

Abstract
The COVID‐19 pandemic has represented an unprecedented challenge for the humanity, and scientists around the world provided a huge effort to elucidate critical aspects in the fight against the pathogen, useful in designing public health strategies, vaccines and therapeutic approaches. One of the first pieces of evidence characterizing the SARS‐CoV‐2 infection has been its breadth of clinical presentation, ranging from asymptomatic to severe/deadly disease, and the indication of the key role played by the immune response in influencing disease severity. This review is aimed at summarizing what the SARS‐CoV‐2 infection taught us about the immune response, highlighting its features of a double‐edged sword mediating both protective and pathogenic processes. We will discuss the protective role of soluble and cellular innate immunity and the detrimental power of a hyper‐inflammation‐shaped immune response, resulting in tissue injury and immunothrombotic events. We will review the importance of B‐ and T‐cell immunity in reducing the clinical severity and their ability to cross‐recognize viral variants.
Keywords: COVID‐19, cross‐immunity, cytokine storm, immunopathogenesis, protective immunity
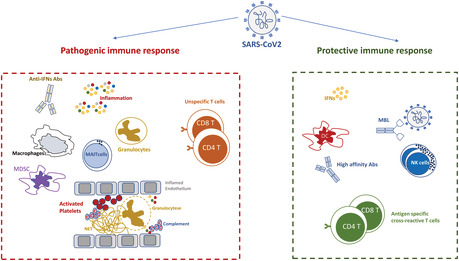
The immune response during SARS‐CoV2 infection represents a double‐edged sword. An unbalanced innate immunity induced a severe inflammation, leading to suppressor cell expansion, platelets activation, vascular damage, specific immune response paralysis and massive activation of unspecific T cells. In contrast, effective innate immunity prevents the amplification of the inflammatory response and allows the differentiation of antibody‐producing plasmacells, memory B cells, effector and memory cross‐reactive T cells, overall mediating a highly efficient, protective immune response. This fine balance between harmful and protective immunity has strongly accelerated the development of new therapeutical immune‐mediated strategies.
INTRODUCTION
SARS‐CoV‐2 infection is characterized by multifaced immunological responses which strongly shapes the disease progression and severity. The activation kinetics, the functional profile, the balance between inflammatory and regulatory signals and the coordination among all the immunological players represents essential elements to orchestrate a protective rather than harmful response. In this scenario, the first processes occurring in the lung seems to have the potential to drive all the subsequent events.
THE INNATE IMMUNITY PLAYS A KEY ROLE IN EARLY PROTECTION
In asymptomatic or pauci‐symptomatic patients infected by SARS‐CoV‐2, the quick control of the infection is probably largely mediated by the innate immune response [1]. Innate immune response includes both soluble and cellular arms, collaborating to fight the infection as first line of defence. The soluble humoral arm consists of pattern recognition molecules (PRMs), including collectins (e.g., mannose‐binding lectin, MBL), ficolins (e.g., pentraxin 3, PTX3) and C1q. These molecules are considered ancestors of antibodies (ante‐antibodies), as they recognize microbes by less specific mechanisms, but mediate virus inhibition through mechanisms shared with antibodies, including agglutination, neutralization, activation of the complement cascade and opsonization‐mediated phagocytosis [2]. In COVID‐19, PTX3 recognizes the viral nucleocapsid protein and its plasma concentrations represents a strong and independent factor predicting the risk of death in individuals with COVID‐19 [3]. Moreover, collectins are able to bind the envelope glycoproteins of several viruses, resulting in opsonization, agglutination, inhibition of viral fusion and entry or complement activation, generally leading to inhibition of infection [4]. A protective role of MBL towards severe COVID‐19 has been recently proposed [5] (Figure 1a); MBL binds trimeric Spike protein in a glycan‐dependent manner and inhibits SARS‐CoV‐2 infection by triggering the lectin pathway of complement activation. MBL can recognize trimeric Spike from different SARS‐CoV‐2 variants of concern (VOC), this finding highlighting its wide binding capability. Of note, MBL2 genetic variants impacting on MBL protein abundance are associated with COVID‐19 severity [5]. These recent findings can have potentially important translational implications, as MBL has been safely administered to individuals with cystic fibrosis and chronic lung infections [6, 7].
FIGURE 1.
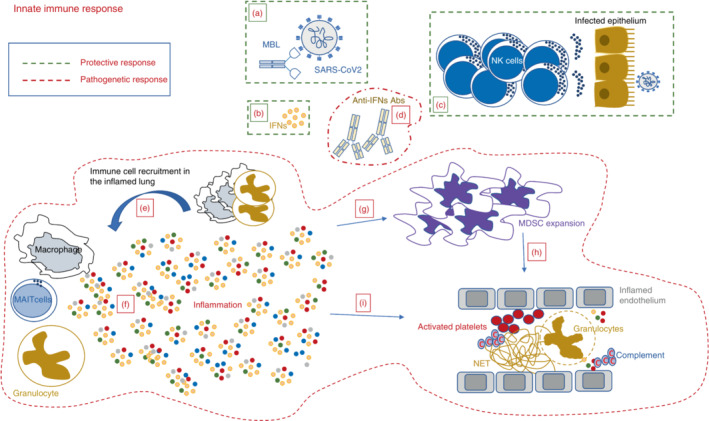
Protective and pathogenic pathways of the innate immune response during SARS‐CoV‐2 infection. Protective innate response: surrounded by a green hatched line, includes both soluble (a and b) and cellular components (d). (a) Mannose‐binding lectin (MBL) binds to trimeric Spike, thus inhibiting viral infection; (b) Early activation of type‐I IFN response exerts antiviral effect and prevents severe clinical manifestations; (d) Early expansion of cytotoxic NK cells contributes to viral clearance and associates with mild clinical presentation. Pathogenic innate response: surrounded by a red hatched line, includes both soluble (c, e and f) and cellular components (g–i). (c) Antibody anti‐type‐I IFN block the IFN antiviral activity and associates with a severe clinical outcome; (e) Lung resident myeloid and MAIT cells produce huge amounts of inflammatory cytokines and chemokines that, in turn, massively recruit macrophages and neutrophils from the peripheral blood, thus exacerbating the cytokine storm; (f) The huge amount of inflammatory mediators reaches the bloodstream, inducing a cascade of events contributing to the host injury; (g) The inflammatory storm induces a massive expansion of myeloid derived suppressor cells (MDSC) that can reduce the inflammation but strongly impair the antigen‐specific T‐cell response; (h) Expanded MDSC participate to platelet activation by reducing the plasmatic arginine. (i) The inflammatory storm triggers several detrimental pathways as complement activation, hypercoagulation, endothelial damage, arterial and venous embolism with NET formation
Toll‐Like receptors (TLRs) are innate immune receptors recognizing pathogen‐associated molecular patterns (PAMPs) and triggering the production of type I interferons and pro‐inflammatory cytokines to fight the infections. The early activation of the innate immune system via TLRs by SARS‐CoV‐2 may contribute to the viral clearance [8]. Nevertheless, a dysregulated TLR responses with an over activation of their intracellular pathways may lead to overproduction of inflammatory molecules inducing tissue damage [9].
SARS‐CoV‐2 is a poor inducer of type‐I IFNs both in vitro and in vivo, and several virus‐encoded proteins are known to target proteins of type‐I IFN pathway, thus inhibiting both IFN production [10] and response [11, 12, 13, 14]. As reported also for other coronavirus infection [15], the kinetics and the amount of type‐I IFN response strongly contribute to the definition of disease severity, as early and potent IFN production is associated with reduced viral titres and mild clinical disease (Figure 1b). Accordingly, several studies showed that patients with mild to moderate COVID‐19 show an effective type‐I IFN response both in the lung and in the peripheral blood. In contrast, patients with severe/critical COVID‐19 manifestations showed a suppressed expression of IFNs and a parallel increase in tissue and systemic inflammation [16, 17].
A pathogenic role of auto‐antibodies (auto‐Abs) directed against IFNs has been recently proposed to play a major role in severe COVID‐19 (Figure 1c). These autoantibodies were rarely found in healthy donors (<0.3%) and in SARS‐CoV‐2‐infected asymptomatic patients [18]. By contrast, at least 10%–15% of individuals with severe COVID‐19 showed auto‐Abs neutralizing type‐I IFNs that were already present before the infection. The prevalence of auto‐Abs increases significantly with age and in individuals who died from COVID‐19 [18]. The functional role of auto‐Abs neutralizing type‐I IFNs has been recently described, reporting the impaired activation of interferon‐stimulated genes (ISGs) in myeloid cells from critical cases, including those producing anti‐IFN‐I autoantibodies [19]. The failure of the type‐I ISG response early in the disease course was associated with autoantibodies to type‐1 IFNs in critical patients. These findings provide the biological basis for possible tailored IFN therapy; as a matter of fact, several studies, aimed at defining the effectiveness of IFN therapy in COVID‐19 patients, highlight the early time of infection as the critical window for IFN administration [20].
Innate immune cells include a broad range of myeloid and lymphoid cell types (e.g., neutrophils, monocytes, NK, dendritic cells, ILC, etc). Common to the majority of these cell types is that they originate from the haematopoietic system, lack somatically recombined antigen‐receptors and conventional immunological memory, and exert prompt antimicrobial or tissue‐protective functions [21]. A protective role of the early innate immune response was also confirmed by the analysis of NK cells (Figure 1d). A correlation between a rapid resolution of SARS‐CoV‐2 infection and high numbers of circulating NK cells at the beginning of the symptoms has been observed. Accordingly, mild patients show a significant and early increase of perforin and granzyme B, which is an early sign of NK‐cell activation that is observed also in other viral infections [22]. Moreover, NK cells from patients with mild COVID‐19 showed an increased production of interferon‐γ (IFN‐γ), whereas NK cells from patients with severe COVID‐19 produced only low levels of IFN‐γ and TNF‐α. In contrast, severe COVID‐19 was characterized by a drastic decrease of both CD56dim and CD56bright NK cells [23, 24] as well as by a reduction of their cytotoxic profile [25, 26], as a consequence of the significant reduction of T‐bet transcription factor [26]. NK cells from patients with mild COVID‐19 express high level of T‐bet, efficiently kill SARS‐CoV‐2 infected cells and produce antiviral ctyokines [24]. A main role of transforming growth factor‐β (TGF‐β) in reducing the antiviral activity of NK cells has been recently reported in severe COVID‐19 [24]. The level of TGFβ increases in severe COVID‐19 patients to curtail excessive inflammatory response, but, on the other hands, impairs the NK‐mediated antiviral response. TGF‐β completely abrogates the NK‐cell‐mediated control of SARS‐CoV‐2 replication in vitro, and is able to block NK function (cytotoxicity and cytokine production) in vivo.
Dendritic cells (DC) are professional Antigen Presenting Cells (APC) that play a key role in activating both innate and adaptive immune responses. They recognize viruses by pattern recognition receptors and activate intracellular pathways able to induce antiviral molecule release. Moreover, the antigen processing and presentation to T cells allows the induction of the adaptive specific T‐cell response. During severe SARS‐CoV‐2 infection, the percentage of DC subsets decrease [27], and their functionality, in terms of IFN‐α production and antigen presentation potential, is impaired [27, 28, 29].
THE HARMFUL POTENTIAL OF THE IMMUNE RESPONSE
The kinetic and the balance of the immune response to SARS‐CoV‐2 infection is strictly related to the definition of clinical severity. In patients with severe COVID‐19 several immunopathological processes take over, contributing to tissue damage and immune paralysis. Among these, the early and uncontrolled cytokine storm, the increased frequency of neutrophils and suppressor cells, the lymphocyte depletion and their activation and functional exhaustion represent main players.
Cytokine storm (Figure 1e,f) : Several studies have demonstrated that the large majority of pathogenic mechanisms induced by SARS‐CoV‐2 infection is driven by the host response rather than by the direct damage caused by the virus. The first steps of the host response seem to be crucial to avoid a cascade of domino events leading to uncontrolled hyper‐inflammation and tissue damage. Several clinical studies showed that the early impaired production of type‐I IFNs observed in critical cases is paralleled by an excessive production of inflammatory cytokines [30, 31]. This ‘cytokines storm’ is not new in the history of host/pathogen interactions, as it was described to occur during other highly pathogenic viral infections, such as Ebola, and is associated with tissue damage and immune paralysis [32]. A huge uncontrolled cytokine release can be the results of the synergistic interactions among several pathways including TLRs, nucleotide‐binding oligomerization domain‐like receptors (NLRs), resulting in innate immune cells hyper activation [33].
In severe COVID‐19 patients, the systemic cytokine profile was similar to the one observed in cytokine release syndromes, such as macrophage activation syndrome, with increased production of cytokines including IL‐1β, IL‐2, IL‐4, IL‐6, IL‐7, IL‐8, IL‐9, IL‐10, IL‐18, G‐CSF, IP‐10, MCP‐1, MCP‐3, MIP‐1A, IFN‐γ, and TNF‐α, IFN‐β, IFN‐γ, and TNF‐α and of inflammatory chemokines including CCL2, CCL3, CCL7, CXCL9 and CXCL10 [34, 35, 36, 37]. This inflammatory environment represents a hallmark of COVID‐19 progression, as systemic levels of IL‐1β, IL‐6 and IL‐8 at the time of hospitalization are strong and independent predictors of patient survival [31]. The huge release of cytokines and chemokines in the infected lungs massively recruits other inflammatory myeloid cells, thus exacerbating the tissue damage and contributing to the development of acute respiratory distress syndrome (ARDS) [38]. To neutralize the overproduced inflammatory factors, therapeutic monoclonal antibodies (mAbs) that target cytokines have been introduced and showed improved outcomes and lower rates of mortality [39]. Of note, the persistence of systemic inflammation (e.g., IL‐1β, IL‐6 and TNF‐a) after viral clearance has been associated with the clinical manifestations of long COVID syndrome [40, 41], confirming a main role of dysregulated immunity in sustaining clinical symptoms.
Innate immune cells: Mucosal‐associated invariant T (MAIT) cells are innate‐like T cells involved in mucosal immunity and protection against viral infections. Nevertheless, in COVID‐19 patients, several evidences suggest that they may participate in the deleterious inflammation affecting severe, long‐term hospitalized patients with COVID‐19 [42]. MAIT cell frequency is strongly reduced and their activation phenotype has been associated with a broad activation of adaptive T cells and with elevated levels of cytokines and chemokines [43].
Changes in the myeloid compartment have been observed in blood and lung of patients with COVID‐19. In severe COVID‐19, the number of classical monocytes decrease, while intermediate and non‐classical pro‐inflammatory monocytes increase [44]. Similarly, broncho‐alveolar hyperinflammatory monocytes were observed in severe COVID‐19 patients [45, 46], indicating their involvement in tissue damage. The ratio of pro‐ to anti‐inflammatory macrophages at the sites of injury strongly influences the course of the pathological process in lungs, and this balance represents a potential therapeutic target [47].
Expansion of suppressor cells (Figure 1g,h) : The inflamed environment (IL‐1β, IL‐6, IL‐8 and TNF‐α) is correlated with a massive expansion of myeloid‐derived suppressor cells (MDSCs), that are immature or mature myeloid cells endowed with regulatory functions able to inhibit T‐cell functions [48]. The highest frequency of MDSCs has been observed in fatal cases of COVID‐19 [49, 50], where they account for as much as 90% of circulating PBMC [51], suggesting their detrimental role. Accordingly, in severe/critical patients, expanded MDSCs inhibit antigen‐specific T‐cell response (both proliferation and cytokine production capabilities) through TGF‐β‐ and inducible nitric oxide synthase (iNOS)‐mediated mechanisms, possibly reducing the overall antiviral immunity. Moreover, MDSC expansion was paralleled by a high Arginase‐1 activity and plasmatic L‐arginine depletion [52, 53, 54]. The deprivation of L‐arginine induces the decrease of CD3ζ, a key molecule in the T‐cell receptor (TCR) signalling, thus inhibiting T‐cell activity. Expanded MDSCs also increase platelet activation, highlighting a new interplay between suppressor cells and platelet function. Indeed, MDSC frequency directly correlates with platelet activation in COVID‐19, and purified MDSCs from COVID‐19 patients were able to induce platelet activation by reducing L‐arginine [54].
Altogether, these data suggest that MDSCs may contribute to the SARS‐CoV‐2‐induced immunopathogenesis. Accordingly, the frequency of MDSCs at time of hospital admission has been found to be an independent factor associated with the fatal outcome of the disease [50], thus representing a possible biomarker and therapeutic target in COVID‐19 pneumonia and associated disease [55].
Vascular damage (Figure 1i) : SARS‐CoV‐2 infection is characterized by a high prevalence of thrombotic complications. Consolidated clinical evidences demonstrate a high incidence (up to 30%) of venous thromboembolism (VTE) among COVID‐19 patients in the intensive care units [56, 57] which is confirmed also by a large COVID‐19 autopsy study reporting pulmonary embolism (PE) in 21% of the patients and deep vein thrombosis (DVT) in 40% of them [58]. Overall, the incidence of VTE in COVID‐19 is at least threefold higher than that reported with other viral respiratory infections [59].
The complement system is part of the innate immunity, being beneficial in the initial response to the virus infections; however, its sustained and long‐lasting activation may contribute to uncontrolled response and tissue damage. SARS‐CoV‐2 triggers direct or indirect activation of extracellular and/or intracellular complement pathways that can lead to hyperinflammation and thrombosis [60]. These data suggest that the hyperactivation of the complement and coagulation systems represents a main player of the clinical syndrome of COVID‐19, opening the door to new possible therapeutic approach focused on its inhibition [61]. Indeed, therapeutic antibodies directed against complement factors showed some efficacy in the treatment of severe COVID‐19 [62].
During SARS‐CoV‐2 infection, following the initial localized thrombo‐inflammatory response, systemic hypercoagulability becomes prominent. Although the exact mechanisms of hypercoagulability occurring during SARS‐CoV‐2 infection remains to be fully elucidated, the damage to the vascular endothelium could be induced by both direct viral damage and severe inflammatory environment. Internalization of SARS‐CoV‐2 receptor, angiotensin converting enzyme 2 (ACE2) on endothelial cells, increases angiotensin II levels, thus causing vasoconstriction and release of inflammatory and pro‐thrombotic substances, including von Willebrand factor (VWF), P‐selectin, factor VIII and Angiopoietin 2. These factors are all involved in the pathogenesis of thrombo‐inflammation in COVID‐19, therefore contributing to ARDS and other organ dysfunctions. The Spike protein of SARS‐CoV‐2 can cause direct damage of endothelial cells, manifested by the impaired mitochondrial function and endothelial nitric oxide synthase (eNOS) activity, as well as downregulation of ACE2, which may further aggravate endothelial dysfunction [63, 64]. On the other hand, the cytokine storm destroys endothelial cells by inducing the release of plasminogen activator inhibitor‐1 PAI‐1 [65], promoting degradation of the endothelial barrier [66] and by downregulating the expression of Kruppel Like Factor 2 (KLF2) to induce adhesion and infiltration of monocytes/macrophages [67]. Moreover, a conserved mechanism of host defence known as immunothrombosis occurs, leading to intravascular clot formation in small and larger vessels [68].
SARS‐CoV‐2 infection has been characterized by increased neutrophil number, which is associated with disease severity and clinical prognosis [69, 70]. In post‐mortem examinations of COVID‐19 patients, neutrophil extravasation has been observed in lung, myocardium and liver [71]. As a result, neutrophils and their effector mechanisms, such as degranulation, oxidative burst and NETosis, are important mediators of COVID‐19 immunopathogenesis. The dynamic changes in the number, percentages of neutrophils and neutrophil granular proteins represent markers of disease severity [72] and it has been suggested that exacerbated inflammation induced by neutrophils may be controlled by an expansion of eosinophils [73]. NETosis is a special form of neutrophil‐programmed cell death, characterized by the extrusion of DNA, histones and antimicrobial proteins known as neutrophil extracellular traps (NETs). NETs have antimicrobial function and are induced by a range of microbial stimuli and by proinflammatory mediators [74]. It has been shown that SARS‐CoV‐2 can directly induce NETosis and NET release by neutrophils depending on ACE2, serine protease, virus replication and peptidyl arginine deiminase‐4 (PAD‐4) [75]. Furthermore, soluble factors in the plasma of COVID‐19 patients, such as IL‐8, IL‐1b, CXCL8 and RANTES, also mediate NET formation [76, 77]. Another route of SARS‐CoV‐2‐induced NET production is platelet activation that can enhance this process by interacting with neutrophils through toll‐like receptor 4 (TLR4), and platelet factor 4 (PF4) [76, 78], causing coagulopathy and thrombosis. Moreover, cell free‐DNA from human neutrophils triggers thrombin generation by binding to factor XII [79]. In addition, complement stimulates tissue factor production and interacts with the platelet/NETs/thrombin axis, contributing to COVID‐19 immunothrombosis. Accordingly, neutrophil produced NETs are major elements of vascular thrombi in COVID‐19 patients, confirming the role of NETs in the pathogenesis of COVID‐19 [80]. Endothelial dysfunction, inflammation, immunothrombosis and decreased antithrombotic molecules (e.g., nitric oxid and reactive oxygen species) are intricately linked and are all involved in the increased risk of thromboembolism events.
Unbalanced adaptive T‐cell response (Figure 2D–G) : Adaptive T‐cell responses develop early after infection and correlate with protection, as a higher specific T‐cell response was associated to mild disease course [81]. T cells recognize several viral proteins with the largest proportion of SARS‐CoV‐2 specific T cells recognizing peptides from proteins N, S and ORF‐1 in patients with a mild course [81, 82]. Highly functional, virus‐specific cellular immune response was also observed in asymptomatic patients [83] and in the absence of seroconversion [84]. The SARS‐CoV‐2‐specific T‐cell memory in COVID‐19 convalescent patients persisted at least 10 months with successful development of stem cell‐like memory T cells [85].
FIGURE 2.
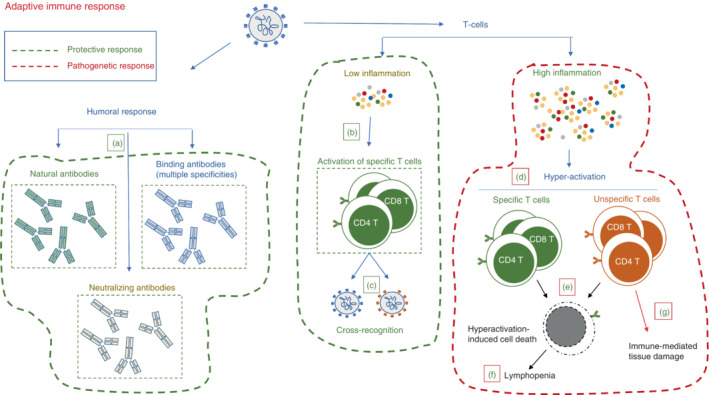
Protective and pathogenic pathways of the T‐cell response during SARS‐CoV‐2 infection. Protective T‐cell response: surrounded by a green hatched line. (a) A low inflammatory environment allows a well‐balanced differentiation of antigen‐specific T cells, contributing to viral clearance and to antibody production; (b) Antigen‐specific T cells induced by natural infection or vaccination have a broad range of viral strain recognition, including different VOCs. Pathogenic T‐cell response: surrounded by a red hatched line, includes cellular components (d–g). (c) A huge inflammatory environment induces hyperactivation of both specific and unspecific T cells; (d) The strong production of inflammatory cytokines leads to overexpression of apoptotic processes inducing lymphocyte death; (e) Lymphocyte death is associated with lymphopenia, one the main markers of severe/fatal COVID‐19; (f) Bystander‐activated unspecific T cells contribute to tissue damage.
In severe COVID‐19, a completely subverted immune response has been described, mainly driven by the strong and persistent inflammatory storm (Figure 2d,e). Indeed, the huge inflammatory environment triggers a deep dysregulation of the activation and differentiation of both innate and adaptive immune response with excessive T‐cell activation and high expression of T‐cell inhibitory and senescence‐associated molecules (PD‐1, TIM3, LAG3, CTLA4, NKG2A, CD39 and CD57) [86, 87]. This immune perturbation was strongly correlated with a more severe clinical course [88]. Single cell RNA seq confirmed excessively activated CD8 T cells in severe disease, which displayed exhausted phenotypes and diminished function of antigen recognition [86, 89]. A similar dysregulated profile was described also in other infections, such as Ebola [90], and was associated to ‘immune paralysis’, persisting overtime [91].
In severe COVID‐19, more than 50% of circulating CD8 T cells have been shown to express activation markers [86, 92], highlighting that unspecific T‐cell activation occurs and can contribute to tissue damage. A bystander activation of virus‐unspecific T cells has been also shown in other viral infections, and seems to be mainly driven by cytokine environment (e.g., type‐I IFN, IL‐18 and IL‐15) and toll‐like receptor agonist, contributing to host injury [32, 93]. Among virus‐specific T cells, in severe COVID‐19 patients a significant skewing of T‐cell activation towards TH17 functional phenotype has been shown, and it can participate in recruiting and activating neutrophils in the lung, thus contributing to the tissue damage [94] (Figure 2g).
A dysregulation of adaptive immune response has been also associated to prolonged symptom duration after viral clearance (e.g., long COVID syndrome), linking a dysregulated immune response to persistent clinical sequelae. Individuals with long COVID syndrome showed an increase overtime of the IgG avidity to SARS‐CoV‐2 spike protein and of the antigen‐specific T‐cell response magnitudes in CD4 and circulating T follicular helper cell populations, suggesting the persistence of viral antigens [95]. Moreover, patients suffering of lung sequelae after SARS‐CoV‐2 pneumonia were characterized by a persistent dysregulated respiratory CD8+ T‐cell response [96].
Severe patients are also characterized by a dramatic lymphopenia (Figure 2f), which is correlated with the plasmatic level of inflammatory mediators (IL‐6, TNF‐a) [97]. The dramatic decrease of circulating T cells can be due to several mechanisms, including their massive recruitment into inflamed tissues and the use of steroid treatment; in patients infected with SARS‐CoV and SARS‐CoV‐2 a significant T‐cell depletion from the secondary lymphoid organs has been documented [98]. The potential mechanisms responsible for T‐cell depletion are still poorly understood, but inflammation‐driven cell death processes and cell cycling arrest can play a major role. Serum levels of TNF, IL‐6 and IL‐10 negatively correlated with T‐cell numbers [97], and higher expression of p53 and p21 have been shown in COVID‐19 patients as compared to healthy controls [99]. Accordingly, higher levels of CD95 expression on T cells, as well as of sFasL in the plasma of COVID‐19 patients were associated with higher levels of caspase activation and with the propensity of T cells to die; in addition, they correlated positively with the extent of T‐cell lymphopenia and with higher levels of CXCL10, both markers of disease severity [100]. A role of apoptotic mechanisms in the pathogenesis of COVID‐19 has been finally demonstrated in autoptic tissues, where high levels of lymphocyte apoptosis in the spleens and lymph nodes was associated with increased expression of the FAS death receptor, suggesting that activation‐induced cell death (AICD) may be involved in T‐cell lymphopenia [98].
Overall, in COVID‐19 patients, a dramatic inflammatory storm triggers concomitant aspects of immune inhibition, including unbalanced activation, exhaustion and depletion of T lymphocytes, resulting in the paradox that immune response, instead of being protective, contributes to the tissue damage and disease progression. Co‐existing co‐morbidities, such as cardiovascular disease, cancer, diabetes, chronic renal failure, pulmonary fibrosis and others, has been associated with more severe outcomes through the modulation of host–viral interactions and immune responses. The specific mechanisms are not well defined, but the low grade of persistent inflammation characterizing several comorbidities may play a central role in worsening the COVID‐19 outcome [101].
PROTECTIVE B‐CELL RESPONSE
Because of their high specificity and affinity, antibodies precisely and effectively neutralize and eliminate viruses (Figure 2a). The generation of antibodies specific for a virus never encountered before, such as SARS‐CoV‐2, is a lengthy process occurring in the germinal centres. Here, activated B cells proliferate and refine their antibodies by introducing somatic mutations in the antigen‐binding part (VH region). Mutated antibodies are then selected for their increased affinity to the antigen. Two weeks are necessary for the completion of the germinal centre reaction and the release of its products: memory B cells and plasmablasts expressing a variety of specific antibodies, including those with neutralizing ability [102]. Because of the long time required for the germinal centre reaction, in the initial phase of the infection, B cells participate in the immune defence only through pre‐formed natural or cross‐reactive antibodies. These low‐affinity antibodies act in concert with the innate immune system to limit the infection [103].
The extraordinary activation of the innate immune system is responsible for severe collateral damages in COVID‐19 (Figure 3b). The cytokines produced in response to the virus also impair the formation of germinal centres, thus jeopardizing the function of the adaptive B‐cell response in severe and fatal COVID‐19 [104]. Extra‐follicular B‐cell activation leads to the generation of B cells, able to produce neutralizing antibodies, but also associated to autoimmunity and inflammation [105]. B‐cell depletion and loss of memory B cells has been reported in patients with severe COVID‐19 [106].
FIGURE 3.
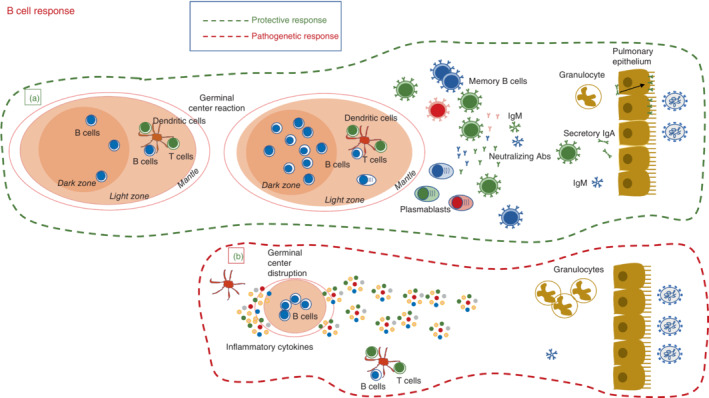
Protective and pathogenic pathways of the adaptive B‐cell response during SARS‐CoV‐2 infection. Protective B‐cell response: surrounded by a green hatched line. (a) In mild COVID‐19 (bottom), the germinal centre reaction generates memory B cells with different affinities for the virus, short‐lived plasmablasts (PB), and long‐lived plasma cells that home to the bone marrow. A fraction of the memory B cells migrate to the site of viral invasion and become resident memory B cells secreting IgA antibodies for local protection. A similar mechanism of protection is generated by vaccination. In breakthrough infections, vaccine‐induced memory B cells rapidly migrate to mucosal sites for local defence. Pathogenic B‐cell response: surrounded by a red hatched line. (b) The increase of inflammatory cytokines disrupts the architecture of the germinal centres by dislocating follicular T cells. The B‐cell response occurs at extrafollicular sites, where B cell remodelling and antigen‐based selection is impaired. At mucosal sites, natural/cross‐reactive antibodies try to limit viral invasion.
The B‐cell response is more physiological in patients with moderate and mild disease (Figure 3a). Serum antibodies become detectable around two weeks after the diagnosis, but memory B cells persist for months, as expected by the kinetics and persistence of the immune response to a novel pathogen [107, 108]. While specific antibodies rapidly decline in the serum of patients recovered from COVID‐19, memory B cells continue to increase in numbers and improve their affinity by acquiring and selecting additional somatic mutations [109]. The antigen persistence in the germinal centres may explain the continuous remodelling of memory B cells. The decline of serum antibodies, occurring a few weeks after the infection, is due to the death of short‐lived plasma blasts that are generated by the full‐blown germinal centre response. Only a few short‐lived plasma blasts become long‐lived plasma cells with the function of maintaining a persistent antibody level that limits the initial infection; meanwhile, memory B cells are activated and produce high levels of antibodies to prevent a new disease caused by an already experienced pathogen [110]. Long‐lived plasma cells are detectable in the bone marrow of individuals who had COVID‐19 [111]. The question of why, if the infection generates all the elements of immunological memory (memory B cells, long‐lived plasma cells and high‐affinity antibodies), reinfection is possible has generated considerable insecurity. The emergence of new variants weakens the effect of the stringent selection of high affinity antibodies against the previously encountered viruses, as demonstrated by the inefficacy of certain Wuhan‐specific monoclonal antibodies against the VOC Omicron. Luckily, the response of B cells is not monoclonal and memory B cells are rapidly able to modify and adapt their antibodies [112]. The solidity and duration of disease‐induced immune memory is demonstrated by observation that previously infected individuals are protected from re‐infection during a follow‐up of 20 months, without evidence of waning immunity [113]. Although reinfection with the same SARS‐CoV‐2 strain is a rare event, emerging variants may infect COVID‐19 convalescent subjects and cause a disease of variable severity [114, 115].
The nasopharyngeal mucosa is the site of entry of SARS‐CoV‐2, and mucosal immunity has been demonstrated during the illness and in convalescence [116, 117]. At mucosal sites, the most abundant antibody is dimeric IgA that is locally produced in response to infection. Dimeric IgA has an enhanced neutralization capacity compared to monomeric antibodies and plays an important role in local protection [118]. In contrast to natural infection, parenterally administered vaccines do not generate mucosal immunity [92]. The establishment of resident memory B cells in the lung requires local antigen encounter and serum antibodies reach mucosal sites in small amounts by transudation. Breakthrough infections in vaccinated individuals may result from lack of local protection. Immune memory established by vaccination, however, prevents severe disease by the combined action of memory T and B cells that migrate to the infection site, thus turning systemic immunity into local protection [119]. Next‐generation COVID‐19 vaccines for mucosal delivery, now in development, may be able to provide local protection against infection [120]. The combination of systemic and mucosal immunity may result in a more effective individual protection and reduce viral growth and contagion.
Despite the success of COVID‐19 vaccination efforts, there is still a need of preventive and therapeutic options for fragile populations, who may have an inadequate response to vaccination. Neutralizing mAbs therapy represents a critical tool for protecting these individuals against severe COVID‐19 and have been used successfully for this purpose [121, 122, 123].
PROTECTIVE T‐CELL RESPONSE
T cells are crucial components of the antiviral immune response. Although they are not able to prevent the infection, CD4+ T cells are needed for the generation of protective antibody responses and to support the maturation of CD8+ T cells. Moreover, T cells recognize linear determinants of viral epitopes with a broader range than antibodies, including protein regions not prone to viral mutation‐driven escape from antibodies.
T‐cell response plays a major role in the protective immune response to SARS‐CoV‐2 [124, 125, 126]. T cells recognize several viral proteins with the largest proportion of SARS‐CoV‐2 specific T cells in patients with a mild course recognizing peptides from proteins N, S and ORF‐1 [81, 82]. A greater CD8+ T‐cell response in blood and highly clonally expanded CD8+ T cells in bronchoalveolar lavage have been observed in convalescent patients who experienced mild or moderate disease as compared with severe disease [46, 127], and CD8 T cells showed partial protective immunity in the context of suboptimal antibody titres in a non‐human primate model [128]. Accordingly, an association of optimal and polyfunctional T‐cell responses with asymptomatic/mild disease following primary infection has been proposed and was supported by more recent evidence of protection conferred by SARS‐CoV‐2‐specific memory T cells on secondary exposure [83]. Of note, persons exposed to SARS‐CoV‐2 may develop virus‐specific T‐cell responses without detectable circulating antibodies, due to abortive infection, to effective and rapid viral clearance by innate immunity and T cells or to a very low level of antibodies quickly disappeared [126, 129].
Most convalescent patients display a durable SARSCoV‐2‐specific memory T‐cell response after COVID‐19, persisting for several months regardless of the severity of COVID‐19. CD4 and CD8 T cells displayed specificity both to structural and non‐structural proteins and showed polyfunctional properties [108, 130, 131]. Of note, COVID‐19 induces the development of antigen‐specific stem cell‐like memory cells, characterized by a good self‐renewal capacity and multipotency that can contribute to the maintenance of protection overtime [85]. Recently, studies on lung and nasal tissue from SARS‐CoV‐2 convalescent individuals have identified tissue‐resident memory T cells with functional reactivity against several SARSCoV‐2 proteins that persist for at least 2–10 months after infection [132, 133]. Both circulating and tissue resident SARS‐Cov‐2 specific T cells can participate in providing protection against severe disease.
A key feature of SARS‐CoV‐2 specific T cells is their cross‐reactivity, that is, their ability to recognize antigens from different viral variants that instead can escape the humoral neutralization response (Figure 2c). The ability of T cells to recognize several viral variants lies in their ability to recognize short linear peptides presented in the context of MHC molecules, a mechanism which is less affected by viral point mutations. Moreover, differently from neutralizing antibody which are exclusively directed against RBD domain, effective T cells can recognize both internal and external viral proteins. As an example, the multiple mutation profile in the Spike protein of the VOC Omicron significantly reduced the neutralization titre of vaccine‐ or infection‐induced antibodies [134, 135, 136, 137]. By contrast, the extent of Omicron cross‐reactive T cells activity was similar for Beta, Delta and Omicron variants, indicating minimal escape at the T‐cell level [138, 139]. Several reports confirmed that natural infection and vaccination are able to induce effective cross‐reactive CD4 and CD8 T cells, characterized by an extensive immune coverage against the Omicron variant [138, 139, 140]. The median relative frequencies of SARS‐CoV‐2 Spike‐specific CD4+ T cells that cross‐recognized Omicron in previously infected or vaccinated individuals were 84% and 91%, respectively, and the corresponding median relative frequencies for SARS‐CoV‐2 Spike‐specific CD8+ T cells were 70% and 92%, respectively. On the other hand, Omicron infections were able to induce T‐cell responses to ancestral Spike, Nucleocapsid and Membrane proteins comparable to those observed in patients hospitalized in previous pandemic waves dominated by the ancestral, Beta or Delta variants [140]. More detailed analysis revealed that SARS‐CoV‐2 Spike‐reactive CD4+ and CD8+ T cells were functionally and phenotypically similar in response to the ancestral strain or Omicron variant and showed a polyfunctional profile [139, 141].
The broad recognition capability of T cells has also been described in other viral infections; for instance, a broad T‐cell reactivity against different influenza viral strains has been widely described and represents a hallmark of influenza virus infection. Both in mice model and in humans, strong influenza‐specific CD8+ T cells correlates with lower viral load and decreased disease severity [142]. Furthermore, CD8+ T cells primed with circulating seasonal influenza A virus (IAV) strains cross‐react with peptides derived from seasonal variants, H1N1pdm, as well as avian H7N9 and H5N1 IAV strains [143].
Overall, the ability of T cells induced by both vaccination and natural infection to recognize VOC, including Omicron, can potentially balance the lack of neutralizing antibodies in preventing or limiting severe COVID‐19.
CROSS‐REACTIVE IMMUNITY TO HUMAN COMMON COLD CORONAVIRUSES
Several recent studies have reported the existence of variable levels of SARS‐CoV‐2‐specific antibodies in unexposed individuals, mainly targeting nucleocapsid (N) and the spike subunit S2 [144, 145]. Accordingly, SARS‐CoV‐2‐reactive T cells were also detected in unexposed individuals, suggesting cross‐reactive T‐cell recognition between circulating common cold coronaviruses and SARS‐CoV‐2 [82, 146].
Whether prior immunity to endemic, human common cold coronaviruses (hCCCoVs) may impact susceptibility or clinical severity of SARS‐CoV‐2 infection still remains an unsolved question. Some evidence suggests a protective role of previous immunity to hCCCoVs, reporting that recent endemic coronavirus infection is associated with less‐severe COVID‐19 [147]. Accordingly, a higher frequency of cross‐reactive memory T cells associates with protection against SARS‐CoV‐2 infection in COVID‐19 contacts who remained PCR negative despite high exposure [126].
In contrast, other studies suggest a pathogenic role of pre‐existing immunity. Pre‐pandemic SARS‐CoV‐2 cross‐reactive antibodies can be boosted upon SARS‐CoV‐2 infection, but they are not associated with protection [148], may hinder the development of effective immunity against SARS‐CoV‐2 [149] and may correlate with disease severity [150]. Accordingly, cross‐reactive T cells display low functional avidity and only contributed a small proportion of the overall Spike‐specific CD4+ T cells in COVID‐19 convalescent individuals, suggesting their minor role to SARS‐CoV‐2 responses in vivo [151, 152, 153]. Finally, other studies suggest that cross‐reactive immunity may trigger immunopathogenesis in COVID‐19 patients, by inducing an early massive unconventional antibody responses and facilitating viral entry by antibody‐dependent enhancement (ADE) mechanisms [154].
Further studies focused on controlled challenge studies in small animal models and human adults are needed to finally determine the impact of cross‐reactive immunity on clinical outcomes in COVID‐19 patients.
CONCLUSIONS
This review focalizes on the double edged role of immune response in the delicate balance between protection and pathogenesis during SARS‐CoV‐2 infection and summarizes the role of the main immune players (Tables 1 and 2). The kinetics of activation, the strength of the single responses and the integration among innate and adaptive signals may define the overall fate of the SARS‐CoV‐2 disease severity. The extraordinary effort on COVID‐19 research allowed us to shed light on critical aspects of the immune response, defining the balance between protective and harmful response that can be useful for approaching also other viral infections. The soluble and cellular innate arms represent the main early drivers of protection, able and define the subsequent fate of disease. An ineffective innate immunity is associated to a chain of events, starting with severe inflammation and leading to a complex network of pathogenic immune‐mediated processes. The huge inflammatory response induces the expansion of suppressor cells mediating the paralysis of specific immune response and massive platelets activation. The inflammation and sustained platelet activation drives in turn the vascular damage and thrombotic complications. Finally, the cytokine storm induces the massive activation of both specific and unspecific T cells, contributing to tissue damage and lymphopenia. In contrast, effective innate immunity prevents the amplification of the inflammatory response and allows the differentiation of antibody‐producing plasma cells, memory B cells, effector and memory cross‐reactive T cells, overall mediating a highly efficient, protective immune response.
TABLE 1.
Immune response to SARS‐CoV‐2: Protective functions
| Immune players | Role | References |
|---|---|---|
| MBL, PTX3 | Antiviral activity | [3, 4, 5, 6, 7] |
| Type‐I IFNs | Associated with mild disease | [10, 11, 12, 13, 14, 15, 16, 17] |
| NK cells | Number and function associated with mild disease | [22, 23, 24, 25, 26] |
| DC | Antigen presentation and type‐I IFN production | [27, 28, 29] |
| High affinity antibody | Associated with recovery | [106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123] |
| Specific T cells | Highly functional, virus‐specific cellular immune response associated with mild disease and recovery | [46, 81, 82, 83, 84, 85, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141] |
TABLE 2.
Immune response to SARS‐CoV‐2: Pathogenic functions
| Immune players | Role | References |
|---|---|---|
| Anti‐IFN antibodies | Associated with severe disease | [18, 19, 20] |
| Cytokine storm | Associated with severe disease and long‐COVID | [32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 65, 66] |
| MAIT cells | Associated with severe disease | [42, 43] |
| Monocytes | Associated with severe disease | [44, 45, 46, 47] |
| MDSC | Associated with severe disease and predictive of death | [49, 50, 51, 52, 53, 54, 55] |
| Complement activation | Associated with thrombosis | [60, 61, 62] |
| Neutrophils—NET | Associated with coagulopathy and thrombosis | [69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80] |
| Adaptive T cells | Massive activation/exhaustion of T cells associated with severe diseases and long COVID | [81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96] |
| Lymphopenia | Associated with severe disease and predictive of death | [97, 98, 99, 100] |
The COVID‐19 is an immune‐mediated disease that has strongly accelerated the development of new therapeutic approaches targeting the immune response. The well‐defined harmful potential of the immune response in severe COVID‐19 and the identification of the main players driving tissue damages stimulated the use of new immune‐based approaches (e.g., mAbs targeting/neutralizing cytokines, inflammatory mediators and complement factors). Finally, although different immunological biomarkers have been proposed as predictor of COVID‐19 worsening or protection, a robust prognostic biomarker is still lacking and desirable for a better and timely treatment of COVID‐19.
Global vaccination programs have changed the natural history of COVID‐19 and saved around several millions of lives thanks to the protective capacity of the adaptive immune response [155]. Vaccines generate SARS‐CoV‐2 specific serum antibodies and memory T and B cells able to react upon infection and limit viral spreading, thus preventing severe disease and death. Vaccines are, however, unable to prevent contagion and infection, because of the continuous emergence of highly infective and antigenically distinct viral variants. Universal vaccines against Coronavirus could be instrumental [156] to stop the ongoing pandemic and prevent future outbreaks.
AUTHOR CONTRIBUTIONS
Conceptualization: Chiara Agrati, Rita Carsetti, Alessandra Sacchi, Veronica Bordoni, Concetta Quintarelli. Supervision: Franco Locatelli, Giuseppe Ippolito, Maria R. Capobianchi. Writing—original draft: Chiara Agrati, Rita Carsetti, Veronica Bordoni, Alessandra Sacchi. Writing—review and editing: Concetta Quintarelli, Franco Locatelli, Giuseppe Ippolito, Maria R. Capobianchi. All authors have read and agreed to the published version of the manuscript.
FUNDING INFORMATION
This work was supported by Ministero della Salute (Ricerca Corrente Linea 1) and by Fondazione Roma (generous liberal donations funding COVID‐19 research).
CONFLICT OF INTEREST
The authors declare no conflict of interests.
Agrati C, Carsetti R, Bordoni V, Sacchi A, Quintarelli C, Locatelli F, et al. The immune response as a double‐edged sword: The lesson learnt during the COVID‐19 pandemic. Immunology. 2022. 10.1111/imm.13564
Funding information Fondazione Roma; Ministero della Salute
Contributor Information
Chiara Agrati, Email: chiara.agrati@inmi.it.
Veronica Bordoni, Email: veronica.bordani@inmi.it.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Long QX, Tang XJ, Shi KL, Li Q, Deng HJ, Jea Y. Clinical and immunological assessment of asymptomatic SARS‐CoV‐2 infections. Nat Med. 2020;26(8):1200–4. [DOI] [PubMed] [Google Scholar]
- 2. Bottazzi B, Doni A, Garlanda C, Mantovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol. 2010;28:157–83. [DOI] [PubMed] [Google Scholar]
- 3. Brunetta E, Folci M, Bottazzi B, de Santis M, Gritti G, Protti A, et al. Macrophage expression and prognostic significance of the long pentraxin PTX3 in COVID‐19. Nat Immunol. 2021;22:1–24. [DOI] [PubMed] [Google Scholar]
- 4. Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol. 2022;21:547–78. [DOI] [PubMed] [Google Scholar]
- 5. Stravalaci M, Pagani I, Paraboschi EM, Pedotti M, Doni A, Scavello F, et al. Recognition and inhibition of SARS‐CoV‐2 by humoral innate immunity pattern recognition molecules. Nat Immunol. 2022;23:275–86. [DOI] [PubMed] [Google Scholar]
- 6. Garred P, Pressler T, Lanng S, Madsen HO, Moser C, Laursen I, et al. Mannose‐binding lectin (MBL) therapy in an MBL‐deficient patient with severe cystic fibrosis lung disease. Pediatr Pulmonol. 2002;33(3):201–7. [DOI] [PubMed] [Google Scholar]
- 7. Jensenius JC, Jensen PH, McGuire K, Larsen JL, Thiel S. Recombinant mannan‐binding lectin (MBL) for therapy. Biochem Soc Trans. 2003;31:763–7. [DOI] [PubMed] [Google Scholar]
- 8. Bortolotti D, Gentili V, Rizzo S, Schiuma G, Beltrami S, Strazzabosco G, et al. TLR3 and TLR7 RNA sensor activation during SARS‐CoV‐2 infection. Microorganisms. 2021;9(9):1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sariol A, Perlman S. SARS‐CoV‐2 takes its toll. Nat Immunol. 2021;22:801–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Banerjee AK, Blanco MR, Bruce EA, Honson DD, Chen LM, Chow A, et al. SARS‐CoV‐2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell. 2020;183(5):1325–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Palermo E, Di Carlo D, Sgarbanti M, Hiscott J. Type I interferons in COVID‐19 pathogenesis. Biology. 2021;829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lowery SA, Sariol A, Perlman S. Innate immune and inflammatory responses to SARS‐CoV‐2: implications for COVID‐19. Cell Host Microbe. 2021;29:1052–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen DY, Khan N, Close BJ, Goel RK, Blum B, Tavares AH, et al. SARS‐CoV‐2 disrupts proximal elements in the JAK‐STAT pathway. J Virol. 2021;95:e0086221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miorin L, Kehrer T, Sanchez‐Aparicio MT, Zhang K, Cohen P, Patel RS, et al. SARS‐CoV‐2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci. 2020;117(45):28344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Channappanavar R, Fehr AR, Zheng J, Wohlford‐Lenane C, Abrahante JE, Mack M, et al. IFN‐I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. 2019;129(9):3625–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science. 2020;369:718–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Santinelli L, De Girolamo G, Borrazzo C, Vassalini P, Pinacchio C, Cavallari EN, et al. Alteration of type I interferon response is associated with subclinical atherosclerosis in virologically suppressed HIV‐1‐infected male patients. J Med Virol. 2021;93(8):4930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life‐threatening COVID‐19. Science. 2020;370(6515):eabd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van der Wijst MGP, Vazquez SE, Hartoularos GC, Bastard P, Grant T, Bueno R, et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID‐19. Sci Transl Med. 2021;13(612):eabh2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sodeifian F, Nikfarjam M, Kian N, Mohamed K, Rezaei N. The role of type I interferon in the treatment of COVID‐19. J Med Virol. 2022;94(1):63–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gasteiger G, D'Osualdo A, Schubert DA, Weber A, Bruscia EM, Hartl D, et al. Cellular innate immunity: an old game with new players. J Innate Immunity. 2017;9:111–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, et al. T‐bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20(4):477–94. [DOI] [PubMed] [Google Scholar]
- 23. Carsetti R, Zaffina S, Piano Mortari E, Terreri S, Corrente F, Capponi C, et al. Different innate and adaptive immune responses to SARS‐CoV‐2 infection of asymptomatic, mild, and severe cases. Front Immunol. 2020;11:610300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Witkowski M, Tizian C, Ferreira‐Gomes M, Niemeyer D, Jones TC, Heinrich F, et al. Untimely TGFβ responses in COVID‐19 limit antiviral functions of NK cells. Nature. 2021;600:295–301. [DOI] [PubMed] [Google Scholar]
- 25. Bordoni V, Sacchi A, Cimini E, Notari S, Grassi G, Tartaglia E, et al. An inflammatory profile correlates with decreased frequency of cytotoxic cells in coronavirus disease 2019. Clin Infect Dis. 2020;71:2272–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Narni‐Mancinelli E, Vivier E. Clues that natural killer cells help to control COVID. Nature. 2021;600:226–7. [DOI] [PubMed] [Google Scholar]
- 27. Zhou R, To KKW, Wong YC, Liu L, Zhou B, Li X, et al. Acute SARS‐CoV‐2 infection impairs dendritic cell and T cell responses. Immunity. 2020;53(4):864–877.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arunachalam PS, Wimmers F, Ka Pun Mok C, Perera RAPM, Scott M, Hagan T, et al. Systems biological assessment of immunity to mild versus severe COVID‐19 infection in humans. Science. 2020;369(6508):1210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang T, Yang J, Deng H, Chen D, Yang XP, Tang ZH. Depletion and dysfunction of dendritic cells: understanding SARS‐CoV‐2 infection. Front Immunol. 2022;13:843342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Del Valle DM, Kim‐Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID‐19 severity and survival. Nat Med. 2020;26(10):1636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Falasca L, Agrati C, Petrosillo N, Di Caro A, Capobianchi MR, Ippolito G, et al. Molecular mechanisms of ebola virus pathogenesis: focus on cell death. Cell Death Differ. 2015;22:1250–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Root‐Bernstein R. Innate receptor activation patterns involving TLR and NLR synergisms in COVID‐19, ALI/ARDS and sepsis cytokine storms: a review and model making novel predictions and therapeutic suggestions. Int J Mol Sci. 2021;22(4):2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID‐19. Nat Immunol. 2020;584(7821):463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wuang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abers MS, Delmonte OM, Ricotta EE, Fintzi J, Fink DL, Almeida De Jesus AA, et al. An immune‐based biomarker signature is associated with mortality in COVID‐19 patients. JCI Insight. 2021;6(1):e144455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Montaldo C, Messina F, Abbate I, Antonioli M, Bordoni V, Aiello A, et al. Multi‐omics approach to COVID‐19: a domain‐based literature review. J Transl Med. 2021;19:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘cytokine storm’ in COVID‐19. J Infect. 2020;80(6):607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. RECOVERY Collaborative Group . Tocilizumab in patients admitted to hospital with COVID‐19 (RECOVERY): a randomised, controlled, open‐label, platform trial. Lancet. 2021;397:1637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Acosta‐Ampudia Y, Monsalve DM, Rojas M, Rodríguez Y, Zapata E, Ramírez‐Santana C, et al. Persistent autoimmune activation and proinflammatory state in post‐coronavirus disease 2019 syndrome. J Infect Dis. 2022;225(12):2155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schultheiß C, Willscher E, Paschold L, Gottschick C, Klee B, Henkes SS, et al. The IL‐1β, IL‐6, and TNF cytokine triad is associated with post‐acute sequelae of COVID‐19. Cell Rep Med. 2022;3(6):100663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Flament H, Rouland M, Beaudoin L, Toubal A, Bertrand L, Lebourgeois S, et al. Outcome of SARS‐CoV‐2 infection is linked to MAIT cell activation and cytotoxicity. Nat Immunol. 2021;22:322–35. [DOI] [PubMed] [Google Scholar]
- 43. Youngs J, Provine NM, Lim N, Sharpe HR, Amini A, Chen YL, et al. Identification of immune correlates of fatal outcomes in critically ill COVID‐19 patients. PLoS Pathog. 2021;17:e1009804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee ACY, et al. Attenuated interferon and proinflammatory response in SARS‐CoV‐2‐infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis. 2020;222(5):734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wauters E, Van Mol P, Dinkarnath Garg A, Jansen S, Van Herck Y, Vanderbeke L, et al. Discriminating mild from critical COVID‐19 by innate and adaptive immune single‐cell profiling of bronchoalveolar lavages. Cell Res. 2021;31(3):272–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single‐cell landscape of bronchoalveolar immune cells in patients with COVID‐19. Nat Med. 2020;26(6):842–4. [DOI] [PubMed] [Google Scholar]
- 47. Kosyreva A, Dzhalilova D, Lokhonina A, Vishnyakova P, Fatkhudinov T. The role of macrophages in the pathogenesis of SARS‐CoV‐2‐associated acute respiratory distress syndrome. Front Immunol. 2021;2:682871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Agrati C, Sacchi A, Bordoni V, Cimini E, Notari S, Grassi G, et al. Expansion of myeloid‐derived suppressor cells in patients with severe coronavirus disease (COVID‐19). Cell Death Differ. 2020;27:3196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Falck‐Jones S, Vangeti S, Yu M, Falck‐Jones R, Cagigi A, Badolati I, et al. Functional monocytic myeloid‐derived suppressor cells increase in blood but not airways and predict COVID‐19 severity. J Clin Invest. 2021;131(6):e144734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sacchi A, Grassi G, Bordoni V, Lorenzini P, Cimini E, Casetti R, et al. Early expansion of myeloid‐derived suppressor cells inhibits SARS‐CoV‐2 specific T‐cell response and may predict fatal COVID‐19 outcome. Cell Death Dis. 2020;11:921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Young MR, Newby M, Wepsic HT. Hematopoiesis and suppressor bone marrow cells in mice bearing large metastatic Lewis lung carcinoma tumors. Cancer Res. 1987;47(1):100–5. [PubMed] [Google Scholar]
- 52. Scheurer J, Kitt K, Huber HJ, Fundel‐Clemens K, Pflanz S, Debatin KM, et al. Graft‐versus‐host disease prevention by in vitro‐generated myeloid‐derived suppressor cells is exclusively mediated by the CD11b+CD11c+ MDSC subpopulation. Front Immunol. 2021;12:754316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reizine F, Lesouhaitier M, Gregoire M, Pinceaux K, Gacouin A, Maamar A, et al. SARS‐CoV‐2‐induced ARDS associates with MDSC expansion, lymphocyte dysfunction, and arginine shortage. J Clin Immunol. 2021;41(3):515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sacchi A, Grassi G, Notari S, Gili S, Bordoni V, Tartaglia E, et al. Expansion of myeloid derived suppressor cells contributes to platelet activation by L‐arginine deprivation during SARS‐CoV‐2 infection. Cell. 2021;10:2111. 10.3390/cells10082111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rowlands M, Segal F, Hartl D. Myeloid‐derived suppressor cells as a potential biomarker and therapeutic target in COVID‐19. Front Immunol. 2019;12:697405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kollias A, Kyriakoulis KG, Dimakakos E, Poulakou G, Stergiou GS, Syrigos K. Thromboembolic risk and anticoagulant therapy in COVID‐19 patients: emerging evidence and call for action. Br J Haematol. 2020;10:1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. 2020;18(6):1421–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Edler C, Schröder AS, Aepfelbacher M, Fitzek A, Heinemann A, Heinrich F, et al. Dying with SARS‐CoV‐2 infection‐an autopsy study of the first consecutive 80 cases in Hamburg, Germany. Int J Leg Med. 2020;134(4):1275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Smilowitz NR, Subashchandran V, Yuriditsky E, Horowitz JM, Reynolds HR, Hochman JS, et al. Thrombosis in hospitalized patients with viral respiratory infections versus COVID‐19. Am Heart J. 2021;231:93–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iffah R, Gavins FNE. Thromboinflammation in coronavirus disease 2019: the clotthickens. Br J Pharmacol. 2022;179(10):2100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Afzali B, Noris M, Lambrecht BN, Kemper C. The state of complement in COVID‐19. Nat Rev Immunol. 2022;22(2):77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vlaar APJ, de Bruin S, Busch M, Timmermans SAMEG, van Zeggeren IE, Koning R, et al. Anti‐C5a antibody IFX‐1 (vilobelimab) treatment versus best supportive care for patients with severe COVID‐19 (PANAMO): an exploratory, open‐label, phase 2 randomised controlled trial. Lancet Rheumatol. 2020;2(12):e764–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Saleh J, Peyssonnaux C, Singh KK, Edeas M. Mitochondria and microbiota dysfunction in COVID‐19 pathogenesis. Mitochondrion. 2020;54:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H, et al. SARS‐CoV‐2 spike protein impairs endothelial function via downregulation of ACE 2. Circ Res. 2021;128(9):1323–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, et al. IL‐6 trans‐signaling induces plasminogen activator inhibitor‐1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U S A. 2020;117(36):22351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sims JT, Krishnan V, Chang CY, Engle SM, Casalini G, Rodgers GH, et al. Characterization of the cytokine storm reflects hyperinflammatory endothelial dysfunction in COVID‐19. J Allergy Clin Immunol. 2021;147(1):107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xu S, Liu Y, Ding Y, Luo S, Zheng X, Wu X, et al. The zinc finger transcription factor, KLF2, protects against COVID‐19 associated endothelial dysfunction. Signal transduction and targeted. Therapy. 2021;6:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID‐19. Nat Rev Immunol. 2021;21(5):319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, et al. COVID‐19 severity correlates with airway epithelium‐immune cell interactions identified by single‐cell analysis. Nat Biotechnol. 2020;38(8):970–9. [DOI] [PubMed] [Google Scholar]
- 70. Giamarellos‐Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID‐19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992–1000.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bian XW, COVID‐19 Pathology Team . Autopsy of COVID‐19 patients in China. Natl Sci Rev. 2020;7(9):1414–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rajamanickam A, Kumar NP, Nancy A, Selvaraj N, Munisankar S, Renji RM, et al. Dynamic changes in neutrophil counts and neutrophil granular protein levels in convalescent COVID‐19 patients. Arch Clin Biomed Res. 2022;6(2):378–89. [Google Scholar]
- 73. Cortés‐Vieyra R, Gutiérrez‐Castellanos S, Álvarez‐Aguilar C, Baizabal‐Aguirre VM, Nuñez‐Anita RE, Rocha‐López AG, et al. Behavior of eosinophil counts in recovered and deceased COVID‐19 patients over the course of the disease. Viruses. 2021;13(9):1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Veras FP, Cornejo Pontelli M, Meirelles Silva C, Toller‐Kawahisa JE, de Lima M, Carvalho Nascimento D, et al. SARS‐CoV‐2‐triggered neutrophil extracellular traps mediate COVID‐19 pathology. J Exp Med. 2020;217(12):e20201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID‐19 acute respiratory distress syndrome. Blood. 2020;136(10):1169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Park JH, Lee HK. Re‐analysis of single cell transcriptome reveals that the NR3C1‐CXCL8‐neutrophil axis determines the severity of COVID‐19. Front Immunol. 2020;11:2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID‐19. Blood. 2020;136(11):1317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Englert H, Rangaswamy C, Deppermann C, Sperhake JP, Krisp C, Schreier D, et al. Defective NET clearance contributes to sustained FXII activation in COVID‐19‐associated pulmonary thrombo‐inflammation. EBioMedicine. 2021;67:103382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dolhnikoff M, Duarte‐Neto AN, de Almeida Monteiro RA, Ferraz da Silva LF, de Oliveira EP, Nascimento Saldiva PH, et al. Pathological evidence of pulmonary thrombotic phenomena in severe COVID‐19. J Thromb Haemost. 2020;18(6):1517–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lafon E, Diem G, Witting C, Zaderer V, Bellmann‐Weiler RM, Reindl M, et al. Potent SARS‐CoV‐2‐specific T cell immunity and low anaphylatoxin levels correlate with mild disease progression in COVID‐19 patients. Front Immunol. 2021;12:684014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to SARS‐CoV‐2 coronavirus in humans with COVID‐19 disease and unexposed individuals. Cell. 2020;181(7):1489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Le Bert N, Clapham HE, Tan AT, Chia WN, Tham CHL, Lim JM, et al. Highly functional virus‐specific cellular immune response in asymptomatic SARS‐CoV‐2 infection. J Exp Med. 2021;218(5):e20202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kilpeläinen A, Jimenez‐Moyano E, Blanch‐Lombarte O, Ouchi D, Peña R, Quirant‐Sanchez B, et al. Skewed cellular distribution and low activation of functional T‐cell responses in SARS‐CoV‐2 non‐seroconvertors. Front Immunol. 2021;13:815041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jung JH, Rha MS, Sa M, Choi HK, Jeon JH, Seok H, et al. SARS‐CoV‐2‐specific T cell memory is sustained in COVID‐19 convalescent patients for 10 months with successful development of stem cell‐like memory T cells. Nat Commun. 2021;12:4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Song JW, Zhang C, Fan X, Meng FP, Xu Z, Xia P, et al. Immunological and inflammatory profiles in mild and severe cases of COVID‐19. Nat Commun. 2020;11(1):3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zheng HY, Zhang M, Yang CX, Zhang N, Wang XC, Yang XP, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID‐19 patients. Cell Mol Immunol. 2020;17(5):541–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kuri‐Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, et al. Comprehensive mapping of immune perturbations associated with severe COVID‐19. Sci Immunol. 2020;5(49):eabd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liu Y, Pan Y, Hu Z, Wu M, Wang C, Feng Z, et al. Thymosin alpha 1 reduces the mortality of severe coronavirus disease 2019 by restoration of lymphocytopenia and reversion of exhausted T cells. Clin Infect Dis. 2020;71(16):2150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ruibal P, Oestereich L, Lüdtke A, Becker‐Ziaja B, Wozniak DM, Kerber R, et al. Unique human immune signature of ebola virus disease in Guinea. Nature. 2016;533(7601):100–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Agrati C, Castilletti C, Casetti R, Sacchi A, Falasca L, Turchi F, et al. Longitudinal characterization of dysfunctional T cell‐activation during human acute ebola infection. Cell Death Dis. 2016;7(3):e2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, Lund FE, et al. The establishment of resident memory B cells in the lung requires local antigen encounter. Nat Immunol. 2019;20:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim TS, Shin EC. The activation of bystander CD8+ T cells and their roles in viral infection. Exp Mol Med. 2019;51:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. De Biasi S, Meschiari M, Gibellini L, Bellinazzi C, Borella R, Fidanza L, et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID‐19 pneumonia. Nat Commun. 2020;11:3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Files JK, Sarkar S, Fram TR, Boppana S, Sterrett S, Qin K, et al. Duration of post‐COVID‐19 symptoms is associated with sustained SARS‐CoV‐2‐specific immune responses. JCI Insight. 2021;6(15):e151544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cheon IS, Li C, Son YM, Goplen NP, Wu Y, Cassmann T, et al. Immune signatures underlying post‐acute COVID‐19 lung sequelae. Sci Immunol. 2021;6(65):eabk1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). Front Immunol. 2020;11:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Feng Z, Diao B, Wang R, Wang G, Wang C, Tan Y, et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) directly decimates human spleens and lymph nodes. medRxiv. 2020. [Google Scholar]
- 99. Bordoni V, Tartaglia E, Sacchi A, Fimia GM, Cimini E, Casetti R, et al. The unbalanced p53/SIRT1 axis may impact lymphocyte homeostasis in COVID‐19 patients. Int J Infect Dis. 2021;105:49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. André S, Picard M, Cezar R, Roux‐Dalvai F, Alleaume‐Butaux A, Soundaramourty C, et al. T cell apoptosis characterizes severe COVID‐19 disease. Cell Death Differ. 2022;22:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bigdelou B, Sepand MR, Najafikhoshnoo S, Negrete JAT, Sharaf M, Ho JQ, et al. COVID‐19 and preexisting comorbidities: risks, synergies, and clinical outcomes. Front Immunol. 2022;13:890517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Allen CDC, Okada T, Cyster JG. Germinal‐center organization and cellular dynamics. Immunity. 2007;27(2):190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ochsenbein AF, Fehr T, Lutz C, Suter M, Brombacher F, Hengartner H, et al. Control of early viral and bacterial distribution and disease by natural antibodies. Science. 1999;286854479:2156–9. [DOI] [PubMed] [Google Scholar]
- 104. Kaneko N, Kuo HH, Boucau J, Farmer JR, Allard‐Chamard H, Mahajan VS, et al. Loss of bcl‐6‐expressing T follicular helper cells and germinal centers in COVID‐19. Cell. 2020;183(1):143–157.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Woodruff MC, Ramonell RP, Nguyen DC, Cashman KS, Saini AS, Haddad NS, et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID‐19. Nat Immunol. 2020;21(12):1506–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Çölkesen F, Kepenek Kurt E, Vatansev H, Korkmaz C, Çölkesen F, Yücel F, et al. Memory B cells and serum immunoglobulins are associated with disease severity and mortality in patients with COVID‐19. Postgrad Med J. 2021;0:1–7. [DOI] [PubMed] [Google Scholar]
- 107. Rajamanickam A, Pavan Kumar N, Nancy A, Selvaraj N, Munisankar S, Renji RM, et al. Recovery of memory B‐cell subsets and persistence of antibodies in convalescent COVID‐19 patients. Am J Trop Med Hyg. 2021;105(5):1255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE, et al. Immunological memory to SARS‐CoV‐2 assessed for up to 8 months after infection. Science. 2021;371:eabf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gaebler C, Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Tokuyama M, et al. Evolution of antibody immunity to SARS‐CoV‐2. Nature. 2021;591(7851):639–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Pollard AJ, Bijker EM. A guide to vaccinology: from basic principles to new developments. Nat Rev Immunol. 2021;21(2):83–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Turner JS, Kim W, Kalaidina E, Goss CW, Rauseo AM, Schmitz AJ, et al. SARS‐CoV‐2 infection induces long‐lived bone marrow plasma cells in humans. Nature. 2021;595(7867):421–5. [DOI] [PubMed] [Google Scholar]
- 112. Grimsholm O, Piano Mortari E, Davydov AN, Shugay M, Obraztsova AS, Bocci C, et al. The interplay between CD27dull and CD27bright B cells ensures the flexibility, stability, and resilience of human B cell memory. Cell Rep. 2020;30(9):2963–77. [DOI] [PubMed] [Google Scholar]
- 113. Nordström P, Ballin M, Nordström A. Risk of SARS‐CoV‐2 reinfection and COVID‐19 hospitalisation in individuals with natural and hybrid immunity: a retrospective, total population cohort study in Sweden. Lancet Infect Dis. 2022;22(26):781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Nguyen NN, Houhamdi L, Hoang VT, Delerce J, Delorme L. SARS‐CoV‐2 reinfection and COVID‐19 severity. Emerg Microbes Infect. 2022;11(1):894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wang J, Kaperak C, Sato T, Sakuraba A. COVID‐19 reinfection: a rapid systematic review of case reports and case series. J Invest Med. 2021;69(6):1253–5. [DOI] [PubMed] [Google Scholar]
- 116. Wright PF, Prevost‐Reilly AC, Natarajan H, Brickley EB, Connor RI, Wieland‐Alter WF, et al. Longitudinal systemic and mucosal immune responses to SARS‐CoV‐2 infection. J Infect Dis. 2022;jiac065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rybkina K, Davis‐Porada J, Farber DL. Tissue immunity to SARS‐CoV‐2: role in protection and immunopathology. Immunol Rev. 2022;309:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Viant C, Gaebler C, et al. Enhanced SARS‐CoV‐2 neutralization by dimeric IgA. Sci Transl Med. 2021;13(577):eabf1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Terreri S, Piano Mortari E, Vinci MR, Russo C, Alteri C, Albano CC, et al. Persistent B cell memory after SARS‐CoV‐2 vaccination is functional during breakthrough infections. Cell Host Microbe. 2022;30(3):400–408.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Afkhami S, D'Agostino MR, Zhang A, Stacey HD, Marzok A, Kang A, et al. Respiratory mucosal delivery of next‐generation COVID‐19 vaccine provides robust protection against both ancestral and variant strains of SARS‐CoV‐2. Cell. 2022;185(5):896–915.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN‐COV2, a neutralizing antibody cocktail, in outpatients with covid‐19. N Engl J Med. 2021;384(3):238–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, et al. SARS‐CoV‐2 neutralizing antibody LY‐CoV555 in outpatients with covid‐19. N Engl J Med. 2021;384(3):229–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Gottlieb RL, Nirula A, Chen P, Boscia J, Heller B, Morris J, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID‐19: a randomized clinical trial. JAMA. 2021;325(7):632–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Tan ATT, Linster M, Tan CW, Le Bert N, Chia WN, Kunasegaran K, et al. Early induction of functional SARS‐CoV‐2‐specific T cells associates with rapid viral clearance and mild disease in COVID‐19 patients. Cell Rep. 2021;34:108728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wyllie D, Jones HE, Mulchandani R, Trickey A, Taylor‐Phillips S, Brooks T, et al. SARS‐CoV‐2 responsive T cell numbers and anti‐spike IgG levels are both associated with protection from COVID‐19: a prospective cohort study in keyworkers. MedRxiv. 2020. [Google Scholar]
- 126. Kundu R, Narean JS, Wang L, Fenn J, Pillay T, Derqui Fernandez N, et al. Cross‐reactive memory T cells associate with protection against SARS‐CoV‐2 infection in COVID‐19 contacts. Nat Commun. 2022;13:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Peng Y, Mentzer AJ, Liu G, Yao X, Yin Z, Dong D, et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS‐CoV‐2 in UK convalescent individuals following COVID‐19. Nat Immunol. 2020;21:1336–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. McMahan K, Yu J, Mercado NB, Loos C, Tostanoski LH, Chandrashekar A, et al. Correlates of protection against SARS‐CoV‐2 in rhesus macaques. Nature. 2021;590:630–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Gallais F, Velay A, Nazon C, Wendling MJ, Partisani ML, Sibilia J, et al. Intrafamilial exposure to SARS‐CoV‐2 associated with cellular immune response without seroconversion, France. Emerg Infect Dis. 2021;27(1):113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rajamanickam A, Kumar NP, Pandiaraj AN, Selvaraj N, Munisankar S, Renji RM, et al. Characterization of memory T cell subsets and common γ‐chain cytokines in convalescent COVID‐19 individuals. J Leukoc Biol. 2022;112(1):201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gurevich M, Zilkha‐Falb R, Sonis P, Magalashvili D, Menascu S, Flechter S, et al. SARS‐CoV‐2 memory B and T cell profiles in mild COVID‐19 convalescent patients. Int J Infect Dis. 2022;115:208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Roukens AHE, Pothast CR, Konig M, Huisman W, Dalebout T, Tak T, et al. Prolonged activation of nasal immune cell populations and development of tissue‐resident SARS‐CoV‐2‐specific CD8(+) T cell responses following COVID‐19. Nat Immunol. 2022;23:23–32. [DOI] [PubMed] [Google Scholar]
- 133. Grau‐Expósito J, Sánchez‐Gaona N, Massana N, Suppi M, Astorga‐Gamaza A, Perea D, et al. Peripheral and lung resident memory T cell responses against SARS‐CoV‐2. Nat Commun. 2021;12(1):3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Cameroni E, Bowen JE, Rosen LE, Saliba C, Zepeda SK, Culap K, et al. Broadly neutralizing antibodies overcome SARS‐CoV‐2 omicron antigenic shift. Nature. 2022;602(7898):664–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Cao Y, Wang J, Jian F, Xiao T, Song W, Yisimayi A, et al. Omicron escapes the majority of existing SARS‐CoV‐2 neutralizing antibodies. Nature. 2022;602(7898):657–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Andrews N, Stowe J, Kirsebom F, Toffa S, Rickeard T, Gallagher E, et al. Covid‐19 vaccine effectiveness against the omicron (B.1.1.529) variant. New Engl J Med. 2022;1532–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Cele S, Jackson L, Khoury DS, Khan K, Moyo‐Gwete T, Tegally H, et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature. 2022;602:654–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Riou C, Keeton R, Moyo‐Gwete T, Hermanus T, Kgagudi P, Baguma R, et al. Escape from recognition of SARS‐CoV‐2 variant spike epitopes but overall preservation of T cell immunity. Sci Transl Med. 2022;14(631):eabj6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Gao Y, Ca C, Grifoni A, Müller TR, Niessl J, Olofsson A, et al. Ancestral SARS‐CoV‐2‐specific T cells cross‐recognize the omicron variant. Nat Med. 2022;28:472–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Keeton R, Tincho MB, Ngomti A, Baguma R, Benede N, Suzuki A, et al. T cell responses to SARS‐CoV‐2 spike cross‐recognize omicron. Nature. 2022;603(7901):488–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Liu J, Chandrashekar A, Sellers D, Barrett J, Jacob‐Dolan C, Lifton M, et al. Vaccines elicit highly conserved cellular immunity to SARS‐CoV‐2 omicron. Nature. 2022;603(7901):493–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. McMichael AJ, Gotch FM, Noble GR, Beare PA. Cytotoxic T‐cell immunity to influenza. N Engl J Med. 1983;309:13–7. [DOI] [PubMed] [Google Scholar]
- 143. Grant EJ, Josephs TM, Loh L, Clemens EB, Sant S, Bharadwaj M, et al. Broad CD8+ T cell cross‐recognition of distinct influenza a strains in humans. Nat Commun. 2018;9:5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. de Assis RR, Jain A, Nakajima R, Jasinskas A, Felgner J, Obiero JM, et al. Analysis of SARS‐CoV‐2 antibodies in COVID‐19 convalescent blood using a coronavirus antigen microarray. Nat Commun. 2021;12:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Ng KW, Faulkner N, Cornish GH, Rosa A, Harvey R, Hussain S, et al. Preexisting and de novo humoral immunity to SARS‐CoV‐2 in humans. Science. 2020;370:1339–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Mateus J, Grifoni A, Tarke A, Sidney J, Ramirez SI, Dan JM, et al. Selective and cross‐reactive SARS‐CoV‐2 T cell epitopes in unexposed humans. Science. 2020;370:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Sagar M, Reifler K, Rossi M, Miller NS, Sinha P, White LF, et al. Recent endemic coronavirus infection is associated with less‐severe COVID‐19. J Clin Invest. 2021;131(1):e143380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Anderson EM, Goodwin EC, Verma A, Arevalo CP, Bolton MJ, Weirick ME, et al. Seasonal human coronavirus antibodies are boosted upon SARS‐CoV‐2 infection but not associated with protection. Cell. 2021;184:1858–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lin CY, Wolf J, Brice DC, Sun Y, Locke M, Cherry S, et al. Pre‐existing humoral immunity to human common cold coronaviruses negatively impacts the protective SARS‐CoV‐2 antibody response. Cell Host Microbe. 2022;30(1):83–96.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Guo L, Wang Y, Kang L, Hu Y, Wang L, Zhong J, et al. Cross‐reactive antibody against human coronavirus OC43 spike protein correlates with disease severity in COVID‐19 patients: a retrospective study. Emerg Microbes Infect. 2021;10(1):664–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Bacher P, Rosati E, Esser D, Martini GR, Saggau C, Schiminsky E, et al. Low‐avidity CD4+ T cell responses to SARS‐CoV‐2 in unexposed individuals and humans with severe COVID‐19. Immunity. 2020;53(6):1258–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Johansson AM, Malhotra U, Kim YG, Gomez R, Krist MP, Wald A, et al. Cross‐reactive and mono‐reactive SARS‐CoV‐2 CD4+ T cells in prepandemic and COVID‐19 convalescent individuals. PLoS Pathog. 2021;17(12):e1010203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Dykema AG, Zhang B, Woldemeskel BA, Garliss CC, Cheung LS, Choudhury D, et al. Functional characterization of CD4+ T cell receptors crossreactive for SARS‐CoV‐2 and endemic coronaviruses. J Clin Invest. 2021;131(10):e146922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Beretta A, Cranage M, Zipeto D. Is cross‐reactive immunity triggering COVID‐19 immunopathogenesis? Front Immunol. 2020;11:567710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Watson OJ, Barnsley G, Toor J, Hogan AB, Winskill P, Ghani AC. Global impact of the first year of COVID‐19 vaccination: a mathematical modelling study. Lancet Infect Dis. 2022;3099(22):00320–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Morens DM, Taubenberger JK, Fauci AS. Universal coronavirus vaccines—an urgent need. N Engl J Med. 2022;386(4):297–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.