

Summary:
The last influenza pandemic in 2009 emerged from swine and surveillance of swine influenza is important for pandemic preparedness. Movement of swine during husbandry, trade or marketing for slaughter provide opportunities for transfer and genetic reassortment of swine influenza viruses. Over 90% of the swine slaughtered at the central swine abattoir in Hong Kong are imported from farms located in multiple provinces in mainland China. There is opportunity for virus cross-infection during this transport and slaughter process. Of the 26,980 swabs collected in the slaughterhouse in Hong Kong from 5th January 2012 to 15th December 2016, we analyzed sequence data on influenza A (H3N2) virus isolates (n = 174) in conjunction with date of sampling and originating farm. Molecular epidemiology provided evidence of virus cross-infection between swine originating from different farms during transport. The findings are also suggestive of a virus lineage persisting in a swine farm for over 2 years, although the lack of information on management practices at farm-level means that alternative explanations cannot be excluded We used virus serology and isolation data from 4,226 pairs of linked serum and swabs collected from the same pig at slaughter from swine originating from Guangdong Province to compare the force of infection (FOI) during transport and within farms. The mean weekly FOI during transport was λt = 0.0286 (95% CI = 0.0211–0.0391) while the weekly FOI in farms was λf = 0.0089 (95% CI = 0.0084–0.0095), assuming a possible exposure duration in farm of 28 weeks, suggesting increased force of infection during the transport process. Pigs sourced from farms with high seroprevalence were found to be a significant risk factor (adjusted OR = 2.24, p-value = 0.015) for infection of imported pigs during transport by multivariable logistic regression analysis, whereas pigs with HAI titer of ≥1:40 were associated with a substantial reduction in infection risk by 67% (p-value = 0.012). Transport may increase virus cross-infection rates and provide opportunities for virus reassortment potentially increasing zoonotic risk to those involved in the transportation and slaughtering processes.
Keywords: influenza A virus, H3N2, swine, value chains, transmission dynamics, force of infection
Introduction
Emerging infectious diseases continue to be a major threat to public health. Most emerging or re-emerging infectious diseases are caused by RNA viruses and are zoonotic in origin, arising from domestic livestock or wildlife (Allen, 2017). Understanding viral transmission dynamics in livestock production and trade pathways can identify potential critical control points that may be amenable to disease control interventions (Leung, 2012).
Swine are an intermediate host in the pathway of pandemic emergence because they express receptors for both avian and human influenza virus in their upper respiratory tract (Ito, 1998). Swine have been thus considered as a “mixing-vessel” for the emergence of pandemic viruses (Scholtissek, 1985). The 2009 H1N1 pandemic virus arose through reassortment of gene segments from Eurasian avian-like H1N1 and North American Triple Reassortant swine viruses (Smith, 2009). The two precursor viruses were likely introduced into Mexico via importation of swine, possibly as breeder-stock (Mena, 2016). Swine husbandry, trading, marketing and slaughter provide opportunities for the emergence of novel reassortants and zoonotic infections through exposure of humans to swine influenza viruses. An example is H3N2 variant (H3N2v) influenza viruses causing over 300 infections in humans with exposure to swine at agricultural fairs in USA (Jhung, 2013). In Asia, the high densities and close proximity between swine, humans, poultry and wild birds provide an even greater threat for interspecies transmission. Surveillance of influenza viruses in swine in Asia is therefore of importance for pandemic preparedness (Kress, 2020; Vijaykrishna, 2011).
In Hong Kong, over 90% of the swine slaughtered at its abattoirs are imported from mainland China with an annual importation of about 1.45 million swine in 2017 (Centre for Food Safety, 2019). Live pigs for consumption are transported in trucks from the source farms in multiple provinces to a transfer house at the Shenzhen-Hong Kong border where they are held for inspection overnight, and are then transported by trucks or lorries to a centralized abattoir in Hong Kong. Along this transport chain, there is opportunity for virus transmission between consignments of pigs originating from different farms, facilitated by the stress of transport and the densely packed environment. Pigs may also be held for hours to days in holding pens at the abattoir, providing further opportunities for cross infection (details in supporting information, Supplementary figure 1).
A previous study on the transmission dynamics of H1-subtype swine influenza viruses (SwIVs) in China (Strelioff, 2013) analyzed virological and serological data to infer the transmission risk over time. However, the interaction and cross-reactivity between different lineages of H1 SwIVs may have affected interpretation of the serology results from the hemagglutination inhibition (HAI) test. There was a lack of epidemiological information of imported swine population, such as source farm or province and length of stay at abattoir.
Multiple swine influenza viruses (SwIV) are enzootic in swine in China, including classical swine (CS), Eurasian avian-like swine (EA), triple-reassortant swine (TRIG) and pandemic H1N1/2009 (H1N1pdm09) lineages (Vijaykrishna, 2011). Infection with one H1 virus lineage could broaden cross-reactive antibody to other H1-subtype SwIVs (Perera, 2011) and hence confound interpretation of sero-epidemiology of H1 swine viruses. There was only one monophyletic H3 lineage (Binh Duong (BD)-like H3N2 lineage) circulating in China since 2010 allowing serology to accurately estimate past infection (Liang, 2014).
The objective of the current study was to understand the transmission dynamics of swine H3N2 viruses at the farm, transport and abattoir stage of the production chain. Focusing on swine H3N2 viruses allowed us to interpret serological data in the context of molecular epidemiology to infer transmission dynamics within the swine production system supplying swine for slaughter in Hong Kong. The force of infection (FOI), defined as the instantaneous rate at which a susceptible pig becomes infected, was used to quantify the transmission potential (Jit, 2011). We compared the risk of infection during transportation and in the farm setting by estimating the FOI and identified the role of epidemiological factors, such as transport distance and the length of stay at slaughterhouse.
Materials and Methods
Surveillance and sample collection
The Sheung Shui Slaughterhouse is the largest slaughterhouse in Asia, able to accommodate 12,000 pigs and slaughter up to 5,000 pigs per day. From 5th January 2012 to 15th December 2016, our surveillance team collected serum and nasal swabs from the same animal (hereafter referred to as linked serum and swab pairs) from randomly selected pigs immediately after slaughter, 50 linked serum and swab pairs being collected at each visit, which was increased to 75 linked serum and swab pairs since 19th November 2015. Tracheal swabs were also collected at post-mortem inspection. Information of the originating province and farm was derived from pig tattoo identification numbers. We also recorded the arrival date of the slaughtered pigs, from which the length of stay at slaughterhouse was derived. Sampling was conducted twice per month to allow identification of any potential seasonal pattern.
Virus isolation and molecular screening
Swab samples were inoculated into Madin Darby canine kidney (MDCK) cells grown in 24-well plates in minimum essential medium (MEM) supplemented with 2 μg/ml tosyl phenylalanyl chloromethyl ketone-treated tryspin (TPCK-trypsin) for virus propagation on the same day and replaced with fresh culture medium on the next day. On day 3 post-inoculation (DPI), viral growth was determined by appearance of cytopathic effects (CPE) in the MDCK cells and by a hemagglutination assay (HA) of cell supernatants using 0.5% turkey red blood cells (TRBCs). The cultures which gave positive results on CPE and HA were further tested using a rapid antigen detection test, Directigen™ EZ Flu A+B (Becton, Dickinson and Company, New Jersey, USA) to confirm influenza A virus infection. Positive cultures were then passaged on MDCK cells again for a second passage for virus isolation. Positive virus isolates were aliquoted and stored at −80°C.
Viral RNA was extracted from cell culture isolates using QIAmp Viral RNA Mini Kit (QIAGEN, Hilden, Germany). The presence of SwIVs was confirmed by a one-step reverse transcription PCR (RT-PCR) targeting the conserved influenza A matrix gene using PrimerScript II High Fidelity RT-PCR Kit (Takara, Japan) (Fouchier, 2000). We adapted a duplex two-step RT-PCR approach for HA and NA subtyping. Complementary DNA (cDNA) was synthesized by reverse transcription using SuperScript III Reverse Transcriptase (Invitrogen™) with Uni12 primer (Hoffmann, 2001). Gene amplification of HA and NA gene segment was performed using AmpliTaq Gold DNA Polymerase (Thermo Fisher Scientific, Sweden) with subtype-specific primers (Supplementary table 1). PCR products were analysed with 2% agarose gel electrophoresis and virus subtypes were determined.
Serological assays
Serum was extracted from clotted blood samples and stored at −20°C. Hemagglutination inhibition (HAI) assays were performed on each of the 6,532 serum samples collected, with a representative swine H3 influenza virus strain: A/Swine/Hong Kong/4348/2016 (BD-like H3N2). HAI tests were performed following WHO recommendations for animal influenza diagnosis (World Health Organization, 2011). Serum samples were first treated overnight at 37°C with receptor destroying enzyme (RDE) (Denka Seiken Co Ltd., Tokyo, Japan) and were heat-inactivated for 30 min at 56°C next morning. Serum samples were then serially diluted two-fold (1:10 to 1:1,280) in 96-well plates and 25 μL of virus antigen with four hemagglutination units was added to an equal volume of each serum dilution in duplicate. After 1h incubation, 50μL of 0.5% TRBCs was added to each well and incubated for 30 min. The HAI titer was defined as the highest serum dilution that inhibited complete hemagglutination. For calculation of the geometric mean titer (GMT), serum samples with a titer of <1:10 was assigned a value of 5 and those with ≥1:1,280 were assigned a value of 1:1,280.
Statistical analysis
We fitted a logistic regression model to predict isolation of influenza A(H3N2) subtype, considering factors such as length of stay at slaughterhouse of the live pigs, transport distance from originating farms to Hong Kong, HAI titer against the reference H3N2 strain (A/swine/Hong Kong/4348/2016) of individual pigs, and H3 seroprevalence of the specific originating farms.
The H3 seroprevalence of each originating farm was calculated as the proportion of seropositive (HAI titer ≥1:40) samples in 2012–2016 and only those samples collected from major source farms (≥20 samples over the study period) were analysed (n = 5,832) (Figure 1). We stratified the individual anti-H3 HAI titer level into sero-negative (<1:40) and sero-positive (≥1:40).
Figure 1.
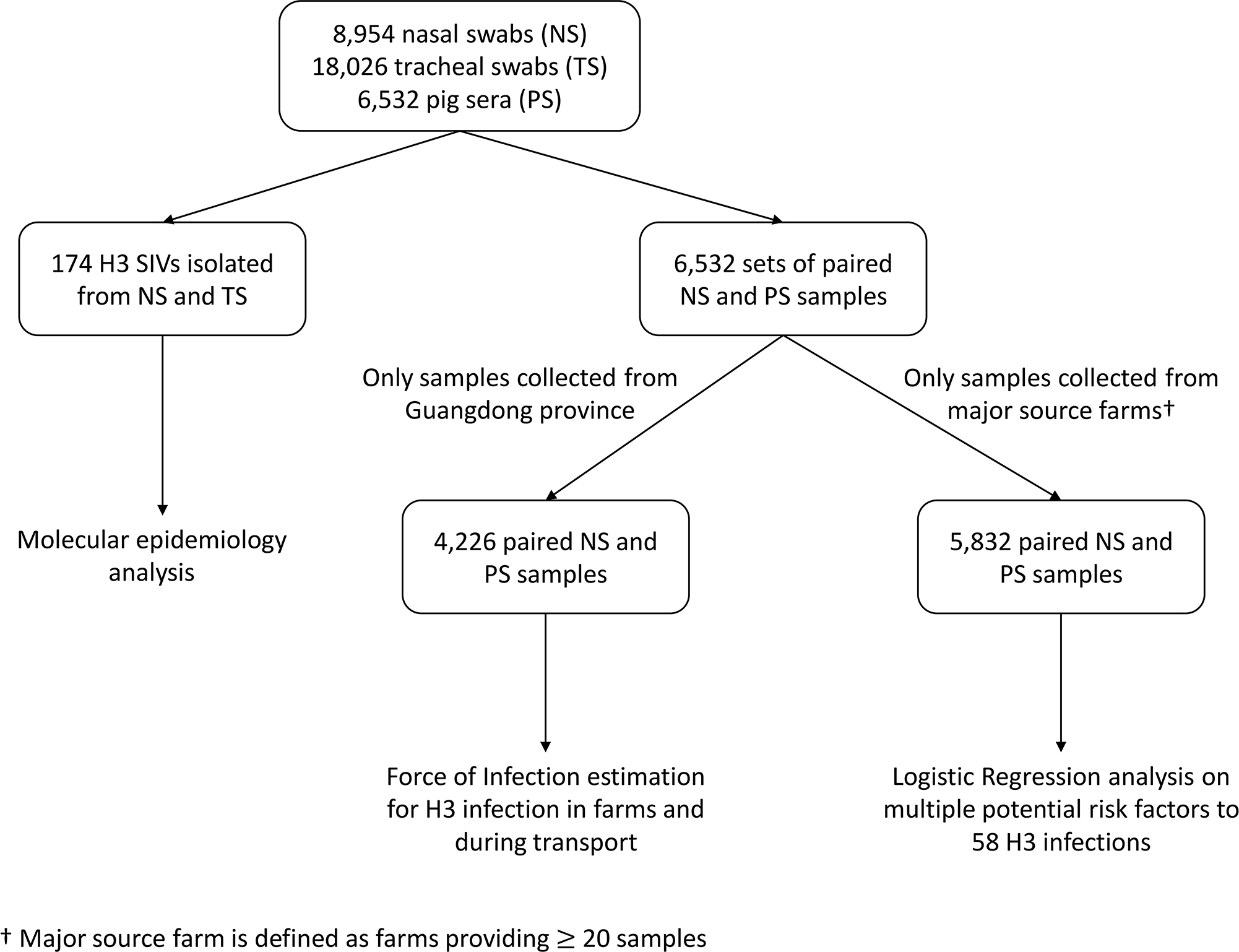
Sample collection and plan of analyses.
The slaughtered pigs originated from more than 150 farms. While we had the location of each farm to the level of the Province, exact geo-spatial farm locations were not available. We estimated the transport distance based on the originating province and stratified data into 3 groups: a) nearest, from Shenzhen, Zhuhai and other cities of Guangdong Provinces (within 150km from Hong Kong approximately); b) more distant, from Guangxi, Hainan, Hunan and Jiangxi Provinces (approximately 500–700 km), and c) most distant, from Hubei, Hebei, Henan and Zhejiang Provinces (approximately >900 km) (Supplementary figure 2). All factors were tested in the multivariable analysis. P value <0.05 was considered statistically significant. All statistical analyses were conducted using R version 4.0.3 (R Foundation for Statistical Computing, Vienna, Austria).
We further estimated the force of infection (FOI) in farms and during transportation of swine respectively, based on the analysis of virological and serological results on H3-subtype SwIVs in the subset where linked serum and swab pairs were available. Prior to importation of live pigs to Hong Kong, pigs from distant provinces were gathered by local traders but without clear records. Therefore, the exact duration of transportation of individual pigs were not available. Hence, we performed the FOI analysis based on pigs from Guangdong Province only (n = 4,226, 65% of the total samples) (Figure 1), the province adjacent to Hong Kong, to minimise discrepancies between actual and assumed exposure duration.
Viral isolation and serological data from linked serum and swab pairs were utilized to deduce the infection history of individual pigs. Previous studies had reported that pigs would begin shedding virus in nasal swabs by 1–3 days post-exposure and virus shedding would last for 4–5 days (Janke, 2013), whereas circulating antibodies become detectable 10–14 days after infection (Detmer, 2013). The time interval between virus shedding period and seroconversion allowed us to deduce where infections had taken place. Specifically, we defined the number of pigs which have experienced 4 possible infection histories: n1, naïve population (negative virological and serological test results); n2, recent primary infection (positive virological and negative serological test results); n3, previous infection (negative virological and positive serological test results); and n4, recent secondary or re-infection (positive virological and serological test results). It is uncertain where infections have taken place for swine with recent secondary or re-infection (n4), and their infection risks were likely substantially lower for seropositive swine (n3 and n4). Hence we estimated the probability of infection during transport (pt) from the virus isolation rate at the abattoir among swine naïve to H3N2 prior to transportation i.e. sero-negative to H3 antigens, so pt = n2 / (n1+n2). The probability of infection in farm (pf) was estimated by the proportion of seropositive samples, i.e., pf = (n3+n4) / (n1+n2+n3+n4), assuming recently infected pigs have not yet seroconverted.
The relationship between FOI and probability of infection p is given by:
where t1, t2 are the start and end times of the exposure, λ(t) is the FOI from infectious pigs in farms or transport system. The probability of infection during transport depends on the duration of travelling time for imported pigs. We assumed the exposure duration related to transport (time before slaughtering, including transportation, inspection and holding) is 3 days. The ages of the pigs sampled were 28 to 36 weeks old (no individual data was recorded) and pigs in farms were assumed to have 4 to 8 weeks of protection by maternal antibodies (Markowska-Daniel, 2011). A previous study on maternally derived antibodies suggested that the maternally acquired immunity may only mask clinical illness of the pigs but not protecting them from infection (Loeffen, 2003). Therefore, we considered possible exposure durations of 20, 28 and 36 weeks for the estimation of FOIs in farm. The standard error and hence 95% confidence interval of the probability of infection p was based on binomial distribution. The 95% CI for FOI was obtained accordingly based on the relationship between p and FOI above.
Whole-genome sequencing and assembly
The whole-genome sequence of each influenza virus isolate was obtained by using the Genome Sequencer Junior (Roche 454) or ABI 3730 genetic analyzer (Applied Biosystems) as previously described (Vijaykrishna, 2011, Liang, 2014). Original viral genetic sequence reads that shared >90% sequence identity in a 40-nucleotide overlapping region were assembled into contigs using Lasergene, version 9.0 (DNAStar).
Phylogenetic analysis and molecular epidemiology
Molecular epidemiology was used to define evidence of virus clustering and thus the occurrence of swine-to-swine transmission. Analysis was conducted on full genomes of all H3N2 swine influenza virus isolates sequenced in our study (Figure 1). The influenza virus sequences were analysed together with other existing Asian H3-subtype swine influenza virus sequences retrieved from the NCBI Influenza Virus Resource (Supplementary table 2) (Bao, 2008) and GISAID (Supplementary table 3) (Shu, 2017). All Asian H3 SwIVs with a complete set of segments in full-length were included and identical sequences were collapsed. Each genome segment was aligned separately using MAFFT version 7 (Katoh, 2013) and alignments were then trimmed to coding regions. The resultant data sets contained a total of 353 full genome sequence sets.
The coding regions of gene segments PB2, PB1, PA, HA, NP, NA, MP and NS were concatenated using Geneious 2019.2.1 (http://www.geneious.com). Multiple alignment for the concatenated whole genomes was performed with MAFFT version 7. We inferred the maximum-likelihood phylogenetic trees using IQ-TREE (Nguyen, 2014) and the built-in ModelFinder (Kalyaanamoorthy, 2017) to determine the best-fit substitution model. Branch supports were obtained using approximate likelihood ratio test (aLRT) (Guindon, 2010) to assess the robustness of the phylogenetic tree. Branch support aLRT statistics were shown at major nodes with values larger or equal to 0.8.
Results
Virus isolation in pigs
During the surveillance period from 2012 to 2016, a total of 8,954 nasal swabs (NS), 18,026 tracheal swabs (TS) and 6,532 pig sera were collected from pigs originated from 505 pig farms in 11 provinces in Mainland China and Hong Kong. The median number of swab samples collected per visit from the Sheung Shui slaughterhouse was 226 (range 102–291). H1N1 (n = 278), H3N2 (n = 174) and H1N2 (n = 51) subtype viruses were isolated. Only H3N2 viruses are further analysed in this study. There was a significantly higher influenza A H3-subtype virus isolation rate in nasal swabs, compared with tracheal swabs (positive rate = 1.61%) (p = 0.014).
Phylogenetic analysis and molecular epidemiology
Whole-genome sequencing and phylogenetic analysis were carried out for all 174 H3N2 viruses isolated from both nasal and tracheal swab samples during the study period. Together with 179 reference H3 SwIV genome sequences retrieved from the existing databases (GenBank, GISAID), a maximum likelihood phylogenetic tree of 353 concatenated genomes of H3 SwIVs was constructed. All tattoo codes indicating farm of origin and sampling dates of the H3 isolates were labelled in the tree (Figure 3). The 174 sequences of the H3N2 virus isolates can be grouped into 9 clusters, numbered 1–9, of closely related virus sublineages within the concatenated phylogenetic tree. Genetically similar viruses were detected in pigs from multiple farms on the same sampling occasion. For example, all H3N2 viruses collected on 3rd July 2014 formed a subcluster of taxa with closely related evolutionary history (subcluster 5). Interestingly, in subcluster 5, 18 nearly identical H3N2 viruses were isolated at the abattoir from pigs originating from 13 different farms in 3 different provinces (Guangdong, Hunan and Jiangxi). Similarly, in subcluster 4, there were 17 highly related H3N2 viruses isolated from pigs originated from 5 different farms in 2 provinces (Jiangxi and Guangdong) on one sampling date 7th January 2016. A similar phenomenon was found in virus subclusters 6, 7 and 8. It was highly improbable that a genetically identical virus was simultaneously circulating in multiple farms and provinces at the same time period. Thus, the phylogeny was strongly suggestive of a virus from a common source which transmitted to swine from other farms during transportation or while being held within the abattoir prior to slaughter.
Figure 3.
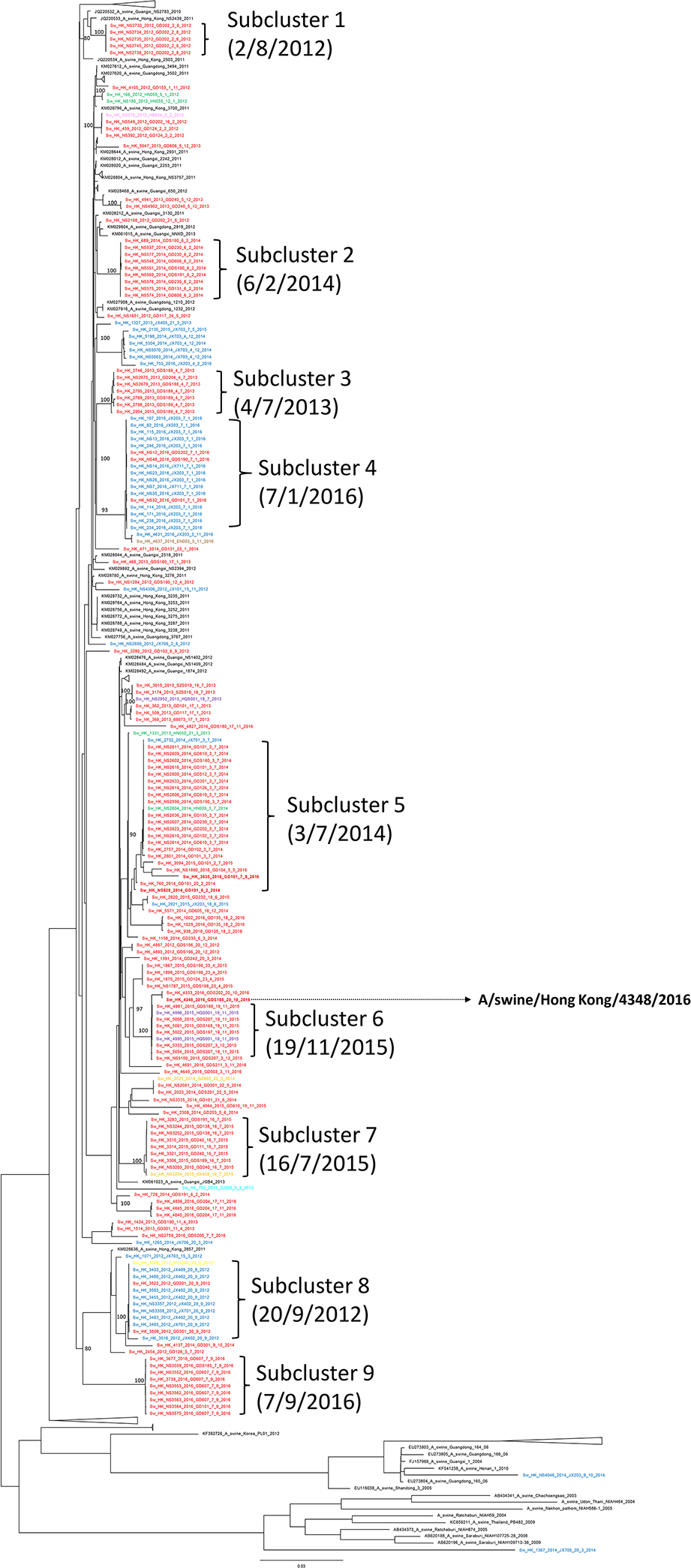
Maximum likelihood phylogenetic tree of 174 concatenated genomes of H3N2 swine influenza viruses in 2012–16. The tree was constructed using IQ-TREE using a GTR+F+R3 substitution model. Scale bar is given in numbers of substitutions per site and branch support aLRT statistics are shown at major nodes with value ≥ 0.8. Strain name is followed by pig tattoo codes and sampling date. Red: sequences from pigs from Guangdong and Shenzhen; Green: sequences from pigs from Hunan; Blue: sequences from pigs from Jiangxi; Orange: sequences from pigs from Guangxi; Cyan: sequences from pigs from Zhejiang; Pink: sequences from pigs from Hubei; Brown: sequences from pigs from Hainan; Yellow: sequences from pigs from Hebei; Black: other representative sequences from GenBank and GISAID. A/swine/Hong Kong/4348/2016 was selected as the representative strain for HAI test in this study.
In addition to clustering by the same sampling date, phylogenetically related virus samples were also clustering by their source farms. Seven samples in subcluster 5 were all originated from farm GD101 but collected on different dates ranging from 6th February 2014 to 7th September 2016 (Figure 3), suggesting that this virus sub-lineage was persisting within the farm for over two years.
Seroprevalence of H3N2 influenza virus in pigs
There was only one monophyletic lineage of H3 SwIVs isolated in our samples, that being the BD-like H3 lineage. H3N2 seroprevalence was assessed in 6,532 sets of linked serum and swab pairs. The HAI seroprevalence of BD-like H3 SwIVs in this subset was 20.5% (1,340/6,532) with HAI titer ≥1:40 against the reference H3 strain (A/swine/Hong Kong/4348/2016). The virus isolation rates and seroprevalence of H3N2 in swine were shown in Figure 2. No clear seasonal pattern was identified from the temporal trend of virus isolation.
Figure 2.
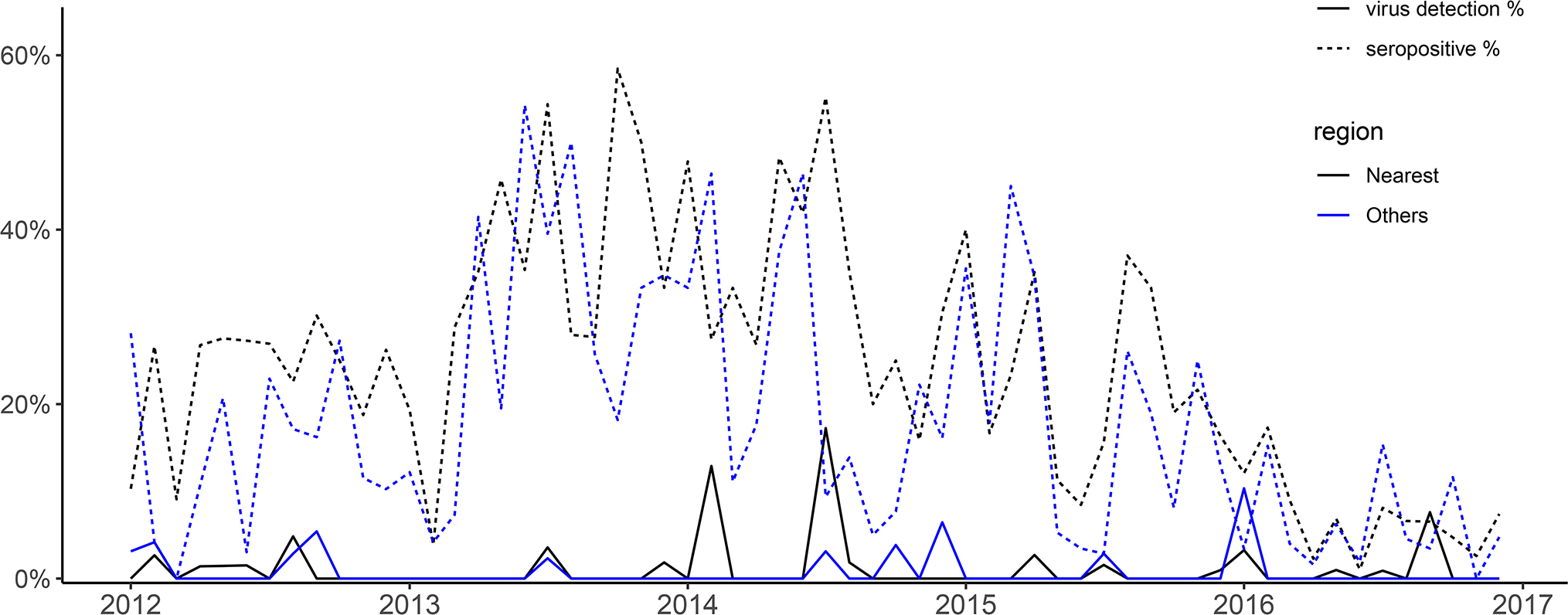
Monthly H3N2 seropositive and virus isolation rate in 6,532 sets of linked serum and swab pairs in 2012–2016, stratified into the nearest region (Guangdong, including Shenzhen and Zhuhai) and other regions. The year ticks at x-axis represent the beginning of years.
Multivariable logistic regression on H3-SwIV
Of the 6,532 linked serum and swab pairs from the same animal collected over the study period, 65% of the swine originated from Guangdong province, followed by Jiangxi (17%), with relatively fewer isolates from other provinces (Table 1). We isolated 63 H3N2 viruses from swabs and H3N2 antibodies were detected in 1,340 pigs (Table 1), with 7 pigs having both virus isolates and antibodies. Samples collected from farms (n = 5,832) which provided ≥20 samples were included in our logistic regression. Multivariable logistic regression was fitted to explain H3-subtype virus infections (Table 2).
Table 1.
Summary of the H3N2 virus isolate and H3 seropositive rates in nasal swab samples from different origins, sorted by transport distance to Hong Kong (by far to near).
| Province of origin | No. of samples | No. of H3 virus isolates | No. (%) H3 seropositive of those tested |
|---|---|---|---|
| Hebei | 298 | 0 | 58 (19.5) |
| Henan | 28 | 1 | 4 (14.3) |
| Hubei | 170 | 1 | 36 (21.2) |
| Zhejiang | 13 | 0 | 1 (7.7) |
| Hunan | 530 | 2 | 108 (20.4) |
| Guangxi | 110 | 1 | 25 (22.7) |
| Hainan | 61 | 0 | 17 (27.9) |
| Jiangxi | 1,072 | 12 | 149 (13.9) |
| Guangdong, other | 4,226 | 46 | 936 (22.1) |
| Zhuhai, Guangdong | 11 | 0 | 3 (27.3) |
| Shenzhen, Guangdong | 13 | 0 | 3 (23.1) |
| Total | 6,532 | 63 (1%) | 1340 (20.5) |
Table 2.
Summary of the influenza A virus isolation rates in samples with different characteristics
| Characteristics | No of samples | No. (%) H3 positive | Adjusted OR | 95% confidence intervals | p-value |
|---|---|---|---|---|---|
| Length of stay at slaughterhouse | |||||
| < 1 day | 3446 | 33 (1.0) | 1 | Reference | |
| 1–2 days | 2183 | 23 (1.1) | 1.30 | 0.76–2.25 | 0.339 |
| >2 days | 203 | 2 (1.0) | 1.20 | 0.28–5.08 | 0.802 |
| Transport distance † | |||||
| Nearest† | 3863 | 45 (1.2) | 1 | Reference | |
| More distant† | 1519 | 12 (0.8) | 0.77 | 0.39–1.15 | 0.449 |
| Most distant† | 450 | 1 (0.2) | 0.18 | 0.02–1.34 | 0.094 |
| Anti-H3 HAI titer | |||||
| Sero-negative (<1:40) | 4625 | 52 (1.1) | 1 | Reference | |
| Sero-positive (≥1:40) | 1207 | 6 (0.5) | 0.33 | 0.14–0.79 | 0.012 |
| Sero-prevalence of source farms | |||||
| Low (<10%) | 1963 | 16 (0.8) | 1 | Reference | |
| Moderate (10–30%) | 2047 | 16 (0.8) | 1.16 | 0.57–2.34 | 0.682 |
| High (>30%) | 1822 | 26 (1.4) | 2.25 | 1.17–4.32 | 0.015 |
| Total | 5832 | 58 (1.0) | |||
Nearest: Guangdong, including Shenzhen and Zhuhai; More distant: Guangxi, Hainan, Hunan, Jiangxi; Most distant: Hubei, Hebei, Henan, Zhejiang.
In the multivariable logistic regression model (Table 2), pigs with anti-H3 HAI titers ≥1:40 were significantly associated (adjusted OR [aOR] = 0.33, p = 0.012) with a lower risk of H3 virus detection in the abattoir (i.e. presumed infection during transportation). This reflected the protection against infection provided by the humoral immune response. Swine from source farms with high H3 seroprevalence (>30%) showed significantly higher H3 virus isolation rates (aOR = 2.24, p-value = 0.015). No significant difference was observed in the virus isolation among pigs with different length of stay at the slaughterhouse and transport distance.
Force of Infection (FOI) in farms and during transport
There were in total 4,226 linked serum and swab pairs collected from swine originating from Guangdong, with 46 (1.09%) H3 viruses isolated and 936 (22.1%) having antibody titers ≥1:40 (sero-positive) to H3 SwIV (Table 1). Among the 3,290 sero-negative samples, there were 40 (1.22%) H3 viruses isolated. The mean weekly FOI λt during transport for swine originating from Guangdong was estimated to be 0.0286 (95% CI = 0.0211–0.0391), while the weekly force of infection in farms were λf = 0.0125 (95% CI = 0.0118–0.0134), 0.0089 (95% CI = 0.0084–0.0095), and 0.0069 (95% CI = 0.0065–0.0074) respectively, assuming exposure durations of 20, 28 and 36 weeks in farms. Sensitivity analysis done with assumption of 1/2/4/5 days for exposure duration related to transport was shown in Supplementary table 4. Hence, the relative FOI during transport was 229–414% of that in farms, indicating a noticeable increased transmission intensity during transportation comparing to that within farms.
Discussion
Swine slaughtered in a large central abattoir in Hong Kong were sourced from farms in multiple provinces of China. We utilized data from linked serum and swab pairs from swine surveillance, together with data on farm of origin and date of sampling to assess the transmission risk in the swine transport and marketing system in China. Our results showed that pigs experienced a higher FOI (229%–414%) per-unit time during transport, compared to that within the farms. This suggested that the industrial scale swine production, transportation and marketing of live pigs provide settings favorable to influenza transmission, though the much longer stay in farms allow more infections over swine’s lifetime. The phylogenetic tree topology of the concatenated genomes also provided clear molecular epidemiological evidence of cross-transmission along the transport chain, between swine originating from different parts of mainland China. Interestingly, the isolation of similar though not identical viruses from the same farm over a two-year period suggested that swine influenza virus lineages may be maintained within a single source farm for over a year. We did not have detailed farm management data to ascertain herd sizes or management practices, in particular whether these large farms are ever completely emptied of pigs, which was unlikely. The likelihood that sequential batches of swine of different age groups are raised on the same farm increases the possibility of a virus to maintain itself within farms with large herd sizes. Our data highlights the desirability of “all-in all-out” production systems for the swine industry.
We found that pigs were more likely to be infected with H3 virus during transportation, if originating farms had a seroprevalence over 30%. The high farm-level seroprevalence implied a high circulation of H3 SwIV in farms. Hence, some pigs from the herd were more likely to be infectious at the beginning of the transport chain and more likely to transmit to others along the route (e.g. inspection or holding at the transfer house and holding within the abattoir). However, the infection risk during transportation for pigs with HAI titer of ≥1:40 reduced substantially by 67%, which is compatible with the protective effect of humoral immune response for humans (Hobson, 1972).
Distance of transportation was not found to be associated with the risk of transmission. Although we observed a lower transmission risk for swine from farms further away from Hong Kong, the difference was not statistically significant (Table 2) due to being confounded by the lower seroprevalence in some provinces. There could be other economic factors which may affect biosecurity measures during transport for these farms. The extent of mixing with other swine during inspection, waiting or holding at the transfer house, quarantine station and abattoir may pose greater transmission risks than that during transportation, with only about 40 pigs in a truck. Hence, the relative contribution of transmission risk during transportation may be less, even with a long trip from source farms to abattoir, than that associated with the mixing taking place at the quarantine stations and other common transport hubs. The isolation rate did not increase significantly for pigs staying longer times in the abattoir in Hong Kong which may imply that transmission within the abattoir was not a major driver of cross-infection. This is possibly related to the short time pigs were held in the abattoir prior to slaughter which may not provide adequate time for transmission and incubation period for there to be detectable virus at slaughter. The emergence of African swine fever in China since 2018 has led to a further shortening of the period of pigs being held in the abattoir in Hong Kong prior to slaughter, likely further reducing risk of cross-infection within the abattoir. The increased FOI during transportation may be facilitated by the high-density and high-stress environment at the transfer houses at the Shenzhen-Hong Kong border.
There were several limitations in our study. First, the actual duration and distance of transportation duration and distance for the live pigs were not available. We used province as a proxy for duration of transport. We could not rule out impacts arising from other activities related to swine trading. Secondly, while pigs from farms not previously infected with H3N2 virus got infected during the transport to the abattoir, we did not have unequivocal evidence that new viruses were introduced back to farms, although this could occur through contaminated trucks. Third, our estimates focused on H3-subtype SwIV only, giving us a smaller sample size. The serological cross-reactivity between multiple co-circulating H1 viruses make such an analysis difficult to interpret with H1 swine viruses. We chose to study this H3-subtype SwIV lineage because it was the only H3 subtype swine viruses detected in our study area during the course of this study. It is noted however, that if other H3-lineage viruses were circulating in our study region during this period, it may affect our serological data. Finally, our study deduced the infection history of individual pigs based on surveillance in the slaughterhouse only. Since it is not known when infection in the pigs would have happened in the production stages, we could only estimate the FOI based on the assumption of the possible exposure durations at farm. The actual prevalence of infectious virus in individual farms or transfer house were not available, which prevented us from estimating the transmission risk at different points of transportation.
In conclusion, we used linked serum and swab pairs collected from a swine slaughterhouse to examine the infection history of the swine from farms and during transportation. We found that transport and holding settings provided a favorable environment for swine-to-swine cross transmission per unit time when compared with farms. However, the longer period of time a pig spends on the farm means that the cumulative risk of acquiring infection in the farm is higher. The risk posed by the transport process was co-infection with new viruses which may provide opportunities for virus reassortment providing some risk to those involved in the transportation and slaughtering processes. There may also be the possibility of viruses being introduced back to naïve source farms via fomites and trucks, although this study was not able to investigate this possibility. Further assessment of the relative contribution of influenza transmission at different processes during transportation is needed to identify high risk area for improving disease control.
Supplementary Material
Acknowledgments:
We acknowledge research funding from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N93021C00016, HHSN266200700005C and HHSN272201400006C and the Theme Based Research grant (T11–712/19-N) from the Research Grants Council of Hong Kong SAR and the Health Medical Research Fund (Ref no: HKS-15-E02) from the Food and Health Bureau of Hong Kong SAR. We acknowledge the Food and Environmental Health Department, Government of the Hong Kong Special Administrative Region, for facilitating our study.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Data availability statement:
All data from this study have been disclosed.
Reference:
- Allen T, Murray KA, Zambrana-Torrelio C, Morse SS, Rondinini C, Di Marco M, Breit N, Olival KJ, Daszak P. Global hotspots and correlates of emerging zoonotic diseases. (2017). Nat Commun. 8(1):1124. doi: 10.1038/s41467-017-00923-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. (2008). The influenza virus resource at the National Center for Biotechnology Information. J Virol. 82(2):596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centre For Food Safety. Slaughterhouses and Disease Surveillance 2019. [Available from: https://www.cfs.gov.hk/english/import/import_sds.html.
- Detmer S, Gramer M, Goyal S, Torremorell M, Torrison J. (2013). Diagnostics and Surveillance for Swine Influenza. In: Richt JA, Webby RJ, editors. Swine Influenza. Berlin, Heidelberg: Springer Berlin Heidelberg. p. 85–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier RA, Bestebroer TM, Herfst S, Van Der Kemp L, Rimmelzwaan GF, Osterhaus AD. (2000). Detection of influenza A viruses from different species by PCR amplification of conserved sequences in the matrix gene. J Clin Microbiol. 38(11):4096–101. doi: 10.1128/JCM.38.11.4096-4101.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hobson D, Curry RL, Beare AS, Ward-Gardner A. (1972). The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J Hyg (Lond). 70(4):767–77. doi: 10.1017/s0022172400022610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. (2001). Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 146(12):2275–89. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- Ito T, Couceiro JN, Kelm S, Baum LG, Krauss S, Castrucci MR, Donatelli I, Kida H, Paulson JC, Webster RG, Kawaoka Y. (1998) Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. J Virol. 72(9):7367–73. doi: 10.1128/JVI.72.9.7367-7373.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke BH. (2013). Clinicopathological Features of Swine Influenza. In: Richt JA, Webby RJ, editors. Swine Influenza. Berlin, Heidelberg: Springer Berlin Heidelberg p. 69–83. [Google Scholar]
- Jhung MA, Epperson S, Biggerstaff M, Allen D, Balish A, Barnes N, Beaudoin A, Berman L, Bidol S, Blanton L, Blythe D, Brammer L, D’Mello T, Danila R, Davis W, de Fijter S, Diorio M, Durand LO, Emery S, Fowler B, Garten R, Grant Y, Greenbaum A, Gubareva L, Havers F, Haupt T, House J, Ibrahim S, Jiang V, Jain S, Jernigan D, Kazmierczak J, Klimov A, Lindstrom S, Longenberger A, Lucas P, Lynfield R, McMorrow M, Moll M, Morin C, Ostroff S, Page SL, Park SY, Peters S, Quinn C, Reed C, Richards S, Scheftel J, Simwale O, Shu B, Soyemi K, Stauffer J, Steffens C, Su S, Torso L, Uyeki TM, Vetter S, Villanueva J, Wong KK, Shaw M, Bresee JS, Cox N, Finelli L. (2013). Outbreak of variant influenza A(H3N2) virus in the United States. Clin Infect Dis. 57(12):1703–12. doi: 10.1093/cid/cit649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jit M, Brisson M (2011). Modelling the epidemiology of infectious diseases for decision analysis: a primer. PharmacoEconomics, 29(5), 371–386. 10.2165/11539960-000000000-00000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress WJ, Mazet JAK, Hebert PDN. (2020). Opinion: Intercepting pandemics through genomics. Proc Natl Acad Sci U S A. 2020 Jun 23;117(25):13852–13855. doi: 10.1073/pnas.2009508117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung Y, Lau E, Zhang L, Guan Y, Cowling B, Peiris JSM. (2012) Avian Influenza and Ban on Overnight Poultry Storage in Live Poultry Markets, Hong Kong. Emerging Infectious Diseases. 18(8):1339–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Lam TT, Fan X, Chen X, Zeng Y, Zhou J, Duan L, Tse M, Chan CH, Li L, Leung TY, Yip CH, Cheung CL, Zhou B, Smith DK, Poon LL, Peiris M, Guan Y, Zhu H. (2014). Expansion of genotypic diversity and establishment of 2009 H1N1 pandemic-origin internal genes in pigs in China. J Virol. 88(18):10864–74. doi: 10.1128/JVI.01327-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffen WL, Heinen PP, Bianchi AT, Hunneman WA, Verheijden JH. (2003). Effect of maternally derived antibodies on the clinical signs and immune response in pigs after primary and secondary infection with an influenza H1N1 virus. Vet Immunol Immunopathol. 20;92(1–2):23–35. doi: 10.1016/s0165-2427(03)00019-9. [DOI] [PubMed] [Google Scholar]
- Markowska-Daniel I, Pomorska-Mól M, Pejsak Z. (2011). The influence of age and maternal antibodies on the postvaccinal response against swine influenza viruses in pigs. Vet Immunol Immunopathol. 142(1–2): 81–6. doi: 10.1016/j.vetimm.2011.03.019. [DOI] [PubMed] [Google Scholar]
- Mena I, Nelson MI, Quezada-Monroy F, Dutta J, Cortes-Fernández R, Lara-Puente JH, Castro-Peralta F, Cunha LF, Trovão NS, Lozano-Dubernard B, Rambaut A, van Bakel H, García-Sastre A. (2016). Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. Elife. 5:e16777. doi: 10.7554/eLife.16777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–74. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RA, Riley S, Ma SK, Zhu HC, Guan Y, Peiris JSM. (2011). Seroconversion to pandemic (H1N1) 2009 virus and cross-reactive immunity to other swine influenza viruses. Emerg Infect Dis. 17(10):1897–9. doi: 10.3201/eid1710.110629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtissek C, Bürger H, Kistner O, Shortridge KF. (1985). The nucleoprotein as a possible major factor in determining host specificity of influenza H3N2 viruses. Virology. 147(2):287–94. doi: 10.1016/0042-6822(85)90131-x. [DOI] [PubMed] [Google Scholar]
- Shu Y, McCauley J. (2017). GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 30;22(13):30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, Peiris JS, Guan Y, Rambaut A. (2009). Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature. 459(7250):1122–5. doi: 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- Strelioff CC, Vijaykrishna D, Riley S, Guan Y, Peiris JSM, Lloyd-Smith JO. (2013). Inferring patterns of influenza transmission in swine from multiple streams of surveillance data. Proc Biol Sci. 280(1762):20130872. doi: 10.1098/rspb.2013.0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijaykrishna D, Smith GJ, Pybus OG, Zhu H, Bhatt S, Poon LL, Riley S, Bahl J, Ma SK, Cheung CL, Perera RA, Chen H, Shortridge KF, Webby RJ, Webster RG, Guan Y, Peiris JSM. (2011). Long-term evolution and transmission dynamics of swine influenza A virus. Nature. 473(7348):519–22. doi: 10.1038/nature10004. [DOI] [PubMed] [Google Scholar]
- World Health Organization. (2011). Manual for the laboratory diagnosis and virological surveillance of influenza. Geneva, Switzerland: World Health Organization. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data from this study have been disclosed.