

Abstract
Natural orthopoxvirus outbreaks such as vaccinia, cowpox, cattlepox and buffalopox continue to cause morbidity in the human population. Monkeypox virus remains a significant agent of morbidity and mortality in Africa. Furthermore, monkeypox virus’s broad host-range and expanding environs make it of particular concern as an emerging human pathogen. Monkeypox virus and variola virus (the etiological agent of smallpox) are both potential agents of bioterrorism. The first line response to orthopoxvirus disease is through vaccination with first-generation and second-generation vaccines, such as Dryvax and ACAM2000. Although these vaccines provide excellent protection, their widespread use is impeded by the high level of adverse events associated with vaccination using live, attenuated virus. It is possible that vaccines could be used in combination with antiviral drugs to reduce the incidence and severity of vaccine-associated adverse events, or as a preventive in individuals with uncertain exposure status or contraindication to vaccination. We have used the intranasal mousepox (ectromelia) model to evaluate the efficacy of vaccination with Dryvax or ACAM2000 in conjunction with treatment using the broad spectrum antiviral, brincidofovir (BCV, CMX001). We found that co-treatment with BCV reduced the severity of vaccination-associated lesion development. Although the immune response to vaccination was quantifiably attenuated, vaccination combined with BCV treatment did not alter the development of full protective immunity, even when administered two days following ectromelia challenge. Studies with a non-replicating vaccine, ACAM3000 (MVA), confirmed that BCV’s mechanism of attenuating the immune response following vaccination with live virus was, as expected, by limiting viral replication and not through inhibition of the immune system. These studies suggest that, in the setting of post-exposure prophylaxis, co-administration of BCV with vaccination should be considered a first response to a smallpox emergency in subjects of uncertain exposure status or as a means of reduction of the incidence and severity of vaccine-associated adverse events.
Keywords: Mousepox, ACAM2000, Dryvax, ACAM3000, MVA, Cidofovir
1. Introduction
Variola virus (VARV), the etiological agent of smallpox, was officially declared eradicated in 1980 following a global vaccination campaign (Fenner et al., 1988). Although stated repositories are maintained in the United States and Russia, smallpox remains a threat to humans as a potential agent of bioterrorism (Buller et al., 2008). Monkeypox virus (MPXV) also remains a significant cause of morbidity/mortality in parts of Africa where mortality approaches 10% in humans, depending on the strain (Parker et al., 2007). The large host-range of MPXV makes eradication via a smallpox-like vaccination campaign virtually impossible (Parker and Buller, 2013). Like VARV, virulent MPXV strains also have key characteristics that would make them attractive vectors for weaponization, including stability in the environment, infection by the respiratory route, long incubation times, and the ability to cause significant morbidity and mortality (Fenner et al., 1988). Finally, orthopoxviruses (OPVs) can readily be created and modified using modern synthetic DNA and recombinant DNA technologies. One such modification is the incorporation of the interleukin-4 (IL4) gene into the genome as this cytokine has been shown to dramatically increase ectromelia (ECTV) virus-induced mortality in vaccinated hosts and hosts receiving antiviral therapy (Chen et al., 2011, Jackson et al., 1998, Jackson et al., 2001, Robbins et al., 2005).
Vaccination with live, replication-competent, vaccinia virus vaccines is highly effective, providing excellent protection in humans and in animal models. These live vaccines are considered the ‘gold standard’ in prophylactic treatment (Handley et al., 2007, Rosenthal et al., 2001). The main disadvantage to these vaccines is their sub-optimal safety profile (Lederman et al., 2009, Lederman et al., 2012); for example, it is postulated that vaccination is contraindicated in about 25% of the US population (Kemper et al., 2002). Safer, non-replicating vaccines, such as ACAM3000 (MVA) and LC16m8, are also available; however, these vaccines often require multiple administrations to achieve 100% protection which reduces their utility in an emergency scenario (Handley et al., 2007, Vollmar et al., 2006).
Because of the diminished threat of VARV and the poor-safety profile of available live vaccines, most governments discontinued routine vaccination decades ago. For this reason, the population lacks herd immunity. In the event of an OPV outbreak, antivirals such as cidofovir (CDV), brincidofovir (BCV) and tecovirimat (ST-246) would likely be employed (Parker et al., 2008a). CDV is an antiviral with activity against dsDNA viruses. While CDV is highly efficacious against OPVs (Israely et al., 2012, Neyts et al., 2004, Quenelle et al., 2003, Smee et al., 2001, Smee et al., 2004), it is limited by its requirement for intravenous administration and its significant risk of nephrotoxicity (De Clercq, 2002). A second antiviral, tecovirimat, has recently been added to the U.S. Strategic National Stockpile for use in a smallpox emergency. Tecovirimat has been shown to be highly efficacious against OPVs in multiple animal models and is well tolerated by humans when administered orally (Berhanu et al., 2009, Duraffour et al., 2007, Grosenbach et al., 2010, Jordan et al., 2008, Mucker et al., 2013, Nalca et al., 2008, Quenelle et al., 2007a, Yang et al., 2005). Unfortunately, antiviral resistance to tecovirimat can be achieved through a single point mutation in the F13L gene (Lederman et al., 2012, Yang et al., 2005), potentially limiting its clinical utility. An investigational antiviral, BCV, is a lipid conjugate of CDV under a development agreement with BARDA as a potential medical countermeasure for smallpox (Ciesla et al., 2003, Hostetler, 2007). BCV is orally bioavailable, has demonstrated efficacy in animal models, and has accumulated a safety database in humans which includes approximately 1000 patients and healthy volunteers with no evidence of dose-limiting nephrotoxicity or hematologic toxicity (Buller et al., 2004, Lederman et al., 2012, Parker et al., 2008b, Parker et al., 2008c, Quenelle et al., 2004, Quenelle et al., 2007b). Unlike tecovirimat, resistance to CDV or BCV is difficult to generate, requiring multiple mutations (Smee et al., 2002, Yang et al., 2005). Both tecovirimat and BCV have been used in emergency situations to treat severe cases of progressive vaccinia – a potentially lethal adverse effect of vaccination (Lederman et al., 2012). BCV is in development for multiple indications leading with prevention of CMV infection in transplant recipients and for treatment of disseminated adenovirus infection, for which the pilot phase of a Phase 3 study is currently enrolling. Both BCV and tecovirimat are in advanced development for treatment of OPVs.
Although current smallpox antivirals offer therapeutic options for treatment, they are not appropriate for long-term prophylactic use. In the event that en masse vaccination is required, most governments will likely rely on first- and second-generation vaccines such as Dryvax and ACAM2000 to vaccinate individuals that do not have a contraindication. In healthy adults, smallpox vaccines are usually administered via scarification with a bifurcated needle. A successful vaccination is described as a ‘take’ and is characterized by the development of a large lesion at the vaccination site (Fenner et al., 1988) which transitions from papule to pustule, followed by scabbing and healing by about six weeks post-vaccination. Typically, all vaccinees will experience at least one mild to moderate adverse event associated with vaccination; these are typically: pruritus (>93%), lymph node pain (81%), injection site pain (78%), fatigue (69%), headache (60%), myalgia (59%), and malaise (58%). In one study, about 10% of vaccinees experienced more severe adverse events (Frey et al., 2007). The lesion sheds live virus and must remain sequestered while healing; in addition, patients must be closely monitored for any vaccine-associated adverse events. It may be possible to limit the severity of vaccination by co-administering an antiviral at the time of vaccination. The use of cidofovir in conjunction with vaccination was evaluated previously and found to successfully reduce viral load and vaccination side-effects while maintaining the development of protective immunity against subsequent challenge (Israely et al., 2012). The efficacy of co-administration of tecovirimat with vaccine has also been studied in mice (Grosenbach et al., 2008). In these studies it was found that tecovirimat did not compromise the development of protective immunity following vaccination, and although tecovirimat did not reduce the severity of lesion formation following vaccination with Dryvax, it did reduce lesion severity when a more virulent VACV vaccine was used (strain Western Reserve) (Grosenbach et al., 2008). Similar studies in non-human primates also revealed that tecovirimat did not compromise protective immunity (Silvera et al., 2009). Interestingly, further studies reported that tecovirimat reduced lesion severity and time to lesion resolution without compromising protection in several immunocompromised murine strains (Berhanu et al., 2010).
In this report, we have evaluated the concurrent administration of Dryvax, ACAM2000 and ACAM3000 vaccines with BCV in mice. We observed that BCV facilitated healing of lesions without compromising vaccine protection, although BCV did alter the immune response compared to vaccination alone.
2. Materials and methods
2.1. Cells and viruses
BSC-1 cells (ATCC CCL 26) were grown in DMEM (Lonza, Basel, Switzerland) containing 10% fetal calf serum (FCS) (HyClone III, Logan, UT), 2 mM l-glutamine (GIBCO, Grand Island, NY), 100 U/ml penicillin (GIBCO, Grand Island, NY), and 100 μg/ml streptomycin (GIBCO, Grand Island, NY). A plaque-purified isolate of the MOS strain of ECTV (ATCC VR-1374) designated MOS-3-P2, was propagated in an African green monkey kidney cell line, BSC-1 (Chen et al., 1992). ECTV was purified through a sucrose cushion as described elsewhere (Moss and Earl, 1998). Virus infectivity was estimated as described previously (Wallace and Buller, 1985). Briefly, virus suspensions were serially diluted in PBS + 1% FCS (Fetal Clone II, HyClone), absorbed to monolayers for 1 h at 37 °C, and overlaid with a suspension of 1% carboxyl methyl cellulose in DMEM + 5% FCS. After 4 days at 37 °C, virus plaques were visualized and virus inactivated by the addition to each well of a 0.3% crystal violet/10% formalin solution.
2.2. Animals
Four to six week old female A and C57BL/6 mice were obtained from the National Cancer Institute, Frederick MD., or Jackson Laboratories, Bar Harbor ME., housed in filter-top microisolator cages and fed commercial mouse chow and water ad libitum. The randomized mice were housed in an animal biosafety level 3 containment area, with sample sizes indicated in the tables. Animal husbandry and experimental procedures were approved by the Institutional Animal Care and Use Committee. Mice were monitored every day until the termination of the experiment.
2.3. Antivirals
BCV was a gift from Chimerix Inc., (Durham, NC). BCV was freshly prepared prior to each experiment by dissolution in sterile, distilled water. BCV was administered by oral gavage in a volume of 0.1 ml/20 g mouse. Formulated drug was stored at 4 °C. CDV (Vistide) was a gift from Gilead Sciences (Foster City, CA) and was diluted in sterile saline and injected IP in a volume of 0.1 ml/20 g mouse. Doses and regimens are described in the figure legends and were determined based on the evolution of the BCV/ectromelia model in each mouse strain.
2.4. Vaccines
For Dryvax and ACAM2000 (gifts from the CDC, Atlanta, GA), mice were vaccinated with the indicated dose in a volume of 2.5 μl (the volume that fills the bifurcated needle) by scarification at the base of the tail with 15 punctures of the bifurcated needle (Precisions Medical Products, Denver, PA). Dryvax and ACAM2000 were provided at doses of approximately 2.5 × 108 and 2 × 108 PFU/ml, respectively; these doses provide neat vaccination doses of approximately 2.5 × 105 and 2 × 105 PFU/mouse, respectively. ACAM3000 (a gift from the NIAID-NIH, Bethesda, MD) was provided at a dose of 2 × 108 PFU/ml and was injected in 0.1 ml between the skin and underlying layers of tissue in the scapular region on the backs of mice.
2.5. Virus challenge
Mice were anesthetized with 0.1 ml/10 g body weight of ketamine HCl (6 mg/ml) and xylazine (0.5 mg/ml) by intraperitoneal injections. Anesthetized mice were placed in a dorsally recumbent position with their bodies angled so that the anterior end was raised 45° from the surface; a plastic mouse holder was used to ensure conformity (Esteban et al., 2012). ECTV was diluted in PBS without Ca2+ and Mg2+ to the required concentration and slowly loaded into each nare (5 μl/nare). Mice were subsequently left in situ for 2–3 min before being returned to their cages.
2.6. Statistics
T-tests were used to compare means between groups of mice and to determine the mean time to death. Mortality rates were compared using the Fisher’s exact test. Blinded lesion pictures were measured qualitatively using a scoring system ranging 0–4 in severity. P values <0.05 were considered statistically significant.
3. Results
3.1. BCV and CDV facilitate lesion healing following vaccination with Dryvax
To determine if BCV or CDV accelerate the healing of the primary vaccination lesion, A-strain mice were vaccinated with 250 PFU or 2500 PFU of Dryvax. These doses are approximately 3.5 to 35-fold higher than the Dryvax dose given to humans (2.5 × 105 PFU/person) based on PFU/bodyweight (assuming 70 kg human weight and 20 g mouse weight). Following vaccination, mice were treated at Day 0 with BCV or cidofovir (CDV) by oral gavage and intraperitoneal injection, respectively. Lesions were scored based on erythema, edema and eschar formation. At the 2500 PFU dose of Dryvax (Fig. 1 A), there was no statistical difference between mice treated with CDV and those not receiving treatment, however, mice treated with BCV had significantly lower lesion scores on Days 9, 14 and 16 when compared to mice not receiving treatment (P < 0.05). When comparing between CDV- and BCV-treated mice, we found that BCV-treated mice had significantly lower lesion scores on day 9 and 14 (P < 0.05). For the 250 PFU Dryvax groups (Fig. 1B), we found that both CDV- and BCV-treated mice had significantly lower lesions scores compared to mice not receiving treatment (P < 0.05); however, no differences in lesion scores were identified when comparing CDV-treated mice with BCV-treated mice. These data suggest that both BCV and CDV facilitate healing, but that BCV has greater efficacy than CDV in this model at the doses evaluated. Fig. S1 (Supplemental) shows example photographs of lesions on the tails of CDV-treated and BCV-treated mice at Days 7 and 9 post vaccination.
Fig. 1.
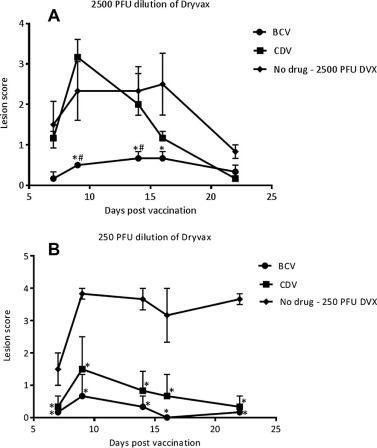
BCV and CDV reduce lesion severity following vaccination with Dryvax in mice. A-strain mice were vaccinated at the base of the tail with either (A) 2500 PFU or (B) 250 PFU of Dryvax. Following vaccination, mice were treated orally with a 10 mg/kg dose of BCV given at Day 0 followed by 4 mg/kg doses given at Day 1, 2, 3, and 4. CDV was used as a control and 12.5 mg/kg of CDV was given by intraperitoneal injection on Day 0, 1, 2, 3, and 4 post vaccination. Representative experiment of N = 5 mice. Error bars indicate SEM. Significance compared to no drug is indicated by ∗. Significance compared between BCV and CDV is indicated by a #.
3.2. Co-administration of BCV and Dryvax does not diminish protective immunity against a lethal ECTV challenge
We next examined whether co-administration of BCV with Dryvax vaccination affected the generation of protective immunity against a lethal, heterologous ECTV challenge. At Day 0, A-strain mice were vaccinated with decreasing doses of Dryvax vaccine ranging from neat (2.5 × 105 PFU/mouse) to 1:625 (400 PFU/mouse) dilutions. Concurrently, these mice received a BCV regimen starting at Day 0 of 10 mg/kg followed by 2.5 mg/kg given every other day until Day 14. Prior to ECTV challenge, blood samples for ELISAs obtained from each mouse demonstrated a statistically significant decrease in the antibody response in mice administered BCV compared to those receiving vehicle (P = 0.0001) (Fig. 2 ). At Day 50 (36 days post last BCV dosing), mice were challenged with ECTV (Day 0 relative to challenge, rtc) via the intranasal (IN) route. Mice in cage 2 received Dryvax-vehicle (Vac-Veh) and BCV-vehicle (Anti-Veh) and experienced 100% mortality with a mean day of death of 8.3 ± 0.2 rtc (Table 1 ). Mice in cage 3 received Vac-Veh and BCV; these mice also experienced 100% mortality. All mice that were vaccinated with Dryvax (groups 4–13) were protected against subsequent challenge (P ⩽ 0.002). Weight-change was measured from Day 0 rtc and mice receiving Vac-Veh (cages 2 and 3) lost weight rapidly at a similar rate (Fig. 3 A). When comparing cages that received Dryvax + Anti-Veh with cages receiving Dryvax + BCV, we found that in all cases the mice receiving the latter regimen lost more weight; however, these differences were only significant (P < 0.05) in the Dryvax-neat and Dryvax-1:5 dilution groups at Days 10, 12, 14, and 15 rtc (Fig. 3 A and B). No differences in weight were observed when comparing the Dryvax-1:125 and Dryvax-1:625 groups (Fig. 3C). These data indicate that Dryvax can be diluted to at least 1:625 and still provide significant protection against death with no statistical difference between Dryvax vaccinated mice receiving vehicle or BCV; however protection, as measured by morbidity (weight-change), is superior when vaccination is used without BCV therapy.
Fig. 2.
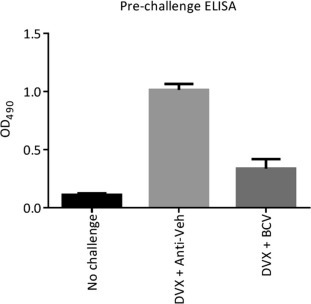
Co-administration of BCV and Dryvax does not diminish the antibody response against a lethal ECTV challenge in mice. A-strain mice were vaccinated with decreasing concentrations (from neat to 1:625 dilution) of Dryvax vaccine at Day 0 and treated by oral gavage with 10 mg/kg of BCV on day 0 followed by 2.5 mg/kg treatments on Day 2, 4, 6, 8, 10, 12, and 14. Prior to challenge, blood was obtained from each animal to perform an ELISA. At Day 50 (day 0 rtc) mice were challenged with 20 PFU (10× LD50) of ECTV via the IN route. Neat dilutions of Dryvax are shown. Representative experiment of N = 2 with 10 mice/group. Error bars indicate SEM.
Table 1.
Protection against a lethal ECTV challenge in A-strain mice vaccinated with Dryvax (DVX) in conjunction with BCV or vehicle (Anti-Veh).a At T = 0 days, A-strain mice were vaccinated with decreasing doses of Dryvax (DVX)c and administered BCV via oral gavage starting at T = 0 days (10 mg/kg) followed by 2.5 mg/kg doses on T = 2, 4, 6, 8, 10, 12, and 14 days. At T = 50 days, mice were challenged via the IN route with 20 PFU (10× LD50) of ECTV.
| Group | Cage | # of mice | DVXb dilution | BCVb | ECTV (T = 50 days) | Day of death (rtc)b | MTD ± SEMb | Mortality (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 10 | N/A | N/A | − | 0 | ||
| 2 |
2 | 10 | Veh |
Veh | + |
8,8,8,8,8,8,8,9,9,9 | 8.3 ± 0.2 | 100 |
| 3 | 10 | + | 8,8,8,8,8,9,9,9,9,9 | 8.5 ± 0.2 | 100 | |||
| 3 |
4 | 10 | Neatc |
Veh | + |
0∗ | ||
| 5 | 10 | + | 10 | 10 | 10∗ | |||
| 4 |
6 | 10 | 1:5c |
Veh | + |
0∗ | ||
| 7 | 10 | + | 0∗ | |||||
| 5 |
8 | 10 | 1:25c |
Veh | + |
0∗ | ||
| 9 | 10 | + | 0∗ | |||||
| 6 |
10 | 10 | 1:125c |
Veh | + |
0∗ | ||
| 11 | 10 | + | 0∗ | |||||
| 7 | 12 | 10 | 1:625c | Veh | + | 12 | 12 | 10∗ |
| 13 | 10 | + | 9,9,10 | 9.3 ± 0.3∗ | 30∗ | |||
P < 0.05 compared to controls.
Protocol is described in the legend of Fig. 2.
DVX, Dryvax; BCV, Brincidofovir; MTD, mean time to death; SEM, standard error of mean; rtc, relative to challenge.
PFU/mouse dilutions are 2.5 × 105 (for Neat), 5 × 104 (for 1:5), 1 × 104 (for 1:25), 2 × 103 (for 1:125), and 400 for (1:625).
Fig. 3.
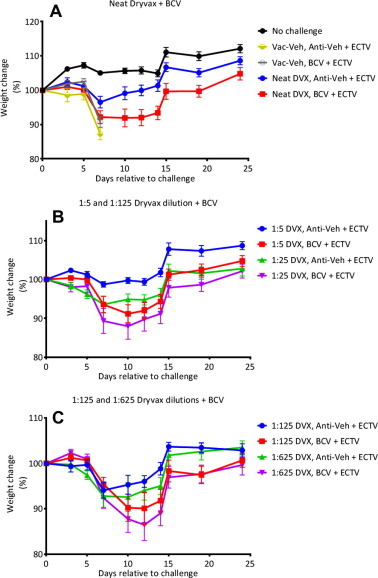
Weight-change of ECTV-challenged mice vaccinated with graded doses of vaccine in the presence and absence of BCV. Mice were treated as indicated in the legend of Fig. 2. At Day 50 (Day 0 relative to challenge) mice were challenged with 20 PFU (10× LD50) of ECTV via the IN route and monitored for morbidity as indicated by weight-change. Representative experiment of N = 2 with 10 mice/group. Error bars indicate SEM.
3.3. Co-administration of BCV and Dryvax protects A-strain mice when given up to two days post challenge
Given our previous finding that lesion severity is reduced when Dryvax vaccination is accompanied by BCV therapy and protection is not compromised, we sought to ascertain if post-exposure Dryvax vaccination could protect against lethal challenge both with and without simultaneous administration of BCV. To this end, we treated groups of A-strain mice with a Dryvax + Anti-Veh or a Dryvax + BCV regimen (Table 2 ). The combined Dryvax + BCV regimen was initiated on Day −4 (prophylactic treatment), Day 0, Day 2, or Day 4 rtc and an ECTV challenge was administered via the IN route on Day 0. Control animals experienced 100% mortality with a mean time of death (MTD) of 8.6 ± 0.4 with rapid weight-loss from Day 5 rtc (group 1) (Fig. 4 ). Dryvax + the Anti-Veh regimen, the BCV regimen, or both Dryvax + BCV regimen protected against a lethal ECTV challenge when given 4 days prior to challenge (P ⩽ 0.0004, groups 2–4). The Dryvax-only regimen (group 5) did not protect when given on the day of challenge (Day 0); however, the BCV-only regimen (group 7) and the Dryvax + BCV regimen (group 6) both protected mice from ECTV disease related mortality (P ⩽ 0.0004), thus indicating that the BCV provided the protection and not the vaccination with Dryvax. As expected, mice administered Dryvax-only at Day 2 (group 8) were not protected; however, protection was provided when the Dryvax + BCV regimen was used (group 9, P = 0.02). The highest level of Day 2 protection (100% survival) was afforded by the BCV-only regimen. At Day 4, no combinations were completely protective, including the BCV-only regimen (P = 0.2).
Table 2.
Protection against a lethal ECTV challenge when a combination of Dryvax (DVX) and BCV are given as pre- and post-exposure prophylaxis to A-strain mice.a Mice were administered Dryvax (2.5 × 105 PFU/mouse) and co-administered an initial dose of BCV (or vehicle) via oral gavage at 10 mg/kg on T = −4, 0, 2, or 4 days followed by 2.5 mg/kg doses of BCV given on Days 2, 4, 6, 8, 10, 12, and 14 following the initial dose. At T = 0 days, mice were challenged via the IN route with 40 PFU (20× LD50) of ECTV.
| Group | # of mice | Vaccine |
Antiviral |
Treatment (T=, day) | ECTV | Day of Death (rtc)b | MTD ± SEMb | Mortality (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| DVXb | Veh | BCVb | Veh | |||||||
| 1 | 10 | + | + | −4 | + | 7,7,8,8,8,9,9,10,10,10 | 8.6 ± 0.4 | 100 | ||
| 2 | 10 | + | + | −4 | + | 42 | 42 | 10⁎ | ||
| 3 | 10 | + | + | −4 | + | 0⁎ | ||||
| 4 | 10 | + | + | −4 | + | 26,27 | 26.5 ± 0.5⁎ | 20⁎ | ||
| 5 | 10 | + | + | 0 | + | 8,8,8,8,8,8,8,8,9,13 | 8.6 ± 0.5 | 100 | ||
| 6 | 10 | + | + | 0 | + | 0⁎ | ||||
| 7 | 10 | + | + | 0 | + | 53,15 | 34 ± 19⁎ | 20⁎ | ||
| 8 | 10 | + | + | 2 | + | 7,7,8,8,8,8,8,8,9,9, | 8 ± 0.2 | 100 | ||
| 9 | 10 | + | + | 2 | + | 9,10,10,10,10 | 9.8 ± 0.2⁎ | 50⁎ | ||
| 10 | 10 | + | + | 2 | + | 0⁎ | ||||
| 11 | 10 | + | + | 4 | + | 7,7,8,8,8,8,8,8,8,9 | 7.9 ± 0.2 | 100 | ||
| 12 | 10 | + | + | 4 | + | 10,10,12,12,12,14,15,29 | 14.3 ± 2.2⁎ | 80 | ||
| 13 | 10 | + | + | 4 | + | 9,11,11,11,12,12,13,20 | 12.4 ± 1.2⁎ | 80 | ||
| 14 | 10 | + | + | 0 | − | 0⁎ | ||||
| 15 | 10 | + | + | 0 | − | 0⁎ | ||||
| 16 | 10 | + | + | 0 | − | 0⁎ | ||||
| 17 | 10 | + | + | 0 | − | 0⁎ | ||||
| 18 | 5 | N/A | N/A | N/A | N/A | N/A | N/A | 0⁎ | ||
P < 0.05 compared to controls.
Protocol is described in the legend of Fig. 4.
DVX, Dryvax; BCV, Brincidofovir; MTD, mean time to death; SEM, standard error of mean; rtc, relative to challenge.
Fig. 4.
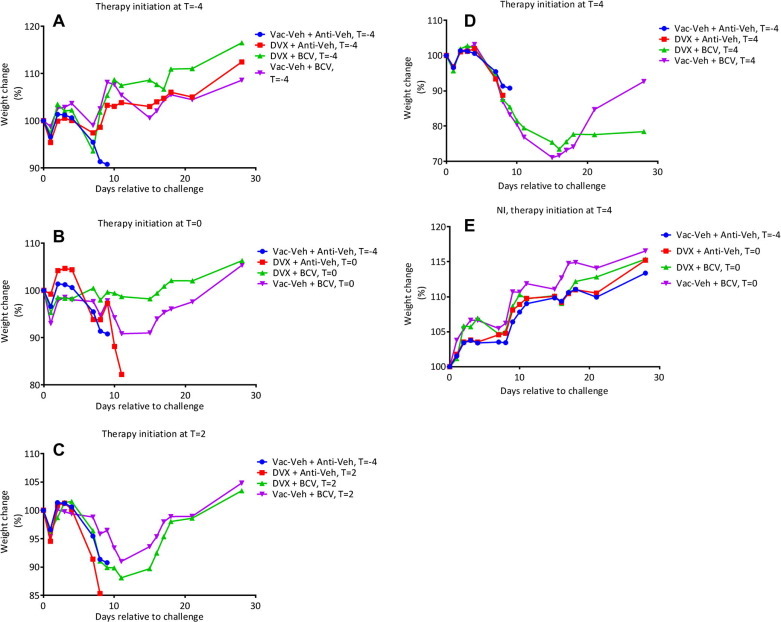
Co-administration of BCV and Dryvax protects A-strain mice from a lethal ectromelia virus challenge when given up to 2 days post-challenge and protects against weight-loss when given at Day -4 or Day 0. Groups of A-strain mice were vaccinated at the base of the tail with Dryvax and/or co-administered 10 mg/kg of BCV via oral gavage starting on Day -4 (prophylactic treatment), Day 0, 2, or 4 p.i., 2.5 mg/kg doses of BCV were given also on Day 2, 4, 6, 8, 10, 12, and 14 following the initial BCV treatment. A 40 PFU (20× LD50) ECTV challenge was administered via the IN route on Day 0. Mice were weighed daily as was mortality and time to death. 10 mice/group.
Morbidity was measured by weight-change. As expected, un-challenged control mice gained weight (groups 14–17) throughout the study. Weight-change data at Day 7 indicated that in the Day −4 groups the BCV-only regimen was slightly superior to the regimens involving Dryvax (Fig. 4); however, this was only significant when comparing against the Dryvax + BCV group (group 4 Vs. group 3, P = 0.006). Supporting the mortality data, we found that the Dryvax-only groups experienced the most weight-loss when the regimen was given on the day of challenge (Day 0, group 5) or post exposure (Day 2, group 8). For Day 2 groups, the BCV-only mice (group 13) experienced slightly less weight-loss than the Dryvax + BCV mice (group 12); however, weight-loss was not significant and neither of these groups were protected from mortality. All surviving mice that had been challenged at Day 0 (groups 1–13) were re-challenged at Day 91. Re-challenged mice experienced 100% survival, indicating that a memory response had been achieved, and that treatment with BCV had not prevented the generation of protective immunity.
3.4. Co-administration of BCV with ACAM2000 attenuates the immune response as measured by ELISA, but not the formation of protective immunity in C57BL/6 mice
ACAM2000 is a second-generation smallpox vaccine derived from the same viral seed-stock as first generation vaccines like Dryvax. Unlike Dryvax, which is cultured and harvested in the skin of calves, sheep and rabbits, ACAM2000 does not contain a heterogeneous population of viruses but rather a single VACV clone. This ACAM2000 clone was selected based on its similar virologic phenotype compared to Dryvax. The ACAM2000 vaccine has not been demonstrated to be any safer, or provide enhanced efficacy, compared to Dryvax; indeed, the vaccines are strikingly comparable (Handley et al., 2009). Due to the similarity between Dryvax and ACAM2000, the US Government selected ACAM2000 as a replacement for Dryvax and therefore all future studies used ACAM2000 as Dryvax stocks were destroyed.
We next examined the efficacy of BCV administration with ACAM2000 in the IN C57BL/6 mouse model. The C57BL/6 strain was chosen because it has been shown to more closely recapitulate smallpox in humans compared to A-strain mice (Parker et al., 2009) and we wished to evaluate the BCV + Dryvax/ACAM2000 vaccination regimen in more than one strain. Accordingly, mice were vaccinated with ACAM2000 and received either Anti-Veh or 20 mg/kg of BCV starting at Day −1, Day 0 or Day 1 followed by 4 more 20 mg/kg doses given every-third-day. Mice were bled at Day 21 and Day 50 post-vaccination for measurement of antibody (ELISA) and CD8+ T cell responses; and were challenged at Day 52 (Day 0 rtc) by the intranasal route with ECTV to assess both immune response to vaccination, as well as formation of protective immunity in the presence and absence of BCV.
ELISAs performed at Day 21 and Day 50 revealed that all vaccinated mice were seropositive. Mice receiving BCV consistently had lower ELISA values compared to mice receiving Anti-Veh, but this decrease was only significant when BCV was administered at Day −1 and Day 0 (P < 0.05) (data not shown). There was no statistical difference in IFN-γ secreting CD8 T cells between ACAM2000 vaccinated mice versus mice vaccinated and treated with BCV (data not shown). Following challenge, Anti-Veh and BCV-treated mice (group 2) experienced 89% and 90% mortality, respectively (Table 3 ). No statistical differences in the MTD and weight changes were observed. None of the vaccinated mice (groups 3–5) experienced mortality; however, we did observe that mice treated with BCV at Day −1 and Day 0 lost more weight than their Anti-Veh counterparts from approximately Day 10 (P < 0.05) (Fig. 5 ). These data suggest that BCV treatment given in conjugation with ACAM2000 vaccination does not affect mortality or morbidity when BCV is initiated at Day 1, and that BCV does not affect mortality and only mildly affects morbidity when given at Day 0 or Day −1. In summary, these data confirm our previous findings that suggest that BCV can be given with vaccinia vaccination without impeding vaccination efficacy in this model.
Table 3.
Protection against a lethal ECTV challenge in C57BL/6 mice vaccinated with ACAM2000 in conjunction with BCV or vehicle (Anti-Veh).a Mice were vaccinated with ACAM2000 (2 × 105 PFU/mouse) at Day 0 and received 20 mg/kg of BCV via oral gavage beginning on Day −1, 0 or 1 relative to vaccination. A further 4 doses of 20 mg/kg of BCV were administered every third day relative to the initial dose. At T = 52 days, mice were challenged with 4000 PFU (40× LD50) of ECTV via the IN route.
| Group | # of mice | Treatment 1. (T = 0) |
Treatment 2. |
ECTV (T = 52 days) | Day of death (rtc)b | MTD ± SEMb | Mortality (%) at T = 21 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Vaccine | Vehicle | Dilution | BCVb | Vehicle | T=, Day | ||||||
| 1 | 20 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 0 | ||
| 2 |
19 | + | N/A | + | 0 | + | 8,8,8,8,8,9,9,9,9,9,10,10,10,10,10,10,13 | 9.3 ± 0.3 | 89 | ||
| 20 | + | N/A | + | 0 | + | 8,8,8,8,8,8,8,8,9,9,9,9,10,10,10,10,10,11 | 8.9 ± 0.2 | 90 | |||
| 3 |
18 | + | Neatc | + | −1 | + | 0⁎ | ||||
| 20 | + | Neatc | + | −1 | + | 0⁎ | |||||
| 4 |
20 | + | Neatc | + | 0 | + | 0⁎ | ||||
| 19 | + | Neatc | + | 0 | + | 0⁎ | |||||
| 5 | 20 | + | Neatc | + | 1 | + | 0⁎ | ||||
| 19 | + | Neatc | + | 1 | + | 0⁎ | |||||
P < 0.05 compared to controls.
Protocol is described in the legend of Fig. 5.
BCV, Brincidofovir; rtc, relative to challenge; MTD, mean time to death; SEM, standard error of mean.
PFU/mouse dilutions are 2 × 105 (for Neat).
Fig. 5.
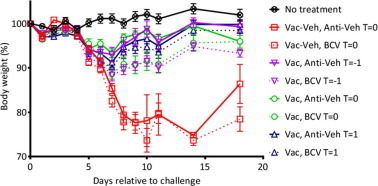
Co-administration of BCV and ACAM2000 protects C57BL/6 mice from a lethal ectromelia virus challenge when given up to 1 day post-challenge but does not protect against weight-loss. C57BL/6 mice were vaccinated with ACAM2000 (2x105 PFU) or vehicle and treated with BCV or Anti-Veh at Day -1, 0 or 1 relative to vaccination. The BCV dosing regimen consisted of a 20 mg/kg oral gavage treatments followed by an additional 4 treatments of 20 mg/kg administered every 3 days for a total of 5 doses. The same regimen was used for vehicle treated animals. Blood was obtained at 21 and 50 days post-vaccination for measurement of antibody (ELISA) and CD8+ T cell responses. Mice were challenged at Day 52 (Day 0 relative to challenge) by the intranasal route with 4000 PFU (40× LD50) of ECTV and weight changes are shown. Representative experiment of N = 2 with 18–20 mice/group. Error bars indicate SEM.
3.5. BCV does not alter the immune response following vaccination with a non-replicating vaccinia vaccine (ACAM3000) in A-strain mice
We have shown that CDV and BCV do not compromise protection against an ECTV challenge when co-administered with Dryvax, although a reduction in the virus-specific antibody response to the vaccine was observed. We next conducted experiments to determine whether the reduced immune response to vaccine was due to BCV limiting viral replication and thereby reducing the antigenic mass of vaccinia virus or due to an effect on the immune system.
To determine whether BCV has a direct impact on the immune system, we examined the effect of BCV on the immunity and protection induced by a single immunization with ACAM3000, a non-replicating smallpox vaccine (Handley et al., 2009). We examined the effect of adding a constant amount of BCV to decreasing doses of ACAM3000 on the generation of a neutralizing antibody response, specific anti-ECTV CD8+ T cell responses, and protection from an intranasal challenge with ECTV.
Mice were vaccinated with 1 × 107, 2 × 106, 4 × 105, and 8 × 104 TCID50 (tissue culture infectious dose) of ACAM3000 in the presence of BCV or Anti-Veh on Day 0. BCV was administered at 20 mg/kg followed by a further 3 doses of 20 mg/kg given at Days 2, 4 and 6 relative to the initial dosing. All mice challenged 52 days later (Day 0 rtc) experienced rapid weight-loss from Day 6 rtc with surviving mice regaining weight from about Days 12 to 16 rtc (Fig. 6 A and B). Mice vaccinated with 1 × 107 and 4 × 105 TCID50 of ACAM3000 and treated with BCV experienced greater weight loss compared to mice receiving Anti-Veh in the same group (P < 0.05 at Days 11–23 p.i. for 1 × 107 and at days 13, 14, 15, and 17 p.i. for 4 × 105). Interestingly, there was no difference in mean weight loss between mice receiving 2 × 106 and 8 × 104 TCID50. As expected, mice receiving Vac-Veh + BCV experienced 100% mortality by Day 8 p.i. A statistically significant decrease in mortality was observed in mice vaccinated with increasing doses of ACAM3000 (1 × 107, 2 × 106 and 4 × 105 PFU; P = ⩽0.02 for cages 3–8; Fig. 6C and D and Table 4 ). No difference in mortality was observed between mice receiving Anti-Veh or BCV within the same group; thus indicating that BCV did not alter protective immunity conferred by the vaccine. The MTD was statistically increased with mice vaccinated with 2 × 106 TCID50 and receiving BCV or Vehicle, and those vaccinated with 4 × 105 TCID50 and receiving BCV. Although mice receiving 8 × 104 TCID50 were not significantly protected, their MTDs were significantly increased (P = 0.008 and 0.005) relative to the respective control. Comparing cages within each group revealed that the BCV regimen did not alter the MTD.
Fig. 6.
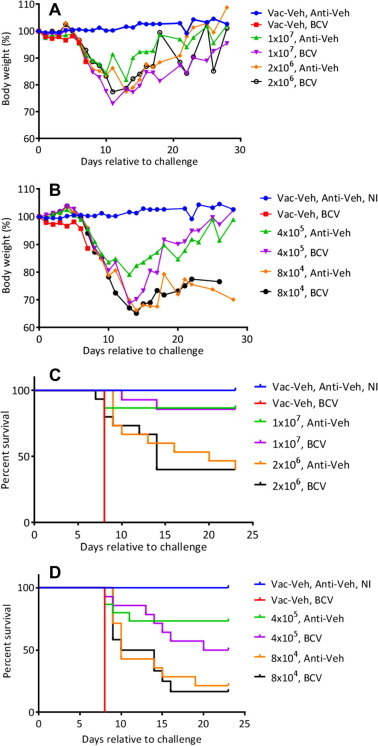
Weight-change and survival of A-strain mice vaccinated with decreasing doses of ACAM3000 in the presence and absence of BCV. Groups of A-strain mice were vaccinated subcutaneously with 1 × 107, 2 × 106, 4 × 105, and 8 × 104 TCID50 of ACAM3000 in the presence of BCV or Anti-Veh. The BCV dosing regimen consisted of an initial 20 mg/kg oral gavage dose followed by 3 further treatments of 20 mg/kg administered on Days 2, 4 and 6, relative to the initial dosing. Mice were bled at Day 50 days post-vaccination for measurement of antibody (ELISA) and were challenged at Day 52 (Day 0 rtc) by the intranasal route with 120 PFU (60× LD50) of ECTV. Mice were weighed daily (Panels A and B) and mortality measured (Panels C and D). Representative experiment of N = 3 with 12–15 mice/group.
Table 4.
Survival of A-strain mice vaccinated with decreasing doses of ACAM3000 in the presences of absence of BCV or vehicle (Anti-Veh).a Mice were vaccinated with 1 × 107, 2 × 106, 4 × 105, or 8 × 104 TCID50 of ACAM3000 at Day 0. At Day 0, 2, 4, and 6 mice also received via oral gavage 20 mg/kg of BCV. At Day 52 (Day 0 rtc), mice were challenged via the IN route with 120 PFU (60× LD50) of ECTV.
| Group | Cage | # of mice | Vaccine treatment (T = 0) |
Antiviral |
ELISA (T = 50, days) | ECTV (T = 52, DAYS) | Time of death (rtc)b | MTD ± SEMb | Mortality (%) at T = 25 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vaccine | Dose | Vehicle | BCVa | Vehicle | ||||||||
| 1 | 1 | 15 | − | N/A | + | + | − | 0 | ||||
| 2 | 2 | 11 | − | N/A | + | + | − | + | 8,8,8,8,8,8,8,8,8,8,8 | 8 ± 0 | 100 | |
| 3 |
3 | 15 | + |
1x107 |
− |
+ | + | + |
8,8 | 8 ± 0 | 13⁎ | |
| 4 | 14 | + | + | 10,14, | 12 ± 2 | 14⁎ | ||||||
| 4 |
5 | 15 | + |
2 × 106 |
− |
+ | − | + |
9,9,9,9,10,13,16,20,23 | 13.1 ± 1.8⁎ | 60⁎ | |
| 6 | 15 | + | − | 7,8,8,9,12,14,14,14,14 | 11.1 ± 1⁎ | 60⁎ | ||||||
| 5 |
7 | 15 | + |
4 × 105 |
− |
+ | − | + |
8,8,9,11 | 9 ± 0.7 | 27⁎ | |
| 8 | 14 | + | − | 8,9,13,14,15,16,20 | 13.6 ± 1.6⁎ | 50⁎ | ||||||
| 6 | 9 | 14 | + | 8 × 104 | − | + | − | + | 9,9,9,9,10,10,10,10,14,15,19 | 11.3 ± 1⁎ | 79 | |
| 10 | 12 | + | − | 9,9,9,9,9,10,14,14,15,16 | 11.4 ± 0.9⁎ | 83 | ||||||
ELISA positive animals at T = 50 post vaccination.
P < 0.05 compared to controls (group 2).
Protocol is described in the legend of Fig. 6.
BCV, Brincidofovir; rtc, relative to challenge; MTD, mean time to death, SEM, standard error of mean.
Anti-OPV antibodies were measured at Day 50 (1/100 dilution) with the intention of comparing vaccinated cages (group 3–6) to the appropriate vehicle controls (cages 1 and 2). We found that mice in group 3, which were vaccinated with the highest ACAM3000 dose, were the only animals that were statistically positive by ELISA (P = 0.003 and 0.009, respectively; Table 4).
Taken together, these data indicate that a single ACAM3000 dose of ⩾4 × 105 PFU is required to provide protection against a lethal ECTV challenge. At the lowest dose of 8 × 104 PFU, protection was not provided but their MTDs were extended significantly. Interestingly, a positive ELISA score cannot be used as a predictor of protection, as groups 4 and 5 were ELISA negative but were statistically protected against challenge. BCV did not alter mortality, ELISA, or MTD data; however, weight-change suggested that in some cases the BCV regimen had an effect on body weight. These data suggest that the reduced immune response when BCV was co-administered with ACAM2000 is due to inhibition of ACAM2000 replication, and thus BCV may be restricting the amount of antigen being made, which therefore corresponds to a lowered immune response.
4. Discussion
The current prophylactic approach to preventing new OPVs infections is to use vaccines similar to those developed by Edward Jenner over 200 years ago. The U.S. now vaccinates individuals at risk of exposure with ACAM2000, which replaced the first-generation Dryvax vaccine; however, the safety profile of ACAM2000 is not significantly improved compared to Dryvax and its efficacy remains untested in naturally transmitted VARV infections in humans. ACAM2000 is considered a second-generation vaccine; these vaccines are derived from the same seed-stock as first generation vaccines. The primary advantage of ACAM2000 compared to its predecessor is simply its modern and sterile method of manufacture. Although first- and second-generation vaccines have been demonstrated to induce a robust immune response with proven efficacy in humans, the high level of associated adverse events limits their utility if en-masse vaccination were ever required; for example, approximately 25% of the US population would be at risk, or would place a close-contact at risk, of adverse events following vaccination (Kemper et al., 2002).
Third-generation vaccines are derived from virus strains that are attenuated and are typically safer than first- and second-generation vaccines. The improved safety profile generally comes from reduced viral replication which also reduces the amount of viral antigen and therefore the immune response. Two third-generation vaccines are of most importance: LC16m8 and ACAM3000. LC16m8 is an attenuated vaccine that is not replication deficient. The vaccine is primarily used in Japan; however, a phase 1/2 clinical trial in the US compared the vaccine to Dryvax and reported a similar number, and severity, of adverse events between both groups, although the vaccine lesion was reportedly smaller with less swelling (Kennedy et al., 2011). ACAM3000 is a highly attenuated, replication-incompetent, virus that was passaged 570 times in chicken embryo fibroblasts which dramatically reduced its virulence and rendered it incapable of replicating in humans. The vaccine has been safely used in more than 2000 humans without any reported significant adverse events (Frey et al., 2007, Vollmar et al., 2006). In animal models, ACAM3000 has demonstrated efficacy against lethal challenges with several different OPVs (Earl et al., 2004, Earl et al., 2008, Garza et al., 2009, Munz et al., 1993). The main limitation of ACAM3000 is that its immunogenicity is several-fold lower than that produced by non-attenuated vaccines which means that it often requires a booster vaccination about 30 days after the initial vaccination. This limitation becomes a significant handicap in the event that rapid vaccination is required, as non-attenuated vaccines typically provide protection within a few days of being administered (Handley et al., 2009, Parker et al., 2008a). Some progress has been made towards developing subunit DNA-, peptide-, and protein-vaccines (Edghill-Smith et al., 2005, Fang et al., 2006, Fogg et al., 2004, Heraud et al., 2006, Hirao et al., 2011, Hooper et al., 2000, Hooper et al., 2004, Pulford et al., 2004, Snyder et al., 2004); however, these vaccines are still in the early stages of development.
Live vaccines using attenuated viruses, like Dryvax and ACAM2000, provide the most robust immune responses to OPVs and have been demonstrated to be efficacious in human and animal models (Fenner et al., 1988, Handley et al., 2007, Handley et al., 2009, Nalca and Zumbrun, 2010). One approach that could be taken is to combine these ‘gold standard’ vaccines with antiviral therapy to reduce the incidence and severity of adverse events associated with vaccination. This approach has been successfully evaluated using both CDV and tecovirimat, both of which reduced vaccine associated side effects without interfering with the development of protective immunity against subsequent OPV challenge (Grosenbach et al., 2008, Israely et al., 2012, Silvera et al., 2009) although this observation was not universal since there is one report in which a combined CDV and vaccine regimen did not result in protective immunity (Wei et al., 2009). In the studies described in this report, we evaluated the efficacy of Dryvax and ACAM2000 smallpox vaccines when administered with a BCV regimen. BCV has been shown to be efficacious against poxvirus infection in several animal models of smallpox (Lanier et al., 2010) and is currently in Phase 3 clinical development for prevention of cytomegalovirus infection in immunocompromised hematopoietic transplant patients and for treatment of disseminated adenovirus disease in immunocompromised individuals. When co-administered with Dryvax vaccination, BCV accelerated the healing process of the lesion compared with vehicle treated mice with efficacy equal to or superior to its parent compound, CDV. This finding is significant because the lesion sheds live virus and is a significant source of auto-inoculation and transmission to unvaccinated individuals. Following a Dryvax + BCV regimen, we found that ECTV challenged mice were equally protected against ECTV-related mortality compared to mice treated with Dryvax alone, however, we did find that the immune response was attenuated as indicated by reduced antibody levels in serum. These findings may explain why mice that received Dryvax alone were more resistant to morbidity following ECTV challenge (as measured by weight-loss). The finding that Dryvax could be diluted several fold and still provide protection when given with or without BCV is consistent with previous studies that have shown that diluting Dryvax 1:10 does not compromise the immune response generated in humans (Frey et al., 2002).
In the event of an act of bioterrorism involving the intentional release of an OPV, public health authorities may not become aware of the attack until after the event. Therefore, one key advantage to any anti-OPV regimen is the ability to protect following exposure to the virus. Several antiviral drugs have been demonstrated to provide post-exposure protection in animal models (Nalca et al., 2008, Parker et al., 2008b, Parker et al., 2008c, Quenelle et al., 2007a, Quenelle et al., 2007c, Smith et al., 2011, Stabenow et al., 2010) and some studies have indicated that smallpox vaccines can also be given post-exposure and still provide protection (Keckler et al., 2013). Others have shown that vaccinating C57BL/6 or BALB/c mice with VACV-Lister (a first-generation smallpox vaccine similar to Dryvax) or ACAM3000 provides post-exposure protection up to 2–3 days post IN ECTV challenge (Paran et al., 2009). Our studies reveal that Dryvax vaccination at the time of exposure, or after exposure, does not protect A-strain mice against a lethal ECTV challenge; importantly however, we found that the dual BCV + Dryvax regimen provided protection when given up to 2 days post challenge with ECTV and provided protection, via vaccination, against subsequent re-challenge. These findings suggest that the BCV + Dryvax regimen may not only reduce vaccine-associated adverse events, but may also provide long-term protection when given post-exposure. Testing the efficacy of these regimens in other mouse strains would be revealing.
The A-strain mouse is highly sensitive to ECTV with low LD50 (<1 PFU) values reported following IN, subcutaneous (SC) and footpad (FP) challenge (Parker et al., 2010). Thus, the A-strain mouse offers a sensitive model to evaluate therapeutic and prophylactic drugs. That said, the C57BL/6 strain more accurately recapitulates the course of VARV and MPXV disease in humans, i.e. an LD50 of about 100 PFU following an IN challenge, resistance to FP and SC challenges, a prolonged disease course, and a more robust immune response to challenge (Parker et al., 2009). We evaluated the efficacy of a dual regimen in C57BL/6 mice with BCV + ACAM2000, where BCV was given at the same time as vaccination, 1 day before, or 1 day after. We found that mice receiving the dual-regimen of vaccine and BCV generated an immune response to vaccination, but this response was reduced compared to mice receiving ACAM2000 alone, except when BCV was given at Day +1 rtc, in which case no difference in antibody levels were observed. Following lethal challenge with ECTV, all mice survived challenge; however, the mice that received BCV at Day 1 after vaccination were better protected against morbidity, as measured by weight-loss. These data suggest that beginning a BCV regimen shortly after vaccination may provide the best protection against a challenge given several weeks post-vaccination in C57BL/6 mice and may be indicative of the clinical utility of a combination therapeutic/vaccine strategy in the event of an outbreak.
In summary, our findings demonstrate that adding BCV to vaccination with Dryvax or ACAM2000 reduces the immune response generated by the vaccine but does not alter the formation of protective immunity following vaccination in mice. The dual-regimen significantly reduced vaccination-associated lesions and facilitated healing; hence, it may be useful to reduce the incidence and severity of adverse events associated with vaccination and accidental exposure. Moreover, humans for whom vaccination is contraindicated could possibly be vaccinated in conjunction with a BCV regimen. Lastly, our findings reveal that the dual-regimen can be administered post-exposure with BCV providing protection against the challenge and the vaccine preventing disease following any subsequent re-challenge in this model. The findings of this research, in whole, are that although there is detectable attenuation of the immune response there is no significant reduction in protective immunity in animals administered a combination regimen of BCV and vaccine compared with mice administered vaccine alone. BCV also reduced the severity of vaccine related events and extended the period of time post-exposure that vaccine remained effective (as measured by prevention of mortality) in mice. Hence, combining BCV with vaccine is a strategy that may be useful in a post-exposure scenario or in immunocompromised patients for whom vaccination would otherwise be contraindicated. Consideration should also be given to developing a combination regimen of BCV in combination with a non-replicating vaccine, such as ACAM3000.
Acknowledgments
This work was supported in part by funding from Contract # N01-AI-15436 from the Virology Branch, DMID, NIAID, NIH and a contract from Chimerix Inc.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.antiviral.2014.08.003.
Appendix A. Supplementary data
BCV and CDV facilitate lesion healing following vaccination with Dryvax in mice. A-strain mice were vaccinated at the base of the tail with 2500 PFU or 250 PFU of Dryvax. Mice were treated with either CDV or BCV and the vaccination lesions were photographed daily. Blinded photographs were randomized and qualitatively scored on a scale of 0–4 (0 = no lesions, 4 = high levels of erythema, edema, and eschar formation for severity of the lesion. Photographs of lesions taken on days 7 and 9 post vaccination are shown. Representative experiment of N = 2 with 5 mice/group.
References
- Berhanu A., King D.S., Mosier S., Jordan R., Jones K.F., Hruby D.E., Grosenbach D.W. ST-246 inhibits in vivo poxvirus dissemination, virus shedding, and systemic disease manifestation. Antimicrob. Agents Chemother. 2009;53:4999–5009. doi: 10.1128/AAC.00678-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berhanu A., King D.S., Mosier S., Jordan R., Jones K.F., Hruby D.E., Grosenbach D.W. Impact of ST-246(R) on ACAM2000 smallpox vaccine reactogenicity, immunogenicity, and protective efficacy in immunodeficient mice. Vaccine. 2010;29:289–303. doi: 10.1016/j.vaccine.2010.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller R.M., Owens G., Schriewer J., Melman L., Beadle J.R., Hostetler K.Y. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology. 2004;318:474–481. doi: 10.1016/j.virol.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Buller R.L., Handley L., Parker S. In: National Institute of Allergy and Infectious Disease: Frontiers in Research. St. Georgiew V., Wester K., McGowan J., editors. Humana Press; 2008. Development of prophylactics and therapeutics against the smallpox and monkeypox biothreat agents; pp. 145–161. [Google Scholar]
- Chen W., Drillien R., Spehner D., Buller R.M. Restricted replication of ectromelia virus in cell culture correlates with mutations in virus-encoded host range gene. Virology. 1992;187:433–442. doi: 10.1016/0042-6822(92)90445-u. [DOI] [PubMed] [Google Scholar]
- Chen N., Bellone C.J., Schriewer J., Owens G., Fredrickson T., Parker S., Buller R.M. Poxvirus interleukin-4 expression overcomes inherent resistance and vaccine-induced immunity: pathogenesis, prophylaxis, and antiviral therapy. Virology. 2011;409:328–337. doi: 10.1016/j.virol.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesla S.L., Trahan J., Wan W.B., Beadle J.R., Aldern K.A., Painter G.R., Hostetler K.Y. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney. Antiviral Res. 2003;59:163–171. doi: 10.1016/s0166-3542(03)00110-4. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Cidofovir in the treatment of poxvirus infections. Antiviral Res. 2002;55:1–13. doi: 10.1016/S0166-3542(02)00008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraffour S., Snoeck R., de Vos R., van Den Oord J.J., Crance J.M., Garin D., Hruby D.E., Jordan R., De Clercq E., Andrei G. Activity of the anti-orthopoxvirus compound ST-246 against vaccinia, cowpox and camelpox viruses in cell monolayers and organotypic raft cultures. Antivir. Ther. 2007;12:1205–1216. [PubMed] [Google Scholar]
- Earl P.L., Americo J.L., Wyatt L.S., Eller L.A., Whitbeck J.C., Cohen G.H., Eisenberg R.J., Hartmann C.J., Jackson D.L., Kulesh D.A., Martinez M.J., Miller D.M., Mucker E.M., Shamblin J.D., Zwiers S.H., Huggins J.W., Jahrling P.B., Moss B. Immunogenicity of a highly attenuated MVA smallpox vaccine and protection against monkeypox. Nature. 2004;428:182–185. doi: 10.1038/nature02331. [DOI] [PubMed] [Google Scholar]
- Earl P.L., Americo J.L., Wyatt L.S., Espenshade O., Bassler J., Gong K., Lin S., Peters E., Rhodes L., Jr., Spano Y.E., Silvera P.M., Moss B. Rapid protection in a monkeypox model by a single injection of a replication-deficient vaccinia virus. Proc. Natl. Acad. Sci. USA. 2008;105:10889–10894. doi: 10.1073/pnas.0804985105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edghill-Smith Y., Golding H., Manischewitz J., King L.R., Scott D., Bray M., Nalca A., Hooper J.W., Whitehouse C.A., Schmitz J.E., Reimann K.A., Franchini G. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat. Med. 2005;11:740–747. doi: 10.1038/nm1261. [DOI] [PubMed] [Google Scholar]
- Esteban D., Parker S., Schriewer J., Hartzler H., Buller R.M. Mousepox, a small animal model of smallpox. Methods Mol. Biol. 2012;890:177–198. doi: 10.1007/978-1-61779-876-4_11. [DOI] [PubMed] [Google Scholar]
- Fang M., Cheng H., Dai Z., Bu Z., Sigal L.J. Immunization with a single extracellular enveloped virus protein produced in bacteria provides partial protection from a lethal orthopoxvirus infection in a natural host. Virology. 2006;345:231–243. doi: 10.1016/j.virol.2005.09.056. [DOI] [PubMed] [Google Scholar]
- Fenner F., Henderson D.A., Arita I., Jezek Z., Ladnyi I.D. World Health Organisation; Geneva: 1988. Smallpox and its eradication. [Google Scholar]
- Fogg C., Lustig S., Whitbeck J.C., Eisenberg R.J., Cohen G.H., Moss B. Protective immunity to vaccinia virus induced by vaccination with multiple recombinant outer membrane proteins of intracellular and extracellular virions. J. Virol. 2004;78:10230–10237. doi: 10.1128/JVI.78.19.10230-10237.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S.E., Couch R.B., Tacket C.O., Treanor J.J., Wolff M., Newman F.K., Atmar R.L., Edelman R., Nolan C.M., Belshe R.B. Clinical responses to undiluted and diluted smallpox vaccine. N. Engl. J. Med. 2002;346:1265–1274. doi: 10.1056/NEJMoa020534. [DOI] [PubMed] [Google Scholar]
- Frey S.E., Newman F.K., Kennedy J.S., Sobek V., Ennis F.A., Hill H., Yan L.K., Chaplin P., Vollmar J., Chaitman B.R., Belshe R.B. Clinical and immunologic responses to multiple doses of IMVAMUNE (Modified Vaccinia Ankara) followed by Dryvax challenge. Vaccine. 2007;25:8562–8573. doi: 10.1016/j.vaccine.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza N.L., Hatkin J.M., Livingston V., Nichols D.K., Chaplin P.J., Volkmann A., Fisher D., Nalca A. Evaluation of the efficacy of modified vaccinia Ankara (MVA)/IMVAMUNE against aerosolized rabbitpox virus in a rabbit model. Vaccine. 2009;27:5496–5504. doi: 10.1016/j.vaccine.2009.06.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosenbach D.W., Jordan R., King D.S., Berhanu A., Warren T.K., Kirkwood-Watts D.L., Tyavanagimatt S., Tan Y., Wilson R.L., Jones K.F., Hruby D.E. Immune responses to the smallpox vaccine given in combination with ST-246, a small-molecule inhibitor of poxvirus dissemination. Vaccine. 2008;26:933–946. doi: 10.1016/j.vaccine.2007.11.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosenbach D.W., Berhanu A., King D.S., Mosier S., Jones K.F., Jordan R.A., Bolken T.C., Hruby D.E. Efficacy of ST-246 versus lethal poxvirus challenge in immunodeficient mice. Proc. Natl. Acad. Sci. USA. 2010;107:838–843. doi: 10.1073/pnas.0912134107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley L.M., Mackey J.P., Buller R.M.L., Bellone C.J. Birkhauser; Basel: 2007. Orthopoxvirus Vaccines and Vaccination. pp. 329–353. [Google Scholar]
- Handley L., Buller R.M., Frey S.E., Bellone C., Parker S. The new ACAM2000 vaccine and other therapies to control orthopoxvirus outbreaks and bioterror attacks. Expert Rev. Vaccines. 2009;8:841–850. doi: 10.1586/erv.09.55. [DOI] [PubMed] [Google Scholar]
- Heraud J.M., Edghill-Smith Y., Ayala V., Kalisz I., Parrino J., Kalyanaraman V.S., Manischewitz J., King L.R., Hryniewicz A., Trindade C.J., Hassett M., Tsai W.P., Venzon D., Nalca A., Vaccari M., Silvera P., Bray M., Graham B.S., Golding H., Hooper J.W., Franchini G. Subunit recombinant vaccine protects against monkeypox. J. Immunol. 2006;177:2552–2564. doi: 10.4049/jimmunol.177.4.2552. [DOI] [PubMed] [Google Scholar]
- Hirao L.A., Draghia-Akli R., Prigge J.T., Yang M., Satishchandran A., Wu L., Hammarlund E., Khan A.S., Babas T., Rhodes L., Silvera P., Slifka M., Sardesai N.Y., Weiner D.B. Multivalent smallpox DNA vaccine delivered by intradermal electroporation drives protective immunity in nonhuman primates against lethal monkeypox challenge. J. Infect. Dis. 2011;203:95–102. doi: 10.1093/infdis/jiq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper J.W., Custer D.M., Schmaljohn C.S., Schmaljohn A.L. DNA vaccination with vaccinia virus L1R and A33R genes protects mice against a lethal poxvirus challenge. Virology. 2000;266:329–339. doi: 10.1006/viro.1999.0096. [DOI] [PubMed] [Google Scholar]
- Hooper J.W., Thompson E., Wilhelmsen C., Zimmerman M., Ichou M.A., Steffen S.E., Schmaljohn C.S., Schmaljohn A.L., Jahrling P.B. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J. Virol. 2004;78:4433–4443. doi: 10.1128/JVI.78.9.4433-4443.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetler K.Y. In: De Clercq E., editor. vol. 5. Elsevir; 2007. Synthesis and antiviral evaluation of broad spectrum, orally active analogs of cidofovir and other acyclic nucleoside phosphonates; pp. 167–184. (Advances in Antiviral Drug Design). [Google Scholar]
- Israely T., Paran N., Lustig S., Erez N., Politi B., Shafferman A., Melamed S. A single cidofovir treatment rescues animals at progressive stages of lethal orthopoxvirus disease. Virol. J. 2012;9:119. doi: 10.1186/1743-422X-9-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson R.J., Maguire D.J., Hinds L.A., Ramshaw I.A. Infertility in mice induced by a recombinant ectromelia virus expressing mouse zona pellucida glycoprotein 3. Biol. Reprod. 1998;58:152–159. doi: 10.1095/biolreprod58.1.152. [DOI] [PubMed] [Google Scholar]
- Jackson R.J., Ramsay A.J., Christensen C.D., Beaton S., Hall D.F., Ramshaw I.A. Expression of mouse interleukin-4 by a recombinant ectromelia virus suppresses cytolytic lymphocyte responses and overcomes genetic resistance to mousepox. J. Virol. 2001;75:1205–1210. doi: 10.1128/JVI.75.3.1205-1210.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan R., Tien D., Bolken T.C., Jones K.F., Shanthakumar T.R., Strasser J., Frimm A., Corrado M.L., Strome P.G., Hruby D.E. Single-dose safety and pharmacokinetics of ST-246, a novel orthopoxvirus egress inhibitor. Antimicrob. Agents Chemother. 2008;52(5):1721–1727. doi: 10.1128/AAC.01303-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keckler M.S., Reynolds M.G., Damon I.K., Karem K.L. The effects of post-exposure smallpox vaccination on clinical disease presentation: addressing the data gaps between historical epidemiology and modern surrogate model data. Vaccine. 2013;31:5192–5201. doi: 10.1016/j.vaccine.2013.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper A.R., Davis M.M., Freed G.L. Expected adverse events in a mass smallpox vaccination campaign. Eff. Clin. Pract. 2002;5:84–90. [PubMed] [Google Scholar]
- Kennedy J.S., Gurwith M., Dekker C.L., Frey S.E., Edwards K.M., Kenner J., Lock M., Empig C., Morikawa S., Saijo M., Yokote H., Karem K., Damon I., Perlroth M., Greenberg R.N. Safety and immunogenicity of LC16m8, an attenuated smallpox vaccine in vaccinia-naive adults. J. Infect. Dis. 2011;204:1395–1402. doi: 10.1093/infdis/jir527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier R., Trost L., Tippin T., Lampert B., Robertson A., Foster S., Rose M., Painter W., O’Mahony R., Almond M., Painter G. Development of CMX001 for the treatment of poxvirus infections. Viruses. 2010;2:2740–2762. doi: 10.3390/v2122740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederman E., Miramontes R., Openshaw J., Olson V.A., Karem K.L., Marcinak J., Panares R., Staggs W., Allen D., Weber S.G., Vora S., Gerber S.I., Hughes C.M., Regnery R., Collins L., Diaz P.S., Reynolds M.G., Damon I. Eczema vaccinatum resulting from the transmission of vaccinia virus from a smallpox vaccinee: an investigation of potential fomites in the home environment. Vaccine. 2009;27:375–377. doi: 10.1016/j.vaccine.2008.11.019. [DOI] [PubMed] [Google Scholar]
- Lederman E.R., Davidson W., Groff H.L., Smith S.K., Warkentien T., Li Y., Wilkins K.A., Karem K.L., Akondy R.S., Ahmed R., Frace M., Shieh W.J., Zaki S., Hruby D.E., Painter W.P., Bergman K.L., Cohen J.I., Damon I.K. Progressive vaccinia: case description and laboratory-guided therapy with vaccinia immune globulin, ST-246, and CMX001. J. Infect. Dis. 2012;206:1372–1385. doi: 10.1093/infdis/jis510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B., Earl P.L. Current Protocols in Molecular Biology. Wiley; 1998. Expression of proteins in mammalian cells using vaccinia cirus vectors. Overview of the vaccinia virus expression system; pp. 16.15.11–16.15.15. [Google Scholar]
- Mucker E.M., Goff A.J., Shamblin J.D., Grosenbach D.W., Damon I.K., Mehal J.M., Holman R.C., Carroll D., Gallardo N., Olson V.A., Clemmons C.J., Hudson P., Hruby D.E. Efficacy of tecovirimat (ST-246) in nonhuman primates infected with variola virus (Smallpox) Antimicrob. Agents Chemother. 2013;57:6246–6253. doi: 10.1128/AAC.00977-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz E., Linckh S., Renner-Muller I.C., Reimann M. The effectiveness of immunization with vaccinia virus type “MVA” against an infection with cowpox virus type “OPV 85” in rabbits. Zentralbl. Veterinarmed. B. 1993;40:131–140. [PubMed] [Google Scholar]
- Nalca A., Zumbrun E.E. ACAM2000: the new smallpox vaccine for United States Strategic National Stockpile. Drug Des. Dev. Ther. 2010;4:71–79. doi: 10.2147/dddt.s3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalca A., Hatkin J.M., Garza N.L., Nichols D.K., Norris S.W., Hruby D.E., Jordan R. Evaluation of orally delivered ST-246 as postexposure prophylactic and antiviral therapeutic in an aerosolized rabbitpox rabbit model. Antiviral Res. 2008;79:121–127. doi: 10.1016/j.antiviral.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Neyts J., Leyssen P., Verbeken E., De Clercq E. Efficacy of cidofovir in a murine model of disseminated progressive vaccinia. Antimicrob. Agents Chemother. 2004;48:2267–2273. doi: 10.1128/AAC.48.6.2267-2273.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paran N., Suezer Y., Lustig S., Israely T., Schwantes A., Melamed S., Katz L., Preuss T., Hanschmann K.M., Kalinke U., Erez N., Levin R., Velan B., Lower J., Shafferman A., Sutter G. Postexposure immunization with modified vaccinia virus Ankara or conventional Lister vaccine provides solid protection in a murine model of human smallpox. J. Infect. Dis. 2009;199:39–48. doi: 10.1086/595565. [DOI] [PubMed] [Google Scholar]
- Parker S., Buller R.M. A review of experimental and natural infections of animals with monkeypox virus between 1958 and 2012. Future Virol. 2013;8:129–157. doi: 10.2217/fvl.12.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S., Nuara A., Buller R.M., Schultz D.A. Human monkeypox: an emerging zoonotic disease. Future Microbiol. 2007;2:17–34. doi: 10.2217/17460913.2.1.17. [DOI] [PubMed] [Google Scholar]
- Parker S., Handley L., Buller R.M. Therapeutic and prophylactic drugs to treat orthopoxvirus infections. Future Virol. 2008;3:595–612. doi: 10.2217/17460794.3.6.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S., Schriewer J., Oberle C., Robertson A., Lanier R., Painter G., Buller R.M. Using biomarkers to stage disease progression in a lethal mousepox model treated with CMX001. Antivir. Ther. 2008;13:863–873. [PMC free article] [PubMed] [Google Scholar]
- Parker S., Touchette E., Oberle C., Almond M., Robertson A., Trost L.C., Lampert B., Painter G., Buller R.M. Efficacy of therapeutic intervention with an oral ether–lipid analogue of cidofovir (CMX001) in a lethal mousepox model. Antiviral Res. 2008;77:39–49. doi: 10.1016/j.antiviral.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S., Siddiqui A.M., Oberle C., Hembrador E., Lanier R., Painter G., Robertson A., Buller R.M. Mousepox in the C57BL/6 strain provides an improved model for evaluating anti-poxvirus therapies. Virology. 2009;385:11–21. doi: 10.1016/j.virol.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S., Siddiqui A.M., Painter G., Schriewer J., Buller R.M. Ectromelia virus infections of mice as a model to support the licensure of anti-orthopoxvirus therapeutics. Viruses. 2010;2:1918–1932. doi: 10.3390/v2091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulford D.J., Gates A., Bridge S.H., Robinson J.H., Ulaeto D. Differential efficacy of vaccinia virus envelope proteins administered by DNA immunisation in protection of BALB/c mice from a lethal intranasal poxvirus challenge. Vaccine. 2004;22:3358–3366. doi: 10.1016/j.vaccine.2004.02.034. [DOI] [PubMed] [Google Scholar]
- Quenelle D.C., Collins D.J., Kern E.R. Efficacy of multiple- or single-dose cidofovir against vaccinia and cowpox virus infections in mice. Antimicrob. Agents Chemother. 2003;47:3275–3280. doi: 10.1128/AAC.47.10.3275-3280.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle D.C., Collins D.J., Wan W.B., Beadle J.R., Hostetler K.Y., Kern E.R. Oral treatment of cowpox and vaccinia virus infections in mice with ether lipid esters of cidofovir. Antimicrob. Agents Chemother. 2004;48:404–412. doi: 10.1128/AAC.48.2.404-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle D.C., Buller R.M., Parker S., Keith K.A., Hruby D.E., Jordan R., Kern E.R. Efficacy of delayed treatment with ST-246 given orally against systemic orthopoxvirus infections in mice. Antimicrob. Agents Chemother. 2007;51:689–695. doi: 10.1128/AAC.00879-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle D.C., Collins D.J., Herrod B.P., Keith K.A., Trahan J., Beadle J.R., Hostetler K.Y., Kern E.R. Effect of oral treatment with hexadecyloxypropyl-[(S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine] [(S)-HPMPA] or octadecyloxyethyl-(S)-HPMPA on cowpox or vaccinia virus infections in mice. Antimicrob. Agents Chemother. 2007;51:3940–3947. doi: 10.1128/AAC.00184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenelle D.C., Prichard M.N., Keith K.A., Hruby D.E., Jordan R., Painter G.R., Robertson A., Kern E.R. Synergistic efficacy of the combination of ST-246 with CMX001 against orthopoxviruses. Antimicrob. Agents Chemother. 2007;51:4118–4124. doi: 10.1128/AAC.00762-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins S.J., Jackson R.J., Fenner F., Beaton S., Medveczky J., Ramshaw I.A., Ramsay A.J. The efficacy of cidofovir treatment of mice infected with ectromelia (mousepox) virus encoding interleukin-4. Antiviral Res. 2005;66:1–7. doi: 10.1016/j.antiviral.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Rosenthal S.R., Merchlinsky M., Kleppinger C., Goldenthal K.L. Developing new smallpox vaccines. Emerg. Infect. Dis. 2001;7:920–926. doi: 10.3201/eid0706.010602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvera, P.M., Peter, E., Bassler, J., Gong, G., Lin, S., Hebblewaite, D., 2009. Impact of concurrent ST-246 administration on ACAM2000 vaccine efficacy in cynomolgus macaques. In: 7th ASM Biodefense Emerging Dis. Res. Meet.
- Smee D.F., Bailey K.W., Wong M.H., Sidwell R.W. Effects of cidofovir on the pathogenesis of a lethal vaccinia virus respiratory infection in mice. Antiviral Res. 2001;52:55–62. doi: 10.1016/s0166-3542(01)00159-0. [DOI] [PubMed] [Google Scholar]
- Smee D.F., Sidwell R.W., Kefauver D., Bray M., Huggins J.W. Characterization of wild-type and cidofovir-resistant strains of camelpox, cowpox, monkeypox, and vaccinia viruses. Antimicrob. Agents Chemother. 2002;46:1329–1335. doi: 10.1128/AAC.46.5.1329-1335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smee D.F., Bailey K.W., Wong M.H., Wandersee M.K., Sidwell R.W. Topical cidofovir is more effective than is parenteral therapy for treatment of progressive vaccinia in immunocompromised mice. J. Infect. Dis. 2004;190:1132–1139. doi: 10.1086/422696. [DOI] [PubMed] [Google Scholar]
- Smith S.K., Self J., Weiss S., Carroll D., Braden Z., Regnery R.L., Davidson W., Jordan R., Hruby D.E., Damon I.K. Effective antiviral treatment of systemic orthopoxvirus disease: ST-246 treatment of prairie dogs infected with monkeypox virus. J. Virol. 2011;85:9176–9187. doi: 10.1128/JVI.02173-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder J.T., Belyakov I.M., Dzutsev A., Lemonnier F., Berzofsky J.A. Protection against lethal vaccinia virus challenge in HLA-A2 transgenic mice by immunization with a single CD8+ T-cell peptide epitope of vaccinia and variola viruses. J. Virol. 2004;78:7052–7060. doi: 10.1128/JVI.78.13.7052-7060.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabenow J., Buller R.M., Schriewer J., West C., Sagartz J.E., Parker S. A mouse model of lethal infection for evaluating prophylactics and therapeutics against Monkeypox virus. J. Virol. 2010;84:3909–3920. doi: 10.1128/JVI.02012-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmar J., Arndtz N., Eckl K.M., Thomsen T., Petzold B., Mateo L., Schlereth B., Handley A., King L., Hulsemann V., Tzatzaris M., Merkl K., Wulff N., Chaplin P. Safety and immunogenicity of IMVAMUNE, a promising candidate as a third generation smallpox vaccine. Vaccine. 2006;24:2065–2070. doi: 10.1016/j.vaccine.2005.11.022. [DOI] [PubMed] [Google Scholar]
- Wallace G.D., Buller R.M. Kinetics of ectromelia virus (mousepox) transmission and clinical response in C57BL/6j, BALB/cByj and AKR/J inbred mice. Lab. Anim. Sci. 1985;35:41–46. [PubMed] [Google Scholar]
- Wei H., Huang D., Fortman J., Wang R., Shao L., Chen Z.W. Coadministration of cidofovir and smallpox vaccine reduced vaccination side effects but interfered with vaccine-elicited immune responses and immunity to monkeypox. J. Virol. 2009;83:1115–1125. doi: 10.1128/JVI.00984-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G., Pevear D.C., Davies M.H., Collett M.S., Bailey T., Rippen S., Barone L., Burns C., Rhodes G., Tohan S., Huggins J.W., Baker R.O., Buller R.L., Touchette E., Waller K., Schriewer J., Neyts J., DeClercq E., Jones K., Hruby D., Jordan R. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J. Virol. 2005;79:13139–13149. doi: 10.1128/JVI.79.20.13139-13149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
BCV and CDV facilitate lesion healing following vaccination with Dryvax in mice. A-strain mice were vaccinated at the base of the tail with 2500 PFU or 250 PFU of Dryvax. Mice were treated with either CDV or BCV and the vaccination lesions were photographed daily. Blinded photographs were randomized and qualitatively scored on a scale of 0–4 (0 = no lesions, 4 = high levels of erythema, edema, and eschar formation for severity of the lesion. Photographs of lesions taken on days 7 and 9 post vaccination are shown. Representative experiment of N = 2 with 5 mice/group.