

Abstract
Pituitary tumors are generally benign, although in rare cases aggressive pituitary tumors (APTs) and carcinomas present important diagnostic and therapeutic challenges and are associated with a high mortality rate. Almost half of these APTs and carcinomas are corticotroph tumors, suggesting a specific prognosis. Clinical, pathological and molecular prognostic markers are limited and do not allow early management of these tumors. Temozolomide remains the first‐line treatment once a diagnosis of aggressive pituitary tumor or carcinoma has been made. Novel alternative treatments exist, including immune checkpoint inhibitors, which can be used in the case of temozolomide treatment failure. The aim of this review is to present the clinical, pathological and molecular characteristics of aggressive corticotroph tumors and carcinomas, and to describe the results obtained with currently available treatments.
Keywords: aggressive pituitary tumor, corticotroph tumor, Cushing's disease, pituitary carcinoma, temozolomide
This review presents a complete overview of aggressive corticotroph tumors and carcinomas, with a special focus on their histopathology, molecular prognostic factors, and treatment.
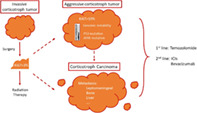
1. INTRODUCTION
Corticotroph tumors represent 4%–8% of all clinically secreting pituitary tumor types. Most of these are microadenomas, responsible for cortisol excess that may be cured after trans‐sphenoidal surgery; however, about 10% of secreting corticotroph tumors are macroadenomas associated with a poor surgical prognosis. 1 Including silent corticotroph adenomas, defined as pituitary tumors without clinical evidence of Cushing's disease, corticotroph tumors overall represent up to 15% of all pituitary neuroendocrine tumors (Pit‐NETs). 2
Aggressive pituitary tumors (APT) are defined as invasive tumors which present with multiple recurrence and do not respond to standard therapies (standard medical treatments such as dopamine agonists and somatostatin analogs, surgery and radiotherapy). 3 These tumors lead to increased morbidity and mortality, and are suspected in the case of unusually rapid or symptomatic tumor growth, despite appropriated treatments including surgery. Pituitary carcinoma (PC) is defined when distant metastases are identified, and is associated with poor survival (median survival <4 years after diagnosis of distant metastasis). 4 The epidemiology of this rare entity is not well‐described; aggressive pituitary tumors seem to represent <2% of macroadenomas, while pituitary carcinomas represent <0.1% of pituitary tumors. Despite the rarity of this clinical situation, all published studies confirm that corticotrophic adenomas are overrepresented in APT and PC, representing 30%–50% of aggressive pituitary tumors or pituitary carcinomas. 5 , 6 This review, which forms part of a special issue on “Update of Cushing's Syndrome: 100 years after Minnie G”, will focus on the clinical, pathological and molecular characteristics of aggressive corticotroph tumors and carcinomas, and describe results obtained with current treatments.
2. CLINICAL CHARACTERISTICS
A recent review of the literature revealed that corticotroph carcinomas represent 34.7% of all published pituitary carcinomas. 7 Most of these cases were case reports; however, recent evaluation of temozolomide efficacy to treat these tumors permits the description of numerous cases. 5 , 8 , 9 At initial diagnosis, no clinical marker can predict the occurrence of metastasis during follow‐up; however, progression from microadenoma to carcinoma is extremely rare. The time from diagnosis to presentation with aggressive behavior, or occurrence of metastasis, is highly variable being from months to years. The diagnosis of a carcinoma usually occurs in the fourth decade and, contrary to what is seen in benign corticotroph tumors, there is no male or female preponderance, suggesting that macrocorticotroph tumors in male may be associated with poor prognosis. The proportion of silent corticotroph adenomas also seems higher than expected, but more importantly the change from initially silent to functional corticotroph tumor seems to be the main prognostic marker that is predictive of malignancy. In such a situation, or in patients with aggressive pituitary tumors and either site‐specific symptoms suggestive of secondary localizations (i.e., back pain, neurological complaints etc.) or discordant biochemical and radiological findings, the European Society of Endocrinology (ESE) guidelines recommend performing an extension assessment to look for metastatic disease. 3 Most secondary tumor localizations are intracranial or spinal, but liver, cervical lymph nodes and bone, and in rare cases, lung, endolymphatic sac, or orbit can be affected. 6
The impact of bilateral adrenalectomy (BADX) on corticotroph tumor progression has been recently reviewed 10 and its implication on corticotroph tumor behavior is uncertain. Indeed, patients with aggressive corticotroph tumors are more prone to undergo BADX and no study has thus far confirmed that loss of feedback inhibition accelerates tumor growth or induces metastasis. These tumors might be either particularly sensitive to loss of feedback inhibition after BADX or may exhibit a distinct intrinsic aggressiveness. In summary, clinical markers in the case of these tumors are limited and should be combined with histopathological or molecular markers.
3. HISTOPATHOLOGY OF CORTICOTROPH AGGRESSIVE TUMORS AND CARCINOMAS
The 2017 World Health Organization (WHO) classification defines corticotroph tumors as neoplasms derived from the T‐PIT‐driven cell lineage. 11 T‐PIT (encoded by TBX19 or T‐Box transcription factor 19 gene) is a critical transcription factor involved in the differentiation of corticotroph cells. Its interaction with other cofactors, such as NeuroD1, is required to activate the pro‐opiomelanocortin (POMC) gene. 12 The detection of T‐PIT by immunohistochemistry is now available and can be used in challenging cases, especially in hormone‐immunonegative tumors. 13 The prototypical example of a corticotroph tumor is the microadenoma that causes Cushing's disease. Histologically, this T‐PIT‐positive tumor is composed of granular basophilic cells showing diffuse cytoplasmic expression of low‐molecular‐weight cytokeratins (LMWCKs) and ACTH. These tumors usually express the somatostatin receptor (SSTR5) which is a target of pasireotide, a somatostatin analog. 14 Corticotroph APTs and carcinomas may also show the same histopathological features (Figures 1A–C and 3A,B).
FIGURE 1.
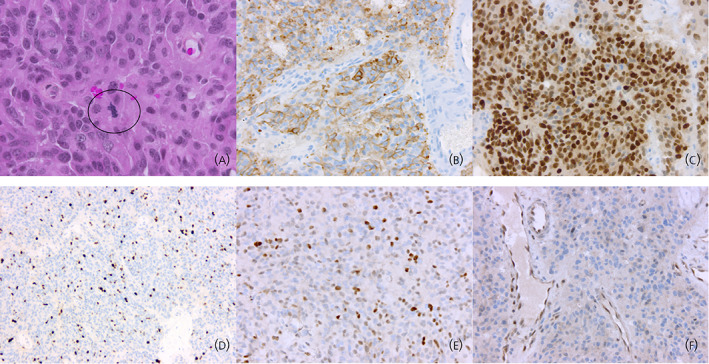
Histopathology of corticotroph carcinomas. (A) Corticotroph carcinomas are composed of basophilic cells. Nuclear atypia may be found but is not a reliable marker of malignancy. Elevated mitotic activity is a common finding but is not observed in all cases (circle: mitosis) (hematoxylin, phloxine, saffron staining, original magnification [OM] × 400). (B) Expression of ACTH in corticotroph carcinomas is variable and may be heterogeneous (ACTH immunohistochemistry, OM × 200). (C) Corticotroph carcinomas express the T‐PIT transcription factor as do all other tumors of the corticotroph lineage (T‐PIT immunohistochemistry, OM × 200). (D) In corticotroph carcinomas, Ki67 proliferation index is usually high, exceeding 3% (in this example, around 15%) (Ki67 immunohistochemistry, OM × 100). (E) Strong nuclear immunopositivity for P53 is suggestive of an aggressive behavior and is in some cases related to TP53 gene mutations (P53 immunohistochemistry, OM × 200). (F) In this example, MGMT nuclear expression is lost in most neoplastic cells (internal positive control: endothelial cells). For some authors, this may be predictive of sensitivity to temozolomide treatment (MGMT immunohistochemistry, OM × 200).
FIGURE 3.
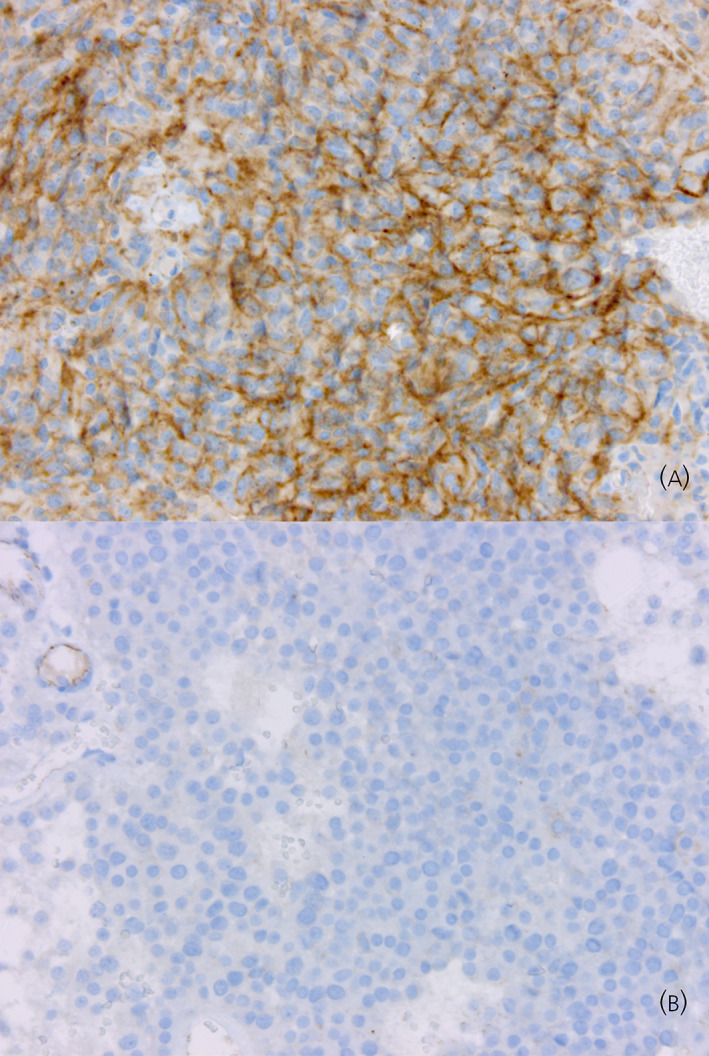
SSTR5 expression in aggressive corticotroph tumors and carcinomas (SSTR5 immunohistochemistry, Original magnification × 200). (A) Strong SSTR5 immunopositivity in a corticotroph carcinoma with Cushing's disease. (B) Absence of SSTR5 immunoexpression in a T‐PIT+/GATA3+ silent corticotroph tumor.
As for other Pit‐NETs, the development of metastases is required to qualify a corticotroph tumor as a “carcinoma”. Identifying corticotroph tumors that may follow an aggressive course solely using histopathology is challenging and controversial. No single histopathological characteristic can accurately predict a malignant or aggressive course. Currently, the use of multiple clinical and pathological parameters is a means to identify those tumors that have a high risk of an unfavorable course.
In pituitary carcinomas overall, the mitotic count is usually greater than 2 per 10 high‐power fields 5 and the median Ki67 proliferation index 10% 5 (Figure 1D). P53 immunopositivity is typically more prevalent in pituitary carcinomas and does not significantly differ from what is seen in APTs 5 (Figure 1E). In corticotroph carcinomas, mitotic activity and Ki67 proliferation index are typically higher in the metastatic lesion than in the primary tumor. 15 P53 immunoreactivity also follows this trend. These observations have led to the proposal that pituitary tumors with increased mitotic activity, elevated Ki67 proliferation index (>3%), and diffuse P53 immunoreactivity should be termed “atypical adenomas” in earlier WHO classifications. However, this definition was not precise enough to allow for easy and reproducible use, and no clear clinicopathological correlation emerged from this concept. Consequently, the term “atypical adenoma” was abandoned in the last WHO classification in 2017.
Some authors have developed a “mixed” clinicopathological classification to overcome these difficulties. In the French five‐tiered classification, two criteria are considered: invasiveness assessed by MRI and peroperative surgical observation (1: noninvasive/2: invasive) and proliferation assessed by microscopy (a: nonproliferative/b: proliferative). Proliferation markers used include mitotic count, Ki67 proliferation index, and P53 immunostaining. Several studies have shown that this classification appears to be an interesting tool for identify pituitary tumors with a higher risk of recurrence/progression. 16 , 17 , 18 , 19 , 20
Consequently, the ESE guidelines recommend the analysis of KI67 proliferation index in all pituitary tumors 3 and mitotic count and P53 immunostaining should be assessed in tumors harboring a Ki67 index ≥3%.
The 2017 WHO classification recognizes that some types of pituitary tumors may show a more aggressive course. Among corticotroph tumors, silent corticotroph adenomas and Crooke cell adenomas are considered as “high‐risk” adenomas. 11
3.1. Silent corticotroph adenoma
Silent corticotroph pituitary adenomas (SCAs) are either densely granulated (silent subtype 1) or sparsely granulated (silent subtype 2) corticotroph tumors with no clinical or biochemical features of Cushing's disease. 21 , 22 Histologically, the typical appearance of SCA includes a pseudopapillary architecture, sparsely granulated neoplastic cells (Figure 2A), cytoplasmic expression of LMWCKs, and variable expression of ACTH. Some authors have associated SCA with a poorer prognosis and the 2017 WHO classification classifies these tumors as a subtype of “high‐risk” adenoma. 11 , 23 However, in one meta‐analysis of 297 patients, the risk of recurrence of SCA was not significantly higher than that of other nonfunctioning pituitary adenomas. 24 Interestingly, these tumors are characterized by mixed corticotroph and gonadotroph transcriptomic signatures. 25 The gonadotroph nature of these tumors is demonstrated by the expression of GATA3 together with the corticotroph‐lineage specific transcription factor TPIT 25 (Figure 2B,C).
FIGURE 2.
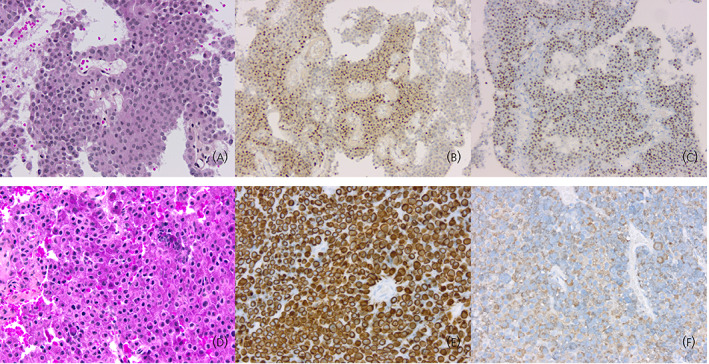
Histotypes of corticotroph aggressive tumors. (A) Silent corticotroph tumors are typically composed of regular cells with chromophobic or slightly basophilic cytoplasm (hematoxylin, phloxine, saffron [HPS] staining, original magnification [OM] × 200). (B) Silent corticotroph tumors express the T‐PIT transcription factor as do all other tumors of the corticotroph lineage (T‐PIT immunohistochemistry, OM × 100). (C) Some silent corticotroph tumors show immunoreactivity for GATA3, a transcription factor also expressed in gonadotroph tumors (GATA3 immunohistochemistry, OM × 100). (D) Crooke cell adenoma is composed of neoplastic cells harboring the ring‐like hyaline change typical of normal corticotroph cells of patients with hypercortisolism (HPS staining, OM × 200). (E) The ring‐like hyaline material corresponds to an accumulation of low‐molecular‐weight cytokeratins (cytokeratin 18 immunohistochemistry, OM × 200). (F) Expression of ACTH in Crooke cell adenomas is typically restricted to the paranuclear and peripheral regions of the cytoplasm (ACTH immunohistochemistry, OM × 200).
3.2. Crooke cell adenoma
Crooke cell adenoma is a very rare subtype of corticotroph tumor. In this tumor, more than 60% of neoplastic cells harbor a ring‐like cytoplasmic accumulation of hyaline material (Figure 2D). 26 This material corresponds to LMWCKs and is typically found in normal corticotroph cells of patients with hypercortisolism (the so‐called “Crooke cells”) (Figure 2E). 27 Crooke cell adenomas are generally macroadenomas associated with Cushing's disease. In comparison to classical macroadenomas with Cushing's disease, they show a higher rate of cavernous sinus invasion but no statistically significant difference regarding the rate of recurrence. 27 Their aggressiveness is thus debatable. Immunoexpression of MGMT in Crooke cell adenomas is mostly weak, in contrast to that seen in classical corticotroph adenomas. 28
Corticotroph tumor progression after bilateral adrenalectomy, known as “Nelson's syndrome”, has a mean prevalence of 43% 10 and is often considered as a severe complication of adrenal surgery in patients with Cushing's disease. In one published study, the pathological characteristics, including mitotic count and Ki67 immunolabelling, were not found to be predictive of tumor progression. 29
4. MOLECULAR PROGNOSTIC FACTORS
A better understanding of the genetic and molecular factors that influence tumor behavior would help clinicians to adapt treatment and follow‐up Pit‐NETs identified as being at risk of aggressiveness. To date there is no strong factor that explains, or is then predictive of corticotroph tumor behavior.
Recent studies have demonstrated that molecular alterations in Pit‐NETs are closely associated with the tumor type. 25 As corticotroph tumors represent the first aggressive Pit‐NETs and present a higher rate of somatic mutations than other types, they are a particular area of interest. Aggressive corticotroph Pit‐NETs are rare and generally develop many years after a Pit‐NET diagnosis, so identification of a strong predictive factor using high power studies are not feasible. Nevertheless, some genetic alterations raise concern, as these have been preferentially found in invasive, large or recurrent tumors, and should thus be further explored.
The most common somatic mutations found in corticotroph Pit‐NETs are UPS8‐activating mutations, first described in 2014. 30 USP8 encodes a deubiquitinating enzyme that impairs EGFR degradation and induces activation of the EGFR pathway and then POMC transcription. USP8 mutations are found in around 40% of Cushing's disease cases 31 and are associated mainly with small and highly secreting tumors. Some studies have reported higher post‐surgical remission of these tumors but an increased risk of recurrence. 32 USP8 mutations have also been exceptionally described in a pituitary carcinoma 33 and do not appear to be associated with aggressive behavior.
USP48 activating mutations have been described in 10%–20% of corticotroph Pit‐NETs and these lead to activation of the Hedgehog signaling pathway via deubiquitination of Gli1. They appear to be associated with smaller tumor size 34 but a recent study of 46 tumors reported 50% rate of invasion in tumors carrying USP48 mutations versus 4% in wild‐type tumors. 35
Some somatic mutations in NR3C1, which encodes the glucocorticoid receptor, have been reported in around 6% of secreting corticotroph tumors. 31 These mutations may reduce glucocorticoid receptor expression, its ability to suppress POMC and ACTH secretion, and induce increased proliferation. 36 An association with aggressiveness was suspected, since it had already been described once in a Nelson syndrome patient who required an emergency craniotomy despite two trans‐sphenoidal surgeries and one radiation treatment, 37 but the association was never confirmed.
A role for impaired expression of CABLES1 in the development of corticotroph tumors has also recently been described. 38 This cell cycle regulator is activated by glucocorticoids in corticotroph cells and its impaired expression has been described in corticotroph tumors. In a recent cohort study, germline missense mutations were found in 2/146 children and somatic missense mutations in 2/35 young adults screened. 39 All were USP8 wild‐type, had large and proliferative tumors, three required a second surgery and one required radiotherapy, so this rare mutation may be associated with a more aggressive pattern.
On rare occasions BRAF mutations have been described in corticotroph Pit‐NETs, inducing an activation of ERK signaling. One study reported BRAF mutations in 16% of Cushing's disease subjects 40 however this high proportion was not later confirmed 34 , 35 and the impact on prognosis is unknown.
Recently, mutations in the tumor suppressor gene PTEN, which induces AKT/PI3K pathway upregulation, was found in a liver metastasis of an ACTH‐secreting carcinoma, 41 and had been previously described in a single case of ACTH carcinoma. 42
Chromosomal alterations and copy number variations (CNV) are quite frequently found in Pit‐NETs, especially in corticotroph tumors. 25 , 43 , 44 Corticotroph tumors present a wide range in the quantity of CNVs, from no alteration to almost the entire genome being altered. The quantity of chromosomal alterations was reported to be associated with invasion in a cohort of 27 pediatric Cushing's disease subjects, 45 and with macroadenomas, silent or Crooke's cell tumors, in another recent study on 22 patients. 33 In our study on 33 cortricotroph tumors, a higher level of CNVs seemed to be associated with macroadenomas, without reaching statistical significance, but was not associated with invasion, with silent tumors, nor with USP8 mutations. 44 Overall, we also failed to find an association between tumor recurrence at 5 years and the quantity of CNVs.
In rare cases, corticotroph tumors can occur as a familial form. Germ‐line mutations of MEN1 and AIP mainly result in PIT‐ 1‐related PitNETs that are resistant to medical treatments. To date, no increased incidence of aggressive tumors, and especially aggressive corticotroph tumors, has been reported in patients harboring these germline mutations. 3 , 46 , 47 However, other germline mutations could lead to more aggressive forms.
A recent study reported three pituitary tumors in 910 patients harboring microsatellite instability due to Lynch syndrome. One patient had a large invasive nonfunctioning macroadenoma (histopathological analysis not available) and one patient developed a corticotroph pituitary carcinoma. 48 More data are required to reach a conclusion on the relationship between an impaired MMR system and aggressive pituitary tumors, but a pituitary tumor which occurs in a patient with Lynch syndrome should be closely monitored.
In a cohort of corticotroph APTs and carcinomas, a loss of nuclear ATRX (alpha thalassemia/mental retardation syndrome X‐linked) immunoreactivity was found in 32% of cases. 49 This lack of ATRX protein expression resulted from loss‐of‐function mutations in the ATRX gene including nonsense mutations, frameshift indels, and large intragenic deletions. 49 In this study, TP53 mutations were frequently associated with ATRX mutations. Mutations in TP53, a tumor suppressor gene with multiple functions, have been described in corticotroph APTs and carcinomas, including in Nelson's syndrome. 34 , 50 Additionally, TP53 mutations may constitute driver events in USP8‐wild‐type corticotroph tumors. 34 In one study, tumors carrying TP53 mutations showed widespread copy number variants compared with tumors carrying USP8 mutations, suggesting higher genomic instability in the former. 33
5. TREATMENT
5.1. Temozolomide
Temozolomide is currently recommended by the European Society of Endocrinology (ESE) guidelines as the first‐line chemotherapy, after the failure of standard treatment options (i.e., surgery, medical treatments, and radiotherapy), for pituitary carcinomas and aggressive pituitary tumors, irrespective of their histological subtype. 3
In the ESE survey, 5 the response to first‐line temozolomide of 73 corticotroph tumors, of which 19 were corticotroph carcinomas, was reported. Of these, 6 (8%) showed radiological complete response, 22 (30%) partial response, and 20 (27%) stable disease, while 25 (34%) progressed under first‐line temozolomide (Figure 4).
FIGURE 4.
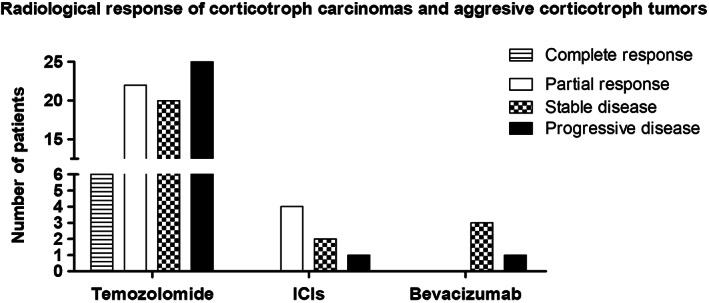
Radiological response of corticotroph carcinomas and aggressive corticotroph tumors treated with temozolomide, immune checkpoint inhibitors (ICIs), and bevacizumab. Derived from data from 5 , 57 , 59 , 60 , 61 , 62 , 63 , 64 , 65
A clinically functioning status, along with concomitant radiotherapy administration and low (<10%) or intermediate (10%–50%) O6‐methylguanine‐DNA methyltransferase (MGMT) immunopositivity, were predictors of a better response to first‐line temozolomide (Figure 2F). 5 , 8 , 51 , 52 Low MGMT also predicted a better overall survival under first‐line temozolomide, and possibly a better response to a second course of treatment. 53 Indeed, it is noteworthy that relapse and/or progression were frequently encountered after an initial response, and that the response to a second course of treatment was less frequently favorable. 5 , 54
Currently, the ESE guidelines recommend a treatment duration of first‐line temozolomide of at least 6 months for responders. 3 Evidence on the use of long‐term temozolomide is still too scarce to enable formal recommendations, but clinical experience points towards the prolonged administration of the drug for as long as it remains effective and well‐tolerated. 55 , 56 In our clinical practice, we administer temozolomide at a standard dosage, as long as it is effective and, if complete response is seen, we then usually reduce dosage to a half‐dose before stopping the drug. 6
The most common adverse events (AEs) reported were fatigue, cytopenia, nausea, and vomiting. Headache, hypotension, edema, abnormal liver function tests, adrenal crisis, and hearing loss, have also been reported. 3 , 5
5.2. Immune checkpoint inhibitors (ICIs)
Seven corticotroph tumors, six of which were corticotroph carcinomas, have so far been treated using ICIs. 57 , 58 , 59 , 60 , 61 The radiological response is summarized in Figure 4.Four functioning corticotroph carcinomas, two that received ipilimumab and nivolumab (followed by nivolumab alone), and two that received pembrolizumab, showed radiological partial response. 57 , 58 , 59 , 61 This radiological partial response was accompanied by biochemical partial response in three of the cases, and by biochemical complete response in one case. 57 , 58 , 59 , 61
One functioning corticotroph carcinoma that was treated with ipilimumab and nivolumab (followed by nivolumab alone) showed radiological stable disease, accompanied by biochemical partial response, 60 while a silent corticotroph carcinoma that received pembrolizumab, showed radiological stable disease, but with clinically relevant tumor growth. 61 Of note, the notion of “clinically relevant tumor growth” defines a tumor growth responsible for new/worsening/imminent neuro‐ophthalmologic signs and symptoms; this notion is independent of the radiological assessment, that is, a “clinically relevant tumor growth” may be seen even in cases with radiological stable disease. 6
The aggressive corticotroph tumor, which was functioning by the time pembrolizumab was administered, showed radiological and biochemical progressive disease. 62
Of note, hypercortisolism was present in two cases, the tumor showing progressive disease and in one tumor showing stable disease, 60 , 62 suggesting endogenous hypercortisolism might indeed lessen the efficacy of ICIs, since hypercortisolism was not noted in any of the responders. This is in line with the known immunosuppressive effect of glucocorticoids. It appears therefore important to try to achieve normal cortisol levels by using, for example, adrenal‐targeted drugs.
AEs included fever, fatigue, rash, myalgia, anorexia, nausea, elevation of liver enzymes, and possibly progressive weight loss and hypophysitis. 57 , 58 , 59 , 60 , 61
In most cancers, PDL1 immunohistochemistry and assessment of the tumor mutational burden (TMB) may be used to predict the response to immunotherapy. Tumors with high PDL1 immunoexpression and TMB would be expected to be the most sensitive to immunotherapy. In the few available cases of corticotroph carcinomas treated by immunotherapy, PDL1 immunoexpression was negative despite partial response or stable disease. 60 , 61 In one case of stable disease, TMB was low. Analysis in larger cohorts is currently lacking to form a conclusion on the relevance of these theranostic markers.
5.3. Bevacizumab
Eight corticotroph tumors, of which five were corticotroph carcinomas, have been treated to date with bevacizumab, an anti‐VEGF monoclonal antibody, alone or in combination with temozolomide (two cases) with the radiological response being available for six cases. 63 , 64 , 65
One functioning corticotroph carcinoma treated concomitantly with bevacizumab, temozolomide, and radiotherapy, showed radiological complete response. The associated biochemical response was not reported. The other corticotroph carcinoma, which was treated concomitantly with bevacizumab and temozolomide, was silent and showed radiological stable disease. 63 , 65
Three corticotroph tumors that were treated with bevacizumab alone showed radiological stable disease, 63 while one progressed. 64 Their responses are shown in Figure 4.Two patients did not present any AEs 65 while for the other cases this information was not available. 63 , 64
5.4. Other treatments
Three corticotroph tumors were treated using everolimus (an mTOR inhibitor), and showed approximately 5 months transient stability in one case, followed by progressive disease, and progressive disease was reported the other two cases. 63 , 64
Two corticotroph tumors were treated with peptide receptor radionuclide therapy, one of which showed radiological progressive disease, while in the second, the radiological response was not evaluated because the patient died due to elevated intracranial pressure shortly after the administration of peptide receptor radionuclide therapy. 63
One corticotroph carcinoma was treated with sunitinib (a multityrosine kinase inhibitor) and showed disease progression. 64
6. CONCLUSION
To conclude, aggressive corticotroph tumors and carcinomas are challenging from their diagnosis to their therapeutic management. Despite limitations, initial pathology can help to identify pituitary tumors with malignant potential; however, clinical follow‐up is still mandatory before initiating chemotherapy. Temozolomide is efficient as first‐line treatment and ICIs may represent a good alternative for resistant corticotroph carcinomas. Despite the recent improvements in our knowledge, identification of early prognostic markers of tumor behavior, tumor response to treatment, and the optimal duration and sequence of treatments are still needed.
This article is part of an update series on the diagnosis and treatment of Cushing's syndrome. 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82
AUTHOR CONTRIBUTIONS
Helene Lasolle: Data curation; writing – original draft; writing – review and editing. Alexandre Vasiljevic: Formal analysis; writing – original draft; writing – review and editing. Emmanuel Jouanneau: Validation; writing – review and editing. Mirela Ilie: Conceptualization; data curation; writing – original draft; writing – review and editing. Gerald Raverot: Conceptualization; data curation; writing – original draft; writing – review and editing.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/jne.13169.
Lasolle H, Vasiljevic A, Jouanneau E, Ilie MD, Raverot G. Aggressive corticotroph tumors and carcinomas. J Neuroendocrinol. 2022;34(8):e13169. doi: 10.1111/jne.13169
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Pivonello R, De Leo M, Cozzolino A, Colao A. The treatment of Cushing's disease. Endocr Rev. 2015;36(4):385‐486. doi: 10.1210/er.2013-1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mete O, Lopes MB. Overview of the 2017 WHO classification of pituitary tumors. Endocr Pathol. 2017;28(3):228‐243. doi: 10.1007/s12022-017-9498-z [DOI] [PubMed] [Google Scholar]
- 3. Raverot G, Burman P, McCormack A, et al. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol. 2018;178(1):G1‐G24. doi: 10.1530/EJE-17-0796 [DOI] [PubMed] [Google Scholar]
- 4. Dekkers OM, Karavitaki N, Pereira AM. The epidemiology of aggressive pituitary tumors (and its challenges). Rev Endocr Metab Disord. 2020;21:209‐212. doi: 10.1007/s11154-020-09556-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McCormack A, Dekkers OM, Petersenn S, et al. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. Eur J Endocrinol. 2018;178(3):265‐276. doi: 10.1530/EJE-17-0933 [DOI] [PubMed] [Google Scholar]
- 6. Raverot G, Ilie MD, Lasolle H, et al. Aggressive pituitary tumours and pituitary carcinomas. Nat Rev Endocrinol. 2021;17:671‐684. doi: 10.1038/s41574-021-00550-w [DOI] [PubMed] [Google Scholar]
- 7. Yoo F, Kuan EC, Heaney AP, Bergsneider M, Wang MB. Corticotrophic pituitary carcinoma with cervical metastases: case series and literature review. Pituitary. 2018;21(3):290‐301. doi: 10.1007/s11102-018-0872-8 [DOI] [PubMed] [Google Scholar]
- 8. Lasolle H, Cortet C, Castinetti F, et al. Temozolomide treatment can improve overall survival in aggressive pituitary tumors and pituitary carcinomas. Eur J Endocrinol. 2017;176(6):769‐777. doi: 10.1530/EJE-16-0979 [DOI] [PubMed] [Google Scholar]
- 9. Elbelt U, Schlaffer SM, Buchfelder M, et al. Efficacy of Temozolomide therapy in patients with aggressive pituitary adenomas and carcinomas‐a German survey. J Clin Endocrinol Metab. 2020;105(3):e660‐e675. doi: 10.1210/clinem/dgz211 [DOI] [PubMed] [Google Scholar]
- 10. Reincke M, Albani A, Assie G, et al. Corticotroph tumor progression after bilateral adrenalectomy (Nelson's syndrome): systematic review and expert consensus recommendations. Eur J Endocrinol. 2021;184(3):P1‐P16. doi: 10.1530/EJE-20-1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. 2017;134(4):521‐535. doi: 10.1007/s00401-017-1769-8 [DOI] [PubMed] [Google Scholar]
- 12. de Moraes DC, Vaisman M, Conceicao FL, Ortiga‐Carvalho TM. Pituitary development: a complex, temporal regulated process dependent on specific transcriptional factors. J Endocrinol. 2012;215(2):239‐245. doi: 10.1530/JOE-12-0229 [DOI] [PubMed] [Google Scholar]
- 13. Villa C, Vasiljevic A, Jaffrain‐Rea ML, et al. A standardised diagnostic approach to pituitary neuroendocrine tumours (PitNETs): a European pituitary pathology group (EPPG) proposal. Virchows Archiv. 2019;475(6):687‐692. doi: 10.1007/s00428-019-02655-0 [DOI] [PubMed] [Google Scholar]
- 14. Castellnou S, Vasiljevic A, Lapras V, et al. SST5 expression and USP8 mutation in functioning and silent corticotroph pituitary tumors. Endocr Connect. 2020;9(3):243‐253. doi: 10.1530/EC-20-0035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gaffey TA, Scheithauer BW, Lloyd RV, et al. Corticotroph carcinoma of the pituitary: a clinicopathological study. Report of four cases. J Neurosurg. 2002;96(2):352‐360. doi: 10.3171/jns.2002.96.2.0352 [DOI] [PubMed] [Google Scholar]
- 16. Trouillas J, Roy P, Sturm N, et al. A new prognostic clinicopathological classification of pituitary adenomas: a multicentric case‐control study of 410 patients with 8 years post‐operative follow‐up. Acta Neuropathol. 2013;126(1):123‐135. doi: 10.1007/s00401-013-1084-y [DOI] [PubMed] [Google Scholar]
- 17. Asioli S, Righi A, Iommi M, et al. Validation of a clinicopathological score for the prediction of post‐surgical evolution of pituitary adenoma: retrospective analysis on 566 patients from a tertiary care Centre. Eur J Endocrinol. 2019;180(2):127‐134. doi: 10.1530/EJE-18-0749 [DOI] [PubMed] [Google Scholar]
- 18. Lelotte J, Mourin A, Fomekong E, Michotte A, Raftopoulos C, Maiter D. Both invasiveness and proliferation criteria predict recurrence of non‐functioning pituitary macroadenomas after surgery: a retrospective analysis of a monocentric cohort of 120 patients. Eur J Endocrinol. 2018;178(3):237‐246. doi: 10.1530/EJE-17-0965 [DOI] [PubMed] [Google Scholar]
- 19. Raverot G, Dantony E, Beauvy J, et al. Risk of recurrence in pituitary neuroendocrine tumors: a prospective study using a five‐tiered classification. J Clin Endocrinol Metab. 2017;102(9):3368‐3374. doi: 10.1210/jc.2017-00773 [DOI] [PubMed] [Google Scholar]
- 20. Guaraldi F, Zoli M, Righi A, et al. A practical algorithm to predict postsurgical recurrence and progression of pituitary neuroendocrine tumours (PitNET)s. Clin Endocrinol (Oxf). 2020;93:36‐43. doi: 10.1111/cen.14197 [DOI] [PubMed] [Google Scholar]
- 21. Ben‐Shlomo A, Cooper O. Silent corticotroph adenomas. Pituitary. 2018;21(2):183‐193. doi: 10.1007/s11102-018-0864-8 [DOI] [PubMed] [Google Scholar]
- 22. Lopez JA, Kleinschmidt‐Demasters Bk B, Sze CI, Woodmansee WW, Lillehei KO. Silent corticotroph adenomas: further clinical and pathological observations. Hum Pathol. 2004;35(9):1137‐1147. [DOI] [PubMed] [Google Scholar]
- 23. Jahangiri A, Wagner JR, Pekmezci M, et al. A comprehensive long‐term retrospective analysis of silent corticotrophic adenomas vs hormone‐negative adenomas. Neurosurgery. 2013;73(1):8‐17; discussion 17‐8. doi: 10.1227/01.neu.0000429858.96652.1e [DOI] [PubMed] [Google Scholar]
- 24. Fountas A, Lavrentaki A, Subramanian A, Toulis KA, Nirantharakumar K, Karavitaki N. Recurrence in silent corticotroph adenomas after primary treatment: a systematic review and meta‐analysis. J Clin Endocrinol Metab. 2019;104(4):1039‐1048. doi: 10.1210/jc.2018-01956 [DOI] [PubMed] [Google Scholar]
- 25. Neou M, Villa C, Armignacco R, et al. Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell. 2020;37(1):123‐134 e5. doi: 10.1016/j.ccell.2019.11.002 [DOI] [PubMed] [Google Scholar]
- 26. George DH, Scheithauer BW, Kovacs K, et al. Crooke's cell adenoma of the pituitary: an aggressive variant of corticotroph adenoma. Am J Surg Pathol. 2003;27(10):1330‐1336. [DOI] [PubMed] [Google Scholar]
- 27. Di Ieva A, Davidson JM, Syro LV, et al. Crooke's cell tumors of the pituitary. Neurosurgery. 2015;76(5):616‐622. doi: 10.1227/NEU.0000000000000657 [DOI] [PubMed] [Google Scholar]
- 28. Takeshita A, Inoshita N, Taguchi M, et al. High incidence of low O(6)‐methylguanine DNA methyltransferase expression in invasive macroadenomas of Cushing's disease. Eur J Endocrinol. 2009;161(4):553‐559. doi: 10.1530/EJE-09-0414 [DOI] [PubMed] [Google Scholar]
- 29. Assie G, Bahurel H, Coste J, et al. Corticotroph tumor progression after adrenalectomy in Cushing's disease: a reappraisal of Nelson's syndrome. J Clin Endocrinol Metab. 2007;92(1):172‐179. doi: 10.1210/jc.2006-1328 [DOI] [PubMed] [Google Scholar]
- 30. Reincke M, Sbiera S, Hayakawa A, et al. Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nat Genet. 2015;47(1):31‐38. doi: 10.1038/ng.3166 [DOI] [PubMed] [Google Scholar]
- 31. Sbiera S, Kunz M, Weigand I, Deutschbein T, Dandekar T, Fassnacht M. The new genetic landscape of Cushing's disease: deubiquitinases in the spotlight. Cancers (Basel). 2019;11(11):1‐14. doi: 10.3390/cancers11111761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Srirangam Nadhamuni V, Korbonits M. Novel insights into pituitary tumorigenesis: genetic and epigenetic mechanisms. Endocr Rev. 2020;41:821‐846. doi: 10.1210/endrev/bnaa006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Uzilov AV, Taik P, Cheesman KC, et al. USP8 and TP53 drivers are associated with CNV in a Corticotroph adenoma cohort enriched for aggressive tumors. J Clin Endocrinol Metab. 2021;106(3):826‐842. doi: 10.1210/clinem/dgaa853 [DOI] [PubMed] [Google Scholar]
- 34. Sbiera S, Perez‐Rivas LG, Taranets L, et al. Driver mutations in USP8 wild‐type Cushing's disease. Neuro Oncol. 2019;21(10):1273‐1283. doi: 10.1093/neuonc/noz109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abraham AP, Pai R, Beno DL, et al. USP8, USP48, and BRAF mutations differ in their genotype‐phenotype correlation in Asian Indian patients with Cushing's disease. Endocrine. 2022;75(2):549‐559. doi: 10.1007/s12020-021-02903-x [DOI] [PubMed] [Google Scholar]
- 36. Miao H, Liu Y, Lu L, et al. Effect of 3 NR3C1 mutations in the pathogenesis of pituitary ACTH adenoma. Endocrinology. 2021;162(11). doi: 10.1210/endocr/bqab167 [DOI] [PubMed] [Google Scholar]
- 37. Karl M, Von Wichert G, Kempter E, et al. Nelson's syndrome associated with a somatic frame shift mutation in the glucocorticoid receptor gene. J Clin Endocrinol Metab. 1996;81(1):124‐129. doi: 10.1210/jcem.81.1.8550738 [DOI] [PubMed] [Google Scholar]
- 38. Roussel‐Gervais A, Couture C, Langlais D, et al. The Cables1 gene in glucocorticoid regulation of pituitary corticotrope growth and Cushing disease. J Clin Endocrinol Metab. 2016;101(2):513‐522. doi: 10.1210/jc.2015-3324 [DOI] [PubMed] [Google Scholar]
- 39. Hernández‐Ramírez LC, Gam R, Valdés N, et al. Loss‐of‐function mutations in the CABLES1 gene are a novel cause of Cushing's disease. Endocr Relat Cancer. 2017;24(8):379‐392. doi: 10.1530/ERC-17-0131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen J, Jian X, Deng S, et al. Identification of recurrent USP48 and BRAF mutations in Cushing's disease. Nat Commun. 2018;9(1):3171. doi: 10.1038/s41467-018-05275-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sumislawski P, Rotermund R, Klose S, et al. ACTH‐secreting pituitary carcinoma with TP53, NF1, ATRX and PTEN mutations Case report and review of the literature. Endocrine. 2022;76:228‐236. doi: 10.1007/s12020-021-02954-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo F, Wang G, Wang F, Xu D, Liu X. Identification of novel genes involved in the pathogenesis of an ACTH‐secreting pituitary carcinoma: a case report and literature review. Front Oncol. 2018;8:510. doi: 10.3389/fonc.2018.00510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bi WL, Horowitz P, Greenwald NF, et al. Landscape of genomic alterations in pituitary adenomas. Clin Cancer Res. 2017;23(7):1841‐1851. doi: 10.1158/1078-0432.CCR-16-0790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lasolle H, Elsensohn M‐H, Wierinckx A, et al. Chromosomal instability in the prediction of pituitary neuroendocrine tumors prognosis. Acta Neuropathol Commun. 2020;8, (1):190. doi: 10.1186/s40478-020-01067-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tatsi C, Pankratz N, Lane J, et al. Large genomic aberrations in corticotropinomas are associated with greater aggressiveness. J Clin Endocrinol Metab. 2019;104(5):1792‐1801. doi: 10.1210/jc.2018-02164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verges B, Boureille F, Goudet P, et al. Pituitary disease in MEN type 1 (MEN1): data from the France‐Belgium MEN1 multicenter study. J Clin Endocrinol Metab. 2002;87(2):457‐465. [DOI] [PubMed] [Google Scholar]
- 47. de Laat JM, Dekkers OM, Pieterman CRC, et al. Long‐term natural course of pituitary tumors in patients with MEN1: results from the DutchMEN1 study group (DMSG). J Clin Endocrinol Metabol. 2015;100(9):3288‐3296. doi: 10.1210/JC.2015-2015 [DOI] [PubMed] [Google Scholar]
- 48. Bengtsson D, Joost P, Aravidis C, et al. Corticotroph pituitary carcinoma in a patient with lynch syndrome (LS) and pituitary tumors in a nationwide LS Cohort. J Clin Endocrinol Metab. 2017;102(11):3928‐3932. doi: 10.1210/jc.2017-01401 [DOI] [PubMed] [Google Scholar]
- 49. Casar‐Borota O, Boldt HB, Engstrom BE, et al. Corticotroph aggressive pituitary tumors and carcinomas frequently harbor ATRX mutations. J Clin Endocrinol Metab. 2021;106(4):1183‐1194. doi: 10.1210/clinem/dgaa749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tanizaki Y, Jin L, Scheithauer BW, Kovacs K, Roncaroli F, Lloyd RV. P53 gene mutations in pituitary carcinomas. Endocr Pathol. 2007;18(4):217‐222. doi: 10.1007/s12022-007-9006-y [DOI] [PubMed] [Google Scholar]
- 51. Bengtsson D, Schroder HD, Andersen M, et al. Long‐term outcome and MGMT as a predictive marker in 24 patients with atypical pituitary adenomas and pituitary carcinomas given treatment with temozolomide. J Clin Endocrinol Metab. 2015;100(4):1689‐1698. doi: 10.1210/jc.2014-4350 [DOI] [PubMed] [Google Scholar]
- 52. Luo M, Tan Y, Chen W, et al. Clinical efficacy of Temozolomide and its predictors in aggressive pituitary tumors and pituitary carcinomas: a systematic review and meta‐analysis. Front Neurol. 2021;12:700007. doi: 10.3389/fneur.2021.700007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bengtsson D, Schrøder HD, Berinder K, et al. Tumoral MGMT content predicts survival in patients with aggressive pituitary tumors and pituitary carcinomas given treatment with temozolomide. Endocrine. 2018;62(3):737‐739. doi: 10.1007/s12020-018-1751-9 [DOI] [PubMed] [Google Scholar]
- 54. Burman P, Lamb L, McCormack A. Temozolomide therapy for aggressive pituitary tumours – current understanding and future perspectives. Rev Endocr Metab Disord. 2020;21(2):263‐276. doi: 10.1007/s11154-020-09551-y [DOI] [PubMed] [Google Scholar]
- 55. Lizzul L, Lombardi G, Barbot M, et al. Long‐course temozolomide in aggressive pituitary adenoma: real‐life experience in two tertiary care centers and review of the literature. Pituitary. 2020;23:359‐366. doi: 10.1007/s11102-020-01040-4 [DOI] [PubMed] [Google Scholar]
- 56. Ilie MD, Jouanneau E, Raverot G. Aggressive pituitary adenomas and carcinomas. Endocrinol Metab Clin North Am. 2020;49(3):505‐515. doi: 10.1016/j.ecl.2020.05.008 [DOI] [PubMed] [Google Scholar]
- 57. Lin AL, Jonsson P, Tabar V, et al. Marked response of a hypermutated ACTH‐secreting pituitary carcinoma to ipilimumab and nivolumab. J Clin Endocrinol Metab. 2018;103(10):3925‐3930. doi: 10.1210/jc.2018-01347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin AL, Tabar V, Young RJ, et al. Synergism of checkpoint inhibitors and peptide receptor radionuclide therapy in the treatment of pituitary carcinoma. J Endocr Soc. 2021;5(10):bvab133. doi: 10.1210/jendso/bvab133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Duhamel C, Ilie MD, Salle H, et al. Immunotherapy in corticotroph and lactotroph aggressive tumors and carcinomas: two case reports and a review of the literature. J Pers Med. 2020;10(3):88. doi: 10.3390/jpm10030088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sol B, de Filette JMK, Awada G, et al. Immune checkpoint inhibitor therapy for ACTH‐secreting pituitary carcinoma: a new emerging treatment? Eur J Endocrinol. 2020;184(1):K1‐K5. doi: 10.1530/EJE-20-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Majd N, Waguespack SG, Janku F, et al. Efficacy of pembrolizumab in patients with pituitary carcinoma: report of four cases from a phase II study. J ImmunoTher Cancer. 2020;8, (2):e001532. doi: 10.1136/jitc-2020-001532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Caccese M, Barbot M, Ceccato F, et al. Rapid disease progression in patient with mismatch‐repair deficiency pituitary ACTH‐secreting adenoma treated with checkpoint inhibitor pembrolizumab. Anticancer Drugs. 2020;31(2):199‐204. doi: 10.1097/CAD.0000000000000856 [DOI] [PubMed] [Google Scholar]
- 63. Ilie MD, Lasolle H, Raverot G. Emerging and novel treatments for pituitary tumors. J Clin Med. 2019;8(8):1‐17. doi: 10.3390/jcm8081107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alshaikh OM, Asa SL, Mete O, Ezzat S. An institutional experience of tumor progression to pituitary carcinoma in a 15‐year cohort of 1055 consecutive pituitary neuroendocrine tumors. Endocr Pathol. 2019;30(2):118‐127. doi: 10.1007/s12022-019-9568-5 [DOI] [PubMed] [Google Scholar]
- 65. Osterhage K, Rotermund R, Droste M, et al. Bevacizumab in aggressive pituitary adenomas – experience with 3 patients. Exp Clin Endocrinol Diabetes. 2021;129(03):178‐185. doi: 10.1055/a-1260-3975 [DOI] [PubMed] [Google Scholar]
- 66. Millar RP, Karavitaki N, Kastelan D. Cushing's syndrome update: 100 years after Minnie G. J Neuroendocrinol. 2022;34(8):e13167. doi: 10.1111/jne.13167 [DOI] [PubMed] [Google Scholar]
- 67. Clayton RN. Cardiovascular complications of Cushings Syndrome: impact on morbidity and mortality. J Neuroendocrinol. 2022;34(8):e13175. doi: 10.1111/jne.13175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Honegger J, Nasi‐Kordhishti I. Surgery and perioperative management of patients with Cushing's disease. J Neuroendocrinol. 2022;34(8):e13177. doi: 10.1111/jne.13177 [DOI] [PubMed] [Google Scholar]
- 69. Balomenaki M, Margaritopoulos D, Vassiliadi DA, Tsagarakis S. Diagnostic workup of Cushing’s syndrome. J Neuroendocrinol. 2022;34(8):e13111. doi: 10.1111/jne.13111 [DOI] [PubMed] [Google Scholar]
- 70. Braun LT, Vogel F, Reincke M. Long‐term morbidity and mortality in patients with Cushing's syndrome. J Neuroendocrinol. 2022;34(8):e13113. doi: 10.1111/jne.13113 [DOI] [PubMed] [Google Scholar]
- 71. Valassi E. Clinical presentation and etiology of Cushing's syndrome: Data from ERCUSYN. J Neuroendocrinol. 2022;34(8):e13114. doi: 10.1111/jne.13114 [DOI] [PubMed] [Google Scholar]
- 72. Hamblin R, Coulden A, Fountas A, Karavitaki N. The diagnosis and management of Cushing's syndrome in pregnancy. J Neuroendocrinol. 2022;34(8):e13118. doi: 10.1111/jne.13118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Castinetti F. Medical management of Cushing's disease: When and how? J Neuroendocrinol. 2022;34(8):e13120. doi: 10.1111/jne.13120 [DOI] [PubMed] [Google Scholar]
- 74. Bonneville J‐F, Potorac I, Petrossians P, Tshibanda L, Beckers A. Pituitary MRI in Cushing's disease ‐ an update. J Neuroendocrinol. 2022;34(8):e13123. doi: 10.1111/jne.13123 [DOI] [PubMed] [Google Scholar]
- 75. Losa M, Albano L, Bailo M, Barzaghi LR, Mortini P. Role of radiosurgery in the treatment of Cushing's disease. J Neuroendocrinol. 2022;34(8):e13134. doi: 10.1111/jne.13134 [DOI] [PubMed] [Google Scholar]
- 76. Hayes AR, Grossman AB. Distinguishing Cushing's disease from the ectopic ACTH syndrome: Needles in a haystack or hiding in plain sight? J Neuroendocrinol. 2022;34(8):e13137. doi: 10.1111/jne.13137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dekkers AJ, Amaya JM, van der Meulen M, Biermasz NR, Meijer OC, Pereira AM. Long‐term effects of glucocorticoid excess on the brain. J Neuroendocrinol. 2022;34(8):e13142. doi: 10.1111/jne.13142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Salehidoost R, Korbonits M. Glucose and lipid metabolism abnormalities in Cushing's syndrome. J Neuroendocrinol. 2022;34(8):e13143. doi: 10.1111/jne.13143 [DOI] [PubMed] [Google Scholar]
- 79. Simon J, Theodoropoulou M. Genetics of Cushing's disease. J Neuroendocrinol. 2022;34(8):e13148. doi: 10.1111/jne.13148 [DOI] [PubMed] [Google Scholar]
- 80. Balasko A, Zibar Tomsic K, Kastelan D, Dusek T. Hypothalamic–pituitary–adrenal axis recovery after treatment of Cushing's syndrome. J Neuroendocrinol. 2022;34(8):e13172. doi: 10.1111/jne.13172 [DOI] [PubMed] [Google Scholar]
- 81. Drouin J. The corticotroph cells from early development to tumorigenesis. J Neuroendocrinol. 2022;34(8):e13147. doi: 10.1111/jne.13147 [DOI] [PubMed] [Google Scholar]
- 82. Guignat L, Bertherat J. Long‐term follow‐up and predictors of recurrence of Cushing's disease. J Neuroendocrinol. 2022;34(8):e13186. doi: 10.1111/jne.13186 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.