
Figure 1: Overview of study of induced macrophage phenotypes in NSCLC co-culture system.
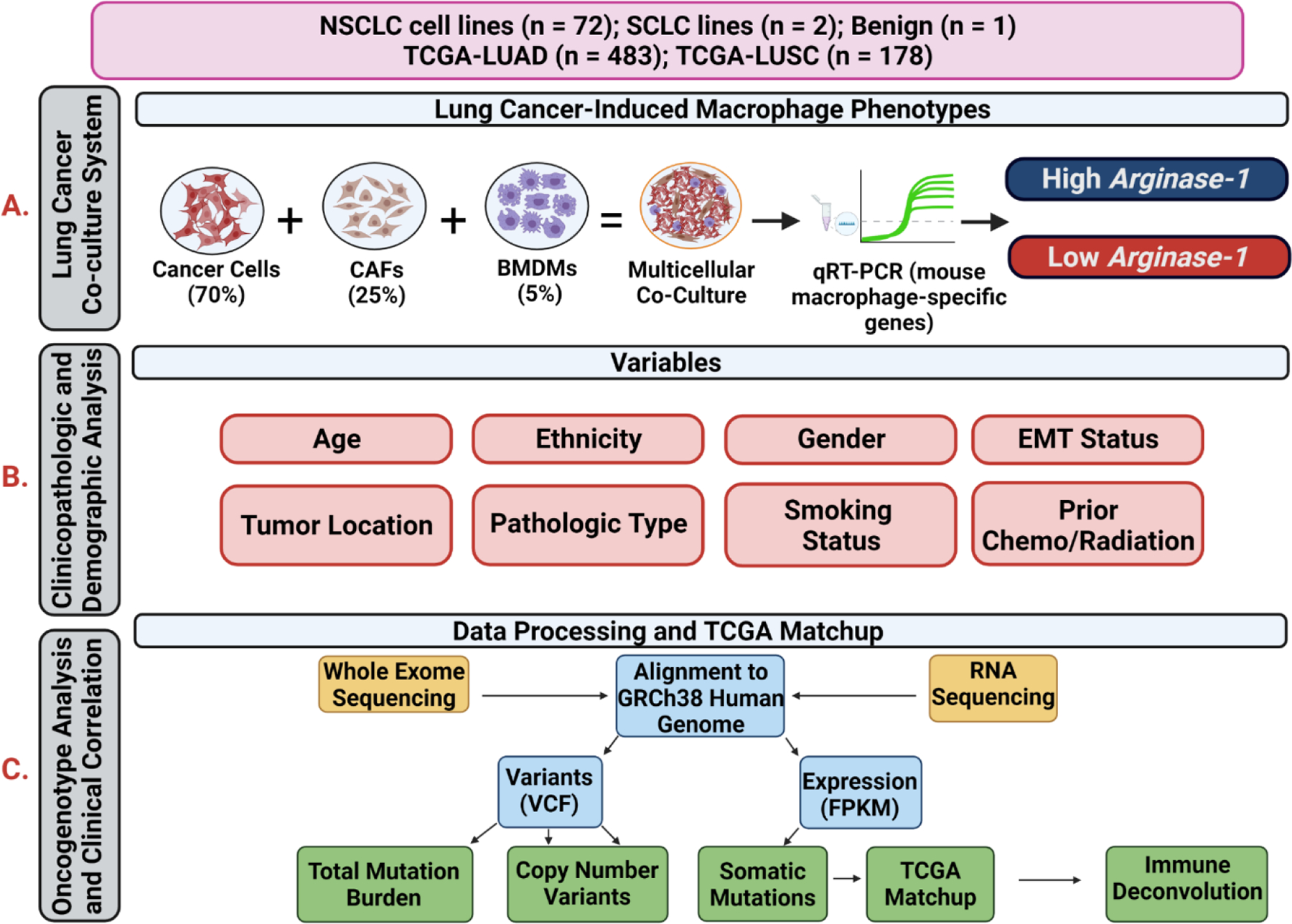
Flowchart showing analytic workflows, integrating molecular and clinicopathologic characteristics, with macrophage phenotypes induced in the lung cancer multicellular co-culture model and the NSCLC cell line information with TCGA datasets. A) Lung Cancer Co-Culture System Assay. We performed co-cultures of NSCLC lines, CAFs, and mouse bone marrow-derived macrophages and assayed by quantitative mRNA expression of mouse macrophage genes by using species-specific primers at 40 hours to determine the induced macrophage phenotype. B) Clinicopathologic and Demographic Analyses. We investigated whether clinicopathologic and demographic data features, or specific mutations or mRNA expression patterns found in the NSCLC lines were correlated with the induced macrophage phenotypes. C) Oncogenotype Analysis and Clinical Correlation. Total mutational burden, copy number variants, and somatic mutation profiling data were abstracted from whole exome and RNAseq analysis from each NSCLC line. These molecular data were “matched” against TCGA molecular data in NSCLC clinical samples by comparing mRNA expression and specific mutations (see Methods) to assign a correlation (ranging from 0.0 to 1.0) between each NSCLC line and TCGA tumor sample. We then performed immune deconvolution using CIBERSORT to estimate immune cell populations in the TCGA bulk tumor data, focusing specifically on M2 and M1 macrophages and tested whether the NSCLC cell line-induced macrophage phenotypes were similar to macrophage phenotypes found in TCGA tumor samples most closely matched to each cell line.