

Abstract
Epigenetics plays a strong role in molecular adaptations to heat by producing a molecular memory of past environmental exposures. Moderate heat, over long periods of time, induces an “adaptive,” epigenetic memory, resulting in a condition of “resilience” to future heat exposures or cross-tolerance to other forms of toxic stress. In contrast, intense, life-threatening heat exposures, such as severe heat stroke, can result in a “maladaptive” epigenetic memory that can place an organism at risk of later health complications. These cellular memories are coded by post-translational modifications of histones on the nucleosomes and/or by changes in DNA methylation. They operate by inducing changes in the level of gene transcription and therefore phenotype. The adaptive response to heat acclimation functions, in part, by facilitating transcription of essential heat shock proteins and exhibits a biphasic short program (maintaining DNA integrity, followed by a long-term consolidation). The latter accelerates acclimation responses after de-acclimation. Though less studied, the maladaptive responses to heat stroke appear to be coded in long lasting DNA methylation changes near the promoter region of genes involved with basic cell function. Whether these memories are also encoded in histone modifications is not yet known. There is considerable evidence that both adaptive and maladaptive epigenetic responses to heat can be inherited, though most evidence comes from lower organisms. Future challenges include understanding the signaling mechanisms responsible and discovering new ways to promote adaptive responses while suppressing maladaptive responses to heat, as all life forms adapt to life on a warming planet.
Keywords: Histone regulation, heat stroke, DNA methylation, environmental physiology
Introduction: Heat and its role in the origins of epigenetics
Almost a century ago, Conrad Hal Waddington made seminal observations that Mendelian genetics could not account entirely for phenotypical outcomes, understanding that critical developmental molecular processes connect genotype with phenotype (Waddington, 1942; Weinhold, 2006; Bollati & Baccarelli, 2010). When studying the developing wing of the common fruit fly (Drosophila), Waddington noticed that the wing changed conformation numerous times prior to its final differentiated form. Using the concept ‘epigenesis’ as fodder, i.e., the process by which an embryo develops from an undifferentiated egg, Waddington coined the term ‘epigenotype’ and ‘epigenetics’ in his seminal paper in 1942 (Waddington, 1942). Thus began the birth of the field of epigenetics (Fig. 1).
Figure 1.

A timeline of some important research breakthroughs in understanding the adaptive and epigenetic responses to exposure to a hyperthermic environment.
However, prior to Waddington, several studies examining the epigenetic phenomenon had already been conducted. These original environmental epigenetic studies used heat to assess phenotype ‘mutations’ in Drosophila, both within a single lifetime and between generations (Plough & Ives, 1934; Jollos, 1934) (Fig. 1). The heat studies were also conducted using fruit fly strains adapted to various environmental backgrounds. What were called ‘mutations’ in response to chronic heat varied between strains. For example, changes were limited in the fruit fly stock from Florida, suggesting that the nature of the heat stimulus, either dry or humid, or the environmental history of the strain elicited different ‘mutations’ (Jollos, 1934). Generational inheritance of these heat-related ‘mutations’ were observed in lines exposed to chronic heat, and spontaneous ‘mutations’ occurred at a higher frequency amongst progeny from heat-exposed lines compared to control lines (Plough & Ives, 1934; Jollos, 1934).
Many of these seminal observations represent the historical origins of our present epigenetic paradigm, as it applies to all members of the animal and plant kingdoms. Only recently were observations made relevant to mammalian models, such as guinea pigs (Weyrich et al., 2016a, 2016b), rats (Tetievsky & Horowitz, 2010; Horowitz, 2014, 2016), and mice (Murray et al., 2021b), as well as in human populations (Bind et al., 2014).
Heat is a logical environmental stimulus to initially characterize epigenetic ‘mutations.’ Because most every organism must endure elevated temperatures at some point in its lifespan. Furthermore, since heat exposure can be life threatening to a population, survival of a species requires the development of evolutionary machinery to adapt to heat over time. As such, dynamic adaptations depend on the nature of the heat exposure, ranging from heat acclimation to negative consequences of heat stroke, as indicated in retrospective epidemiological studies (Wallace et al., 2007; Wang et al., 2019) and original preclinical and clinical research (McClung et al., 2008; Umschweif et al., 2013; Horowitz, 2014; Horowitz et al., 2015; Périard et al., 2015, 2016; Laitano et al., 2020a; Murray et al., 2021b). This spectrum of epigenetic responses to heat represents a classical example of hormesis in physiological adaptation (Berry & López-Martínez, 2020).
The aims of this review are to (1) provide a historical context to how research began in the area of the epigenetic responses to heat exposure and how that relates to physiology. 2) Describe the history of research in the area of the epigenetic mechanisms underlying heat acclimation and re-acclimation and how that has led to recent breakthroughs in our understanding. 3) Discuss the basic biochemistry of DNA methylation, how it affects gene transcription and what is known about DNA methylation changes in response to exposure to heat stroke. 4) Discuss the range of disorders such as heart and kidney disease that appear to only emerge months to years after heat stroke exposure and may reflect underlying epigenetic changes. 5) Discuss what is known about inherited epigenetic responses to heat exposure in man and animals, and 6) suggest considerations and implications for future work in this field.
Historical observations on the role of epigenetics in heat acclimation
There was copious interest in understanding adaptations to environmental heat as the tropics were colonized, including discussions of whether “infectious diseases” were linked to sunstroke, such as “Siriasis” (named after Sirius, with incidence increasing in the hottest month with the sun) or infectious diseases and hygiene conditions, reducing the quality of life in the colonies (Sambon, 1898; Cutler, 1902). Physiological experimentation on acclimation/acclimatization to heat and exercise began at the start of the 20th century with the need to improve the adaptability of humans to work in these conditions (note: acclimation = artificial environment; acclimatization = natural environment) (Fig.1). Suzanne Schneider (Schneider, 2016) wrote an excellent review on experiences in the gold mines of South Africa, and initially focused on the scientific background leading to experimentation to improve performance in a harsh, hot-humid environment. She then elegantly described the evolution of ideas, bringing us to current studies on the role of gene expression plasticity in heat acclimation. In 1977, Pandolf, based on physiological thermoregulatory parameters, reported that heat acclimation has a memory, and noted that re-acclimation following the decay of acclimation is faster than the original acclimation episode (Pandolf et al., 1977; Pandolf, 1998), Fig. 1. He noted that following 9 days of acclimation and acclimation decay of 3, 6, 12 and 18 days, re-acclimation only took 2 days. Similar results have been reported by others (Daanen et al., 2018). Pandolf, concluded that further well-designed and definitive studies on decay of heat acclimation appear necessary.
After attending a lecture that Pandolf gave at the Technion (Haifa, Israel), the necessity of developing an experimental animal model to study heat acclimation (AC) memory became clear. In addition, it became clear that the acclimation machinery likely involved regulation of heat shock proteins (HSPs). Briefly, following, Rittosa’s (1962) discovery of heat shock protein response (Ritossa, 1962) (Fig. 1), Gehring and Wehner (Gehring & Wehner, 1995) made seminal findings on the regulation of HSPs in the Saharan ant (Cataglyphis), which pre-synthesizes significant levels of HSP70 and HSP72 prior to exposure to extremely high ambient temperatures. This discovery promoted the idea of “evolutionary solution” to thermal protection during heat acclimatization through cellular thermotolerance machinery. Moseley, attributed an additional adaptive role of HSPs to animals’ acclimatization via its role in signal transduction as well as being a mediator of danger signals and contributing to other protective responses (Moseley, 1997). The contribution of HSPs to heat acclimatization pushed forward the studies of the role of HSP70/72 in heat acclimation. Maloyan et al., showed that similarly to the Saharan ant, that Long Term Heat Acclimation (LTHA) but not short, elevates HSP72 in rats (Rattus norvegicus) and provides heat resilience and cross-tolerance (Maloyan et al., 1999), while, interestingly, heat acclimation with β-adrenergic blockade suppresses HSP72 production and cytoprotection (Maloyan & Horowitz, 2002). The impact of heat acclimation on the evolvement of cellular cytoprotection via HSPs and its role in immune-protection was demonstrated in peripheral blood mononuclear cells of heat acclimated humans (Yamada et al., 2007). Since these pioneering studies, a plethora of data on the various HSP families and their functions have accumulated, providing additional potential pathways for development of resilience to heat and other kinds of stress exposures.
Based on extensive studies using this model (Horowitz, 2017), we hypothesized that heat acclimation memory originates in “within-life Upstream Epigenetic Machinery” (Fig. 2) Whether this is an inherited phenomenon (in adult mammalian species), remains as yet unknown, but there is some evidence for inheritance in mammals (Weyrich et al., 2016a, 2016b) and other classes of animals depending on the impacted tissue/organ. For example, Skinner suggested next generation inheritance following parental exposure to toxins (Skinner, 2014) and using a C. elegans model, Das et al., established that there is a heritable epigenetic memory to prior heat stress exposures (Das et al., 2021).
Figure 2. A narrative illustration of acclimation-induced memory and cross-tolerance.
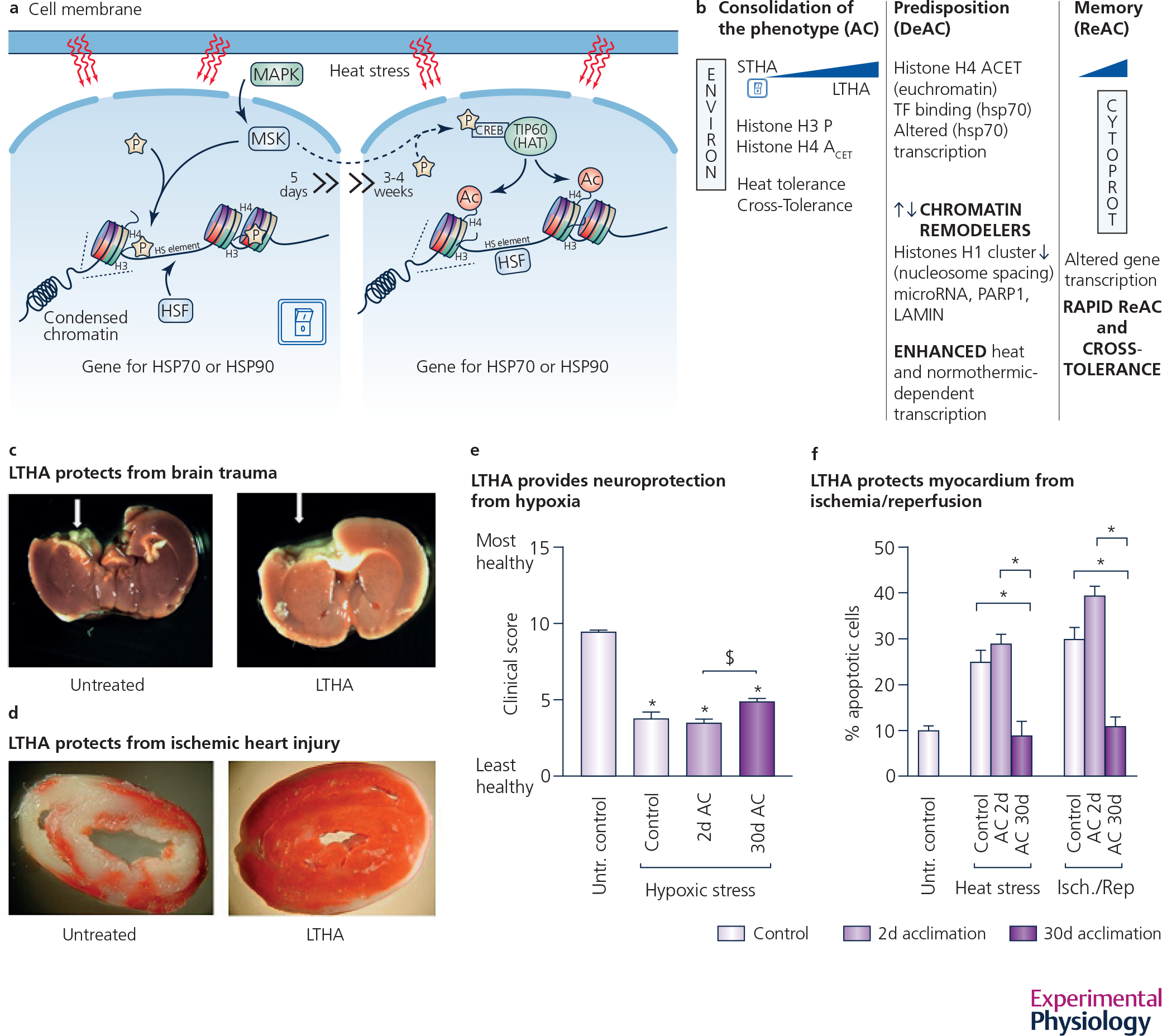
a: Heat acclimation is a bi-phasic process in which ON SWITCH, involving histone H3 phosphorylation leads to acclimatory homeostasis, i.e. long term heat acclimation (LTHA). b: Consolidation of the acclimated phenotype (~ 3 weeks) and its predisposition to rapid re-acclimation. LTHA is characterized by heat tolerance as well as cross-tolerance to other novel stimuli, as depicted in a-d. c: Neuroprotection post TBI (traumatic brain injury), d: Cardioprotection, infarct size post 30’ ischemia, e: Neuroprotection-resilience score (recovery) post 10’ hypoxia in rats (adapted from Yacobi et al. 2014), f: Apoptosis- long (LTHA) but not short (STHA) profoundly diminishes apoptosis in the heart (adapted from Assayag et al. 2010). STHA - short term heat acclimation, LTHA - long term heat acclimation, AC-heat acclimation, DeAC-deacclimation, ReAC-reacclimation, ENVIRON-Environment, CYTOPROT-cytoprotection. All symbols represent P<0.05 signficance. (Assayag et al., 2010; Yacobi et al., 2014; Horowitz et al., 2015; Umschweif et al., 2015).
Given that an inseparable feature of AC is cross-tolerance to novel stressors (i.e., stressors with no history of previous exposure) and that stress induces transcriptional memory, we applied a multidisciplinary approach while working with our young adult rat model, to test AC memory and its faster re-induction (Fig. 2). We were able to: (1) characterize the Heat Acclimated, Deacclimated/Reacclimated (DeAC/ReAC) physiological phenotypes; (2) profile the transcriptional activation of stress-associated genes in order to elucidate whether signaling molecules generate and propagate the “on” (able to reacclimate quickly) state. As a marker of successful acclimation (or ReAC) and cross tolerance, we measured body temperature during heat stress and cardiac infarct size following ischemic insult or rigor contracture in cardiomyocytes (Tetievsky et al., 2008).
In short, physiological evidence from Tc (colonic temperature), measured in response to heat stress and cardio-protection (infarct size) following an ischemic insult demonstrated that following one and two months of DeAC, the protected physiological AC phenotype returns quickly. After only two days of exposure to the original acclimating conditions the rats became reacclimatized vs. the 1 month required to achieve the initial long-term heat acclimation (LTHA) phenotype. We discovered a dichotomy between the genotypic and phenotypic responses. Briefly, despite the return of the physiological phenotype to its preacclimation state, after deacclimation (DeAC) periods, the gene transcripts did not resume their preacclimation levels. There were still chromatin remodeling genes that were transcriptionally active (e.g., HMGP, nucleoplasmin, CREB, SSRP1) implying that chromatin remodeling plays a pivotal role in the transcriptome profile and in preconditioning to enable cytoprotective acclimatory memory. These findings led us to thoroughly investigate the role of post transcriptional events and the epigenetic machinery involved in heat acclimation memory.
Post translational modifications on histones
The transfer of epigenetic information, such as instructions “do not transcribe this gene” or “transcribe this gene if...,” is associated with chromatin remodeling, via molecular and biochemical processes that maintain the chromatin-DNA package in active or silent states (Figure 2A). Using HSP72, HSP90 and HSF1, three essential components in the activation of the heat shock response (HSR), as a prototype model (Tetievsky & Horowitz, 2010), the role of epigenetic machinery as an upstream initiator of heat acclimation memory was validated. “Histone H3 phosphorylation switches on HSF-1 binding to the HSE (heat shock element) with subsequent histone H4 acetylation” inducing a maintained euchromatin state (i.e., open to transcription) throughout AC (phenotype consolidation), DeAC (dormant memory) and ReAC (Figure 2A and B). It is likely that the euchromatin state, with no HSF1-HSE binding during DeAC, leads to rapid ReAC. While the ON SWITCH of AC and H3 phosphorylation are temperature dependent, H4 acetylation is temperature independent (Tetievsky & Horowitz, 2010). Tetievsky et al., employed gene-chips, and provided unequivocal evidence that DeAC is characterised by a “storm” of up/down regulation of transcripts, including chromatin remodelers (e.g., PARP, LAMIN, linker histone) promoting the reacclimated physiological phenotype with a different transcriptome profile (Tetievsky et al., 2014).
An interesting model of heat stress-induced epigenetic preconditioning was established in chicks by intervention in their thermoregulatory center in the hypothalamus via administration of antisense glucocorticoid to the 3rd ventricle, prior to developmental establishment of the thermoregulatory center. This allowed for discrimination between vulnerable and heat resilient phenotypes (Cramer et al., 2015). These findings provide us insight into the diversion of epigenetic responses between adaptive responses to heat resulting in resilience vs. more maladaptive responses that can occur with exposure to severe exertional heat stroke, discussed later in this review.
DNA methylation: its role and the impact of heat
In the field of environmental epigenetics, DNA methylation is arguably the most well-studied and understood epigenetic mark (Weinhold, 2006). In short, DNA methylation is a fluid epigenetic chemical change (i.e., addition of a methyl group) that can be added or removed from cytosines adjacent to guanines (CpGs) on the DNA strand. This modification can affect the expression of genes in response to environmental stimuli, such as heat stress (Figure 3A). Like the response of nucleosomes described previously, these molecular events produce an avenue of altered cellular and physiological function of an organism to adapt within a lifetime, and if altered in germ cells, can be passed through to future generations of cells.
Figure 3. Primary Epigenetic Pathways for DNA Methylation.

a. Left: Loss of methylation by activation of the TET deoxygenases results in a reduction in chromatin packing, greater probability of DNA transcription and stimulation of RNA polymerase (POLII). Right: DNA methyltransferases (DNMTs) methylate cytosines in response to stress and other signals and often lead to suppression of transcription. b. Left: Exposure to severe exertional heat stroke results in significantly differentiated cytosine-p-guanines (CPGs) after 30 days, with a predominance of hypermethylation; right: effects of EHS exposure on changes in differentially methylated regions (DMRs) of > 10 cpgs, with a predominance of hypermethylation, recalculated from (Murray et al., 2021b). c. An example of hypermethylation of a series of genes responsible for NFκB stimulation, resulting in a state of immunosuppression. This pattern of lower transcription of these genes is predictive of observed immunosuppression phenotype in blood leukocytes of the same cells, 30 days after exertional heat stroke (EHS) (Murray et al., 2021b).
DNA methylation has been studied in the context of heat exposure across a wide breadth of experimental animal models from C. elegans (Wan et al., 2021), plants (Gao et al., 2014; Liu et al., 2015, 2021), fish (Metzger & Schulte, 2017), mammals (Weyrich et al., 2016a, 2016b; Del Corvo et al., 2021; Murray et al., 2021b), and chickens (Vinoth et al., 2018), as well as humans (Bind et al., 2014). This body of work suggests that exposure to heat can change the DNA methylome and, in many cases, alter the phenotype in parallel.
DNA methylation can occur on any CpG found throughout the gene body as well as in intergenic regions (Ehrlich & Lacey, 2013). However, there are several locations that result in consistent and reliable relationships between methylation and ensuing gene transcription. Methylation within the promoter region, ~100–1,000, bps directly upstream and ~100 bp downstream from the transcription start site (TSS) (Cooper et al., 2006), has been most often correlated with changes in transcription. Increases in methylation in this region are associated with reductions in transcription (and vice versa) through steric hindrance of transcription factor binding (Becker et al., 1987; Razin & Cedar, 1991) or through changes in hydrophobic and electrostatic interactions (Dantas Machado et al., 2015), Fig. 3A. Methylation status of other gene body locations near the TSS have also been found to have significant correlations with transcription. For example, the first exon (Brenet et al., 2011) is an extremely important site. Briefly, the exonic portion of the gene codes for functional protein (Deutsch & Long, 1999), and similar to the promoter, increased methylation in this region, suppresses transcription (Brenet et al., 2011). Importantly, methylation within the first exon is more significantly correlated with gene silencing than methylation found within other parts of the promoter (Brenet et al., 2011).
A second important region is the first intron (Anastasiadi et al., 2018) a region of the gene between exons that does not code for functional protein (Carmel & Chorev, 2012). Its methylation status has a similar relationship to that of the first exon, i.e., increased methylation inhibits transcription (Anastasiadi et al., 2018). It is thought that the importance of the first intron hinges on the fact that this gene region is rich in ‘enhancers’ (Park et al., 2014; Anastasiadi et al., 2018). Enhancers are potent regulatory elements that transcription factors (or DNA-bending proteins) can bind to and increase gene transcription events (Spitz & Furlong, 2012). Following the binding of a transcriptional element to an enhancer, the chromatin forms a loop to close the DNA gap between the enhancer and the promoter, thus increasing transcriptional probability (Pennacchio et al., 2013). In mammals, these elements can reside as close as 100 bp (the first intron) or as far away as 1,000,000 bp (intergenic regions) from the TSS (Krivega & Dean, 2012; Pennacchio et al., 2013).
DNA methylation responds dynamically to changing environmental stimuli by enzymatic processes of methylation and demethylation (Weinhold, 2006; Bollati & Baccarelli, 2010; Feinberg, 2018). Methylation reactions are governed by a family of enzymes called DNA methyltransferases (DNMTs) (Lyko, 2018), while demethylation reactions are performed by ten-eleven translocases (TETs) (Tahiliani et al., 2009; Wu & Zhang, 2017), Fig. 3A. In mammals, canonical DNMTs are a highly conserved enzymatic family made up of three enzymes: DNMT1, DNMT3A, and DNMT3B (Lyko, 2018). Their chief role is to methylate cytosine nucleotides in response to a variety of stimuli. DNMT1 is responsible for transferring DNA methylation patterns to duplicated DNA during mitosis and meiosis (Gruenbaum et al., 1982; Bestor & Ingram, 1983; Gaysinskaya et al., 2018). This supports persistence of DNA methylation marks during cell replication. Recently, the DNMT1-facilitated maintenance of DNA methylation has been found to be less precise than previously thought, resulting in epimutations that could contribute to transgenerational epigenetic flexibility both within and between generations (Wang et al., 2020). The other two canonical DNMTs, DNMT3A and DNMT3B, are known as de novo methyltransferases and add new methyl groups in response to various kinds of stress (Okano et al., 1999; Lyko, 2018). TETs, on the other hand, catalyze the oxidation and removal of methyl groups from cytosines (Tahiliani et al., 2009; Wu & Zhang, 2017), which depending on location, generally increase the probability of expression of the affected genes.
The regulation of DNMT3 during heat stress has not been studied extensively, but isoforms have been found to be significantly upregulated in heat exposure in zebrafish larvae (Dorts et al., 2016). Inhibition of TETs during heat conditioning, reported in embryonic chicks, alters future physiological responses to thermal stress by ameliorating inflammatory resiliency of the hypothalamus (Rosenberg et al., 2020). These observations need further study, but support the concept that physiological adaptations to heat exposure at various stages are controlled, in part, by regulation of the activity of both DNMTs and TETS.
Heat acclimatization/acclimation leads to a resilient epigenetic phenotype
Initial evidence for the significance of heat acclimation in thermoprotection in humans was discovered in the process of overcoming problems with colonization in hot climates, as discussed above. However, the importance of gene transcription and post-translational histone modifications in this process only emerged much later. In this section we will integrate what is currently known about these molecular responses and describe some of the origins of the “RESILIENT EPIGENETIC PHENOTYPE.”
Thermotolerance achieved by acclimatization (natural) or acclimation (artificial) is characterized by reversible phenotypic changes enabling individuals to match their phenotype to environmental demands throughout life. Although physiological thermoregulatory effectors execute the adaptations, they are controlled by transcriptional changes initiated by the upstream, within-life epigenetic machinery. The heat acclimation memory phenomenon is now well established. The acclimation-cross tolerance phenomenon comes largely from observations in organ systems where heat acclimation enhances ‘ON CALL’ innate cytoprotective pathways, shared by both acclimation and other novel stressors [e.g. stressors with no history of previous exposure to the organism (Horowitz et al., 2015)], Fig. 2. Human studies trying to implement the cross-tolerance machinery upon the integrated responses to a novel stimulus such as hypoxia (Daanen et al., 2018; Sotiridis et al., 2019) have so far, failed to verify this. However, the acclimation protocols used consisted of 10, 90 min sessions of exercise in the heat, whereas cross tolerance requires more time to develop and to consolidate (Arieli et al., 2003; Assayag et al., 2010; Tetievsky & Horowitz, 2010; Horowitz, 2016). This was emphasized by Daanen et al. (Daanen et al., 2018)., who demonstrated the requirement for long HA periods in humans for some adaptive responses such as sweat rates.
The acclimation cross tolerance phenomenon comes from the observation that heat acclimation enhances ‘ON CALL’ innate cytoprotective pathways that are shared by acclimation and other novel stressors [i.e., stressors with no history of previous exposure to the organism; e.g. hypoxia, hyperoxia, TBI, etc.) (Horowitz et al., 2015)]. These mechanisms limit the need for de novo recruitment of cytoprotective pathways upon exposure to such novel stressors and are unique aspects of the epigenetic changes following heatstroke (or heat stress) that are critical for survival, beyond the immediate reduction in core temperature. Acclimation memory/cross tolerance takes time to consolidate (Horowitz, 2016), whereas the physiological derangement that occurs in heat stroke fatalities during acute episodes of heat stress are rapid (though ultimate DNA methylation outcomes also evolve over weeks). Murray et al., (2021) reported that exertional heat stroke leads to long-term epigenetic memory, but in a vulnerable context (Murray et al., 2021b). The differences in epigenetic acclimatory vs. heat stroke memory mechanisms depend on duration (e.g., heat acclimation memory needs time for consolidation), heat dose and location (organ), as well as the reversibility or severity of the initial damage (as summarized in Fig. 4). While post translational histone modifications have been shown to be reversible, less is known about methylation. Kisliouk et al., using their preconditioned chick model, noted elevated methylation levels in a distal part of HSP70 (consensus thermotolerant gene) promoter leading to recruitment of nucleosome remodeling deacetylase chromatin-remodeling complex (NuRD) and higher histone H3 acetylation at the HSP70 promoter in harsh-temperature-conditions vs. controls (Kisliouk et al., 2017). This reflects a heat-stress-related epigenetic memory, with potential use in differentiating between individuals that are resilient or vulnerable to stress (Kisliouk et al., 2017). These findings confirm a heat stress induced epigenetic memory; however, the model was not examined in chicks with a mature thermoregulatory center. Gan et al., also in chicks, arrived at the conclusion that HSP70 may negatively correlate with DNA methylation pattern in the HSP70 promoter, whereas tissue HSP70 level-dependence is unrelated to the DNA methylation pattern (Gan et al., 2013; Wu et al., 2020).
Fig. 4. Summary of differences between maladaptive and adaptive epigenetic responses.
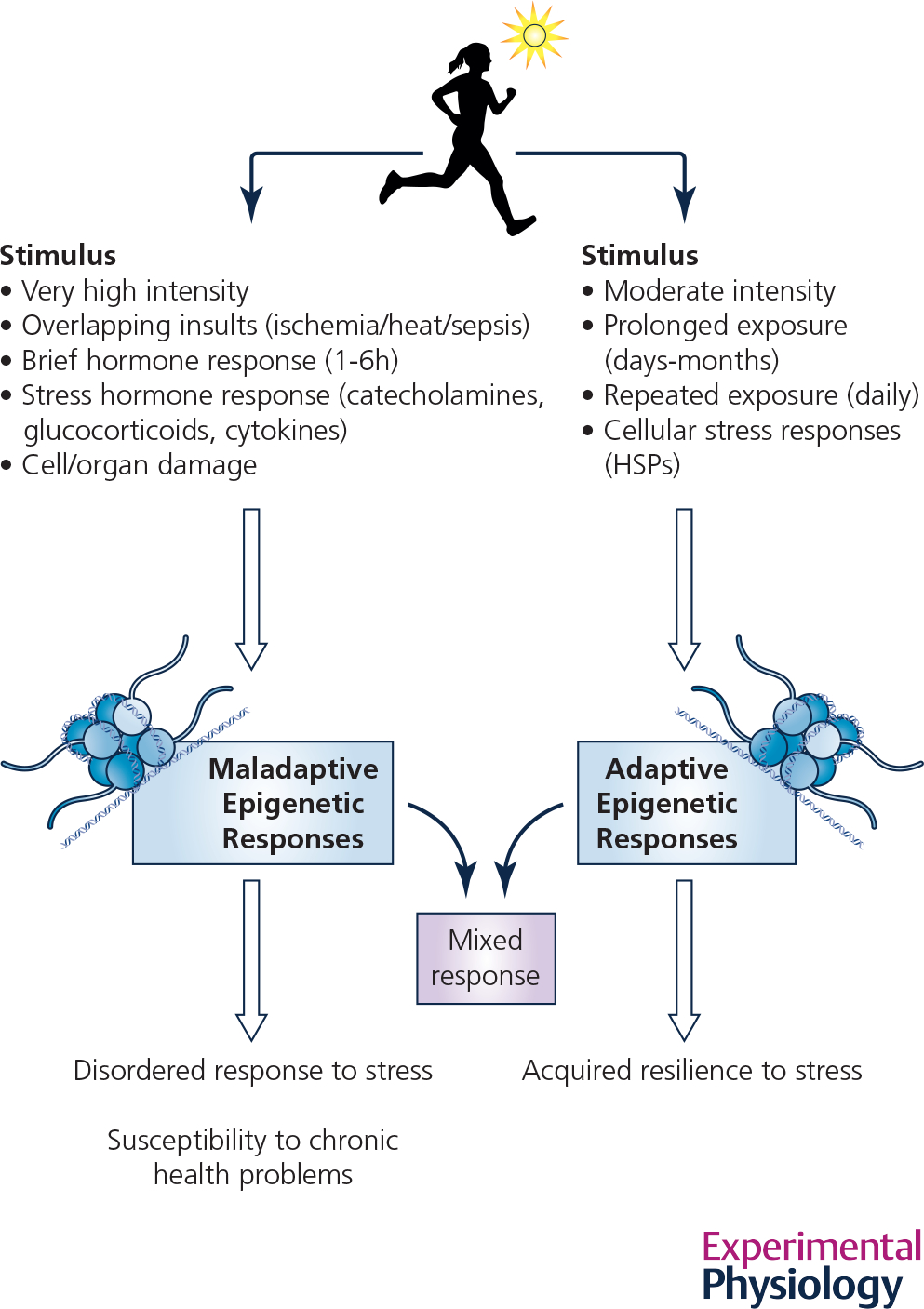
A comparison of the differing conditions that appear to lead to adaptive vs. maladaptive epigenetic changes in response to heat exposure, an example of physiological hormesis.
Despite the abundance of studies on the genetic and epigenetic mechanisms involved in cytoprotection, heat acclimation, and thermotolerance, the scope of each specific protocol is limited in its design. The features emerging after many protocols, differing in issues, e.g., timing of thermoregulatory disruption, stress application (recurrent vs. chronic), or the dose, enable the construction of a scheme that integrates the accumulated knowledge. Notably, acclimation/acclimatization is an in vivo process. It is likely that the epigenetic machinery controls whether the individual will show resilience or experience deleterious effects following stress, not by altering physiological parameters, but rather via transcriptional changes affecting the activity of the controller organs. Yacobi et al. described the outcome of acclimation memory in the frontal cortex and the hippocampus, demonstrating decreased activity in the glutamate gated ion channels in heat acclimated rats, leading to a resilient phenotype (via decreased Ca2+ overload) (Yacobi et al., 2014). Cramer et al., used the chick model, and presented the role of epigenetic machinery in the hypothalamus via activation of the levels of corticotrophin releasing hormone (CRH) in post translational modifications of histones (Cramer et al., 2015, 2019).
Long-term disease risk following heat stroke exposure: a case for epigenetic maladaptation
Recent epidemiological and original research studies have suggested associations between previous exposure to severe heat stroke, or heat injury, and a variety of long-term health consequences, including heart disease (Wallace et al., 2007; Wang et al., 2019; Tseng et al., 2019), cerebral stroke (Wallace et al., 2007; Wang et al., 2019), immunosuppression (Dematte et al., 1998; Davido et al., 2006; Argaud et al., 2007), alterations to skeletal muscle function (Sagui et al., 2015), and chronic kidney disease (Wang et al., 2019, 2019). These delayed outcomes resemble long-term effects of other severe forms of trauma or disease. Though many mechanisms could be responsible, given the potent impact of severe heat exposure, such as EHS, on epigenetic responses in many organisms, a strong candidate must include a retained maladaptive epigenetic memory. Such a memory may confer a loss of “resiliency” to secondary stress exposures later in life and may include earlier onset of disease and/or mortality, as seen following exposure to a high fat diet in utero (Masuyama & Hiramatsu, 2012; Masuyama et al., 2015).
Any long-term health effects of severe heat exposure are difficult to prove or disprove in humans, as only associative data is available. Therefore, relying on preclinical models to help establish a cause-and-effect relationship between exposure to heat stroke and long-term health sequelae is important. Seminal studies of heat stroke in mice have concluded that DNA methylation alterations, including the existence of long-term DNA methylation memories, are clearly evident in response to a single exposure to EHS that impacts many different cell signaling pathways in many types of tissues, including immune cells (Murray et al., 2021b), ventricular cardiomyocytes (Murray et al., 2022), and skeletal muscle (Murray et al., 2019) in mice. However, the challenge is to mechanistically link heat stroke-induced epigenetic changes with alterations to physiology in these models, such as changes in immune responsiveness or cardiovascular and skeletal muscle function. Success in such endeavors could establish preclinical models of heat stroke as translational windows into the underpinnings of long-term clinical health consequences.
In this section, we will review (1) the current associations of long-term health consequences in humans exposed to heat stroke or heat injury, (2) how long-term DNA methylation landscapes change following EHS in a preclinical mouse model, and (3) how such long-term methylation changes may predict alterations in respective phenotype, in mice, reminiscent of the human condition.
Myocardium
Retrospective epidemiological studies have presented strong evidence that exposure to heat stroke or heat illness increases prevalence of all-cause cardiovascular events and mortality (Table I summary). One such study was performed by the US Army (Wallace et al., 2007), in that 17,233 military personnel were included [average age, 24 yo at exposure; matched controls]. During the follow-up period (15±8 years) those patients exposed to heat injury had a 1.4 times greater risk of dying of all causes, with the majority of deaths under the age of 40, and a 1.8 times greater risk of acquiring significant cardiovascular disease. Similar results were reported by Wang et al., who followed 150 heat stroke and 150 heat illness victims with matched controls over 14 years (Wang et al., 2019); average age was 45 yo. The adjusted odds of acquiring a significant cardiovascular disease within 14 years was 3.9 times greater in heat stroke victims. Additionally, there was a 5.5 times greater incidence of cerebral stroke. A final retrospective study was conducted on a cohort in Taiwan that included 628 heat stroke victims (~50 yo at exposure; 1884 controls), with an average follow-up period of 12 years. Heat stroke victims had a 3.5 times greater incidence of developing ischemic heart disease emerging only 2.1 years after heat stroke and a 3.1 times greater incidence of cerebral stroke (Tseng et al., 2019). These results provide strong support for the potential of a molecular linchpin, such as an altered epigenetic landscape, that mediates delayed development of vascular abnormalities following heat stroke exposure.
Table 1.
Long-term epidemiological and preclinical studies investigating association of disease (human) and epigenetic reprogramming and phenotype (mice) following exposure to heat stroke.
| Studies in Humans | Findings | Preclinical Studies | Findings | |
|---|---|---|---|---|
| Cardiovascular |
Wallace et al., 2007
1
Wang et al., 2019 2 |
Increased long-term risk of all-cause cardiovascular events and mortality following heat stroke in US Army1 and Taiwan2 cohorts |
Murray et al., 20228 Laitano et al., 20209 |
• Long-term reprogramming of DNA methylome after 30 days of recovery from EHS8 • Widespread metabolomic changes @ 2 weeks following exposure9 • Some metabolomic changes may be predicted by DNA methylation8 |
| Immune |
Dematte et al., 1998
3
Davido et al., 2006 4 Argaud et al., 2007 5 |
Increased susceptibility of infection in urine, blood, and lungs in those exposed to heat stroke following American3 and French4,5 heat waves | Murray et al., 2021 | • Long-term reprogramming of DNA methylome after 30 days of recovery from EHS • Reduced whole blood response to LPS • Reprogramming of DNA methylation predicted immunotolerance |
| Skeletal Muscle | Sagui et al., 2015 | Dysfunctional Ca2+ handling in muscle biopsies exposed to caffeine & halothane months to years after initial heat stroke exposure as indicated by an increase in sensitivity to both agonists |
Murray et al., 201910 Murray et al., 202111 |
• Long-term reprogramming of DNA methylome after 30 days of recovery from EHS10 • Ex vivo whole muscle less sensitive to caffeine10 • Reduction in long-term satellite cell proliferation11 • Delayed muscle repair11 |
| Kidney |
Wang et al., 2019
6
Tseng et al., 2020 7 |
Increased incidence of chronic kidney6,7 and end-stage renal diseases7 in those exposed to heat stroke | EHS studies are needed | N/A |
EHS = exertional heat stroke
LPS = lipopolysaccharide
In a preclinical mouse model (King et al., 2015; Murray et al., 2021b), exposure to EHS resulted in a substantial DNA methylation memory in ventricular cardiomyocytes after 30 days of recovery Fig 3B. Much of the signal resided within the promoter regions of genes involved in a variety of stress responses (i.e., responses to reactive oxygen species and cellular heat acclimation) and metabolic processes (Murray et al., 2022). Although this does not directly link long-term EHS-induced alterations to DNA methylation with future cardiovascular disease incidence and risk, it does strongly suggest a heart that has acquired modified stress responses that may cause adverse reactions to future stimuli or stress events. Previous studies in the same preclinical EHS mouse model suggest that EHS leads to striking metabolic alterations that mirror early onset heart failure, as well as robust increases in the presence of oxidative stress and inflammation (Laitano et al., 2020a). These changes only emerged after two weeks of recovery. However, at the present, a direct link between the EHS-induced long-term epigenetic and metabolomic alterations has not yet been established.
Immune cells
Following heat waves in both the United States (Dematte et al., 1998) and in Europe (Davido et al., 2006; Argaud et al., 2007), those suffering from heat stroke presented with an increased incidence of infection, that included infections of the blood, urine, and lungs. An increase in infection was found to occur regardless of mortality outcome (Davido et al., 2006).
Following exposure to heat stroke, a large-scale immune response is launched in an attempt to repair organ damage and dysfunction (Bouchama & Knochel, 2002; King et al., 2017; Bouchama et al., 2022); such organ damage may include the disruption of the gut epithelium leading to endotoxin or bacteria leaking into the interstitial space (King et al., 2015, 2021). It has been shown that monocytes and myeloid cells exposed to prolonged lipopolysaccharide (LPS) ex vivo develop a tolerance to endotoxin (Álvarez-Errico et al., 2015; Widdrington et al., 2018). This phenomenon has been shown to be governed by alterations to the DNA methylome (Álvarez-Errico et al., 2015). Therefore, it is plausible that endotoxin released from the gut during and following heat stroke may induce an epigenetically mediated LPS-tolerance in immune cells, a form of immunosuppression.
An equally plausible explanation is the secretion of glucocorticoids in response to heat stress (Brenner et al., 1998; Garcia et al., 2018). Glucocorticoids are potent anti-inflammatory mediators that inhibit the pro-inflammatory NFκB pathways in immune cells through molecular reprogramming (Hermoso & Cidlowski, 2003; Ehrchen et al., 2019). Over several days of recovery from heat stroke, in mice, corticosterone levels are elevated suggesting a persistent and systemic exposure to increased glucocorticoids that may reprogram the epigenome (Murray et al., 2021b). Previous studies in chicks indicate that embryonic heat stress produces an epigenetically mediated cross-tolerance effect on IL-6 secretion in response to in vivo LPS (repressive DNA methylation and histone marks on intron 1 of the IL-6 gene) (Rosenberg et al., 2021). This finding supports glucocorticoid-induced effects of heat stress on long-term immune responsiveness (Rosenberg et al., 2021).
Initial evidence that exposure to EHS induces a long-term suppression of the immune system was observed in mice following a 30-day recovery from EHS. Blood collected from these mice was almost unresponsive to an ex vivo LPS challenge compared to blood from sham controls. In bone marrow progenitor monocytes, this response paralleled repressive DNA methylation marks on regulatory regions of pro-inflammatory genes (Murray et al., 2021b), as summarized in Figure 3C. The fact that these changes were observed in progenitor cells, and that monocytes have extremely short life expectancies, suggests that a distinct DNA methylation memory may be carried across many generations of immune cells during repeated cell division of bone marrow hematopoietic stem cells.
Skeletal Muscle
Dysfunctional Ca2+ handling within skeletal muscle, months to years after exposure to heat stroke was observed in a cohort of French warfighters (Sagui et al., 2015). This phenomenon was evaluated using the malignant hyperthermia (MH) diagnostic test, i.e. the in vitro contracture test (IVCT) (Sagui et al., 2015). MH is a genetic disease by which skeletal muscle exhibits Ca2+ dysregulation in response to caffeine or halothane exposure (Ellis, 1984). Note that this test is not entirely specific to the MH disease, but merely measures the sensitivity to Ca2+ (Heytens et al., 1992). This report is probably not consistent with the large number of soldiers having actual genetic MH (Laitano et al., 2020b), but rather that there could be underlying molecular changes, e.g., by epigenetics or other mechanisms, that alters their muscle phenotype over the long term.
In mice, substantial long-term alterations to gastrocnemius DNA methylation has been observed after recovering from EHS for 30 days (Murray et al., 2019). Some of the changes resided within transcriptional regulatory regions of genes involved in skeletal muscle Ca2+ handling. When sensitivity to caffeine was assessed on whole soleus muscle during IVCT testing, there was a significant reduction in caffeine sensitivity, the opposite of the report in humans that resembled MH (Sagui et al., 2015). Furthermore, isolated satellite cells (muscle fiber stem cells) exhibited significant reductions in the capacity to proliferate, and changes in protein showed decreased Myh7 protein (slow, oxidative myosin heavy chain 7) and increases in RNA for slow-oxidative isoforms, including Myh7 (Murray et al., 2021a). The altered skeletal muscle phenotype, after 30 days of recovery from heat stroke, suggests long term effects of EHS on skeletal muscle that may or may not be of epigenetic origin, but may be consistent with methylation patterns observed.
Kidney
Following exposure to heat stroke, individuals frequently acquire acute kidney injury (AKI). Such injury seems to predispose individuals to delayed development of chronic kidney disease (CKD). AKI can arise from secondary effects of rhabdomyolysis, particularly from accumulation of myoglobin in the kidney (Petejova & Martinek, 2014), inflammatory responses, and secondary responses to reductions in perfusion and dehydration during heat stroke (Tseng et al., 2020). Several studies have analyzed the incidence of late-developing CKD in heat stroke victims. Following exposure to heat stroke, there is a 4.4 times greater risk of acquiring all stages of CKD (average age, 43 yo at exposure) with onset delayed by 4.2 years (Tseng et al., 2020). Interestingly, the risk of acquiring ‘severe’ forms of CKD and end stage renal failure (ESRD) were much higher. For Stage 5 CKD, the risk was 11.1 times greater than controls, while ESRD was 9.1 times greater (Tseng et al., 2020). A second study reported a 3 times greater incidence of CKD in heat stroke victims compared to a control population consisting of other non-heat-related illnesses (Wang et al., 2019).
The mechanisms for acquiring CKD long after exposure to heat stroke are not fully understood and may or may not be mediated by epigenetic mechanisms. However, studies assessing the role of DNA methylation in either diseased or injured kidneys have suggested underlying epigenetic mechanisms and associations. Such studies include diabetic kidney disease (Gluck et al., 2019), and the development of CKD following AKI (Zager & Johnson, 2009; Naito et al., 2009; Rodríguez-Romo et al., 2015).
Inherited epigenetic memory of previous heat exposure
There are a number of environmentally induced epigenetic responses that result in adaptive or maladaptive phenotypes that appear to be inherited in future generations, but the evidence in humans is nonexistent. One of the examples most often cited for an inherited epigenetic environmental exposure in humans is the severe caloric restriction seen during the “Dutch Famine,” where expectant mothers (F0) in the Netherlands during World War II were exposed to conditions of starvation over a five-month period. Their offspring (F1) often suffered from higher rates of obesity, diabetes, heart disease and schizophrenia (Bleker et al., 2021). This observation has been confirmed in other world populations (Cao-Lei et al., 2020; Bleker et al., 2021). Furthermore, there have been reports that the F2 male offspring had a significantly greater incidence of obesity (Painter et al., 2008; Veenendaal et al., 2013). Though unique DNA methylation differences in peripheral blood cells have been shown to be present as long as sixty years after the starvation period, none of these observations necessarily represent transgenerational inheritance of epigenetic marks, because the fetus (F1) and possibly the gonads of that fetus (F2) could have also been exposed to the original stress (Cao-Lei et al., 2020). Therefore, using phenotype to define transgenerational epigenetic inheritance is clouded by the possibility of confounding ‘indirect’ mechanisms that impact offspring but are not of epigenetic origin. For example, the altered nurturing behavior of nutritionally stressed F0 mothers can affect the health of subsequent generations (Lacal & Ventura, 2018).
Though we have little information in humans regarding inherited traits of heat exposure, there is considerable evidence in animals. For example, in brine shrimp (Daphnia magna) exposure of parthogenic females to repeated non-lethal heat shocks results in several generations of offspring with phenotypic traits of heat tolerance, overlapping global DNA methylation changes across generations and sustained HSP elevations (Norouzitallab et al., 2014). This effect is adaptive to heat and results in cross-tolerance to other insults such as toxic metal exposure (Pestana et al., 2016), much like the cross tolerance discussed previously. In the nematode (C. elegans), a HSP90 response to being raised at the elevated temperature of 25°C during development can be passed on by both mother and father to as much as 14 generations (Klosin et al., 2017). Another example of transgenerational effects can be seen in male guinea pigs allowed to adapt to two months of increased temperature acclimation, resulting in an inherited DNA methylation landscape, portions of which are transmitted to the F2 offspring (Weyrich et al., 2016b). Interestingly, the transgenerational effects were carried in the male. Though we don’t know if it can also be carried in the female, it is carried in the female in other species such as C elegans (Das et al., 2021).
Animal models and in vitro human cells have also been useful in showing transgenerational inheritance of ‘maladaptive’ epigenetic changes to heat exposure. As discussed previously, these maladaptive changes are generally in response to high levels of exposure for brief periods (Fig. 4), whereas adaptive responses are more often associated with more moderate exposures over longer time periods. For example, in fruit fly larvae (Drosophila melanogaster), when exposed to intense, but sub-lethal heat stress for 1 hour at 37°C, epigenetic-dependent heterochromatin disruption occurs that is passed on to at least 3 generations of offspring (Seong et al., 2011). Such remodeling can have drastic effects on gene replication and inhibition of DNA repair processes that ultimately leads to reduced resilience and adverse outcomes. Mechanisms for this maladaptive response are still poorly understood but may lie in hyperthermia-induced posttranslational modifications of histones, such as deacetylation of H4K16ac, which suppresses normal DNA repair pathways (Chakraborty et al., 2022), or HSF-1–induced methylation of H3K9me2, which can lead to both improved stress responses in some conditions but disordered stress responses in others, depending on the duration of exposure to heat (Das et al., 2021). Delayed effects of heat stress in cell culture experiments have been associated with persistent DNA strand breaks, which can accumulate and ultimately lead to a state of cellular senescence (Velichko et al., 2015). One can imagine that compared to a prolonged exposure to moderate heat, where the normal processes of acclimation and adaptation are active, brief but severe exposures to life-threatening hyperthermia are more likely to result in conditions resembling other forms of traumatic stress. The epigenetic machinery may be alternatively activated by cell injury or exposure to stress hormones such as high glucocorticoids. Compared to the slow steady-state molecular responses of prolonged, moderate heat, these kinds of intense stress events are more likely to result in non-targeted epigenetic changes and loss of normal homeostatic regulation (Fig 4).
An additional outcome of epigenetic changes that can be inherited has rarely been discussed but has important implications for evolution, i.e., the increased tendency for single nucleotide mutations to arise in methylated cytosines vs. unmethylated cytosines or other nucleotides (Holliday & Grigg, 1993; Zhou et al., 2020). This effect is particularly potent in germ-cells (Zhou et al., 2020). This means that presumably permanent and inheritable changes in the DNA sequence of germ cells could result from environmentally induced epigenetic responses. Furthermore, the process of inherited epigenetic flexibility (Wang et al., 2020), i.e., epimutations manifest through stochastic changes to the methylation status of a given CpG, could contribute to changes in species survival, much like basic Mendelian genetics, in the face of changing environmental challenges, such as increases in acute and chronic heat attributed to global warming (Rossati, 2016; Ryu et al., 2018; McCaw et al., 2020). If we couple these observations with the knowledge that spontaneous mutation rates are exponentially elevated during exposure to temperatures exceeding species-specific, normal core temperatures (Waldvogel & Pfenninger, 2021) then a scenario emerges in which long-term or repeated extreme temperature exposures could accelerate the rate of Darwinian evolutionary change over generations. This lays another level of complexity on the original Darwinian vs. Lamarckian debates over the influence of environment upon species evolution.
Challenges and Future Opportunities
As our world enters an era of rapidly changing climate, it is of value to consider that the molecular epigenetic programs that help to create our adaptive capacity are also adapting over time, hopefully to the benefit of our species. Though far from proven, some of the resulting changes in epigenetic memory within germ cells could be inherited by our progeny, helping our species to survive. On the other hand, since many parts of the world in this era live in a rarified air-conditioned climate, with rising global temperatures, it becomes more likely that susceptible individuals will experience brief, intense heat exposures that could result in long-term negative medical consequences later in life, though this possibility is suggested only by association and still unproven.
Can our epigenetic machinery help us adapt? Can it be optimized therapeutically? Much work needs to be done to link the molecular epigenetic observations with actual phenotype, particularly with respect to DNA methylation changes, a difficult challenge. Can we find predictable epigenetic marks in an accessible sample in humans that can tell us whether an individual has fully recovered from heat stroke or is sufficiently acclimatized to protect them from subsequent heat stroke or injury? Can we identify how epigenetic programming affects different heat-sensitive organ systems and over what different time frames and exposure paradigms? At a basic science level, much is needed to understand how DNA methylation is linked to histone/nucleosome regulation and vice-versa, which is poorly understood, and how specific topographical patterns of DNA methylation changes define transcriptional or translational responsiveness of a given gene or to a given stimulus.
The field of environmental epigenetics has only emerged over the past few decades, but it gives us the capacity for the first time to get a glimpse into a large number of fascinating and complex riddles underlying physiological adaptation to such a primordial stimulus as increased temperature. For example, we have discovered that tissues and cells can ‘learn’ from experience. We have learned that different phenotypes, in some ways learn similarly but in other ways respond uniquely for their phenotype. We have learned that some memories can be transferred to future generations of cells from stem cells within the same organism and in some cases to future progeny from gametes. Additionally, within-life upstream epigenetics machinery, leading to adaptive memory of the individual, may provide protection from novel stressors arising in a changing environment, which may be critical for survival in a future harsh settings. This difficult area of research is still in its infancy but has much to offer in deepening our understanding of the molecular basis of physiologic adaptation to environmental change.
New Findings.
What is the topic of this review?
This review outlines the history of research on epigenetic adaptations to heat exposure. The perspective taken is that adaptations reflect properties of hormesis, where low, repeated doses of heat induce adaptation (acclimation/acclimatization); whereas, brief, life-threatening exposures can induce maladaptive responses.
What advances does it highlight?
The epigenetic mechanisms underlying acclimation/acclimatization comprise specific molecular programs on histones that transcriptionally regulate heat shock proteins and protect the organism from subsequent heat exposures, even after long delays. The epigenetic signaling underlying maladaptive responses may rely, in part, on extensive DNA-methylation changes that are sustained over time and may contribute to later health challenges.
Acknowledgements
The research was supported by contracts from the U.S. Department of Defense, BA180078 (T.L. Clanton), with supplemental support from the B.K. and Betty Stevens Endowment at the University of Florida (T.L. Clanton) and Israel Science Foundation Grant 321/06 (M. Horowitz)). K.O. Murray is supported by the training grant 5T32DK007135–46 (University of Colorado, R.J. Johnson).
References
- Álvarez-Errico D, Vento-Tormo R, Sieweke M & Ballestar E (2015). Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol 15, 7–17. [DOI] [PubMed] [Google Scholar]
- Anastasiadi D, Esteve-Codina A & Piferrer F (2018). Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics & Chromatin 11, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaud L, Ferry T, Le Q-H, Marfisi A, Ciorba D, Achache P, Ducluzeau R & Robert D (2007). Short- and long-term outcomes of heatstroke following the 2003 heat wave in Lyon, France. Arch Intern Med 167, 2177–2183. [DOI] [PubMed] [Google Scholar]
- Arieli Y, Eynan M, Gancz H, Arieli R & Kashi Y (2003). Heat acclimation prolongs the time to central nervous system oxygen toxicity in the rat. Possible involvement of HSP72. Brain Res 962, 15–20. [DOI] [PubMed] [Google Scholar]
- Assayag M, Gerstenblith G, Stern MD & Horowitz M (2010). Long- but not short-term heat acclimation produces an apoptosis-resistant cardiac phenotype: a lesson from heat stress and ischemic/reperfusion insults. Cell Stress Chaperones 15, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker PB, Ruppert S & Schütz G (1987). Genomic footprinting reveals cell type-specific DNA binding of ubiquitous factors. Cell 51, 435–443. [DOI] [PubMed] [Google Scholar]
- Berry R & López-Martínez G (2020). A dose of experimental hormesis: When mild stress protects and improves animal performance. Comp Biochem Physiol A Mol Integr Physiol 242, 110658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor TH & Ingram VM (1983). Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. PNAS 80, 5559–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bind M-A, Zanobetti A, Gasparrini A, Peters A, Coull B, Baccarelli A, Tarantini L, Koutrakis P, Vokonas P & Schwartz J (2014). Effects of temperature and relative humidity on DNA methylation. Epidemiology 25, 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleker LS, de Rooij SR, Painter RC, Ravelli AC & Roseboom TJ (2021). Cohort profile: the Dutch famine birth cohort (DFBC)- a prospective birth cohort study in the Netherlands. BMJ Open 11, e042078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollati V & Baccarelli A (2010). Environmental epigenetics. Heredity 105, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchama A, Abuyassin B, Lehe C, Laitano O, Jay O, O’Connor FG & Leon LR (2022). Classic and exertional heatstroke. Nat Rev Dis Primers 8, 8. [DOI] [PubMed] [Google Scholar]
- Bouchama A & Knochel JP (2002). Heat Stroke. New England Journal of Medicine 346, 1978–1988. [DOI] [PubMed] [Google Scholar]
- Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND & Scandura JM (2011). DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One 6, e14524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner I, Shek PN, Zamecnik J & Shephard RJ (1998). Stress hormones and the immunological responses to heat and exercise. Int J Sports Med 19, 130–143. [DOI] [PubMed] [Google Scholar]
- Cao-Lei L, de Rooij SR, King S, Matthews SG, Metz GAS, Roseboom TJ & Szyf M (2020). Prenatal stress and epigenetics. Neuroscience & Biobehavioral Reviews 117, 198–210. [DOI] [PubMed] [Google Scholar]
- Carmel L & Chorev M (2012). The Function of Introns. Frontiers in Genetics 3, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S et al. (2022). Heat-Induced SIRT1-Mediated H4K16ac Deacetylation Impairs Resection and SMARCAD1 Recruitment to Double Strand Breaks. Social Science Research Network, Rochester, NY. Available at: https://papers.ssrn.com/abstract=4007582 [Accessed March 21, 2022]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SJ, Trinklein ND, Anton ED, Nguyen L & Myers RM (2006). Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res 16, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer T, Kisliouk T, Yeshurun S & Meiri N (2015). The balance between stress resilience and vulnerability is regulated by corticotropin-releasing hormone during the critical postnatal period for sensory development. Developmental Neurobiology 75, 842–853. [DOI] [PubMed] [Google Scholar]
- Cramer T, Rosenberg T, Kisliouk T & Meiri N (2019). Early-life epigenetic changes along the corticotropin-releasing hormone (CRH) gene influence resilience or vulnerability to heat stress later in life. Molecular Psychiatry 24, 1013–1026. [DOI] [PubMed] [Google Scholar]
- Cutler JE (1902). Tropical Acclimatization. American Anthropologist 4, 421–440. [Google Scholar]
- Daanen HAM, Racinais S & Périard JD (2018). Heat Acclimation Decay and Re-Induction: A Systematic Review and Meta-Analysis. Sports Med 48, 409–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas Machado AC, Zhou T, Rao S, Goel P, Rastogi C, Lazarovici A, Bussemaker HJ & Rohs R (2015). Evolving insights on how cytosine methylation affects protein–DNA binding. Brief Funct Genomics 14, 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Min S & Prahlad V (2021). Gene bookmarking by the heat shock transcription factor programs the insulin-like signaling pathway. Mol Cell 81, 4843–4860.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davido A, Patzak A, Dart TS, Sadier MP, Méraud P, Masmoudi R, Sembach N & Cao TH (2006). Risk factors for heat related death during the August 2003 heat wave in Paris, France, in patients evaluated at the emergency department of the Hôpital Européen Georges Pompidou. Emergency medicine journal : EMJ 23, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Corvo M, Lazzari B, Capra E, Zavarez L, Milanesi M, Utsunomiya YT, Utsunomiya ATH, Stella A, de Paula Nogueira G, Garcia JF & Ajmone-Marsan P (2021). Methylome Patterns of Cattle Adaptation to Heat Stress. Frontiers in Genetics 12, 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dematte JE, O’Mara K, Buescher J, Whitney CG, Forsythe S, McNamee T, Adiga RB & Ndukwu IM (1998). Near-fatal heat stroke during the 1995 heat wave in Chicago. Ann Intern Med 129, 173–181. [DOI] [PubMed] [Google Scholar]
- Deutsch M & Long M (1999). Intron-exon structures of eukaryotic model organisms. Nucleic Acids Res 27, 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorts J, Falisse E, Schoofs E, Flamion E, Kestemont P & Silvestre F (2016). DNA methyltransferases and stress-related genes expression in zebrafish larvae after exposure to heat and copper during reprogramming of DNA methylation. Sci Rep 6, 34254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrchen JM, Roth J & Barczyk-Kahlert K (2019). More Than Suppression: Glucocorticoid Action on Monocytes and Macrophages. Front Immunol; DOI: 10.3389/fimmu.2019.02028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M & Lacey M (2013). DNA methylation and differentiation: silencing, upregulation and modulation of gene expression. Epigenomics 5, 553–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis F (1984). A protocol for the investigation of malignant hyperpyrexia (MH) susceptibility. The European Malignant Hyperpyrexia Group. Br J Anaesth 56, 1267–1269. [DOI] [PubMed] [Google Scholar]
- Feinberg AP (2018). The Key Role of Epigenetics in Human Disease Prevention and Mitigation. New England Journal of Medicine 378, 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan JK, Zhang DX, He DL, Zhang XQ, Chen ZY & Luo QB (2013). Promoter methylation negatively correlated with mRNA expression but not tissue differential expression after heat stress. Genet Mol Res 12, 809–819. [DOI] [PubMed] [Google Scholar]
- Gao G, Li J, Li H, Li F, Xu K, Yan G, Chen B, Qiao J & Wu X (2014). Comparison of the heat stress induced variations in DNA methylation between heat-tolerant and heat-sensitive rapeseed seedlings. Breed Sci 64, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia CK, Mattingly AJ, Robinson GP, Laitano O, King MA, Dineen SM, Leon LR & Clanton TL (2018). Sex-dependent responses to exertional heat stroke in mice. J Appl Physiol 125, 841–849. [DOI] [PubMed] [Google Scholar]
- Gaysinskaya V, Miller BF, De Luca C, van der Heijden GW, Hansen KD & Bortvin A (2018). Transient reduction of DNA methylation at the onset of meiosis in male mice. Epigenetics & Chromatin 11, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring WJ & Wehner R (1995). Heat shock protein synthesis and thermotolerance in Cataglyphis, an ant from the Sahara desert. Proc Natl Acad Sci U S A 92, 2994–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluck C, Qiu C, Han SY, Palmer M, Park J, Ko Y-A, Guan Y, Sheng X, Hanson RL, Huang J, Chen Y, Park ASD, Izquierdo MC, Mantzaris I, Verma A, Pullman J, Li H & Susztak K (2019). Kidney cytosine methylation changes improve renal function decline estimation in patients with diabetic kidney disease. Nat Commun 10, 2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum Y, Cedar H & Razin A (1982). Substrate and sequence specificity of a eukaryotic DNA methylase. Nature 295, 620–622. [DOI] [PubMed] [Google Scholar]
- Hermoso MA & Cidlowski JA (2003). Putting the brake on inflammatory responses: the role of glucocorticoids. IUBMB Life 55, 497–504. [DOI] [PubMed] [Google Scholar]
- Heytens L, Martin JJ, Van de kelft E & Bossaert LL (1992). IN VITRO CONTRACTURE TESTS IN PATIENTS WITH VARIOUS NEUROMUSCULAR DISEASES. British Journal of Anaesthesia 68, 72–75. [DOI] [PubMed] [Google Scholar]
- Holliday R & Grigg GW (1993). DNA methylation and mutation. Mutat Res 285, 61–67. [DOI] [PubMed] [Google Scholar]
- Horowitz M (2014). Heat acclimation, epigenetics, and cytoprotection memory. Compr Physiol 4, 199–230. [DOI] [PubMed] [Google Scholar]
- Horowitz M (2016). Epigenetics and cytoprotection with heat acclimation. J Appl Physiol 120, 702–710. [DOI] [PubMed] [Google Scholar]
- Horowitz M (2017). Heat Acclimation-Mediated Cross-Tolerance: Origins in within-Life Epigenetics? Front Physiol; DOI: 10.3389/fphys.2017.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz M, Umschweif G, Yacobi A & Shohami E (2015). Molecular programs induced by heat acclimation confer neuroprotection against TBI and hypoxic insults via cross-tolerance mechanisms. Frontiers in Neuroscience. Available at: 10.3389/fnins.2015.00256 [Accessed February 21, 2022]. [DOI] [PMC free article] [PubMed]
- Jollos V (1934). Inherited changes produced by heat-treatment in Drosophila melanogaster. Genetica 16, 476–494. [Google Scholar]
- King MA, Leon LR, Morse DA & Clanton TL (2017). Unique cytokine and chemokine responses to exertional heat stroke in mice. J Appl Physiol 122, 296–306. [DOI] [PubMed] [Google Scholar]
- King MA, Leon LR, Mustico DL, Haines JM & Clanton TL (2015). Biomarkers of multiorgan injury in a preclinical model of exertional heat stroke. J Appl Physiol 118, 1207–1220. [DOI] [PubMed] [Google Scholar]
- King MA, Rollo I & Baker LB (2021). Nutritional considerations to counteract gastrointestinal permeability during exertional heat stress. Journal of Applied Physiology 130, 1754–1765. [DOI] [PubMed] [Google Scholar]
- Kisliouk T, Cramer T & Meiri N (2017). Methyl CpG level at distal part of heat-shock protein promoter HSP70 exhibits epigenetic memory for heat stress by modulating recruitment of POU2F1-associated nucleosome-remodeling deacetylase (NuRD) complex. J Neurochem 141, 358–372. [DOI] [PubMed] [Google Scholar]
- Klosin A, Casas E, Hidalgo-Carcedo C, Vavouri T & Lehner B (2017). Transgenerational transmission of environmental information in C. elegans. Science 356, 320–323. [DOI] [PubMed] [Google Scholar]
- Krivega I & Dean A (2012). Enhancer and promoter interactions — long distance calls. Curr Opin Genet Dev 22, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacal I & Ventura R (2018). Epigenetic Inheritance: Concepts, Mechanisms and Perspectives. Frontiers in Molecular Neuroscience. Available at: 10.3389/fnmol.2018.00292 [Accessed February 8, 2022]. [DOI] [PMC free article] [PubMed]
- Laitano O, Garcia CK, Mattingly AJ, Robinson GP, Murray KO, King MA, Ingram B, Ramamoorthy S, Leon LR & Clanton TL (2020a). Delayed metabolic dysfunction in myocardium following exertional heat stroke in mice. The Journal of Physiology 598, 967–985. [DOI] [PubMed] [Google Scholar]
- Laitano O, Murray KO & Leon LR (2020b). Overlapping Mechanisms of Exertional Heat Stroke and Malignant Hyperthermia: Evidence vs. Conjecture. Sports Med 50, 1581–1592. [DOI] [PubMed] [Google Scholar]
- Liu J, Feng L, Li J & He Z (2015). Genetic and epigenetic control of plant heat responses. Frontiers in Plant Science 6, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, de Jonge J, Trejo-Arellano MS, Santos-González J, Köhler C & Hennig L (2021). Role of H1 and DNA methylation in selective regulation of transposable elements during heat stress. New Phytologist 229, 2238–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F (2018). The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet 19, 81–92. [DOI] [PubMed] [Google Scholar]
- Maloyan A & Horowitz M (2002). beta-Adrenergic signaling and thyroid hormones affect HSP72 expression during heat acclimation. J Appl Physiol (1985) 93, 107–115. [DOI] [PubMed] [Google Scholar]
- Maloyan A, Palmon A & Horowitz M (1999). Heat acclimation increases the basal HSP72 level and alters its production dynamics during heat stress. Am J Physiol 276, R1506–1515. [DOI] [PubMed] [Google Scholar]
- Masuyama H & Hiramatsu Y (2012). Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology 153, 2823–2830. [DOI] [PubMed] [Google Scholar]
- Masuyama H, Mitsui T, Nobumoto E & Hiramatsu Y (2015). The Effects of High-Fat Diet Exposure In Utero on the Obesogenic and Diabetogenic Traits Through Epigenetic Changes in Adiponectin and Leptin Gene Expression for Multiple Generations in Female Mice. Endocrinology 156, 2482–2491. [DOI] [PubMed] [Google Scholar]
- McCaw BA, Stevenson TJ & Lancaster LT (2020). Epigenetic Responses to Temperature and Climate. Integrative and Comparative Biology 60, 1469–1480. [DOI] [PubMed] [Google Scholar]
- McClung JP, Hasday JD, He J-R, Montain SJ, Cheuvront SN, Sawka MN & Singh IS (2008). Exercise-heat acclimation in humans alters baseline levels and ex vivo heat inducibility of HSP72 and HSP90 in peripheral blood mononuclear cells. Am J Physiol Regul Integr Comp Physiol 294, R185–191. [DOI] [PubMed] [Google Scholar]
- Metzger DCH & Schulte PM (2017). Persistent and plastic effects of temperature on DNA methylation across the genome of threespine stickleback ( Gasterosteus aculeatus ). Proc R Soc B 284, 20171667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley PL (1997). Heat shock proteins and heat adaptation of the whole organism. J Appl Physiol (1985) 83, 1413–1417. [DOI] [PubMed] [Google Scholar]
- Murray K, Laitano O, Salyers Z, Thome T, Robinson G, Gambino B, Ryan T & Clanton T (2021a). Exertional Heat Stroke Causes Long-Term Satellite Cell Dysfunction and Delayed Muscle Repair. The FASEB Journal; DOI: 10.1096/fasebj.2021.35.S1.04087. [DOI] [Google Scholar]
- Murray K, Sheikh L, Laitano O, Iwaniec J, Garcia C, Robinson G, Hammamieh R, Campbell R, Yang R & Clanton T (2019). Epigenetic Memory and Phenotype Change Observed in Mouse Skeletal Muscle 30 Days after Exertional Heat Stroke. The FASEB Journal 33, 842.5–842.5. [Google Scholar]
- Murray KO, Brant JO, Iwaniec JD, Sheikh LH, de Carvalho L, Garcia CK, Robinson GP, Alzahrani JM, Riva A, Laitano O, Kladde MP & Clanton TL (2021b). Exertional heat stroke leads to concurrent long-term epigenetic memory, immunosuppression and altered heat shock response in female mice. The Journal of Physiology 599, 119–141. [DOI] [PubMed] [Google Scholar]
- Murray KO, Brant JO, Kladde MP & Clanton TL (2022). Long-term myocardial DNA methylation and metabolomic responses to exertional heat stroke in mice. FASEB J. [Google Scholar]
- Naito M, Zager RA & Bomsztyk K (2009). BRG1 Increases Transcription of Proinflammatory Genes in Renal Ischemia. J Am Soc Nephrol 20, 1787–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norouzitallab P, Baruah K, Vandegehuchte M, Van Stappen G, Catania F, Vanden Bussche J, Vanhaecke L, Sorgeloos P & Bossier P (2014). Environmental heat stress induces epigenetic transgenerational inheritance of robustness in parthenogenetic Artemia model. FASEB J 28, 3552–3563. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA & Li E (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DIW & Roseboom TJ (2008). Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG 115, 1243–1249. [DOI] [PubMed] [Google Scholar]
- Pandolf KB (1998). Time course of heat acclimation and its decay. Int J Sports Med 19 Suppl 2, S157–160. [DOI] [PubMed] [Google Scholar]
- Pandolf KB, Burse RL & Goldman RF (1977). Role of physical fitness in heat acclimatisation, decay and reinduction. Ergonomics 20, 399–408. [DOI] [PubMed] [Google Scholar]
- Park SG, Hannenhalli S & Choi SS (2014). Conservation in first introns is positively associated with the number of exons within genes and the presence of regulatory epigenetic signals. BMC Genomics 15, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Bickmore W, Dean A, Nobrega MA & Bejerano G (2013). Enhancers: five essential questions. Nat Rev Genet 14, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Périard JD, Racinais S & Sawka MN (2015). Adaptations and mechanisms of human heat acclimation: Applications for competitive athletes and sports. Scandinavian Journal of Medicine & Science in Sports 25, 20–38. [DOI] [PubMed] [Google Scholar]
- Périard JD, Travers GJS, Racinais S & Sawka MN (2016). Cardiovascular adaptations supporting human exercise-heat acclimation. Autonomic Neuroscience 196, 52–62. [DOI] [PubMed] [Google Scholar]
- Pestana JLT, Novais SC, Norouzitallab P, Vandegehuchte MB, Bossier P & De Schamphelaere KAC (2016). Non-lethal heat shock increases tolerance to metal exposure in brine shrimp. Environ Res 151, 663–670. [DOI] [PubMed] [Google Scholar]
- Petejova N & Martinek A (2014). Acute kidney injury due to rhabdomyolysis and renal replacement therapy: a critical review. Crit Care 18, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plough HH & Ives PT (1934). Heat Induced Mutations in Drosophila. Proc Natl Acad Sci USA 20, 268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin A & Cedar H (1991). DNA methylation and gene expression. Microbiol Rev 55, 451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritossa F (1962). A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 18, 571–573. [Google Scholar]
- Rodríguez-Romo R, Berman N, Gómez A & Bobadilla NA (2015). Epigenetic regulation in the acute kidney injury to chronic kidney disease transition. Nephrology 20, 736–743. [DOI] [PubMed] [Google Scholar]
- Rosenberg T, Kisliouk T, Ben-Nun O, Cramer T & Meiri N (2021). Cross-tolerance: embryonic heat conditioning induces inflammatory resilience by affecting different layers of epigenetic mechanisms regulating IL6 expression later in life. Epigenetics 16, 228–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg T, Kisliouk T, Cramer T, Shinder D, Druyan S & Meiri N (2020). Embryonic Heat Conditioning Induces TET-Dependent Cross-Tolerance to Hypothalamic Inflammation Later in Life. Frontiers in Genetics 11, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossati A (2016). Global Warming and Its Health Impact. Int J Occup Environ Med 8, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu T, Veilleux HD, Donelson JM, Munday PL & Ravasi T (2018). The epigenetic landscape of transgenerational acclimation to ocean warming. Nature Clim Change 8, 504–509. [Google Scholar]
- Sagui E, Montigon C, Abriat A, Jouvion A, Duron-Martinaud S, Canini F, Zagnoli F, Bendahan D, Figarella-Branger D, Brégigeon M & Brosset C (2015). Is there a link between exertional heat stroke and susceptibility to malignant hyperthermia? PLoS ONE 10, e0135496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambon LW (1898). The ETIOLOGY of SUNSTROKE (SIRIASIS): NOT HEAT FEVER but an INFECTIOUS DISEASE. Br Med J 1, 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider SM (2016). Heat acclimation: Gold mines and genes. Temperature (Austin) 3, 527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong K-H, Li D, Shimizu H, Nakamura R & Ishii S (2011). Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell 145, 1049–1061. [DOI] [PubMed] [Google Scholar]
- Skinner MK (2014). Environmental stress and epigenetic transgenerational inheritance. BMC Med; DOI: 10.1186/s12916-014-0153-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiridis A, Debevec T, Ciuha U, Eiken O & Mekjavic IB (2019). Heat acclimation does not affect maximal aerobic power in thermoneutral normoxic or hypoxic conditions. Experimental Physiology 104, 345–358. [DOI] [PubMed] [Google Scholar]
- Spitz F & Furlong EEM (2012). Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L & Rao A (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetievsky A, Assayag M, Ben-Hamo R, Efroni S, Cohen G, Abbas A & Horowitz M (2014). Heat acclimation memory: do the kinetics of the deacclimated transcriptome predispose to rapid reacclimation and cytoprotection? Journal of Applied Physiology 117, 1262–1277. [DOI] [PubMed] [Google Scholar]
- Tetievsky A, Cohen O, Eli-Berchoer L, Gerstenblith G, Stern MD, Wapinski I, Friedman N & Horowitz M (2008). Physiological and molecular evidence of heat acclimation memory: a lesson from thermal responses and ischemic cross-tolerance in the heart. Physiological Genomics 34, 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetievsky A & Horowitz M (2010). Posttranslational modifications in histones underlie heat acclimation-mediated cytoprotective memory. J Appl Physiol (1985) 109, 1552–1561. [DOI] [PubMed] [Google Scholar]
- Tseng M-F, Chou C-L, Chung C-H, Chen Y-K, Chien W-C, Feng C-H & Chu P (2020). Risk of chronic kidney disease in patients with heat injury: A nationwide longitudinal cohort study in Taiwan. PLoS ONE 15, e0235607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng M-F, Chou C-L, Chung C-H, Chien W-C, Chen Y-K, Yang H-C & Chu P (2019). Association between heat stroke and ischemic heart disease: A national longitudinal cohort study in Taiwan. Eur J Intern Med 59, 97–103. [DOI] [PubMed] [Google Scholar]
- Umschweif G, Alexandrovich AG, Trembovler V, Horowitz M & Shohami E (2013). Hypoxia-inducible factor 1 is essential for spontaneous recovery from traumatic brain injury and is a key mediator of heat acclimation induced neuroprotection. J Cereb Blood Flow Metab 33, 524–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umschweif G, Shein NA, Alexandrovich AG, Trembovler V, Horowitz M & Shohami E (2015). Heat acclimation provides sustained improvement in functional recovery and attenuates apoptosis after traumatic brain injury. J Cereb Blood Flow Metab 35, 1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenendaal MVE, Painter RC, de Rooij SR, Bossuyt PMM, van der Post J a. M, Gluckman PD, Hanson MA & Roseboom TJ (2013). Transgenerational effects of prenatal exposure to the 1944–45 Dutch famine. BJOG 120, 548–553. [DOI] [PubMed] [Google Scholar]
- Velichko AK, Petrova NV, Razin SV & Kantidze OL (2015). Mechanism of heat stress-induced cellular senescence elucidates the exclusive vulnerability of early S-phase cells to mild genotoxic stress. Nucleic Acids Res 43, 6309–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinoth A, Thirunalasundari T, Shanmugam M, Uthrakumar A, Suji S & Rajkumar U (2018). Evaluation of DNA methylation and mRNA expression of heat shock proteins in thermal manipulated chicken. Cell Stress Chaperones 23, 235–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH (1942). The epigenotype. Endeavour 1, 18–20. [Google Scholar]
- Waldvogel A-M & Pfenninger M (2021). Temperature dependence of spontaneous mutation rates. Genome Res 31, 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace RF, Kriebel D, Punnett L, Wegman DH & Amoroso PJ (2007). Prior heat illness hospitalization and risk of early death. Environ Res 104, 290–295. [DOI] [PubMed] [Google Scholar]
- Wan Q-L, Meng X, Dai W, Luo Z, Wang C, Fu X, Yang J, Ye Q & Zhou Q (2021). N6-methyldeoxyadenine and histone methylation mediate transgenerational survival advantages induced by hormetic heat stress. Science Advances 7, eabc3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J-C, Chien W-C, Chu P, Chung C-H, Lin C-Y & Tsai S-H (2019). The association between heat stroke and subsequent cardiovascular diseases. PLoS ONE 14, e0211386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Yu G, Ming X, Xia W, Xu X, Zhang Y, Zhang W, Li Y, Huang C, Xie H, Zhu B & Xie W (2020). Imprecise DNMT1 activity coupled with neighbor-guided correction enables robust yet flexible epigenetic inheritance. Nat Genet 52, 828–839. [DOI] [PubMed] [Google Scholar]
- Weinhold B (2006). Epigenetics: The Science of Change. Environ Health Perspect 114, A160–A167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich A, Benz S, Karl S, Jeschek M, Jewgenow K & Fickel J (2016a). Paternal heat exposure causes DNA methylation and gene expression changes of Stat3 in Wild guinea pig sons. Ecology and Evolution 6, 2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich A, Lenz D, Jeschek M, Chung TH, Rübensam K, Göritz F, Jewgenow K & Fickel J (2016b). Paternal intergenerational epigenetic response to heat exposure in male Wild guinea pigs. Mol Ecol 25, 1729–1740. [DOI] [PubMed] [Google Scholar]
- Widdrington JD, Gomez-Duran A, Pyle A, Ruchaud-Sparagano M-H, Scott J, Baudouin SV, Rostron AJ, Lovat PE, Chinnery PF & Simpson AJ (2018). Exposure of Monocytic Cells to Lipopolysaccharide Induces Coordinated Endotoxin Tolerance, Mitochondrial Biogenesis, Mitophagy, and Antioxidant Defenses. Frontiers in Immunology. Available at: 10.3389/fimmu.2018.02217 [Accessed February 7, 2022]. [DOI] [PMC free article] [PubMed]
- Wu J, Zhang W & Li C (2020). Recent Advances in Genetic and Epigenetic Modulation of Animal Exposure to High Temperature. Frontiers in Genetics; DOI: 10.3389/fgene.2020.00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X & Zhang Y (2017). TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18, 517–534. [DOI] [PubMed] [Google Scholar]
- Yacobi A, Stern Bach Y & Horowitz M (2014). The protective effect of heat acclimation from hypoxic damage in the brain involves changes in the expression of glutamate receptors. Temperature (Austin) 1, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada PM, Amorim FT, Moseley P, Robergs R & Schneider SM (2007). Effect of heat acclimation on heat shock protein 72 and interleukin-10 in humans. J Appl Physiol (1985) 103, 1196–1204. [DOI] [PubMed] [Google Scholar]
- Zager RA & Johnson ACM (2009). Renal ischemia-reperfusion injury upregulates histone-modifying enzyme systems and alters histone expression at proinflammatory/profibrotic genes. Am J Physiol Renal Physiol 296, F1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, He F, Pu W, Gu X, Wang J & Su Z (2020). The Impact of DNA Methylation Dynamics on the Mutation Rate During Human Germline Development. G3 Genes|Genomes|Genetics 10, 3337–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]