investigated further.
Figure 8.

Diagram of the suggested mechanism by which deficiency of peroxisomal NUDT7 induces NAFLD
Limitations of the study
Our study demonstrates that heterozygous deficiency of a peroxisomal gene, Nudt7, could be responsible for NAFLD through DNL-PPAR PPARγ activation and liraglutide markedly prevented NAFLD induced by NUDT7 deficiency. The limitation of this study is the number of patients used in this study. Further investigation with a large pool of patients to verify Nudt7 as biomarker and potent therapeutic and clinical application for NAFLD will be warranted.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ACOX1 | Cusabio | Cat# PA617998HA01HU |
| H3K4me3 | Abcam | Cat# ab8580; RRID:AB_306649 |
| NUDT7 | Youngin Frontier | N/A |
| PPARγ | Abcam | Cat# ab41928; RRID:AB_777392 |
| FABP4 | Abcam | Cat# ab92501; RRID:AB_10562486 |
| FASN | Cell signaling technology | Cat# 3180S; RRID:AB_2100796 |
| PMP70 | Abcam | Cat# ab211533 |
| SCD1 | Abcam | Cat# ab19862; RRID:AB_445179 |
| H3K4me2 | Cell signaling technology | Cat#9725; RRID:AB_10205451 |
| H3K27me2 | Cell signaling technology | Cat#9755; RRID:AB_659841 |
| H3K27me3 | Abcam | Cat#ab6002; RRID:AB_305237 |
| Histone H3 | Cell signaling technology | Cat#4499; RRID:AB_10544537 |
| Chemicals, peptides, and recombinant proteins | ||
| CellMask™ Orange Plasma membrane Stain | Thermo Fisher Scientific | C10045 |
| Oil red O | Sigma-Aldrich | Cat# O0625s |
| BODIPY493/503 | Thermo Fisher Scientific | Cat# D3922 |
| BODIPY581/591 | Thermo Fisher Scientific | Cat# D3861 |
| Filipin | Sigma-Aldrich | Cat# SAE0088 |
| 3rd generation packaging system | ABM | Cat# LV053 |
| Lentifectin | ABM | Cat# G074 |
| Lenti-X Concentrator | Clontech | Cat# PT4421-2 |
| Critical commercial assays | ||
| Nuclear/Cytosol Extraction Kit | BioVision | K266-100 |
| EZ-Triglyceride Quantification Assay Kit | DoGen | Cat# DG-TGC100 |
| EZ-Free Fatty Acid Assay Kit | DoGen | Cat # DG-FFA100 |
| EZ-Total Cholesterol Assay Kit | DoGen | Cat# DG-TSC100 |
| Quick Cell Proliferation Colorimetric Assay Kit | BioVision | Cat# K302-2500 |
| Catalase Activity Colorimetric/Fluorometric Assay Kit | BioVision | Cat# K773-100 |
| ChIP-IT Express Kit | Active Motif | Cat# 53009 |
| Experimental models: Cell lines | ||
| HEK-293T | ATCC | CRL-3216 |
| Hepatocyte | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Nudt7+/−: C57BL/6N | Song et al. (2018, NComm) | N/A |
| Mouse: Catalse−/−: C57BL/6N | Generously donated by Goo Taeg Oh, Ewha Womans University | N/A |
| Oligonucleotides | ||
| See Table S2 for primer sequences. | ||
| Software and algorithms | ||
| RStudio | RStudio | https://github.com/rstudio/rstudio |
| QIAGEN’s Ingenuity Pathway Analysis algorithm (IPA) | QIAGEN | www.qiagen.com/ingenuity |
| Gene Set Enrichment Analysis (GSEA) | Subramanian, Tamayo, et al. (2005, PNAS) and and Mootha, Lindgren, et al. (2003, Nature Genetics) | www.gsea-msigdb.org/gsea |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eun-Jung Jin (jineunjung@wku.ac.kr).
Materials availability
Materials in this study will be made available from the lead contact with a material transfer agreement.
Experimental model and subject details
Ethical approval
Liver tissues were obtained from 22 patients who underwent bariatric and metabolic surgery at Keimyung University, Dongsan Hospital, between January 2019 and December 2019. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was reviewed and approved by the Institutional Review Board of Keimyung University Dongsan Hospital (IRB No. 2015-01-015). Animal studies were performed following approval from the Wonkwang University Animal Care and Use Committee and were in compliance with institutional guidelines (#WKU18-23). Sample size for animal studies was determined by statistical analysis of variance and on the basis our experience with similar studies. The sample size (n) for each experimental group is indicated in the figure legends and is between three and six mice per group. All of the in vivo experiments were replicated at least three times by two experimentalists independently. All histological and IHC samples were blinded before the images were taken and quantified by trained liver pathologists.
Animals
NUDT7-knockout (KO) mice on a C57BL/6 background were generated by TALEN-mediated gene targeting [31]. {Song, 2018 #19}Twelve-month-old male C57BL/6N (Nudt7+/+) and NUDT7 KO (Nudt7+/−) mice or Catalase KO (Cat−/−) were fed normal chow diet (NCD) ad-libitum or high-fat diet (HFD; 60% kcal from fat; Research Diets Inc.) for the indicated times. Nudt7+/− mice were fed a HFD for 8 weeks to induce NAFLD. Next, liraglutide (200 μg/kg) or an equal volume of sterile saline was intraperitoneal (i.p.) injected for 4 weeks. All animals were fasted for 12 h (overnight) before scarification, and blood was collected to isolate the serum for biochemical analysis.
Method details
Indirect calorimetry
Water and food intake, VO2, VCO2, and Heat were analyzed using an Oxymax system (Columbus Instruments). Mice were placed in the chambers at 23°C with free access to food and water and acclimated for more than 50 min before measurement.
Lentiviral constructs packaging and tail vein injection
Mock-control, Nudt7 and shAcox1-containing lentiviruses were produced using the 3rd generation packaging system (ABM, Richmond, BC, Canada). Lentivirus plasmids were transfected into HEK-293T cells using Lentifectin (ABM) in DMEM serum free medium (ThermoFisher Scientific) and cultured overnight. The lentiviral particle containing supernatant was concentrated using a Lenti-X Concentrator (Clontech) and stored at −80°C. For in-vivo delivery, concentrated lentivirus supernatant was injected into the tail vein twice a week for 6 weeks.
Histological analysis
Paraffin-embedded tissues were sliced into 5 μm sections and stained with hematoxylin and eosin (H&E). Plasma membrane was stained with CellMask Orange (Thermo Fisher Scientific). Frozen liver sections were cut into 5 μm slices and stained with 0.3% Oil red O (Sigma-Aldrich) in 60% isopropanol or 0.25 mg/mL filipin. For immunohistochemistry (IHC), sections were incubated overnight at 4°C (1:200 dilution) in a humidified chamber with the following primary antibodies; ACOX1 (Cusabio), H3K4me3 (Abcam), NUDT7 (Youngin Frontier), PPARγ (Abcam), FABP4(Abcam), FASN (Cell signaling technology), PMP70 (Abcam), and SCD1 (Abcam). Positive staining was visualized using the Liquid DAB Substrate Chromogen System (Dako) combined with counterstaining with hematoxylin. Histological images were acquired with a light microscope (Leica).
Cell culture
Hepatocytes were isolated using collagenase perfusion technique from 10-week-old Nudt7+/+ and Nudt7+/− mice and cultured in DMEM high glucose medium (ThermoFisher Scientific) supplemented with 10% FBS (ThermoFisher Scientific) and 100 U/ml penicillin-streptomycin (ThermoFisher Scientific).
Immunoblotting
Mouse tissues or cells were homogenized with RIPA buffer (Cell Signaling Technology) supplemented with phenylmethylsulfonyl fluoride (PMSF) protease inhibitor (Sigma-Aldrich), and nuclear lysates were extracted using a Nuclear/Cytosol Extraction Kit (BioVision). Proteins (40 μg) were separated on a sodium dodecyl sulfate (SDS)-polyacrylamide gel, transferred to a nitrocellulose membrane (GE Healthcare), and then incubated with the following primary antibodies; H3K4me2 (Cell Signaling Technology), H3K4me3 (Abcam), H3K27me2 (Cell Signaling Technology), H3K27me3 (Abcam), and histone H3 (Cell Signaling Technology). The blotted nitrocellulose membranes were developed using horseradish peroxidase-conjugated secondary antibodies (α-Rabbit or α-Mouse, 1:2500 dilution) and target proteins were detected and visualized using either an enhanced chemiluminescence (ECL) system (ThermoFisher Scientific).
Lipid and cholesterol accumulation assay
Cells were stained with 2 μM BODIPY493/503 (ThermoFisher Scientific) or 5 μM Lipid ROS (BODIPY581/591; ThermoFisher Scientific) or 50 μg/mL filipin (Sigma-Aldrich) to evaluate lipid accumulation, and images were captured with an Imaging System (ThermoFisher Scientific).
Mouse liver and serum analysis
Hepatic or serum TG and FFA were measured using a EZ-Triglyceride Quantification Assay Kit (DoGen) or EZ-Free Fatty Acid Assay Kit (DoGen) or EZ-Total Cholesterol Assay Kit (DoGen) according to the manufacturer’s instructions. Serum aspartate transaminase (AST) and alanine transferase (ALT) were obtained from the Seoul Clinical Laboratories.
Cell proliferation and catalase activity assay
Cell proliferation and catalase activity were analyzed using Quick Cell Proliferation Colorimetric Assay Kit (BioVision) and Catalase Activity Colorimetric/Fluorometric Assay Kit (BioVision) according to the manufacturer’s instructions.
qRT-PCR and peroxisomal gene profiling
Total RNA was isolated using RNAiso Plus (TaKaRa) according to the manufacturer’s instructions. Then, 1 μg RNA was reverse transcribed using the 5X All-In-One RT MasterMix (Abm). Each target gene amplified with the StepOne Plus Real-Time PCR System (Applied Biosystems) using specific primers listed in Table S2 and relative expression level of each gene was normalized with Rn18s. The 42 peroxisomal genes were determined by qRT-PCR. Heatmap images and visualized using the R studio.
RNA sequencing
RNA sequencing was performed using the Illumina HiSeq 4000 System. Libraries were quantified using qRT-PCR and their quality was analyzed using the Agilent 2100 Bioanalyzer (Agilent Technologies). Raw counts were generated by calculating the fragments per kilobase of transcript per million mapped reads (FPKM) of each sample using the Cufflinks software. Data were transformed logarithmically and normalized using the quantile normalization technique.
Chromatin immunoprecipitation (ChIP) assay
The H3K4me3-ChIP assay was performed using a ChIP-IT Express Kit (Active Motif). The genomic DNA was sheared using components of the ChIP-IT Express Kit (Active Motif) and 20 μg of genomic DNA was immunoprecipitated with 3 μg of H3K4me3 antibody (Abcam) and Protein G-coupled magnetic beads (Active Motif). The bead-bound chromatin was eluted with 1% SDS and 0.1 M NaHCO3 and purified using phenol-chloroform. Promoter sequences for qRT-PCR were obtained from the Eukaryotic Promoter Database (http://epd.vital-it.ch) and specific primers listed in Table S2.
Pathway analysis using GSEA and IPA
Microarray data of GSE33814 (steatosis and steatohepatitis patient liver), GSE49541 (mild NASH and advanced NASH patient liver), and GSE39549 (HFD fed mouse liver) from Gene Expression Omnibus (GEO) and RNA sequencing data of 12 month-old chow fed Nudt7+/− or Nudt7+/+ mouse liver were analyzed in this study. Differentially expressed genes (DEGs) was analyzed using QIAGEN’s Ingenuity Pathway Analysis algorithm (IPA, QIAGEN, www.qiagen.com/ingenuity) or Gene Set Enrichment Analysis (GSEA, http: www. broadinstitute.org/gsea).
Histology analysis and IHC of human samples
Formalin-fixed paraffin-embedded (FFPE) block specimens from intraoperative liver biopsy during bariatric surgery were obtained. All the H&E stained slides were reviewed by an experienced pathologist (HWL) who was blinded to the clinicopathological features or clinical outcome. The severity of NAFLD was evaluated histologic features according to a NAFLD activity score (NAS): steatosis, lobular inflammation, hepatocyte ballooning, and fibrosis (Kleiner et al., 2005).
Protein expressions were assessed by an immunohistochemical study. IHC staining was performed using an automated immunostainer, Ventana BenchMark XT (Ventana Medical Systems, Tucson, AZ, USA) with an UltraView kit, according to the manufacturer’s protocol. NUDT7 (Ab Frontier), PPARγ (Abcam) and H3K4me3 (Abcam) were applied as the primary antibodies, after which the samples were incubated at 37°C for 32 min followed by standard Ventana signal amplification, and hematoxylin and a bluing reagent counterstaining, consecutively. After the autostainer process, the slides were mounted and examined by light microscopy. Positive staining of NUDT7 was indicated by a prominent brownish pigmentation in the cytoplasm, and PPARγ and H3K4me3 were in the nucleus. Negative controls were obtained by omitting the specific primary antibodies of the same species. Positive and negative controls stained appropriately. Expressions were assessed according to staining intensity and scored from 0 to 3 as follows: 0 (no), 1 (weak), 2 (moderate), and 3 (strong). Staining score 0 to 1 considered low expression and score 2 to 3 considered high, respectively.
Quantification and statistical analysis
Results are expressed as the mean ± standard error of mean (SEM). Significant differences between two groups were analyzed by Student’s t-test. Differences among three or more groups were compared by one-way or two-way ANOVA followed by Tukey’s multiple comparisons test. p ≤ 0.05 was considered statistically significant.
Acknowledgments
The biospecimens and data used for this study were provided by the Biobank of Keimyung University Dongsan Hospital Biobank, a member of the Korea Biobank Network. This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI22C0729).
Author contributions
Conceptualization: E-J.J.; Data curation: J.S. and K.S.; Formal analysis: J.S.; Funding acquisition: E-J.J. and J.S.; Investigation: J.S., I-J.B., S.P., J.O., D.K., M.K.K., H.W.L., and B.K.J.; Writing original draft: E-J.J.
Declaration of interests
The authors declare that they have no competing interests.
Published: October 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105135.
Supplemental information
Data and code availability
-
•
Data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original codes.
-
•
Any additional information available from the lead contact upon request.
References
- Benedict M., Zhang X. Non-alcoholic fatty liver disease: an expanded review. World J. Hepatol. 2017;9:715–732. doi: 10.4254/wjh.v9.i16.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M.C., Clair H.B., Hardesty J.E., Falkner K.C., Feng W., Clark B.J., Sidey J., Shi H., Aqel B.A., McClain C.J., Prough R.A. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta. 2016;1859:1083–1099. doi: 10.1016/j.bbagrm.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao L., Marcus-Samuels B., Mason M.M., Moitra J., Vinson C., Arioglu E., Gavrilova O., Reitman M.L. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J. Clin. Invest. 2000;106:1221–1228. doi: 10.1172/JCI11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decara J., Arrabal S., Beiroa D., Rivera P., Vargas A., Serrano A., Pavón F.J., Ballesteros J., Dieguez C., Nogueiras R., et al. Antiobesity efficacy of GLP-1 receptor agonist liraglutide is associated with peripheral tissue-specific modulation of lipid metabolic regulators. Biofactors. 2016;42:600–611. doi: 10.1002/biof.1295. [DOI] [PubMed] [Google Scholar]
- Demarquoy J., Le Borgne F. Crosstalk between mitochondria and peroxisomes. World J. Biol. Chem. 2015;6:301–309. doi: 10.4331/wjbc.v6.i4.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doege H., Baillie R.A., Ortegon A.M., Tsang B., Wu Q., Punreddy S., Hirsch D., Watson N., Gimeno R.E., Stahl A. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis. Gastroenterology. 2006;130:1245–1258. doi: 10.1053/j.gastro.2006.02.006. [DOI] [PubMed] [Google Scholar]
- El Hajj H.I., Vluggens A., Andreoletti P., Ragot K., Mandard S., Kersten S., Waterham H.R., Lizard G., Wanders R.J.A., Reddy J.K., Cherkaoui-Malki M. The inflammatory response in acyl-CoA oxidase 1 deficiency (pseudoneonatal adrenoleukodystrophy) Endocrinology. 2012;153:2568–2575. doi: 10.1210/en.2012-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E., Sullivan S., Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell G.C., Larter C.Z. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- Fernandez M.A., Albor C., Ingelmo-Torres M., Nixon S.J., Ferguson C., Kurzchalia T., Tebar F., Enrich C., Parton R.G., Pol A. Pol, Caveolin-1 is essential for liver regeneration. Science. 2006;313:1628–1632. doi: 10.1126/science.1130773. [DOI] [PubMed] [Google Scholar]
- Gao M., Ma Y., Alsaggar M., Liu D. Dual outcomes of rosiglitazone treatment on fatty liver. AAPS J. 2016;18:1023–1031. doi: 10.1208/s12248-016-9919-9. [DOI] [PubMed] [Google Scholar]
- Gavrilova O., Haluzik M., Matsusue K., Cutson J.J., Johnson L., Dietz K.R., Nicol C.J., Vinson C., Gonzalez F.J., Reitman M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- Gong Z., Tas E., Yakar S., Muzumdar R. Hepatic lipid metabolism and non-alcoholic fatty liver disease in aging. Mol. Cell. Endocrinol. 2017;455:115–130. doi: 10.1016/j.mce.2016.12.022. [DOI] [PubMed] [Google Scholar]
- Gross B., Pawlak M., Lefebvre P., Staels B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017;13:36–49. doi: 10.1038/nrendo.2016.135. [DOI] [PubMed] [Google Scholar]
- Hashimoto T., Fujita T., Usuda N., Cook W., Qi C., Peters J.M., Gonzalez F.J., Yeldandi A.V., Rao M.S., Reddy J.K. Peroxisomal and mitochondrial fatty acid beta-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor alpha and peroxisomal fatty acyl-CoA oxidase. Genotype correlation with fatty liver phenotype. J. Biol. Chem. 1999;274:19228–19236. doi: 10.1074/jbc.274.27.19228. [DOI] [PubMed] [Google Scholar]
- Hu D., Gao X., Morgan M.A., Herz H.M., Smith E.R., Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell Biol. 2013;33:4745–4754. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui T.Y., Olivier L.M., Kang S., Davis R.A. Microsomal triglyceride transfer protein is essential for hepatic secretion of apoB-100 and apoB-48 but not triglyceride. J. Lipid Res. 2002;43:785–793. [PubMed] [Google Scholar]
- Ipsen D.H., Rolin B., Rakipovski G., Skovsted G.F., Madsen A., Kolstrup S., Schou-Pedersen A.M., Skat-Rørdam J., Lykkesfeldt J., Tveden-Nyborg P. Liraglutide decreases hepatic inflammation and injury in advanced lean non-alcoholic steatohepatitis. Basic Clin. Pharmacol. Toxicol. 2018;123:704–713. doi: 10.1111/bcpt.13082. [DOI] [PubMed] [Google Scholar]
- Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W., Ferrell L.D., Liu Y.C., Torbenson M.S., Unalp-Arida A., et al. Nonalcoholic Steatohepatitis Clinical Research, Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- Kojima M., Takahashi H., Kuwashiro T., Tanaka K., Mori H., Ozaki I., Kitajima Y., Matsuda Y., Ashida K., Eguchi Y., Anzai K. Glucagon-like peptide-1 receptor agonist prevented the progression of hepatocellular carcinoma in a mouse model of nonalcoholic steatohepatitis. Int. J. Mol. Sci. 2020;21:E5722. doi: 10.3390/ijms21165722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo S.H. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013;19:210–215. doi: 10.3350/cmh.2013.19.3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasmi L., McLennan A.G. The mouse Nudt7 gene encodes a peroxisomal nudix hydrolase specific for coenzyme A and its derivatives. Biochem. J. 2001;357:33–38. doi: 10.1042/0264-6021:3570033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Hong S.W., Rhee E.J., Lee W.Y. GLP-1 receptor agonist and non-alcoholic fatty liver disease. Diabetes Metab. J. 2012;36:262–267. doi: 10.4093/dmj.2012.36.4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.S., Pineau T., Drago J., Lee E.J., Owens J.W., Kroetz D.L., Fernandez-Salguero P.M., Westphal H., Gonzalez F.J. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodhi I.J., Semenkovich C.F. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. 2014;19:380–392. doi: 10.1016/j.cmet.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv X., Dong Y., Hu L., Lu F., Zhou C., Qin S. Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) for the management of nonalcoholic fatty liver disease (NAFLD): a systematic review. Endocrinol. Diabetes Metab. 2020;3:e00163. doi: 10.1002/edm2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusue K., Haluzik M., Lambert G., Yim S.H., Gavrilova O., Ward J.M., Brewer B., Jr., Reitman M.L., Gonzalez F.J. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Invest. 2003;111:737–747. doi: 10.1172/JCI17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Salvador E., Lopez-Parra M., Garcia-Alonso V., Titos E., Martinez-Clemente M., Gonzalez-Periz A., Lopez-Vicario C., Barak Y., Arroyo V., Claria J. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011;25:2538–2550. doi: 10.1096/fj.10-173716. [DOI] [PubMed] [Google Scholar]
- Moreno-Fernandez M.E., Giles D.A., Stankiewicz T.E., Sheridan R., Karns R., Cappelletti M., Lampe K., Mukherjee R., Sina C., Sallese A., et al. Peroxisomal beta-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI Insight. 2018;3:93626. doi: 10.1172/jci.insight.93626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso G., Gambino R., Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD) Prog. Lipid Res. 2009;48:1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- Onuma H., Inukai K., Kitahara A., Moriya R., Nishida S., Tanaka T., Katsuta H., Takahashi K., Sumitani Y., Hosaka T., Ishida H. The glucagon-like peptide 1 receptor agonist enhances intrinsic peroxisome proliferator-activated receptor γ activity in endothelial cells. Biochem. Biophys. Res. Commun. 2014;451:339–344. doi: 10.1016/j.bbrc.2014.07.136. [DOI] [PubMed] [Google Scholar]
- Park H., He A., Tan M., Johnson J.M., Dean J.M., Pietka T.A., Chen Y., Zhang X., Hsu F.F., Razani B., et al. Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J. Clin. Invest. 2019;129:694–711. doi: 10.1172/JCI120606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raney A.K., Johnson J.L., Palmer C.N., McLachlan A. Members of the nuclear receptor superfamily regulate transcription from the hepatitis B virus nucleocapsid promoter. J. Virol. 1997;71:1058–1071. doi: 10.1128/jvi.71.2.1058-1071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector R.S., Thyfault J.P., Wei Y., Ibdah J.A. Non-alcoholic fatty liver disease and the metabolic syndrome: an update. World J. Gastroenterol. 2008;14:185–192. doi: 10.3748/wjg.14.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolo A.P., Teodoro J.S., Palmeira C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012;52:59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Rose N.R., Klose R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta. 2014;1839:1362–1372. doi: 10.1016/j.bbagrm.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader M., Costello J., Godinho L.F., Islinger M. Peroxisome-mitochondria interplay and disease. J. Inherit. Metab. Dis. 2015;38:681–702. doi: 10.1007/s10545-015-9819-7. [DOI] [PubMed] [Google Scholar]
- Shai N., Schuldiner M., Zalckvar E. No peroxisome is an island - peroxisome contact sites. Biochim. Biophys. Acta. 2016;1863:1061–1069. doi: 10.1016/j.bbamcr.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Black S.M. Carnitine homeostasis, mitochondrial function, and cardiovascular disease, drug discovery today. Drug Discov. Today Dis. Mech. 2009;6:e31–e39. doi: 10.1016/j.ddmec.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimano H., Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol. 2017;13:710–730. doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- Song J., Baek I.J., Chun C.H., Jin E.J. Dysregulation of the NUDT7-PGAM1 axis is responsible for chondrocyte death during osteoarthritis pathogenesis. Nat. Commun. 2018;9:3427. doi: 10.1038/s41467-018-05787-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strable M.S., Ntambi J.M. Genetic control of de novo lipogenesis: role in diet-induced obesity. Crit. Rev. Biochem. Mol. Biol. 2010;45:199–214. doi: 10.3109/10409231003667500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svegliati-Baroni G., Saccomanno S., Rychlicki C., Agostinelli L., De Minicis S., Candelaresi C., Faraci G., Pacetti D., Vivarelli M., Nicolini D., et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31:1285–1297. doi: 10.1111/j.1478-3231.2011.02462.x. [DOI] [PubMed] [Google Scholar]
- Tailleux A., Wouters K., Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim. Biophys. Acta. 2012;1821:809–818. doi: 10.1016/j.bbalip.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Tamura S., Shimomura I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J. Clin. Invest. 2005;115:1139–1142. doi: 10.1172/JCI24930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahli W., Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012;23:351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Wang G., Bonkovsky H.L., de Lemos A., Burczynski F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015;56:2238–2247. doi: 10.1194/jlr.R056705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerbacka J., Kolak M., Kiviluoto T., Arkkila P., Sirén J., Hamsten A., Fisher R.M., Yki-Järvinen H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes. 2007;56:2759–2765. doi: 10.2337/db07-0156. [DOI] [PubMed] [Google Scholar]
- Wolf Greenstein A., Majumdar N., Yang P., Subbaiah P.V., Kineman R.D., Cordoba-Chacon J. Hepatocyte-specific, PPARgamma-regulated mechanisms to promote steatosis in adult mice. J. Endocrinol. 2017;232:107–121. doi: 10.1530/JOE-16-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Xiao X., Zheng J., Li M., Yu M., Ping F., Wang T., Wang X. Liraglutide protects cardiac function in diabetic rats through the PPARalpha pathway. Biosci. Rep. 2018;38 doi: 10.1042/BSR20180059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original codes.
-
•
Any additional information available from the lead contact upon request.