

Abstract
Cellular heterogeneity is commonly investigated using single-cell genomics and transcriptomics to investigate biological questions such as disease mechanism, therapeutic screening, and genomic and transcriptomic diversity between cellular populations and subpopulations at the cellular level. Single-cell mass spectrometry (MS)-based proteomics enables the high-throughput examination of protein expression at the single-cell level with wide applicability, and with spatial and temporal resolution, applicable to the study of cellular development, disease, effect of treatment, etc. The study of single-cell proteomics has lagged behind genomics and transcriptomics largely because proteins from single-cell samples cannot be amplified as DNA and RNA can using well established techniques such as PCR. Therefore, analytical methods must be robust, reproducible, and sensitive enough to detect the very small amount of protein within a single cell. To this end, nearly every step of the proteomics process has been extensively altered and improved to facilitate the proteomics analysis of single cells including cell counting and sorting, lysis, protein digestion, sample cleanup, separation, MS data acquisition, and data analysis. Here, we have reviewed recent advances in single-cell protein separation using nano reversed phase liquid chromatography (nRPLC) and capillary electrophoresis (CE) to inform application driven selection of separation techniques in the laboratory setting.
Keywords: Single-cell proteomics, reversed-phase liquid chromatography, capillary electrophoresis mass spectrometry
Introduction
Single-cell genomics and transcriptomics has enlightened the scientific community to the genetic variation between individuals, within organ tissue, and as a result of disease states.1 The further development of mass spectrometry (MS)-based single-cell bioanalysis (e.g., proteomics and metabolomics) techniques holds even more promise for the opportunity to interrogate biological phenotypes at the single-cell level. Standard bioanalytical methods are designed to analyze cellular biomolecules from lysates made from blending thousands of cells to interrogate average cellular expression.2 These bulk cell analysis techniques can obscure the cellular variation of unique cellular phenotypes as well as their responses to environmental changes, disease progression, and therapeutic treatment.
The advantage that the development of single-cell proteomics techniques will afford is the ability to analyze phenotypical cellular variation with spatial and temporal resolution. Spatial resolution of cellular protein expression is valuable to the study of tissue morphologies,3 localization of diseased tissues, and observation of the effect of environmental or therapeutic treatment on diseased vs. healthy tissue.4 Furthermore, temporal resolution of single-cell proteomics has the potential to track cellular development,5 disease progression,6 and observe response to stimuli such as disease treatment.7 However, analysis of the extremely limited sample available in a single cell is challenging for a myriad of reasons including low amounts of analyte with high dynamic range, inefficient small volume sample preparation and handling techniques, inadequate separation methods, and low throughput. These challenges required the overhaul of nearly every step of the MS-based proteomics methodology.
The state of single-cell proteomics has been recently reviewed with respect to current technologies.8–11 Additionally, an excellent review that discusses sample preparation, separation, and MS analysis of mass-limited samples indicates potential application to single-cell proteomics.12 This perspective will discuss developments and applications of single-cell MS-based proteomics with a particular focus on the separation methodology (i.e., nano reversed phase liquid chromatography (nRPLC) and capillary electrophoresis (CE)) (Figure 1 and Table 1). Furthermore, we will discuss the future of this field and how further improvements and developments with regard to sample preparation and separation may impact the feasibility of single-cell proteomics analysis.
Figure 1:
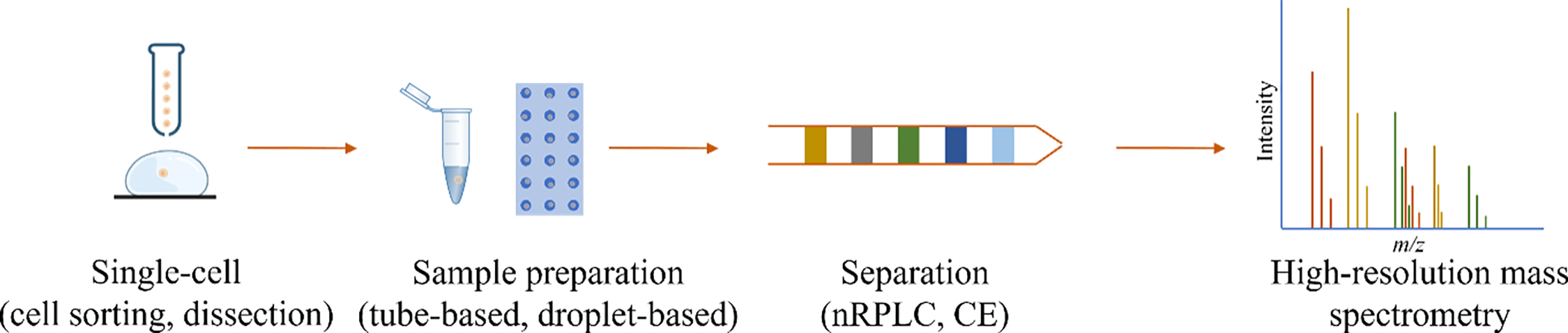
General workflow for MS-based single-cell proteomics.
Table 1:
Comparison of single-cell applications including selected publications that utilize single-cell sample preparation methods.
| Quantification Method | Separation | Cell type | Sample handling | Sample loading amount | CE/LC column | MS | Protein ID count |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Isobaric labeling with carrier/boosting | CE | Neurons (C57Bl6/J mice) | Tube-based | 0.001 ng (1 pg, ~0.25% of the total cellular proteome) + 0.1 ng carrier/boosting peptides | 100 cm fused silica capillary (90/20 μm o.d./i.d.) | Q Exactive Plus MS | 15722 |
|
| |||||||
| RPLC | Murine (C10, epithelial cells; RAW, macrophage cells; SVEC, endothelial cells) | Droplet-based | 1 cell + l50 ng carrier/boosting peptides | In-house packed 50 cm C18 column (30 μm i.d., 50 nL/min) | Fusion Lumos Tribrid MS | 140853 | |
|
| |||||||
| Acute myeloid leukemia (AML) cell (MOLM-14, K562, and CMK) | Droplet-based | 1 cell + 10 ng carrier/boosting peptides | In-house packed 30 cm C18 column (50 μm i.d., 100 nL/min) | Fusion Lumos Tribrid MS | 192651 | ||
|
| |||||||
| Acute myeloid leukemia (AML) cell (MOLM-14, K562, and CMK) | Droplet-based | 1 cell + 10 ng carrier/boosting peptides | In-house packed 60 cm C18 column (50 μm i.d., 150 nL/min) | Fusion Lumos Tribrid MS | ~180044 | ||
|
| |||||||
| OCI-AML8227 cell | Droplet-based | 1 cell + 500 cell digest as carrier/boosting channel | A 15 cm C18 column (ThermoFisher EasySpray ES804A, 100 nL/min) | Exploris480 with FAIMS Pro | ~100040 | ||
|
| |||||||
| Murine (C10, epithelial cells; RAW, macrophage cells; SVEC, endothelial cells) | Droplet-based | 1 cell + 10 ng carrier/boosting peptides | A 25 cm C18 column (Waters, 50 μm i.d., 100 nL/min) | Q Exactive Plus MS | ~150031 | ||
|
| |||||||
| U-937 (monocytes) cell | Tube-based | 1 cell + 200 cell digest as carrier/boosting channel | 25 cm C18 column (AUR2-25075C18A, 200 nL/min) | Q Exactive MS | ~100050 | ||
|
| |||||||
| Isobaric labeling without carrier/boosting | CE | Frog (Xenopus) Embryo | Tube-based | 20 ng | 85-cm fused silica capillary (110/40 μm o.d./i.d.) | Orbitrap Fusion Tribrid MS | 500–80016 |
|
| |||||||
| Frog (Xenopus) Embryo | Tube-based | 16 ng | 85 cm fused silica capillary (110/40 μm o.d./i.d.) | Orbitrap Fusion Tribrid MS | 43817 | ||
|
| |||||||
| Frog (Xenopus) Embryo | Tube-based | ~5 ng | 90 cm fused silica capillary (110/40 μm o.d./i.d.) | Q Exactive Plus MS | ~750–80020 | ||
|
| |||||||
| CE | Frog (Xenopus) Embryo | Tube-based | ~5 ng | 90 cm fused silica capillary (110/40 μm o.d./i.d.) | Q Exactive Plus MS | 72283 | |
|
| |||||||
| Label free quantification | Human Oocytes | Tube-based | 1 cell (~100 pg) | 20 cm C18 column (nanoAcquity, 300 nL/min) | Orbitrap Velos Pro MS | ~45084 | |
|
| |||||||
| RPLC | Frog (Xenopus) Embryo | Tube-based | 200–800 ng | 10 cm C18 column (Waters, 100 μm i.d., 700 nL/min) | Q-Exactive HF MS | 644–146618 | |
|
|
|||||||
| HeLa | Droplet-based | 1 cell (~0.15 ng) | 50 cm C18 column (30 μm i.d., 60 nL/min) | Orbitrap Fusion Lumos Tribrid MS | 66985 | ||
|
| |||||||
| HeLa | Droplet-based | 10 cells (~1.5 ng) | In-house packed 70 cm C18 column (30 μm i.d., 60 nL/min) | Orbitrap Fusion Lumos Tribrid MS | ~300029 | ||
|
| |||||||
| Frog (Xenopus) Embryo | Tube-based | ~40ng | In-house packed 20 cm C18 column (75 μm i.d., 300 nL/min) | Q-Exactive HF-X Hybrid Quadrupole-Orbitrap MS | ∼165021 | ||
|
| |||||||
| Chick utricle E15 (Hair cell) | Droplet-based | 1 cell | In-house packed 50 cm C18 column (30 μm i.d., 60 nL/min) | Orbitrap Fusion Lumos Tribrid MS | 50–7586 | ||
|
| |||||||
| HeLa | Droplet-based | 1 cell (~0.15 ng) | In-house packed 50 cm C18 column (20 μm i.d., 20 nL/min) | Orbitrap Eclipse Tribrid MS | 105681 | ||
|
| |||||||
| MCF10A (MCF7) breast cancer cell | Tube-based | 1 cell | In-house packed 70 cm C18 column (50 μm i.d., 150 nL/min) | Q Exactive Plus MS | 38487 | ||
|
| |||||||
| HeLa | Droplet-based | 1 cell | In-house packed 45 cm C18 column (75 μm i.d., 300 nL/min) | Trapped ion mobility spectrometry quadrupole time-of-flight MS | 127988 | ||
|
| |||||||
| Human lung adenocarcinoma cell (PC-9) | Chip-based | 1 cell | A 25 cm C18 column (Waters, nanoEase, 75 μm i.d., 300 nL/min) | Orbitrap Eclipse MS | ~150042 | ||
|
| |||||||
| HeLa | Droplet-based | 1 cell | In-house packed 10 cm C18 column (50 μm i.d., 80 nL/min) | Orbitrap Exploris 480 MS | 98643 | ||
|
| |||||||
| Label free quantification | RPLC | Hela | Droplet-based | 1 cell | In-house packed 25 cm C18 column (50 μm i.d., 100 nL/min) | Orbitrap Fusion Lumos Tribrid MS with FAIMS pro | 121282 |
|
| |||||||
| Frog (Xenopus) Embryo | Tube-based | ~200 ng | In-house packed C18 column (75 μm i.d., 315 nL/min) | Orbitrap hybrid Fusion Lumos MS | 346889 | ||
Single-cell Proteomics Sample Preparation
Early demonstrations of single-cell MS proteomics analysis were performed on single red blood cells to observe hemoglobin in healthy cells and those effected by sickle cell disease using matrix assisted laser desorption/ionization (MALDI). High-throughput proteomics workflows have since been applied to deepen proteome coverage of single cells. Protein preparation for high-throughput proteomics generally consist of a series of steps including bulk cell lysis, protein extraction, proteolysis (denaturation, reduction, alkylation, and digestion), desalting, separation, MS data acquisition, and data processing.2 Efforts have been made to adapt existing bulk cell lysis and protein extraction techniques (i.e., tube-based methods that are conducted in Eppendorf tubes) to processing limited mass samples and miniaturization of high-throughput proteomics workflow has been demonstrated to outperform traditional workflows for mass limited samples.13–15 Some of the first iterations of high-throughput proteomics analysis to single cells were performed on Frog (Xenopus laevis) embryos16–19 which contain relatively large cells with high protein content (approximately 1 μg of yolk-free protein at the 16-cell stage that decreases as embryonic development proceeds to higher number cell stages20–21). These experiments utilized dissection to isolate cells and a miniaturized, tube-based, bottom-up workflow for lysis, protein extraction, and digestion. Additionally, more recent proteomics analysis of these relatively large cells has been performed via the direct penetration of the cells to aspirate cytosol for further downstream processing.20, 22 Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS) was also developed initially as a tube-based method in which cells are lysed by sonication to avoid the addition of non-MS compatible reagents.23–24 Samples were then TMT labeled and combined with a carrier proteome to increase identification numbers.
Tube-based sample preparation typically used for bulk cell sample preparation has been demonstrated to cause sample loss due to adhesion to surfaces, desalting, and speed-vac drying which disproportionately effect low sample quantities.25 Nonspecific adsorption has even been recently reviewed in the literature and was discussed as a primary inhibitor to application of single-cell proteomics analysis.26 Thus, application of typical bottom-up sample preparation techniques are not suitable for extremely small sample amounts in human somatic cells, (e.g., hundreds of picograms).25, 27 Furthermore, consistent sample processing on such a small scale is susceptible to human error when conducted by hand and can be time consuming leading to low throughput. Therefore, as an alternative to tube-based sample processing, ultra-low-volume (e.g., nL range) sample preparation techniques have been developed to enable processing of single-cell analytes with reduced sample loss.
One of the most promising areas of advancement for single-cell proteomics is droplet-based sample preparation including Oil-Air-Droplet (OAD)28, nanodroplet Processing in One pot for Trace Samples (nanoPOTS)29, automated Preparation in One pot for Trace Samples (autoPOTS)30, nested nanoPOTS (N2)31, and nano-ProteOmic sample Preparation (nPOP).32 These methods utilize automated sample handling platforms for droplet-based cell lysis and digestion in small sample volumes (approximately 200 and 550 nL for nano/autoPOTS and OAD, respectively). The N2 chip and nPOP method further scale down the sample volume to 30 nL31 and 20 nL32, respectively. m(n)POP was further integrated into the SCoPE-MS workflow resulting in SCoPE-2.33 As an alternative to the droplet sample preparation approach, the Zhang lab introduced the integrated Proteomic Analysis Device for single-cell proteomics (iPAD-1).34 Unlike the droplet-based approaches, the iPAD-1 directly aspirates a single cell into a capillary and performs in-capillary lysis and protein digestion in 2 nL volume and the whole volume can then be loaded for sample analysis.
In addition to the sample preparation methods for single cells discussed, some new techniques have emerged that have not yet been applied to single-cell proteomics but demonstrate promise in the analysis of very small numbers of cells. For example, adaptations of the microreactor-based platforms including single-pot solid-phase-enhanced sample preparation (SP3)35 and filter-aided sample preparation (FASP) method have been introduced by Yang et. al. as the nanoparticle-aided nanoreactor for nanoproteomics (Nano3)36, Zhang et. al. as MICRO-FASP37, and Kostas et. al. as on-microsolid-phase extraction tip (OmSET)38. These methods show promise as low volume sample preparation methods with limited sample loss for mass limited samples. Additionally, Burns et. al. developed a platform that utilizes an automated liquid handler to prepare cells for bottom-up proteomics directly in a 384-well plate used to culture the cells. While this method is not yet directly applicable to single cell analysis, it allows high throughput proteomics for drug screening with minimal sample loss.39
To enhance the throughput and precision of these single-cell sample preparation platforms, dedicated platforms have been implemented for cell sorting to isolate single cells onto the sample preparation chips such as fluorescence activated cell sorting (FACS) and CellenONE (SCIENION).24, 32–33, 40 Additionally, CellenONE can integrate cell sorting and nano-drop based sample prep for a completely automated sample preparation methodology as initially demonstrated by Woo et. al.31
Decreased volume of sample preparation is preferable to single-cell analysis because the decrease in sample volume limits exposure to surfaces to decrease protein adsorption.25 Minimal protein loss is critical to single-cell analysis in which the protein is very limited. Furthermore, automation of the sample preparation platforms, particularly for exceedingly small volumes of sample, increases sample throughput and limits sample to sample variation.
Nanoscale Reversed phase liquid chromatography (nRPLC)
For high-throughput proteomics of biological samples, high sample complexity and dynamic range complicates MS spectra obscuring peptide identifications. Single-cell proteomics is further complicated due to the small amount of protein contained within a single cell as most high-resolution mass spectrometers need many copies of a protein for fragmentation and ionization to result in identification of peptides. Separation has been implemented to decrease sample complexity and concentrate peptides prior to MS analysis to increase proteome coverage, particularly for low abundance proteins. nRPLC is the most popularly implemented separation technique for single-cell analysis because it is highly sensitive with high resolution and can utilize MS compatible buffers to facilitate direct coupling with MS via nanoESI interfaces.
After single-cell sample preparation (either tube-based or droplet-based) that typically incorporate one of two types of quantitation, label-free or isobaric chemical tag labeling, the sample volume is commonly scaled up from nL to μL and transferred to a tube for nRPLC sample injection. Online solid-phase extraction columns for concentration and/or sample cleanup increase the separation efficiency and sensitivity followed by application of the LC gradient for separation and elution.
Label-free quantitation is a simple method for relative quantitation of single-cell proteins that benefits from limited sample preparation, low cost, and wide applicability (not limited to lab cultured samples like isotopic labeling).41 Notably, label-free quantitation and data independent acquisition were applied to the analysis of 10 single human lung adenocarcinoma (PC-9) cells (processed using a chip-based sample preparation method, SciProChip) for average identification of approximately 1500 protein groups/cell.42
Generally speaking, the primary drawback of label-free quantitation (aside from run-to-run variation) is limited throughput. To increase throughput, the Kelly group developed an LC configuration that utilized two parallel subsystems to alternate sample separation and data acquisition with the support functions of the system such as sample loading, desalting, and regeneration to maximize instrument utilization.43 With this platform, they identified ~1000 protein groups/cell using a 30-minute gradient resulting in the capability to analyze 48 samples per day. Additionally, Williams et. al. developed an autosampler for the nanoPOTS system to allow automated sampling of single cells to further increase throughput.44,43 Another drawback of single-cell label-free quantitation is the prevalence of missed identifications between runs due to inadequate MS peptide identification based on MSn fragmentation.45 The number of quantifiable proteins from label-free datasets can be increased when protein libraries from larger numbers of cells are combined using accurate mass and time tag (AMT)46 comparisons or algorithms such as MaxQuant’s Match-Between-Runs (MBR).47–48
Generally, the advantage of isobaric chemical tag labeling (tandem-mass-tag, TMT; or isobaric tags for relative and absolute quantitation, iTRAQ) is multiplexing to increase throughput and eliminate run-to-run variation.49 For single-cell proteomics, isobaric chemical tag labeling also has the advantage of increasing the total amount of protein that can be injected to boost signal and increase the number of identified proteins. To this end a series of techniques including the SCoPE23–24, 33, 50 and nanoPOTS29, 31, 44 related methods and Improved Boosting to Amplify Signal with Isobaric Labeling (iBASIL)51–52 that utilize carrier proteomes or booster channels, samples that are labeled that contain higher masses of proteins than the single cell, are multiplexed with single-cell samples to increase the amount of protein injected for nRPLC analysis.51, 53 In one study, the iBASIL approach led to the identification and quantification of 1500 proteins from 3 different myeloid leukemia cell lines.51 While TMT labeling with carrier or boosting proteomes increase the number of identified protein groups, the accuracy of the quantitation can be effected by precursor co-isolation54, isotopic impurities55, and batch effects56. Specific to single-cell analysis using carrier proteomes, quantitative accuracy was dependent on the ratio of carrier to sample which limits the amount of protein that can be added for the carrier/booster.57 Overall, the advantages of nRPLC single-cell analysis include the ability to automate the sample preparation and analysis platform for decreased sample variation and the ability of nRPLC to incorporate online SPE sample cleanup and concentration for more sensitive detection.
To improve the proteome coverage, miniaturization of nRPLC column is used as a strategy to increase the efficiency of nRPLC separation and ESI-MS sensitivity for single-cell level peptide analysis. Shen and co-workers detected ~190-fold more mass features upon decreasing self-packed LC column inner diameter from 74.5 μm to 14.5 μm using 100 ng yeast tryptic digest.58 Zhu and co-workers demonstrated a 32% increase in peptide identifications using 30-μm-i.d. columns compared with standard 75-μm-i.d. columns using 10 ng tryptic peptides.59 Recently, Cong and co-workers achieved a ~41% increase in protein group identification using an ultranarrow-bore (20 μm i.d.) LC column compared with a 30 μm i.d. column for nanoPOTS-prepared single HeLa cells.60 Overall, decreasing the inner diameter of the columns can increase protein identification; however, the improvement is only incremental and the relatively large sample volume and dilutions required for nRPLC analysis can still lead to sample loss and decreased sensitivity. Application of methods beyond the packed LC column, such as open tubular61 or micropillar array columns62, may also be explored for increased nRPLC sensitivity.
Capillary Electrophoresis (CE)
Capillary electrophoresis (CE) coupled with mass spectrometry is advantageous for the analysis of single cells due to the highly efficient separation63 of extremely small (low nL) sample volumes64 with ultra-low detection limit (zmol)65. However, CE separation has been less frequently implemented for single-cell proteomics than nRPLC, with the most popular targets being large, nonhuman cell types such as mouse neurons22 and embryonic cells from frog16–18, 20 and zebrafish20. Typically, sample preparation for single-cell CE-MS analysis was done via dissection66 to isolate single cells into tubes followed by a miniaturized version of the traditional bottom-up proteomics sample preparation workflow2 to minimize sample loss. Subsequently, a portion of the extracted proteins were injected for CE-MS analysis, and 3–4 technical replicates were performed. Further adaptation of these techniques led to direct sampling of cytoplasm from living cells followed by protein digestion and CE-MS analysis.20
Using label-free CE-MS methods, the Nemes group analyzed 16 ng of cytoplasmic protein digest from biological triplicate runs of three different Frog embryo cells resulting in the identification of 438 protein groups.17 Application of TMT labeling to the same system allowed for the identification of 1709 protein groups from 3 biological replicates using 20 ng of protein.16 As an impressive application of this technology, the Nemes group sampled cytoplasm directly from living Xenopus laevis embryos and zebrafish embryos at different development stages to compare protein expression throughout development.20 They were able to identify 750–800 protein groups from 5 ng of protein in 16-cell Xenopus laevis embryo using label-free quantitation.20 In what is, to our knowledge, the smallest amount of protein analyzed by CE-MS single-cell proteomics to date, the Nemes group analyzed 1 pg of protein digest from single mouse neurons to quantify 157 proteins using TMT labeling and a carrier proteome.22
Overall, CE-MS has been demonstrated to be a powerful method for single-cell proteomics. Label-free and isobaric chemical tag labeling techniques have resulted in identification of hundreds to more than 1000 protein groups utilizing low nanogram levels of sample performed primarily with tube-based sample preparation methods (Table 1). While application of CE separation to human single cells has lagged behind LC-based single cell analysis, CE-MS/MS analysis has been applied to the analysis mass limited human lysate. For example, Johnson et. al. identified 744±127 proteins from 1-cell equivalents of HeLa lysate using their CE-MS/MS platform.67 Application of droplet-based sample preparation methods may further amplify the number of identifications and allow further application of CE separation to human cell lines.
Some common drawbacks associated with CE separation are nonspecific sample adsorption to the bare CE capillary column and unstable electroosmotic flow (EOF) resulting in reduced separation efficiency.68 Application of coatings including covalently bound chemical coatings such as linear polyacrylamide69 or polyethyleneimine70 not only decrease nonspecific sample adsorption but also increase separation efficiency by adjusting EOF; coatings used for CE separation of peptides have been reviewed in the literature.71 Another challenge associated with single-cell CE proteomics is manipulating and loading the exceedingly small sample volumes associated with the small-scale sample preparation. CE can tolerate extremely low injection volumes from low nL to pL so it is not necessary to dilute single-cell samples to high volumes as is generally done with the droplet-based methods21; however, pressure-based sample injection and micropipettes can be limiting regarding the sample volume they are capable of manipulating. Methods such as sample stacking72 or SPME73–74 have been used to increase the sample loading capacity for CE separation and, while these methods have not yet been applied to single cells, they have proven to be useful for single-cell amounts of protein digests and would be valuable to single-cell applications. Furthermore, as an alternative to micropipette-based CE sampling, a microsampling device, Spray-capillary, has been developed that uses the pressure differential from generation of ESI as the driving force for tunable and quantitative ultra-low volume sample injection (e.g., as low as 15 pL/s).75 Furthermore, the device can be used directly for CE separation with no additional sample handling steps.76 Coupling of the spray-capillary CE-MS platform with droplet-based sample preparation without excessive dilution could enable injection of contents from a single cell with minimal sample loss. Since the sample loading requirement for CE-MS analysis is very low, the spray-capillary could also be further applied for multidimensional, single-cell analysis coupled with nRPLC fractionation for deeper single-cell proteome coverage.
Conclusions and Final Perspectives
nRPLC workflows are advantageous due to the SPE loading and high-resolution separation of peptides; CE separation benefits from low detection limit and low mass/volume sample consumption. NanoRPLC and CE can both utilize MS compatible solvent systems for direct coupling with ESI-MS techniques. In both methods, isobaric chemical tag labeling based single-cell sample preparation techniques can be adapted for increased sample injection resulting in higher proteome coverage, increased throughput, and decreased sample-to-sample variation.57, 77 Further optimization of nRPLC columns such as utilizing monolithic columns or nano-open-tubular columns61 can improve the separation efficiency and ESI-MS sensitivity for improving single-cell proteome coverage.58–60, 78 Separation efficiency of CE separation for single-cell proteomics can also be improved by the application of column coatings such as linear polyacrylamide and polyethylenimine69–70 or using online SPME trapping prior to CE separation74. Sample volume (nL level) used for most droplet and tube-based single-cell methods and a lack of ultra-low-volume sample handling methods limit the application of CE due restrictions on sample injection volume.79 Improvements to ultra-low-volume sample handling techniques and further minimization of single-cell sample preparation volume to limit sample loss may broaden the applications of CE separation for more diverse single-cell proteomics. Thus, both nRPLC and CE have room for improvement as they are applied to single-cell proteomics and selection of these methods is largely application driven and will be dependent on future improvements to sample preparation and handling methods as well as optimization of the separation methods themselves.
Aside from improvements to liquid-phase separation, innovative gas phase separation techniques have been applied to single-cell MS analysis to increase instrumental sensitivity including implementation of High field asymmetric waveform ion mobility spectrometry (FAIMS). FAIMS can be applied to high resolution mass spectrometers to filter out chemical noise and interfering ions to improve dynamic range and detection limits.80 Since these instruments have a maximum charge capacity, the presence of +1 charged contaminant species can lead to signal suppression. The first report of the application of FAIMS to single-cell proteomics utilized FAIMS to remove singly charged species and reported the identification of 1056 protein groups from 2912 peptides which demonstrated a 2.3 and 2.0-fold improvement, respectively, over the same analytical setup without FAIMS.81 As a further implementation of FAIMS filtering applied to single-cell proteomics, Transferring Identification based on FAIMS Filtering (TIFF) creates a library of peptides via the repetitive analysis of larger sample amounts using varying FAIMS compensation voltages (CV).82 This library is integrated with precursor mass and elution time as in an AMT or MBR approach for MS1 level matching to single-cell spectra to decrease false discovery rates. TIFF, using a library created using 4 CVs, was applied to the analysis of single HeLa cells and increased the number of proteins identified from an average of 209 to 1,212/cell.82
Overall, the application of nRPLC or CE separation methods to single-cell proteomics is application dependent and requires deep consideration of relevant available methods for successful analysis. While it seems unlikely that any particular method or platform will emerge as vastly superior, the work done in this field has made impressive strides toward the realization of comprehensive single-cell proteomics; perhaps the implementation of novel technologies or combination platforms as discussed here will give rise to new heights in single-cell proteomics.
References
- 1.Aldridge S; Teichmann SA, Single cell transcriptomics comes of age. Nature Communications 2020, 11 (1), 4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y; Fonslow BR; Shan B; Baek M-C; Yates JR, Protein Analysis by Shotgun/Bottom-up Proteomics. Chemical Reviews 2013, 113 (4), 2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathys H; Davila-Velderrain J; Peng Z; Gao F; Mohammadi S; Young JZ; Menon M; He L; Abdurrob F; Jiang X; Martorell AJ; Ransohoff RM; Hafler BP; Bennett DA; Kellis M; Tsai L-H, Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570 (7761), 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor MJ; Lukowski JK; Anderton CR, Spatially Resolved Mass Spectrometry at the Single Cell: Recent Innovations in Proteomics and Metabolomics. J. Am. Soc. Mass Spectrom. 2021, 32 (4), 872–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Onjiko RM; Portero EP; Moody SA; Nemes P, In Situ Microprobe Single-Cell Capillary Electrophoresis Mass Spectrometry: Metabolic Reorganization in Single Differentiating Cells in the Live Vertebrate (Xenopus laevis) Embryo. Analytical Chemistry 2017, 89 (13), 7069–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bach K; Pensa S; Zarocsinceva M; Kania K; Stockis J; Pinaud S; Lazarus KA; Shehata M; Simões BM; Greenhalgh AR; Howell SJ; Clarke RB; Caldas C; Halim TYF; Marioni JC; Khaled WT, Time-resolved single-cell analysis of Brca1 associated mammary tumourigenesis reveals aberrant differentiation of luminal progenitors. Nature Communications 2021, 12 (1), 1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aissa AF; Islam ABMMK; Ariss MM; Go CC; Rader AE; Conrardy RD; Gajda AM; Rubio-Perez C; Valyi-Nagy K; Pasquinelli M; Feldman LE; Green SJ; Lopez-Bigas N; Frolov MV; Benevolenskaya EV, Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nature Communications 2021, 12 (1), 1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly RT, Single-cell proteomics: progress and prospects. Mol. Cell. Proteomics 2020, 19 (11), 1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao X; Weng L; Gao M; Zhang X, Single-cell analysis for proteome and related researches. TrAC Trends in Analytical Chemistry 2019, 120, 115666. [Google Scholar]
- 10.Xie H; Ding X, The Intriguing Landscape of Single-Cell Protein Analysis. Adv. Sci. (Weinheim, Ger.) 2022, 9 (12), 2105932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ctortecka C; Mechtler K, The rise of single-cell proteomics. Anal. Sci. Adv. 2021, 2 (3–4), 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Z; Sun L, Recent technical progress in sample preparation and liquid-phase separation-mass spectrometry for proteomic analysis of mass-limited samples. Analytical Methods 2021, 13 (10), 1214–1225. [DOI] [PubMed] [Google Scholar]
- 13.Sun L; Zhu G; Dovichi NJ, Integrated Capillary Zone Electrophoresis–Electrospray Ionization Tandem Mass Spectrometry System with an Immobilized Trypsin Microreactor for Online Digestion and Analysis of Picogram Amounts of RAW 264.7 Cell Lysate. Analytical Chemistry 2013, 85 (8), 4187–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L; Zhu G; Li Y; Yang P; Dovichi NJ, Coupling Methanol Denaturation, Immobilized Trypsin Digestion, and Accurate Mass and Time Tagging for Liquid-Chromatography-Based Shotgun Proteomics of Low Nanogram Amounts of RAW 264.7 Cell Lysate. Analytical Chemistry 2012, 84 (20), 8715–8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whittal RM; Keller BO; Li L, Nanoliter Chemistry Combined with Mass Spectrometry for Peptide Mapping of Proteins from Single Mammalian Cell Lysates. Analytical Chemistry 1998, 70 (24), 5344–5347. [DOI] [PubMed] [Google Scholar]
- 16.Lombard-Banek C; Moody SA; Nemes P, Single-Cell Mass Spectrometry for Discovery Proteomics: Quantifying Translational Cell Heterogeneity in the 16-Cell Frog (Xenopus) Embryo. Angew. Chem., Int. Ed. 2016, 55 (7), 2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lombard-Banek C; Reddy S; Moody SA; Nemes P, Label-free Quantification of Proteins in Single Embryonic Cells with Neural Fate in the Cleavage-Stage Frog (Xenopus laevis) Embryo using Capillary Electrophoresis Electrospray Ionization High-Resolution Mass Spectrometry (CE-ESI-HRMS). Mol. Cell. Proteomics 2016, 15 (8), 2756–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun L; Dubiak KM; Peuchen EH; Zhang Z; Zhu G; Huber PW; Dovichi NJ, Single Cell Proteomics Using Frog (Xenopus laevis) Blastomeres Isolated from Early Stage Embryos, Which Form a Geometric Progression in Protein Content. Anal. Chem. (Washington, DC, U. S.) 2016, 88 (13), 6653–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun L; Bertke MM; Champion MM; Zhu G; Huber PW; Dovichi NJ, Quantitative proteomics of Xenopus laevis embryos: expression kinetics of nearly 4000 proteins during early development. Scientific Reports 2014, 4 (1), 4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lombard-Banek C; Moody SA; Manzini MC; Nemes P, Microsampling Capillary Electrophoresis Mass Spectrometry Enables Single-Cell Proteomics in Complex Tissues: Developing Cell Clones in Live Xenopus laevis and Zebrafish Embryos. Anal. Chem. (Washington, DC, U. S.) 2019, 91 (7), 4797–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saha-Shah A; Esmaeili M; Sidoli S; Hwang H; Yang J; Klein PS; Garcia BA, Single Cell Proteomics by Data-Independent Acquisition To Study Embryonic Asymmetry in Xenopus laevis. Anal. Chem. (Washington, DC, U. S.) 2019, 91 (14), 8891–8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi SB; Polter AM; Nemes P, Patch-Clamp Proteomics of Single Neurons in Tissue Using Electrophysiology and Subcellular Capillary Electrophoresis Mass Spectrometry. Anal. Chem. (Washington, DC, U. S.) 2022, 94 (3), 1637–1644. [DOI] [PubMed] [Google Scholar]
- 23.Budnik B; Levy E; Harmange G; Slavov N, SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biology 2018, 19 (1), 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Specht H; Harmange G; Perlman DH; Emmott E; Niziolek Z; Budnik B; Slavov N, Automated sample preparation for high-throughput single-cell proteomics. bioRxiv, Syst. Biol. 2018, 1–19. [Google Scholar]
- 25.Wu R; Xing S; Badv M; Didar TF; Lu Y, Step-Wise Assessment and Optimization of Sample Handling Recovery Yield for Nanoproteomic Analysis of 1000 Mammalian Cells. Analytical Chemistry 2019, 91 (16), 10395–10400. [DOI] [PubMed] [Google Scholar]
- 26.Sun B; Kumar S, Protein Adsorption Loss─The Bottleneck of Single-Cell Proteomics. Journal of Proteome Research 2022. [DOI] [PubMed] [Google Scholar]
- 27.Cohen D; Dickerson JA; Whitmore CD; Turner EH; Palcic MM; Hindsgaul O; Dovichi NJ, Chemical Cytometry: Fluorescence-Based Single-Cell Analysis. Annual Review of Analytical Chemistry 2008, 1 (1), 165–190. [DOI] [PubMed] [Google Scholar]
- 28.Li Z-Y; Huang M; Wang X-K; Zhu Y; Li J-S; Wong CCL; Fang Q, Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis. Anal. Chem. (Washington, DC, U. S.) 2018, 90 (8), 5430–5438. [DOI] [PubMed] [Google Scholar]
- 29.Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian W-J; Kelly RT, Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nature Communications 2018, 9 (1), 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang Y; Acor H; McCown MA; Nwosu AJ; Boekweg H; Axtell NB; Truong T; Cong Y; Payne SH; Kelly RT, Fully Automated Sample Processing and Analysis Workflow for Low-Input Proteome Profiling. Analytical Chemistry 2021, 93 (3), 1658–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woo J; Williams SM; Markillie LM; Feng S; Tsai C-F; Aguilera-Vazquez V; Sontag RL; Moore RJ; Hu D; Mehta HS; Cantlon-Bruce J; Liu T; Adkins JN; Smith RD; Clair GC; Pasa-Tolic L; Zhu Y, High-throughput and high-efficiency sample preparation for single-cell proteomics using a nested nanowell chip. Nat. Commun. 2021, 12 (1), 6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leduc A; Huffman RG; Slavov N, Droplet sample preparation for single-cell proteomics applied to the cell cycle. bioRxiv 2021, 2021.04.24.441211. [Google Scholar]
- 33.Petelski AA; Emmott E; Leduc A; Huffman RG; Specht H; Perlman DH; Slavov N, Multiplexed single-cell proteomics using SCoPE2. Nat. Protoc. 2021, 16 (12), 5398–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao X; Wang X; Guan S; Lin H; Yan G; Gao M; Deng C; Zhang X, Integrated Proteome Analysis Device for Fast Single-Cell Protein Profiling. Analytical Chemistry 2018, 90 (23), 14003–14010. [DOI] [PubMed] [Google Scholar]
- 35.Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J, Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology 2014, 10 (10), 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Z; Zhang Z; Chen D; Xu T; Wang Y; Sun L, Nanoparticle-Aided Nanoreactor for Nanoproteomics. Analytical Chemistry 2021, 93 (30), 10568–10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z; Dubiak KM; Huber PW; Dovichi NJ, Miniaturized Filter-Aided Sample Preparation (MICRO-FASP) Method for High Throughput, Ultrasensitive Proteomics Sample Preparation Reveals Proteome Asymmetry in Xenopus laevis Embryos. Analytical Chemistry 2020, 92 (7), 5554–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kostas JC; Greguš M; Schejbal J; Ray S; Ivanov AR, Simple and Efficient Microsolid-Phase Extraction Tip-Based Sample Preparation Workflow to Enable Sensitive Proteomic Profiling of Limited Samples (200 to 10,000 Cells). Journal of Proteome Research 2021, 20 (3), 1676–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burns AP; Zhang Y-Q; Xu T; Wei Z; Yao Q; Fang Y; Cebotaru V; Xia M; Hall MD; Huang R; Simeonov A; LeClair CA; Tao D, A Universal and High-Throughput Proteomics Sample Preparation Platform. Analytical Chemistry 2021, 93 (24), 8423–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schoof EM; Furtwangler B; Uresin N; Rapin N; Savickas S; Gentil C; Lechman E; Keller U. a. d.; Dick JE; Porse BT, Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021, 12 (1), 3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu W; Smith JW; Huang C-M, Mass spectrometry-based label-free quantitative proteomics. J. Biomed. Biotechnol. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gebreyesus ST; Siyal AA; Kitata RB; Chen ES-W; Enkhbayar B; Angata T; Lin K-I; Chen Y-J; Tu H-L, Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry. Nat. Commun. 2022, 13 (1), 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webber KGI; Truong T; Johnston SM; Zapata SE; Liang Y; Davis JM; Buttars AD; Smith FB; Jones HE; Mahoney AC; Carson RH; Nwosu AJ; Heninger JL; Liyu AV; Nordin GP; Zhu Y; Kelly RT, Label-Free Profiling of up to 200 Single-Cell Proteomes per Day Using a Dual-Column Nanoflow Liquid Chromatography Platform. Anal. Chem. (Washington, DC, U. S.) 2022, 94 (15), 6017–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams SM; Liyu AV; Tsai C-F; Moore RJ; Orton DJ; Chrisler WB; Gaffrey MJ; Liu T; Smith RD; Kelly RT; Pasa-Tolic L; Zhu Y, Automated Coupling of Nanodroplet Sample Preparation with Liquid Chromatography-Mass Spectrometry for High-Throughput Single-Cell Proteomics. Anal. Chem. (Washington, DC, U. S.) 2020, 92 (15), 10588–10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jonathon JOB; Harsha PG; Joao AP; Xian C; Joseph GI; Steven PG; Bahjat FQ, The effects of nonignorable missing data on label-free mass spectrometry proteomics experiments. The Annals of Applied Statistics 2018, 12 (4), 2075–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paša-Tolić L; Masselon C; Barry RC; Shen Y; Smith RD, Proteomic analyses using an accurate mass and time tag strategy. BioTechniques 2004, 37 (4), 621–639. [DOI] [PubMed] [Google Scholar]
- 47.Tyanova S; Temu T; Cox J, The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols 2016, 11 (12), 2301–2319. [DOI] [PubMed] [Google Scholar]
- 48.Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M, Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ *. Molecular & Cellular Proteomics 2014, 13 (9), 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rauniyar N; Yates JR, Isobaric Labeling-Based Relative Quantification in Shotgun Proteomics. Journal of Proteome Research 2014, 13 (12), 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Specht H; Emmott E; Petelski AA; Huffman RG; Perlman DH; Serra M; Kharchenko P; Koller A; Slavov N, Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021, 22 (1), 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai C-F; Zhao R; Williams SM; Moore RJ; Schultz K; Chrisler WB; Pasa-Tolic L; Rodland KD; Smith RD; Shi T; Zhu Y; Liu T, An improved boosting to amplify signal with isobaric labeling (iBASIL) strategy for precise quantitative single-cell proteomics. Mol. Cell. Proteomics 2020, 19 (5), 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yi L; Tsai C-F; Dirice E; Swensen AC; Chen J; Shi T; Gritsenko MA; Chu RK; Piehowski PD; Smith RD; Rodland KD; Atkinson MA; Mathews CE; Kulkarni RN; Liu T; Qian W-J, Boosting to Amplify Signal with Isobaric Labeling (BASIL) Strategy for Comprehensive Quantitative Phosphoproteomic Characterization of Small Populations of Cells. Analytical Chemistry 2019, 91 (9), 5794–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dou M; Clair G; Tsai C-F; Xu K; Chrisler WB; Sontag RL; Zhao R; Moore RJ; Liu T; Pasa-Tolic L; Smith RD; Shi T; Adkins JN; Qian W-J; Kelly RT; Ansong C; Zhu Y, High-Throughput Single Cell Proteomics Enabled by Multiplex Isobaric Labeling in a Nanodroplet Sample Preparation Platform. Anal. Chem. (Washington, DC, U. S.) 2019, 91 (20), 13119–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sandberg A; Branca RMM; Lehtiö J; Forshed J, Quantitative accuracy in mass spectrometry based proteomics of complex samples: The impact of labeling and precursor interference. Journal of Proteomics 2014, 96, 133–144. [DOI] [PubMed] [Google Scholar]
- 55.Ow SY; Salim M; Noirel J; Evans C; Rehman I; Wright PC, iTRAQ Underestimation in Simple and Complex Mixtures: “The Good, the Bad and the Ugly”. Journal of Proteome Research 2009, 8 (11), 5347–5355. [DOI] [PubMed] [Google Scholar]
- 56.Brenes A; Hukelmann J; Bensaddek D; Lamond AI, Multibatch TMT Reveals False Positives, Batch Effects and Missing Values*. Molecular & Cellular Proteomics 2019, 18 (10), 1967–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ye Z; Batth TS; Ruther P; Olsen JV, A deeper look at carrier proteome effects for single-cell proteomics. Commun. Biol. 2022, 5 (1), 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen Y; Zhao R; Berger SJ; Anderson GA; Rodriguez N; Smith RD, High-Efficiency Nanoscale Liquid Chromatography Coupled On-Line with Mass Spectrometry Using Nanoelectrospray Ionization for Proteomics. Analytical Chemistry 2002, 74 (16), 4235–4249. [DOI] [PubMed] [Google Scholar]
- 59.Zhu Y; Zhao R; Piehowski PD; Moore RJ; Lim S; Orphan VJ; Paša-Tolić L; Qian W-J; Smith RD; Kelly RT, Subnanogram proteomics: Impact of LC column selection, MS instrumentation and data analysis strategy on proteome coverage for trace samples. International Journal of Mass Spectrometry 2018, 427, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cong Y; Liang Y; Motamedchaboki K; Huguet R; Truong T; Zhao R; Shen Y; Lopez-Ferrer D; Zhu Y; Kelly RT, Improved Single-Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Analytical Chemistry 2020, 92 (3), 2665–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiang P; Zhu Y; Yang Y; Zhao Z; Williams SM; Moore RJ; Kelly RT; Smith RD; Liu S, Picoflow Liquid Chromatography-Mass Spectrometry for Ultrasensitive Bottom-Up Proteomics Using 2-μm-i.d. Open Tubular Columns. Anal. Chem. (Washington, DC, U. S.) 2020, 92 (7), 4711–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stejskal K; Op de Beeck J; Dürnberger G; Jacobs P; Mechtler K, Ultrasensitive NanoLC-MS of Subnanogram Protein Samples Using Second Generation Micropillar Array LC Technology with Orbitrap Exploris 480 and FAIMS PRO. Analytical Chemistry 2021, 93 (25), 8704–8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou W; Zhang B; Liu Y; Wang C; Sun W; Li W; Chen Z, Advances in capillary electrophoresis-mass spectrometry for cell analysis. TrAC Trends in Analytical Chemistry 2019, 117, 316–330. [Google Scholar]
- 64.Hirayama A; Abe H; Yamaguchi N; Tabata S; Tomita M; Soga T, Development of a sheathless CE-ESI-MS interface. ELECTROPHORESIS 2018, 39 (11), 1382–1389. [DOI] [PubMed] [Google Scholar]
- 65.Sun L; Zhu G; Zhao Y; Yan X; Mou S; Dovichi NJ, Ultrasensitive and Fast Bottom-up Analysis of Femtogram Amounts of Complex Proteome Digests. Angewandte Chemie International Edition 2013, 52 (51), 13661–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Au - Grant PA; Au - Herold MB; Au - Moody SA, Blastomere Explants to Test for Cell Fate Commitment During Embryonic Development. JoVE 2013, (71), e4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson KR; Greguš M; Kostas JC; Ivanov AR, Capillary Electrophoresis Coupled to Electrospray Ionization Tandem Mass Spectrometry for Ultra-Sensitive Proteomic Analysis of Limited Samples. Analytical Chemistry 2022, 94 (2), 704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leclercq L; Renard C; Martin M; Cottet H, Quantification of Adsorption and Optimization of Separation of Proteins in Capillary Electrophoresis. Analytical Chemistry 2020, 92 (15), 10743–10750. [DOI] [PubMed] [Google Scholar]
- 69.Zhu G; Sun L; Dovichi NJ, Thermally-initiated free radical polymerization for reproducible production of stable linear polyacrylamide coated capillaries, and their application to proteomic analysis using capillary zone electrophoresis–mass spectrometry. Talanta 2016, 146, 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han X; Wang Y; Aslanian A; Fonslow B; Graczyk B; Davis TN; Yates JR, In-Line Separation by Capillary Electrophoresis Prior to Analysis by Top-Down Mass Spectrometry Enables Sensitive Characterization of Protein Complexes. Journal of Proteome Research 2014, 13 (12), 6078–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hajba L; Guttman A, Recent advances in column coatings for capillary electrophoresis of proteins. TrAC Trends in Analytical Chemistry 2017, 90, 38–44. [Google Scholar]
- 72.Yang Z; Shen X; Chen D; Sun L, Microscale Reversed-Phase Liquid Chromatography/Capillary Zone Electrophoresis-Tandem Mass Spectrometry for Deep and Highly Sensitive Bottom–Up Proteomics: Identification of 7500 Proteins with Five Micrograms of an MCF7 Proteome Digest. Analytical Chemistry 2018, 90 (17), 10479–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Z; Sun L; Zhu G; Cox OF; Huber PW; Dovichi NJ, Nearly 1000 Protein Identifications from 50 ng of Xenopus laevis Zygote Homogenate Using Online Sample Preparation on a Strong Cation Exchange Monolith Based Microreactor Coupled with Capillary Zone Electrophoresis. Analytical Chemistry 2016, 88 (1), 877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y; Fonslow BR; Wong CCL; Nakorchevsky A; Yates JR, Improving the Comprehensiveness and Sensitivity of Sheathless Capillary Electrophoresis–Tandem Mass Spectrometry for Proteomic Analysis. Analytical Chemistry 2012, 84 (20), 8505–8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang L; Wang Z; Cupp-Sutton KA; Smith K; Wu S, Spray-Capillary: An Electrospray-Assisted Device for Quantitative Ultralow-Volume Sample Handling. Analytical Chemistry 2020, 92 (1), 640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang L; Fang M; Cupp-Sutton KA; Wang Z; Smith K; Wu S, Spray-Capillary-Based Capillary Electrophoresis Mass Spectrometry for Metabolite Analysis in Single Cells. Analytical Chemistry 2021, 93 (10), 4479–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ctortecka C; Stejskal K; Krssakova G; Mendjan S; Mechtler K, Quantitative Accuracy and Precision in Multiplexed Single-Cell Proteomics. Anal. Chem. (Washington, DC, U. S.) 2022, 94 (5), 2434–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Røberg-Larsen H; Lundanes E; Nyman TA; Berven FS; Wilson SR, Liquid chromatography, a key tool for the advancement of single-cell omics analysis. Analytica Chimica Acta 2021, 1178, 338551. [DOI] [PubMed] [Google Scholar]
- 79.Tomlinson A; Guzman N; Naylor S, Enhancement of concentration limits of detection in CE and CE-MS: a review of on-line sample extraction, cleanup, analyte preconcentration, and microreactor technology. Journal of capillary electrophoresis 1995, 2, 247–66. [PubMed] [Google Scholar]
- 80.Swearingen KE; Moritz RL, High-field asymmetric waveform ion mobility spectrometry for mass spectrometry-based proteomics. Expert Review of Proteomics 2012, 9 (5), 505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cong Y; Motamedchaboki K; Misal SA; Liang Y; Guise AJ; Truong T; Huguet R; Plowey ED; Zhu Y; Lopez-Ferrer D; Kelly RT, Ultrasensitive single-cell proteomics workflow identifies >1000 protein groups per mammalian cell. Chem. Sci. 2021, 12 (3), 1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Woo J; Clair GC; Williams SM; Feng S; Tsai C-F; Moore RJ; Chrisler WB; Smith RD; Kelly RT; Pasa-Tolic L; Ansong C; Zhu Y, Three-dimensional feature matching improves coverage for single-cell proteomics based on ion mobility filtering. Cell Syst. 2022, 13 (5), 426–434.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shen B; Pade LR; Choi SB; Munoz-Llancao P; Manzini MC; Nemes P, Capillary electrophoresis mass spectrometry for scalable single-cell proteomics. Front. Chem. (Lausanne, Switz.) 2022, 10, 863979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Virant-Klun I; Leicht S; Hughes C; Krijgsveld J, Identification of Maturation-Specific Proteins by Single-Cell Proteomics of Human Oocytes. Mol. Cell. Proteomics 2016, 15 (8), 2616–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu Y; Clair G; Chrisler WB; Shen Y; Zhao R; Shukla AK; Moore RJ; Misra RS; Pryhuber GS; Smith RD; Ansong C; Kelly RT, Proteomic Analysis of Single Mammalian Cells Enabled by Microfluidic Nanodroplet Sample Preparation and Ultrasensitive NanoLC-MS. Angewandte Chemie International Edition 2018, 57 (38), 12370–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhu Y; Scheibinger M; Ellwanger DC; Krey JF; Choi D; Kelly RT; Heller S; Barr-Gillespie PG, Single-cell proteomics reveals changes in expression during hair-cell development. eLife 2019, 8, e50777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsai C-F; Zhang P; Scholten D; Martin K; Wang Y-T; Zhao R; Chrisler WB; Patel DB; Dou M; Jia Y; Reduzzi C; Liu X; Moore RJ; Burnum-Johnson KE; Lin M-H; Hsu C-C; Jacobs JM; Kagan J; Srivastava S; Rodland KD; Steven Wiley H; Qian W-J; Smith RD; Zhu Y; Cristofanilli M; Liu T; Liu H; Shi T, Surfactant-assisted one-pot sample preparation for label-free single-cell proteomics. Commun. Biol. 2021, 4 (1), 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brunner A-D; Thielert M; Vasilopoulou C; Ammar C; Coscia F; Mund A; Hoerning OB; Bache N; Apalategui A; Lubeck M; Richter S; Fischer DS; Raether O; Park MA; Meier F; Theis FJ; Mann M, Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol. 2022, 18 (3), e10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Z; Dubiak KM; Shishkova E; Huber PW; Coon JJ; Dovichi NJ, High-Throughput, Comprehensive Single-Cell Proteomic Analysis of Xenopus laevis Embryos at the 50-Cell Stage Using a Microplate-Based MICROFASP System. Anal. Chem. (Washington, DC, U. S.) 2022, 94 (7), 3254–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]