

Abstract
DNA methylation is an epigenetic modification that has consistently been shown to be linked with a variety of human traits and diseases. Because DNA methylation is dynamic and potentially reversible in nature and can reflect environmental exposures and predict the onset of diseases, it has piqued interest as a potential disease biomarker. DNA methylation patterns are more stable than transcriptomic or proteomic patterns, and they are relatively easy to measure to track exposure to different environments and risk factors. Importantly, technologies for DNA methylation quantification have become increasingly cost effective—accelerating new research in the field—and have enabled the development of novel DNA methylation biomarkers. Quite a few DNA methylation‐based predictors for a number of traits and diseases already exist. Such predictors show potential for being more accurate than self‐reported or measured phenotypes (such as smoking behavior and body mass index) and may even hold potential for applications in clinics.
In this review, we will first discuss the advantages and challenges of DNA methylation biomarkers in general. We will then review the current state and future potential of DNA methylation biomarkers in two human traits that show rather consistent alterations in methylome—obesity and smoking. Lastly, we will briefly speculate about the future prospects of DNA methylation biomarkers, and possible ways to achieve them.
Keywords: biomarker, DNA methylation, epigenetics, obesity, prediction, smoking
Introduction
Epigenetics is a field of science investigating molecular mechanisms with the potential to change gene expression without altering the underlying DNA sequence. The term “epigenetics” was first coined to explain molecular events underlying cellular differentiation—how cells with identical DNA sequences end up with such diverse morphologies and functions [1]. During the past two decades, the focus in epigenetic research has shifted from understanding single epigenetic events to profiling the human epigenome and characterizing the complex and dynamic relationship between the epigenetic landscape and multiple human traits and diseases.
The three main epigenetic modifications are DNA methylation, histone modifications, and noncoding RNAs, out of which DNA methylation is the most widely studied. Since its discovery in the late 1940s [2], DNA methylation has been characterized as the main determining factor for X‐chromosome inactivation, parental imprinting of genes, and silencing of transposable elements. Decades since the first discovery of DNA methylation, the link between DNA methylation and gene expression was observed [3]. Only after the completion of the Human Genome Project in 2003 [4] and the development of high‐throughput DNA sequencing methodologies did the profound role of DNA methylation in many biological processes and traits start to be acknowledged [5, 6, 7]. DNA methylation means the addition of a methyl (CH3) group into a cytosine base (Fig. 1). This reaction is catalyzed by a family of enzymes called DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b) [8, 9]. DNA methylation occurs predominantly in cytosine–guanine dinucleotides (CpGs) [10]. There are more than 28 million CpG sites distributed across the human genome, and the majority of them are generally methylated [11, 12]. Certain genomic regions with a concentrated number of CpG sites are referred to as CpG islands (CGIs). CGIs are usually found in regions involved in transcription initiation, although some are outside of currently known gene promoters [13]. CGIs, unlike most other CpG sites, are usually unmethylated [12, 14, 15]. Traditionally, unmethylated CGIs are associated with gene activity, whereas methylation of these regions links to gene silencing. Two main mechanisms through which methylated cytosines (5mCs) exert their potential in modifying gene activity have been proposed: they may either physically block the binding of transcription factors, resulting in gene silencing, or attract methyl‐binding proteins that can recognize 5mCs and cause changes in gene expression. Nevertheless, a growing body of recent literature has demonstrated that the relationship between DNA methylation and gene expression is far more complex than this canonical view suggests [16, 17]. Methylation of other genomic regions, such as gene bodies, can also influence expression levels [18], in addition to which there are CpG sites that may distally modify the expression of their target genes [19]. Furthermore, it is well known that different epigenetic modifications interact with each other [20, 21, 22, 23], creating an extremely complex signaling network that fine‐tunes gene expression.
Fig. 1.
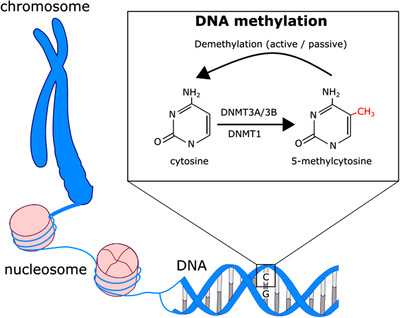
The mechanism of DNA methylation. The basic unit of chromosome is a nucleosome, which consists of a DNA strand and four histone proteins. Methylation of DNA in cytosine bases is a prevalent modification and has an important role in cellular differentiation, transposon silencing, parental imprinting, and regulation of gene expression. DNA methyltransferase (DNMT) enzymes catalyze the methylation reaction. Demethylation can occur passively or actively (mediated by ten‐eleven translocation enzymes).
DNA methylation marks are mostly passed on through cell division [24], but conclusive evidence on transgenerational epigenetic inheritance in humans [25, 26] is still lacking. The heritability of DNA methylation has been estimated between 0.1 and 0.3 [27] when measuring human methylome with a microarray that targets more than 450,000 CpGs (450K). Heritability close to zero means that almost all the trait variability—here, DNA methylation—is due to environmental factors, and heritability close to 1 means that genetic differences among individuals account for most of the trait variability. Up to 45% of the 450K CpGs have been shown to be under direct genetic influence [28]. Such single‐nucleotide polymorphic (SNP) genomic sites that associate with the variation of DNA methylation in specific CpG sites are referred to as methylation quantitative trait loci.
Demethylation occurs in living cells, and it can be either passive or active. In passive demethylation, established methylation marks are not passed onto the daughter cells in cell replication due to the absence or inhibition of DNMTs. Ten‐eleven translocation enzymes mediate active demethylation by oxidizing 5mCs into hydroxymethylated cytosines (5hmCs) [29, 30], which is followed by subsequent reactions and results in unmethylated cytosine bases [31]. It was later shown that hydroxymethylation is a stable modification rather than just an intermediate product in active demethylation reaction [32], and it has been further discovered to contribute to gene regulation. Its functions have been less studied over the years, partly because the most frequently used measuring techniques of DNA methylation can only detect attached methyl groups but not hydroxymethyl groups or cannot differentiate between those two.
Due to the rapid technological progress in the field of epigenetics, a wide range of techniques are emerging to measure DNA methylation. The selection of the most suitable method may depend on several preferences. For instance: is it relevant to use a hypothesis‐based approach or a hypothesis‐generating approach? What is the required level of resolution (average methylation level across the whole genome, methylation status of certain genomic regions, or single CpG sites)? Is it sufficient to examine the methylation of coding regions, or should the method also capture regulatory and intergenic regions?
The most extensively used methods for DNA methylation quantification rely on bisulfite conversion. In bisulfite conversion, DNA is treated with sodium bisulfite, which converts unmethylated cytosines into uracils while leaving 5mC or 5hmCs unchanged. To differentiate between 5mC and 5hmC, an oxidation step is added to the treatment of the DNA sample. This results in the oxidation of 5hmC to 5‐formylcytosine (5fC). Conversion of the newly formed 5fC to uracil by bisulfite treatment allows 5hmC to be discriminated from 5mC. Uracil and cytosine can be differentiated from each other either by sequencing or by microarray technologies. Commonly used cost‐effective technology to identify disease or trait‐associated methylation patterns is the Illumina Infinium BeadChip array. The principle of Infinium arrays is based on genotyping the bisulfite‐converted DNA using site‐specific target probes followed by single‐base extensions to reveal the methylation status of each targeted CpG site (n = 450,000 and 850,000 CpG in 450K and EPIC, respectively). Infinium arrays provide a cost‐effective way to quantify hundreds of thousands of CpG sites at single‐CpG resolution. In addition, a number of algorithms have been developed for data processing, normalization, and analysis, which are often grouped into different pipelines [33, 34, 35, 36, 37, 38]. Despite the increased coverage, only about 3% of the total number of CpGs in the human genome are captured on the EPIC array. If a more extensive characterization of the full methylome is required, sequencing of the bisulfite‐converted DNA is a valid alternative. Sequencing technologies, including whole‐genome bisulfite sequencing (WGBS) and reduced‐representation bisulfite sequencing (RRBS), offer improved coverage compared to the arrays—in theory, up to 100% of the CpG sites in WGBS. However, sequencing is expensive, and the amount of input DNA needed is often higher than in the array‐based detection. In addition, challenges in the sequence alignment and data analyses could result in a much lower coverage than expected in theory. It has been reported that RRBS covers less than 20% and WGBS less than 50% of the human genome [39]. Large‐scale epigenome‐wide association studies (EWAS) using WGBS or RRBS are still uncommon due to the expense and analytical challenges.
New technologies capable of examining the native DNA strand without bisulfite conversion or polymerase chain reaction (PCR) are rapidly emerging. Both the Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) sequencing platforms can provide simultaneous real‐time information on DNA sequence and DNA modifications [40], including methylation and hydroxymethylation. Native DNA sequencing reduces biases that bisulfite conversion may introduce and lowers the required amount of input DNA. High costs and lack of standardized data‐handling methods are still hindering the wide‐scale adoption of ONT, PacBio, or similar methods. Nevertheless, with the increasing availability of high‐performance computational tools for data analysis, in the future, native DNA sequencing technologies may become the gold standard in genomic and epigenomic research and will advance the development of DNA methylation‐based biomarkers.
Biomarkers are biological measures that indicate a presence (or absence) of a physiological condition. However, they are not always involved in disease causation. Biomarkers are used for screening, diagnosis, and prognosis of a disease or other traits, and for monitoring a treatment response or examining the safety of pharmacological compounds. For example, fasting blood sugar for diagnosis of diabetes, troponin‐I to identify coronary ischemia, and prostate‐specific antigen that is used in prostate cancer screening are some of the common biomarkers used in clinical practices. In this review, predictor refers to a biomarker that can predict the likelihood of a clinical condition such as disease onset or recurrence. Examples include BRCA1 and BRCA2, which are genomic variants that increase the risk for breast cancer compared to individuals without these susceptibility genes [41].
Clinically feasible biomarkers should possess several characteristics. First, they should be obtained as noninvasively as possible, which is why most molecular biomarkers have been developed for blood tissue. Second, quantification of biomarkers should be rapid—ideally taking hours rather than weeks—and the interpretation of results should be straightforward. Finally, the costs of measuring a biomarker should be minimal, allowing for frequent monitoring.
There is a clear need and demand for new biomarkers, specifically to identify risks for diseases at earlier stages than most of the current biomarkers are capable of. Diagnosis of many complex traits may be ambiguous due to many related comorbidities or different disease subtypes, which are not well differentiated by the currently available biomarkers. This highlights the need for biomarkers that are more informative about the disease condition and the underlying biological process, aiding in faster and more reliable diagnosis and personalized treatment. Epigenetic biomarkers, namely DNA methylation, can serve as an ideal alternative to traditional biomarkers for several reasons, as discussed below.
The human epigenome has been considered relatively stable across the lifespan [42], with the exception of massive epigenetic reprogramming during early embryogenesis. However, certain methylation sites are shown to react to external signals, such as diet [43], physical activity [44, 45], polluting agents [46], or drugs [47]. Altered methylation can modify gene expression, which in turn translates to changes in cellular pathways and phenotypes. Indeed, numerous studies so far have reported associations between DNA methylation levels and hundreds of human diseases and traits.
The dynamic and reversible nature of DNA methylation—potentially transmitting environmental and internal stimuli into biological functions—makes it a suitable biomarker. As methylation signatures may appear even before any cellular or clinical changes arise, profiling the epigenome has potential in identifying diseases and traits at early stages. This would allow for a rapid clinical or lifestyle‐related intervention to delay or halt the progression of a disease.
Furthermore, DNA methylation profiles can provide a holistic perspective on the whole‐body metabolism and health condition without the need for analyzing multiple biomarker panels. Profiles can be generated by a single blood draw and measurement. This could improve and speed up the diagnosis, and potentially assist in predicting the predisposition for related comorbidities.
From a practical point of view, implementation of DNA methylation profiling does not require completely new infrastructure. Many health‐care facilities already have standardized and efficient blood‐collection protocols in place, which are the basis for quantifying DNA methylation (in biomarker use). Although DNA methylation is highly tissue specific, studies have shown that blood can be used as a surrogate tissue for multiple difficult‐to‐sample tissues (e.g., brain) [48]. In addition, the rapid technological advances in the field of epigenetics allowing for the quantification of DNA methylation in a genome‐wide manner have enabled a swift increase in studies aiming at potential biomarker detection.
Large‐scale epigenome‐wide studies are required to identify trait‐related methylation sites with the potential to be developed into biomarkers. In addition to having a large set of CpG sites with quantified methylation values, a large number of samples are required to uncover reliable relationships between DNA methylation and the trait of interest, as small or moderate effect sizes are expected. Because epigenetic alterations are dynamic, samples must be collected at the time of exposure or at a certain time point based on the research premise. Also, the tissue‐ and cell type–specific nature of epigenetic marks entails additional challenges. We must ensure that the tissue of interest reflects disease‐associated changes in methylation. Measuring methylation in bulk tissue may cause spurious associations due to distinct cell‐type proportions. These challenges add another layer of complexity in translating research findings into clinical practices, as the obtained results cannot be automatically generalized across different ages, ancestries, and tissues. Epigenetic studies require many rounds of replication and validation using different study cohorts to determine the potential of the associated methylation sites as clinical biomarkers.
The ultimate goal in the search for a potential DNA methylation biomarker is to determine a CpG, a set of CpGs, or genomic regions whose methylation status indicates a presence of a trait or predicts a risk or treatment response for a trait. One approach is to first run an exploratory EWAS to identify CpG sites or differentially methylated regions that show significant association with the trait of interest. Then, such trait‐associated methylation markers can be computed into useful weighted scores in another study sample to predict risk for the trait. This kind of approach has been used to construct a score that associates with smoking behavior [49]. However, it may not be suitable for direct risk estimation, as unique score thresholds need to be applied to different populations. Another approach using EWAS hits as the starting point is to apply logistic stepwise regression with forward selection on the identified significant methylation hits, as was done in building a methylation score to distinguish current and former smokers from never smokers [50].
Another strategy—particularly when developing predictors—is to perform feature selection using cross validation rather than significance testing (as in EWAS), as the goal is prediction rather than establishing association/causality. A penalized regression such as LASSO (Least Absolute Shrinkage and Selection Operator) with nested cross validation can be applied to select the best set of CpG sites to reflect the trait of interest. The penalty parameter prevents overfitting and controls the size of the selected feature set. In LASSO regression, the penalty parameter makes the fit to choose only the features (here, methylation probes) that contribute the most to the prediction and shrinks the coefficients of all other probes to zero.
Perhaps the most well‐known tools using such an approach are the various epigenetic clocks. These clocks are based on the discovery that DNA methylation at certain CpG sites changes over the lifespan in a predictable manner [51, 52, 53]. Epigenetic clocks are algorithms developed with elastic net regression to predict age‐related phenotypes from genome‐wide DNA methylation data. The age estimates from these clocks serve as a proxy for accelerated aging [54, 55, 56] and may reflect the lifestyle‐stress burden [57, 58, 59]. The epigenetic clocks have been widely used to estimate chronological age [54, 55], pace of biological aging [60, 61], time to disease, and time to death [58], as well as their associations with various lifestyle factors [59]. EpiSmokEr, a DNA methylation‐based predictor [62], is another example demonstrating the use of LASSO regression and cross validation to select the CpGs that are predictive of smoking.
Some DNA methylation‐based biomarkers already exist in commercial use, mainly in detecting cancer or predicting response to cancer treatment. This is because a significant number of DNA methylation abnormalities arise early in carcinogenesis and are strongly associated with certain types of cancer. For example, based on a study using DNA methylation screening to simultaneously detect different cancer stages and tissues of origin [63], the health‐care company GRAIL currently offers a cancer‐detection kit, which can be obtained through health‐care providers. DNA methylation biomarkers in oncology are beyond the scope of this paper but reviewed elsewhere [64]. The potential of DNA methylation as a biomarker has been recently identified and acknowledged in complex traits and diseases other than cancer. Next, we will discuss two human phenotypes that have been discovered to be strongly associated with DNA methylation—obesity and smoking.
DNA methylation biomarkers in obesity
Obesity is a multifactorial trait characterized by an excessive accumulation of body fat. According to the World Health Organization (WHO), over 1.9 billion adults were overweight in 2016, of which 650 million were obese. Obesity has been described as the pandemic of the 21st century; the prevalence has almost tripled since 1975. The recent growth in prevalence among children and adolescents is especially alarming.
Obesity has a significant genetic background—twin studies have estimated the heritability of obesity to range from 40% to 80% [65]. Only about 6% of the variance in body mass index (BMI) has been captured by polygenic scores; however, the genome‐wide association studies (GWAS) suggest that about 20% of the variance is explained by common genetic variation [66, 67]. The discrepancy between the heritability estimates of twin studies and GWAS is known as the missing heritability. This missing heritability can be partially explained by rare genetic variants, but it has also been attributed to epigenetic mechanisms.
While genetics modify the predisposition for weight gain, they do not alone explain the interindividual variation in obesity. Genetic evolution is too slow to account for the enormous increase in the incidence of obesity since the 1970s. Instead, we need to shift our focus to the other external factors. Modern society strongly promotes weight gain, and on the other hand, prevents weight loss due to unhealthy dietary habits and increased sedentary lifestyle.
It is well known that obesity substantially increases the risk for multiple diseases, including type 2 diabetes (T2D), cardiometabolic disorders, and some cancers [68]. However, the underlying molecular mechanisms remain poorly characterized as the research is hampered by the multifactorial nature of obesity. Obese individuals may display highly dissimilar metabolic characteristics from each other [69]. Hence, it seems that excess weight does not solely explain why some people are more prone to develop related diseases. The factors driving these obesity subtypes and comorbidities need to be elucidated. Not only would this enable the identification of individuals who are at higher risk of becoming overweight, but more importantly would pinpoint those who are at risk of developing obesity‐related metabolic aberrations and secondary diseases. This would be extremely beneficial for the person and would also reduce the huge economic burden that obesity inflicts on society.
Several anthropometric and molecular measures are currently used to assess obesity (Table 1), but each metric comes with serious limitations. The most widely used measure of obesity is BMI (kg/m2), which classifies individuals as underweight (BMI <18), normal weight (18 < BMI <25), overweight (25 < BMI <30), or obese (BMI >30). The majority of GWAS and EWAS investigating obesity apply BMI because it is easily measured and, hence, is often available. While BMI is a very useful measure to detect obesity at the population level, it has serious downsides when applied at the individual level. BMI does not reflect the body composition—that is, whether the body mass is a result of fat content or muscle mass, or where the extra fat has accumulated in the body. Obesity is therefore also diagnosed based on fat mass of 32% and 25% or over in women and men, respectively. As central adiposity (fat in the abdominal area) is considered particularly unhealthy, waist circumference (WC) and waist‐to‐hip ratio (WHR) are better indicators for central obesity than BMI but are more prone to measurement errors. WC of 88 cm and 102 cm in women and men, respectively, are the clinical cut‐offs for central obesity diagnosis.
Table 1.
Overview of potential biomarkers for obesity; their associations with type 2 diabetes (T2D), cardiovascular diseases (CVD), and all‐cause mortality; and implications in clinical practices
| Type | Biomarker | Associations with T2D, CVD, or mortality | Clinical relevance |
|---|---|---|---|
| Anthropometric | Body mass index |
T2D [162] Mortality [164] |
Easy to measure Correlates with fat mass Not a measure of body composition |
| Waist circumference and waist‐to‐hip ratio |
CVD [165] Mortality [166] |
Proxy for abdominal fat | |
| Molecular | Cytokines (e.g., IL‐6, TNF‐α) | CVD [167] | Detects obesity‐related inflammation |
| Adipokines (e.g., leptin, adiponectin) | T2D [168] | Detects obesity‐related insulin resistance (adiponectin) | |
|
Insulin related (insulin, IGF‐1, C‐peptide) |
CVD [169] | Detects obesity‐related hyperinsulinemia | |
| Glucose related (fasting glucose, oral glucose tolerance test, glycated hemoglobin (HbA1c)) | T2D [170] | Detects obesity‐related hyperglycemia | |
| Plasma lipids (e.g., low‐density lipoprotein/high‐density lipoprotein, triglycerides) |
CVD [171] Mortality [172] |
Detects obesity‐related dyslipidemia | |
| C‐reactive protein | CVD [173] | Detects obesity‐related inflammation | |
| Polygenic risk scores | 97 single‐nucleotide polymorphisms (SNPs) | − [66] | Identifies high‐risk individuals |
| 1458 SNPs |
CVD [174] Mortality [174] |
||
| Genome‐wide polygenic score (2,100,302 genetic variants) |
T2D [175] CVD [175] Mortality [175] |
||
| DNA methylation‐based scores |
1109 cytosine–guanine dinucleotides (CpGs) 226 CpGs 400 CpGs |
− [89] Mortality [89] T2D [90] CVD [90] |
Identifies high‐risk individuals, identifies obesity subtypes, and monitors treatment |
In addition to anthropometric measures, blood‐based molecular biomarkers (e.g., blood cholesterol and triglyceride profiles) are used in assessing obesity. Elevated levels of these biomarkers most often indicate a prolonged period of obesity, which has already significantly added to the risk of secondary diseases. The most prevalent and important obesity‐associated adverse health state is metabolic syndrome (MetS). According to the National Cholesterol Education Program Adult Treatment Panel III, MetS is diagnosed when an individual presents with at least three of the following: WC greater than 88 cm in women or 102 cm in men, blood pressure greater than 130/85 mmHg, fasting triglyceride level greater than 150 mg/dl, fasting high‐density lipoprotein (HDL) cholesterol less than 40 mg/dl (men) or 50 mg/dl (women), and fasting blood sugar greater than 100 mg/dl.
Obesity and associated metabolic imbalances result from complex interactions between an unhealthy environment such as dietary habits and sedentary lifestyle, demographics (age and sex), and genotype. The potential molecular mechanism linking these risk factors together may be epigenetic regulation. Unsurprisingly, in order to understand the complex interactions between the environment, lifestyle, and genetics in the development of obesity and related traits, there has been an increased interest in epigenetic studies on obesity and MetS. These studies also provide a promising opportunity to address the demand for the development of reliable, informative, and affordable biomarkers to improve the diagnosis of risks associated with obesity.
To date, a few studies have investigated the epigenetic landscape in human obesity and weight loss in blood (reviewed by Ling C and Rönn [70] and Do [71], and Samblas et al. [72] and Aronica et al. [73], respectively). Some molecular pathways and genes, which could be prioritized when developing novel epigenetic biomarkers for obesity, have systematically been identified (Fig. 2).
Fig. 2.
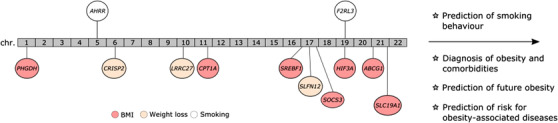
The most common genes with differentially methylated cytosine–guanine dinucleotides (CpGs) in obesity and smoking shown in their respective chromosome of the genome. In addition to studying target genes, recent development in machine learning has enabled the use of the whole epigenome to identify sets of CpGs that can be treated as biomarkers for smoking behavior, obesity, or weight loss. Shown are genes with their methylation being replicated in independent methylation studies (smoking and body mass index [BMI]), and genes from a recent randomized controlled trial [98] of adjusted p < 10‐5 (weight loss). The list of genes may be nonexclusive.
Given the definition of obesity and the availability of study cohorts with BMI measures, it may not be surprising that several studies have consistently highlighted associations between BMI and CpG sites that are located close to genes related to lipid metabolism. Altered methylation in genes ABCG1 [74, 75, 76, 77], SREBF1 [74, 76–78], and CPT1A [74, 77, 79] has been implicated in obesity. Both ABGC1 (ATP binding cassette subfamily G member 1) and SREBF1 (sterol regulatory element binding transcription factor 1) have also been associated with other closely related traits such as dyslipidemia and T2D [80, 81]. Gene expression levels of CPT1A (carnitine palmitoyltransferase 1A) have been previously linked to blood lipid profiles and insulin resistance [82], which correlate with the methylation levels of this gene.
Methylation in genes related to the transport and metabolism of amino acids and other small molecules is affected in obesity. A gene, PHGDH (phosphoglycerate dehydrogenase), involved in serine metabolism, has been implicated in people with high BMI [75, 79]. In addition, methylation of the solute carrier protein (SLC) genes are also associated with BMI [76, 78] and WC [78]. One of the implicated genes in this family is SLC19A1, which regulates intracellular folate concentration [83]. The folate pathway is tightly connected to the methionine cycle, which is the main enzymatic pathway for DNA methylation.
Hypoxia is a common occurrence in obesity [84], and it describes a state of inadequate blood flow to a tissue, resulting in a microenvironment with low levels of oxygen. One of the earliest large‐scale EWAS in 2014 found that increased methylation of three CpGs in the H1F3A gene was significantly associated with BMI [85]. A year later, another study demonstrated a similar effect in children [86]. HIF3A (hypoxia inducible factor 3 subunit alpha) codes for a protein that is one of the main factors regulating cells’ response to hypoxia. However, a number of recent studies have failed to replicate this finding [75, 78, 79], which could be attributable to a variety of factors. One of the proposed explanations is the effect of maternal BMI on the HIF3A methylation in the offspring rather than the offspring's body weight per se [87].
Inflammation is among the various changes in the human body caused by obesity. Lipid accumulation results in adipocyte hypertrophy (increase in size), which is accompanied by infiltration of immune cells and secretion of pro‐inflammatory factors such as cytokines. As a result, obesity is often described as a state of low‐grade inflammation characterized by, for example, elevated levels of C‐reactive protein [88]. Consequently, DNA methylation shows alterations in genomic regions and pathways that are known to be involved in inflammation processes [74, 75]. However, studies have not consistently identified the same set of differentially methylated CpGs and genes. It is possible that each of them has a small effect size and hence fails to reach genome‐wide significance in underpowered studies. Alternatively, these CpGs could be specific to other confounders or obesity‐related comorbidities. Nevertheless, it appears that obesity‐related systemic low‐grade inflammation associates with DNA methylation changes; however, the molecular details and causal relationships are still unclear.
Machine‐learning models have recently emerged alongside the traditional EWAS to discover the methylation landscape in obesity. While the previously described potential biomarkers are based on EWAS findings, a few studies have applied feature selection–based approaches to indicate obesity from methylation data. For example, in one study, researchers used LASSO with cross validation to construct methylation scores for BMI and WHR [89]. Their model performed moderately in discriminating obese individuals from nonobese. Another study used a similar approach to develop a methylation‐based predictor for BMI [90], which was found to be strongly associated with T2D and cardiovascular diseases. These models were developed to indicate current obesity, that is, to discriminate obese people from nonobese. Some attempts have been made to apply DNA methylation as a predictor for future obesity and related diseases, as discussed below.
The majority of the alterations in methylation arise as a consequence of fat accumulation, leading to metabolic disturbances through various routes, as demonstrated by Mendelian randomization analyses [75, 76, 91] and longitudinal studies [92]. Studies on children and adolescents show little evidence on the connection between early life BMI and DNA methylation [93]. Even though addressing the causality of BMI‐associated methylation markers is less relevant in the search of diagnostic biomarkers, the inability to do so might limit the application of these results as a predictor. Fortunately, some encouraging discoveries have been made during the last 5 years. For instance, a methylation risk score of 82 CpGs was found to be strongly predictive of incident T2D [75], yielding better predictions than traditional risk factors such as high BMI. Another recent study showed that the baseline methylation score based on 116 CpGs could predict levels of interleukin‐6, an inflammatory marker elevated in obesity [91]. Further, a population‐based DNA methylation study on habitual diet with a relatively large sample size (n = 6662 European, 2702 African, and 360 Hispanic ancestry) provided evidence for the link between diet‐associated DNA methylation and metabolic disorders as well as all‐cause mortality [94]. The findings additionally supported a causal pathway between diet‐associated DNA methylation and BMI, triglycerides, HDL cholesterol, and T2D. These results indicate a promising role of DNA methylation as a predictor of obesity and associated diseases. However, further well‐powered, multitrait genome‐wide studies on DNA methylation are needed to discover the value of DNA methylation in predicting later‐life outcomes.
In addition to holding great potential for predicting the progression to obesity and obesity‐associated comorbidities [95], DNA methylation may also be an indicator for lifestyle changes, as shown by various intervention studies. DNA methylation associates with both short‐ and long‐term weight loss [96], it differs between responders and nonresponders [97], and it may predict the outcome of the intervention [98]. For example, a clinical study on overfeeding has shown that saturated and polyunsaturated fats induce differential DNA methylation, and that DNA methylation prior to dietary changes may predict weight increase in response to overfeeding [99]. A few other studies have also observed DNA methylation alterations in blood cells and adipose tissue in response to diet quality [94, 100–102], suggesting that DNA methylation may mediate the effects of diet on the risk for chronic diseases, including obesity and T2D. Lifestyle changes in childhood and adolescence are also reflected in DNA methylation [95, 97] and may give us important clues about which genomic regions to target for obesity prevention or treatment. However, most of the lifestyle or weight‐loss intervention studies conducted to date are small and rather short in duration and there is limited reproducibility of the results. Therefore, these findings should be interpreted with caution, and studies with appropriate designs—including larger sample sizes and adequate power—are needed to make stronger conclusions regarding the role of DNA methylation in relation to lifestyle changes.
Regardless of a great deal of evidence on obesity‐related alterations in the human epigenome, some challenges hinder the translation of epigenetic biomarkers in clinical use. Some of these challenges are common to all epigenetic research, as discussed above. However, when investigating DNA methylation in the context of obesity, a few stand out—such as failure in replicating or validating identified associations.
To date, we have identified several methylation sites consistently associated with obesity, but there are still numerous significant findings that have not been successfully replicated in any other studies. This could be due to discrepancies in study cohorts having different underlying confounder characteristics such as age and sex. In addition, some of the CpGs can also be specific to ethnicity. However, there has been limited research on population heterogeneity in obesity‐related methylation sites [75, 91]. Finally, distinct obesity‐related comorbidities or metabolic characteristics may exist among study participants that are not accounted for by BMI, the most commonly studied obesity measure. For instance, a blood methylation study noticed that monozygotic twins discordant for BMI had no differences in their methylome, but when BMI and liver‐fat percentage were considered together, pairs discordant for both BMI and liver fat showed differential methylation in multiple genomic sites [103]. It is likely that different obesity comorbidities exhibit specific DNA methylation profiles. This is also supported by the results obtained from EWAS of traits closely related to BMI, such as T2D [70] or MetS [81]. While some of the significant findings are shared between BMI and related traits (e.g., SHREB and ABCG), a proportion of nonreplicated CpGs may be due to different molecular phenotypes. Although BMI is a general, indirect measure of obesity, it has provided an important starting point for the discovery of associations between increased body fat and DNA methylation. Nevertheless, discrepancies in the results between studies have demonstrated that, in the future, we will need to incorporate additional obesity‐related measures to distinguish DNA methylation signatures amongst obesity comorbidities. This would allow for the identification of obese individuals who are at high risk for secondary diseases such as T2D. To combine the knowledge on methylation profiles in different obesity subtypes and related traits, we need improved phenotyping, standardized study procedures, large prospective cohort studies, and clinical interventions through which the findings can be validated before introducing them to the primary‐care and public‐health sectors.
More comprehensive approaches may help in developing the epigenetic obesity biomarkers for clinical use. Similar to epigenetic clocks as biomarkers of aging, it could be useful to construct such algorithms for obesity and metabolic diseases. This could be achieved by predicting levels of several relevant obesity‐related clinical values and markers (e.g., BMI and those important in the diagnosis of MetS) using genome‐wide methylation data. These DNA methylation biomarkers could then be combined into one score or estimate for an overall biomarker of obesity and metabolic status. Such a methylation score could serve as a more specific biomarker for the metabolic status than current obesity biomarkers. Like epigenetic clocks that capture both chronological age and age‐associated blood measures and therefore yield better prediction for age‐related diseases, a methylation score for obesity could improve the diagnosis of unhealthy obesity and prediction of the risk for obesity‐related diseases.
Integrating epigenomic data with other physiological, lifestyle, and “omics” data holds another promising avenue for future biomarker development [104]. The underlying molecular network of complex traits is likely to be seen in multiple layers of biological data (genomic, epigenomic, transcriptomic, and metabolomic), the integration of which will aid in disentangling the etiology of diseases. For instance, when the methylation score by McCartney et al. was combined with polygenic scores, the model explained 19.7% of the phenotypic variation in BMI and 12.5% when used alone [89]. This is not surprising, because obesity is known to have a genetic background; therefore, incorporation of the polygenic score naturally adds to the prediction accuracy. The most recent and currently the most comprehensive study aimed at developing a predictor for obesity risk combined BMI‐associated SNPs and methylation CpG sites with dietary and lifestyle factors in machine‐learning algorithms [105]. The best‐performing model in the study had an overall accuracy of 70% in predicting current obesity by using 21 SNPs, 230 CpG sites, and 26 dietary factors—such as processed meat, high‐fat dairy, French fries, artificial sweeteners, and alcohol intake. These more comprehensive approaches to developing an obesity biomarker—such as (1) combining multiple methylation sites into scores, (2) using a composite biomarker based on multiple DNA methylation surrogates of clinical markers of obesity, or (3) combining obesity‐associated DNA methylation sites with multiple omics and lifestyle factors—will likely increase the clinical value of obesity biomarkers.
Taken together, it is clear that obesity is highly heritable; however, the eventual weight of an individual is determined by the complex interactions of genetic and environmental factors, such as diet and lifestyle. Epigenetic marks react to the environment, and may reflect (or capture) both the external (e.g., exposure to a certain diet) and internal (e.g., metabolic state) environments of an individual. Therefore, epigenetic markers of obesity have great potential to be of clinical value as obesity and associated disease risk biomarkers. Multiple genetic studies have identified obesity‐associated variants that determine the genetic predisposition to obesity. However, the genetic makeup of an individual cannot be changed, and therefore obesity prevention and treatment strategies target modifiable factors such as diet and physical activity. The clinical value of methylation biomarkers of obesity comes from their ability to precisely predict obesity‐ and weight loss–related outcomes. By the aid of epigenetic markers, we will be able to predict obesity with precision and identify modifiable factors that can change the risk of obesity and associated diseases. Such epigenetic biomarkers will, in the future, have their value as tools to develop effective strategies for personalized prevention and treatment of obesity and associated diseases. Epigenetic markers that have a causal role in obesity or associated diseases could also be targeted for pharmacological treatment.
DNA methylation biomarkers in smoking
Smoking is a significant risk factor for a variety of chronic diseases, as well as a leading cause of preventable mortality, accounting for more than 8 million deaths annually and costing the global economy 1.4 trillion US dollars [106]. Besides the well‐established detrimental effects of smoking with regard to lung cancer, chronic obstructive pulmonary disease, and heart disease, it has also been associated with comorbidities such as tuberculosis, alcoholism, and mental illness [107].
Smoking is a complex behavior and has been regarded as a multifactorial disease that involves both genetic and environmental factors. Smoking behavior encompasses multiple phases, starting with smoking initiation then progressing to nicotine dependency (observed in most smokers), nicotine withdrawal (while attempting to quit smoking), cessation, and relapse [108]. Peer pressure throughout adolescence [109, 110], a positive image of smoking, socioeconomic status, parental smoking, gender, ethnicity, and usage of other substances are the major factors that lead to smoking initiation [111, 112, 113].
In addition to behavioral, physiological, and other environmental factors, smoking behavior is also strongly influenced by genetic and perhaps epigenetic factors. According to family and twin studies, the heritability of smoking behavior is high, with genetic effects increasing from adolescence into adulthood [114]. Thus, genetic differences among individuals have a substantial impact on smoking behavior. Several loci associated with nicotine dependence, cigarettes per day, and smoking cessation have been identified, with nicotinic receptor genes CHRNA5‐CHRNA3‐CHRNB4 at 15q25 [115, 116, 117, 118, 119, 120, 121] and the primary nicotine metabolism gene CYP2A6 at 19q13 [122] being the most significant and consistently replicated hits. However, the observed genotype associations only account for low‐ to moderate‐heritability estimates for each smoking‐related trait [123].
Epigenetic factors including DNA methylation could, on the one hand, partially predispose an individual to smoking behavior or mediate the causal effect of the genotypes on smoking behavior [124, 125]. On the other hand, smoking may lead to alterations in DNA methylation. Numerous studies have shown that smoking has a significant influence on DNA methylation levels. Multiple independent studies on different populations have revealed several smoking‐associated CpGs in the blood methylome [49, 126–134]. The biggest EWAS to date with approximately 16,000 individuals identified almost 19,000 significantly differently methylated CpGs across 7000 genes between current and never smokers [127]. Interestingly, the majority of smoking‐associated CpGs are hypomethylated in current smokers compared to never smokers. Smoking‐associated methylation signals seem rather robust, with many of the studies [128, 132–134] consistently reporting the same top significant CpGs at the AHRR and F2RL3 genes (Fig. 2). AHRR (aryl hydrocarbon receptor repressor), a known tumor suppressor, is involved in the detoxification of harmful components present in cigarette smoke [135]. F2RL3 (coagulation factor II receptor‐like 3 gene) encodes PAR‐4 (thrombin protease‐activated receptor‐4), which plays a crucial role in leukocyte recruitment and is thereby involved in smoking‐induced inflammatory reactions [134]. Hundreds of novel methylation smoking signals have also been identified in lung tissue [136] and whole blood [137] using EPIC array, which targets CpGs across the genome, including enhancer regions.
It has also been demonstrated that methylation levels in former smokers partially reverse after cessation, approaching those of never smokers [126, 128, 130, 134, 138–140]. However, the extent of reversal can be site specific depending on the degree of smoking‐induced methylation changes at a specific CpG site [128, 138]. Furthermore, CpGs with persistent methylation changes have been observed after decades of smoking cessation, implying that smoking has a broader and longer‐lasting impact on the methylome [128, 131, 138].
Despite the tissue‐specific nature of DNA methylation, studies have also reported widespread effects of smoking on methylation. For instance, a comparable overlap of smoking‐associated methylation patterns was found in buccal tissue and whole blood [141]. This suggests that complex behaviors like smoking have broader impacts on methylation at specific CpGs across tissues, resulting in tissue‐shared effects.
The ability to create suitable treatments—ranging from preventative interventions for occasional smokers to cessation therapy for current smokers—depends on having a precise understanding of smoking history. Furthermore, an accurate smoking history can be used to predict long‐term health concerns linked to smoking (e.g., lung cancer). Self‐administered questionnaires have traditionally been used to assess smoking exposure. The most widely used self‐report questionnaires to capture nicotine dependency are the Diagnostic and Statistical Manual of Mental Disorders [142] and the Fagerström Test of Nicotine Dependence [143]. Due to under‐reporting and poor recollection of long‐term smoking history, self‐reported smoking status is prone to mistakes. Furthermore, it does not take passive smoking into consideration.
Nicotine, cotinine, and carbon monoxide (CO) are the most widely used smoking biomarkers. Nicotine is the most active component of the tobacco leaf and is highly addictive in nature, thereby making it extremely difficult to quit smoking permanently [144]. Nicotine's rapid absorption followed by speedy elimination from body tissues—resulting in a short half‐life of about 2 h—limits its effectiveness as a biomarker. Cotinine—a primary metabolite of nicotine—can be used to measure the amount of nicotine absorbed in bodily fluids [145]. However, given the short half‐life of about 16 h, cotinine can only be identified for a few days at most after smoking [146]. Hence, its usefulness is restricted to evaluating recent smoking. Furthermore, the use of nicotine‐replacement therapy, smokeless tobacco, and e‐cigarettes may cause elevated cotinine levels, leading to erroneous evidence of smoking. Exhaled CO can effectively measure recent smoking exposure in smokers [147]. However, limitations of CO include a short half‐life of 5–6 h and its inability to distinguish exposure to nontobacco sources like air pollution. This clearly illustrates the need for a reliable smoking‐exposure indicator that can overcome these shortcomings and reliably quantify current and former smoking.
Self‐reported smoking status and traditional biomarkers like cotinine—which can only quantify short‐term exposure—have been demonstrated to be less reliable than methylation‐based smoking‐status prediction. The methylation status of the genes AHRR [132, 133] and F2RL3 [134] are the most consistently reported smoking‐associated signals, and they have been proposed as potential biomarkers to estimate smoking habits (smoking cessation for F2RL3) [128, 132–134, 148]. Some of these smoking‐associated methylation signals have been shown to be robust across ethnicities [62, 137, 148], demonstrating the possibility of using trans‐ethnic smoking biomarkers to predict smoking status. Demethylation at both AHRR (measured by Illumina BeadChip probe cg05575921) and 6p21.33 (cg06126421) was linked to cardiovascular mortality and a 2.5‐fold increased risk of dying from any cancer [149]. DNA methylation levels at AHRR (cg05575921), F2RL3 (cg03636183), and 6p21.33 (cg06126421) were shown to be predictive of lung‐cancer incidence [150]. A droplet digital PCR (ddPCR) assay measuring methylation at cg05575921 successfully predicted smoking status in adults with varying smoking histories [151]. Also, methylation‐sensitive ddPCR assessments of the same CpG site have been shown to identify current smokers in both whole blood and saliva [152]. These examples demonstrate the effective implementation of methylation biomarkers in clinical settings to reliably assess smoking.
Similar to the polygenic risk score approach, smoking‐associated EWAS findings have been translated into scores that reflect the extent of smoking (Table 2). To determine smoking status, existing DNA methylation‐based approaches employ scores generated from cumulative methylation levels at smoking‐associated CpGs. Smoking score [49] and methylation score [50] are two popular approaches. Smoking score uses an ethnic‐specific threshold to distinguish smokers from never smokers, restricting its universal application, and it requires a separate threshold for each ethnicity. Methylation score was built from cotinine EWAS hits to distinguish current and former smokers from never smokers. It can be used to measure current and long‐term smoking exposure, which is important when assessing the health risks posed by cumulative smoking exposure. However, because a score threshold value must be determined for each dataset and these methods can only conduct binary classifications, they have limited utility. Machine‐learning approaches have also been used to build classifiers to estimate smoking status based on DNA methylation profiles [62, 153]. A smoking‐status classifier [62] developed using whole‐blood data has demonstrated good predictability in buccal tissue samples. A DNA methylation composite score that predicts both smoking status and damage to biological systems—namely, the lungs and gums—has also been proposed [154].
Table 2.
An overview of recent studies with a focus on development of DNA methylation‐based smoking scores or predictors
| Purpose | Methodology | Reference |
|---|---|---|
| Smoking score based on 187 smoking‐associated cytosine–guanine dinucleotides (CpGs) identified in whole blood, can distinguish heavy smokers from nonsmokers (former and never) | A weighted DNA methylation score was calculated using methylation values of 187 CpGs identified by an earlier epigenome‐wide association study (EWAS) [130] as reference values. | [49] |
| A DNA methylation score based on two top smoking‐associated CpGs shown to be predictive of all‐cause, cardiovascular, and cancer mortality | Restricted cubic spline regression | [149] |
| Methylation score based on methylation values of four smoking‐associated CpGs in whole blood; can discriminate current smokers from never smokers, as well as former smokers from never smokers | EWAS followed by stepwise logistic regression with forward selection | [50] |
| A smoking status estimator (EpiSmokEr) that can predict the smoking status of individuals from whole‐blood methylation data | Least Absolute Shrinkage and Selection Operator (LASSO) regression | [62] |
| A DNA methylation smoking score that can classify newborns based on the maternal smoking exposure during pregnancy | EWAS followed by LASSO regression | [160] |
| A prenatal DNA methylation smoking score to predict prenatal exposure to maternal smoking | A weighted DNA methylation score calculated using the methylation values of CpGs identified by an earlier genome‐wide consortium meta‐analysis [176] | [159] |
| A machine‐learning based DNA methylation score that distinguishes individuals exposed to in utero smoke from individuals not exposed to in utero smoke | Elastic net regression | [161] |
Further investigations are required to establish the predictive role of DNA methylation in passive smoking [155] and e‐cigarette usage [156]. For example, methylation at the cg05575921 CpG site most frequently used as a biomarker for tobacco smoke is not suitable alone for assessing the use of e‐cigarettes and smokeless‐tobacco products [157]. Similarly, as discussed above in relation to obesity, combining DNA methylation biomarkers with conventional metabolite biomarkers for smoking may enhance the precision and characterization of smoking behavior. For example, propylene glycol, 2‐cyanoethylmercapturic acid, and anabasine measured from urine have been used together with smoking‐associated cg05575921 methylation biomarker to distinguish combustible‐tobacco users from e‐cigarette and smokeless‐tobacco users [157]. While cigarette smokers were identified by dose‐dependent lowered methylation levels of cg05575921, this CpG site was not affected in smokeless‐tobacco users. However, both groups showed increased urinary anabasine levels [157].
In addition to active and passive smoking in adults, maternal smoking is a crucial research and public‐health topic. The adverse impact of prenatal exposure to maternal smoking during pregnancy is well established [158]. Mounting evidence suggests that DNA methylation can serve as a robust predictor of the long‐term effects of maternal smoking on offspring health [159, 160, 161].
Conclusions and future prospects
Substantial evidence shows that several human traits and diseases are associated with specific DNA methylation signatures, prompting studies to investigate the potential of DNA methylation as a biomarker. DNA methylation is dynamic and reversible in nature and is easy to collect and quantify, making it a viable diagnostic and predictive biomarker. DNA methylation marks can be used to detect the early onset of diseases, aid in diagnosis, reveal environmental exposures, and predict the likelihood of a trait or disease. DNA methylation biomarkers can also be used in conjunction with traditional biomarkers, and their utility can be further enhanced by integrating them with other omics data. The most promising biomarkers or predictors are those that reflect the genetic and environmental causes of complex diseases or traits, and thus may incorporate genomic and epigenomic data with other omics (such as metabolomics, proteomics, transcriptomics, and metabolomics) and lifestyle factors, and moreover, consider their interactions to predict individuals’ health status.
There are no widely used epigenetic biomarkers in clinical settings outside of oncology. The field of epigenetic biomarkers is relatively new and the trait‐associated changes may be difficult to discover due to sample sizes, small effect sizes, cell‐type heterogeneity, confounding factors, phenotype heterogeneity, and lack of replication. Nevertheless, the latest research in recent years has presented compelling evidence on trait‐specific DNA methylation profiles for conditions other than cancer, including obesity and smoking. In addition to the scientific rigor, there is also a need to validate novel findings and further attract industrial stakeholders and policy makers to accelerate the development of methylation biomarkers.
With the rapid advancement of the field of epigenetics and the decreasing costs associated with high‐throughput technologies, the quantification of DNA methylation may soon become a part of routine clinical evaluations. Novel technologies are emerging and—especially those of long‐read native DNA sequencing—can revolutionize the availability of examining individuals’ genomes and epigenomes in research and health care. By offering a comprehensive molecular picture of the clinical condition, DNA methylation profiling provides notable opportunities for health‐care systems that are currently shifting towards personalized medicine.
Conflict of interest
The authors declare no conflict of interest.
Heikkinen A, Bollepalli S, Ollikainen M. The potential of DNA methylation as a biomarker for obesity and smoking. J Intern Med. 2022;292:390–408.
References
- 1. Waddington CH. The epigenotype. Endeavour. 1942;1:18–20. [Google Scholar]
- 2. Hotchkiss RD. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem. 1948;175:315–32. [PubMed] [Google Scholar]
- 3. Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science (80‐). 1975;187:226–32. [PubMed] [Google Scholar]
- 4. National Human Genome Research Institute . The Human Genome Project. https://www.genome.gov/human‐genome‐project (accessed 2 March 2022) (2020).
- 5. Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. [DOI] [PubMed] [Google Scholar]
- 6. Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi‐Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases DNMT3a and DNMT3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. [DOI] [PubMed] [Google Scholar]
- 9. Hermann A, Goyal R, Jeltsch A. The DNMT1 DNA (cytosine‐C5)‐methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem. 2004;279:48350–9. [DOI] [PubMed] [Google Scholar]
- 10. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. [DOI] [PubMed] [Google Scholar]
- 11. Ehrlich M, Gama‐Sosa MA, Huang L‐H, Midgett RM, Kuo KC, Mccune RA, et al. Amount and distribution of 5‐methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Illingworth R, Kerr A, Desousa D, Jørgensen H, Ellis P, Stalker J, et al. A novel CpG island set identifies tissue‐specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zeng J, Nagrajan HK, Yi SV. Fundamental diversity of human CpG islands at multiple biological levels. Epigenetics. 2014;9:483–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spainhour JC, Lim HS, Yi SV, Qiu P. Correlation patterns between DNA methylation and gene expression in The Cancer Genome Atlas. Cancer Inform. 2019;18:1176935119828776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu A, Dai Y, Mendez EF, Hu R, Fries GR, Najera KE, et al. Genome‐wide correlation of DNA methylation and gene expression in postmortem brain tissues of opioid use disorder patients. Int J Neuropsychopharmacol. 2021;24:879–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ball MP, Li JB, Gao Y, Lee J‐H, Leproust EM, Park I‐H, et al. Targeted and genome‐scale strategies reveal gene‐body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013;14:R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rottach A, Leonhardt H, Spada F. DNA methylation‐mediated epigenetic control. J Cell Biochem. 2009;108:43–51. [DOI] [PubMed] [Google Scholar]
- 21. Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–66. [DOI] [PubMed] [Google Scholar]
- 22. Okitsu CY, Hsieh C‐L. DNA methylation dictates histone H3K4 methylation. Mol Cell Biol. 2007;27:2746–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fuso A, Raia T, Orticello M, Lucarelli M. The complex interplay between DNA methylation and miRNAs in gene expression regulation. Biochimie. 2020;173:12–6. [DOI] [PubMed] [Google Scholar]
- 24. Rougier N, Bourc'his D, Gomes DM, Niveleau A, Plachot M, Pàldi A, et al. Chromosome methylation patterns during mammalian preimplantation development. Genes Dev. 1998;12:2108–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mcrae AF, Powell JE, Henders AK, Bowdler L, Hemani G, Shah S, et al. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 2014;15:R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fitz‐James MH, Cavalli G. Molecular mechanisms of transgenerational epigenetic inheritance. Nat Rev Genet. 2022. 10.1038/s41576-021-00438-5 [DOI] [PubMed] [Google Scholar]
- 27. Villicaña S, Bell JT. Genetic impacts on DNA methylation: research findings and future perspectives. Genome Biol. 2021;22:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Min JL, Hemani G, Hannon E, Dekkers KF, Castillo‐Fernandez J, Luijk R, et al. Genomic and phenotypic insights from an atlas of genetic effects on DNA methylation. Nat Genet. 2021;53:1311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Skirmantas K, Nathaniel H. The nuclear DNA base 5‐hydroxymethylcytosine is present in Purkinje neurons and the brain. Science (80‐). 2009;324:929–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mamta T, Peng KK, Yinghua S, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (80‐). 2009;324:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu X, Zhang Y. TET‐mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18:517–34. [DOI] [PubMed] [Google Scholar]
- 32. Bachman M, Uribe‐Lewis S, Yang X, Williams M, Murrell A, Balasubramanian S. 5‐hydroxymethylcytosine is a predominantly stable DNA modification. Nat Chem. 2014;6:1049–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maksimovic J, Gordon L, Oshlack A. SWAN: Subset‐quantile Within Array Normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Triche TJ, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low‐level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013;41:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fortin J‐P, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data‐driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Müller F, Scherer M, Assenov Y, Lutsik P, Walter J, Lengauer T, et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol. 2019;20:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 2017;33:3982–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beck D, Ben Maamar M, Skinner MK. Genome‐wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons. Epigenetics. 2021:1–13. 10.1080/15592294.2021.1924970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ameur A, Kloosterman WP, Hestand MS. Single‐molecule sequencing: towards clinical applications. Trends Biotechnol. 2019;37:72–85. [DOI] [PubMed] [Google Scholar]
- 41. National Cancer Institute . BRCA gene mutations: cancer risk and genetic testing. https://www.cancer.gov/about‐cancer/causes‐prevention/genetics/brca‐fact‐sheet (accessed 14 January 2022) (2020).
- 42. Kanherkar RR, Bhatia‐Dey N, Csoka AB. Epigenetics across the human lifespan. Front Cell Dev Biol. 2014;2:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang FF, Santella RM, Wolff M, Kappil MA, Markowitz SB, Morabia A. White blood cell global methylation and IL‐6 promoter methylation in association with diet and lifestyle risk factors in a cancer‐free population. Epigenetics. 2012;7:606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sailani MR, Halling JF, Møller HD, Lee H, Plomgaard P, Pilegaard H, et al. Lifelong physical activity is associated with promoter hypomethylation of genes involved in metabolism, myogenesis, contractile properties and oxidative stress resistance in aged human skeletal muscle. Sci Rep. 2019;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ling C, Rönn T. Epigenetic adaptation to regular exercise in humans. Drug Discov Today. 2014;19:1015–8. [DOI] [PubMed] [Google Scholar]
- 46. Rider CF, Carlsten C. Air pollution and DNA methylation: effects of exposure in humans. Clin Epigenetics. 2019;11:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ganesan A, Arimondo PB, Rots MG, Jeronimo C, Berdasco M. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenetics. 2019;11:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome‐wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Elliott HR, Tillin T, Mcardle WL, Ho K, Duggirala A, Frayling TM, et al. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenetics. 2014;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang Y, Florath I, Saum K‐U, Brenner H. Self‐reported smoking, serum cotinine, and blood DNA methylation. Environ Res. 2016;146:395–403. [DOI] [PubMed] [Google Scholar]
- 51. Florath I, Butterbach K, Muller H, Bewerunge‐Hudler M, Brenner H. Cross‐sectional and longitudinal changes in DNA methylation with age: an epigenome‐wide analysis revealing over 60 novel age‐associated CpG sites. Hum Mol Genet. 2014;23:1186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Teschendorff AE, Menon U, Gentry‐Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, et al. Age‐dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rakyan VK, Down TA, Maslau S, Andrew T, Yang T‐P, Beyan H, et al. Human aging‐associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome‐wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weidner C, Lin Q, Koch C, Eisele L, Beier F, Ziegler P, et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu Z, Leung D, Thrush K, Zhao W, Ratliff S, Tanaka T, et al. Underlying features of epigenetic aging clocks in vivo and in vitro. Aging Cell. 2020;19:e13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY). 2019;11:303–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018;10:573–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Belsky DW, Caspi A, Arseneault L, Baccarelli A, Corcoran DL, Gao X, et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. Elife. 2020;9:e54870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Belsky DW, Caspi A, Corcoran DL, Sugden K, Poulton R, Arseneault L, et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. Elife. 2022;11:e73420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bollepalli S, Korhonen T, Kaprio J, Anders S, Ollikainen M. EpiSmokEr: a robust classifier to determine smoking status from DNA methylation data. Epigenomics. 2019;11:1469–86. [DOI] [PubMed] [Google Scholar]
- 63. Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Liu MC, et al. Sensitive and specific multi‐cancer detection and localization using methylation signatures in cell‐free DNA. Ann Oncol. 2020;31:745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mancarella D, Plass C. Epigenetic signatures in cancer: proper controls, current challenges and the potential for clinical translation. Genome Med. 2021;13:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Silventoinen K, Jelenkovic A, Sund R, Hur Y‐M, Yokoyama Y, Honda C, et al. Genetic and environmental effects on body mass index from infancy to the onset of adulthood: an individual‐based pooled analysis of 45 twin cohorts participating in the COllaborative project of Development of Anthropometrical measures in Twins (CODATwins). Am J Clin Nutr. 2016;104:371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, et al. Meta‐analysis of genome‐wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet. 2018;27:3641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. National Heart, Lung and Blood Institute . Overweight and obesity. https://www.nhlbi.nih.gov/health‐topics/overweight‐and‐obesity (accessed 26 January 2022) (2022).
- 69. Lin Z, Feng W, Liu Y, Ma C, Arefan D, Zhou D, et al. Machine learning to identify metabolic subtypes of obesity: a multi‐center study. Front Endocrinol. 2021;12:713592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab. 2019;29:1028–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Do WL, Gohar J, Mccullough LE, Galaviz KI, Conneely KN, Narayan KMV. Examining the association between adiposity and DNA methylation: a systematic review and meta‐analysis. Obes Rev. 2021;22:e13319. [DOI] [PubMed] [Google Scholar]
- 72. Samblas M, Milagro FI, Martínez A. DNA methylation markers in obesity, metabolic syndrome, and weight loss. Epigenetics. 2019;14:421–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Aronica L, Levine AJ, Brennan K, Mi J, Gardner C, Haile RW, et al. A systematic review of studies of DNA methylation in the context of a weight loss intervention. Epigenomics. 2017;9:769–87. [DOI] [PubMed] [Google Scholar]
- 74. Demerath EW, Guan W, Grove ML, Aslibekyan S, Mendelson M, Zhou Y‐H, et al. Epigenome‐wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet. 2015;24:4464–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, et al. Epigenome‐wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mendelson MM, Marioni RE, Joehanes R, Liu C, Hedman ÅK, Aslibekyan S, et al. Association of body mass index with DNA methylation and gene expression in blood cells and relations to cardiometabolic disease: a Mendelian randomization approach. PLoS Med. 2017;14:e1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dhana K, Braun KVE, Nano J, Voortman T, Demerath EW, Guan W, et al. An epigenome‐wide association study of obesity‐related traits. Am J Epidemiol. 2018;187:1662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sayols‐Baixeras S, Subirana I, Fernández‐Sanlés A, Sentí M, Lluís‐Ganella C, Marrugat J, et al. DNA methylation and obesity traits: an epigenome‐wide association study. The REGICOR study. Epigenetics. 2017;12:909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aslibekyan S, Demerath EW, Mendelson M, Zhi D, Guan W, Liang L, et al. Epigenome‐wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity (Silver Spring). 2015;23:1493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Willmer T, Johnson R, Louw J, Pheiffer C. Blood‐based DNA methylation biomarkers for type 2 diabetes: potential for clinical applications. Front Endocrinol. 2018;9:744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nuotio M‐L, Pervjakova N, Joensuu A, Karhunen V, Hiekkalinna T, Milani L, et al. An epigenome‐wide association study of metabolic syndrome and its components. Sci Rep. 2020;10:20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Díaz‐Rúa R, Palou A, Oliver P. Cpt1a gene expression in peripheral blood mononuclear cells as an early biomarker of diet‐related metabolic alterations. Food Nutr Res. 2016;60:33554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Day SE, Coletta RL, Kim JY, Garcia LA, Campbell LE, Benjamin TR, et al. Potential epigenetic biomarkers of obesity‐related insulin resistance in human whole‐blood. Epigenetics. 2017;12:254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–11. [DOI] [PubMed] [Google Scholar]
- 85. Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aïssi D, Wahl S, et al. DNA methylation and body‐mass index: a genome‐wide analysis. Lancet. 2014;383:1990–8. [DOI] [PubMed] [Google Scholar]
- 86. Wang S, Song J, Yang Y, Zhang Y, Wang H, Ma J. HIF3A DNA methylation is associated with childhood obesity and ALT. PLoS One. 2015;10:e0145944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Richmond RC, Sharp GC, Ward ME, Fraser A, Lyttleton O, Mcardle WL, et al. DNA methylation and BMI: investigating identified methylation sites at HIF3A in a causal framework. Diabetes. 2016;65:1231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Visser M. Elevated C‐reactive protein levels in overweight and obese adults. JAMA. 1999;282:2131–5. [DOI] [PubMed] [Google Scholar]
- 89. Mccartney DL, Hillary RF, Stevenson AJ, Ritchie SJ, Walker RM, Zhang Q, et al. Epigenetic prediction of complex traits and death. Genome Biol. 2018;19:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hamilton OKL, Zhang Q, Mcrae AF, Walker RM, Morris SW, Redmond P, et al. An epigenetic score for BMI based on DNA methylation correlates with poor physical health and major disease in the Lothian birth cohort. Int J Obes. 2019;43:1795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chen Y, Kassam I, Lau SH, Kooner JS, Wilson R, Peters A, et al. Impact of BMI and waist circumference on epigenome‐wide DNA methylation and identification of epigenetic biomarkers in blood: an EWAS in multi‐ethnic Asian individuals. Clin Epigenetics. 2021;13:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Reed ZE, Suderman MJ, Relton CL, Davis OSP, Hemani G. The association of DNA methylation with body mass index: distinguishing between predictors and biomarkers. Clin Epigenetics. 2020;12:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vehmeijer FOL, Küpers LK, Sharp GC, Salas LA, Lent S, Jima DD, et al. DNA methylation and body mass index from birth to adolescence: meta‐analyses of epigenome‐wide association studies. Genome Med. 2020;12:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ma J, Rebholz CM, Braun KVE, Reynolds LM, Aslibekyan S, Xia R, et al. Whole blood DNA methylation signatures of diet are associated with cardiovascular disease risk factors and all‐cause mortality. Circ Genomic Precis Med. 2020;13:e002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gallardo‐Escribano C, Buonaiuto V, Ruiz‐Moreno MI, Vargas‐Candela A, Vilches‐Perez A, Benitez‐Porres J, et al. Epigenetic approach in obesity: DNA methylation in a prepubertal population which underwent a lifestyle modification. Clin Epigenetics. 2020;12:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bollepalli S, Kaye S, Heinonen S, Kaprio J, Rissanen A, Virtanen KA, et al. Subcutaneous adipose tissue gene expression and DNA methylation respond to both short‐ and long‐term weight loss. Int J Obes. 2018;42:412–23. [DOI] [PubMed] [Google Scholar]
- 97. Moleres A, Campión J, Milagro FI, Marcos A, Campoy C, Garagorri JM, et al. Differential DNA methylation patterns between high and low responders to a weight loss intervention in overweight or obese adolescents: the EVASYON study. FASEB J. 2013;27:2504–12. [DOI] [PubMed] [Google Scholar]
- 98. Keller M, Yaskolka Meir A, Bernhart SH, Gepner Y, Shelef I, Schwarzfuchs D, et al. DNA methylation signature in blood mirrors successful weight‐loss during lifestyle interventions: the CENTRAL trial. Genome Med. 2020;12:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Perfilyev A, Dahlman I, Gillberg L, Rosqvist F, Iggman D, Volkov P, et al. Impact of polyunsaturated and saturated fat overfeeding on the DNA methylation pattern in human adipose tissue: a randomized controlled trial. Am J Clin Nutr. 2017;105:991–1000. [DOI] [PubMed] [Google Scholar]
- 100. Arpón A, Riezu‐Boj JI, Milagro FI, Marti A, Razquin C, Martínez‐González MA, et al. Adherence to Mediterranean diet is associated with methylation changes in inflammation‐related genes in peripheral blood cells. J Physiol Biochem. 2016;73:445–55. [DOI] [PubMed] [Google Scholar]
- 101. Do WL, Whitsel EA, Costeira R, Masachs OM, Le Roy CI, Bell JT, et al. Epigenome‐wide association study of diet quality in the Women's Health Initiative and TwinsUK cohort. Int J Epidemiol. 2021;50:675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Arpón A, Milagro F, Razquin C, Corella D, Estruch R, Fitó M, et al. Impact of consuming extra‐virgin olive oil or nuts within a Mediterranean diet on DNA methylation in peripheral white blood cells within the PREDIMED‐Navarra randomized controlled trial: a role for dietary lipids. Nutrients. 2017;10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ollikainen M, Ismail K, Gervin K, Kyllönen A, Hakkarainen A, Lundbom J, et al. Genome‐wide blood DNA methylation alterations at regulatory elements and heterochromatic regions in monozygotic twins discordant for obesity and liver fat. Clin Epigenetics. 2015;7:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Cazaly E, Saad J, Wang W, Heckman C, Ollikainen M, Tang J. Making sense of the epigenome using data integration approaches. Front Pharmacol. 2019;10:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lee Y‐C, Christensen JJ, Parnell LD, Smith CE, Shao J, Mckeown NM, et al. Using machine learning to predict obesity based on genome‐wide and epigenome‐wide gene–gene and gene–diet interactions. Front Genet. 2022;12:783845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. World Health Organization . WHO report on the global tobacco epidemic 2021: addressing new and emerging products. https://www.who.int/publications/i/item/9789240032095 (accessed 7 January 2022) (2021).
- 107. World Health Organization . WHO report on the global tobacco epidemic 2017: monitoring tobacco use and prevention policies. https://escholarship.org/uc/item/8nw5p0zt (accessed 7 January 2022) (2017).
- 108. Belsky DW, Moffitt TE, Baker TB, Biddle AK, Evans JP, Harrington H, et al. Polygenic risk and the developmental progression to heavy, persistent smoking and nicotine dependence: evidence from a 4‐decade longitudinal study. JAMA Psychiatry. 2013;70:534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Aloise‐Young PA, Graham JW, Hansen WB. Peer influence on smoking initiation during early adolescence: a comparison of group members and group outsiders. J Appl Psychol. 1994;79:281–7. [DOI] [PubMed] [Google Scholar]
- 110. Maxwell KA. Friends: the role of peer influence across adolescent risk behaviors. J Youth Adolesc. 2002;31:267–77. [Google Scholar]
- 111. Buller DB. Understanding factors that influence smoking uptake. Tob Control. 2003;12:iv16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Conrad KM, Flay BR, Hill D. Why children start smoking cigarettes: predictors of onset. Br J Addict. 1992;87:1711–24. [DOI] [PubMed] [Google Scholar]
- 113. Mercken L, Candel M, Willems P, De Vries H. Social influence and selection effects in the context of smoking behavior: changes during early and mid adolescence. Heal Psychol. 2009;28:73–82. [DOI] [PubMed] [Google Scholar]
- 114. Rose RJ, Broms U, Korhonen T, Dick DM, Kaprio J. Genetics of smoking behavior. In: Kim Y‐K, editor. Handbook of behavior genetics. New York: Springer; 2010. p. 411–32. [Google Scholar]
- 115. Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, et al. Sequence variants at CHRNB3‐CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet. 2010;42:448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Chen L‐S, Baker TB, Piper ME, Breslau N, Cannon DS, Doheny KF, et al. Interplay of genetic risk factors (CHRNA5‐CHRNA3‐CHRNB4) and cessation treatments in smoking cessation success. Am J Psychiatry. 2012;169:735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PAF, et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet. 2007;16:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Freathy RM, Ring SM, Shields B, Galobardes B, Knight B, Weedon MN, et al. A common genetic variant in the 15q24 nicotinic acetylcholine receptor gene cluster (CHRNA5‐CHRNA3‐CHRNB4) is associated with a reduced ability of women to quit smoking in pregnancy. Hum Mol Genet. 2009;18:2922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, et al. Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry. 2008;165:1163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Bierut LJ, Madden PAF, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, et al. Novel genes identified in a high‐density genome wide association study for nicotine dependence. Hum Mol Genet. 2007;16:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. The Tobacco and Genetics Consortium . Genome‐wide meta‐analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42:441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Malaiyandi V, Lerman C, Benowitz NL, Jepson C, Patterson F, Tyndale RF. Impact of CYP2A6 genotype on pretreatment smoking behaviour and nicotine levels from and usage of nicotine replacement therapy. Mol Psychiatry. 2006;11:400–9. [DOI] [PubMed] [Google Scholar]
- 123. Erzurumluoglu AM, Liu M, Jackson VE, Barnes DR, Datta G, Melbourne CA, et al. Meta‐analysis of up to 622,409 individuals identifies 40 novel smoking behaviour associated genetic loci. Mol Psychiatry. 2020;25:2392–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gupta R, van Dongen J, Fu Y, Abdellaoui A, Tyndale RF, Velagapudi V, et al. Epigenome‐wide association study of serum cotinine in current smokers reveals novel genetically driven loci. Clin Epigenetics. 2019;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Loukola A, Buchwald J, Gupta R, Palviainen T, Hällfors J, Tikkanen E, et al. A genome‐wide association study of a biomarker of nicotine metabolism. PLOS Genet. 2015;11:e1005498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Tsaprouni LG, Yang T‐P, Bell J, Dick KJ, Kanoni S, Nisbet J, et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics. 2014;9:1382–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9:436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ambatipudi S, Cuenin C, Hernandez‐Vargas H, Ghantous A, Le Calvez‐Kelm F, Kaaks R, et al. Tobacco smoking‐associated genome‐wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8:599–618. [DOI] [PubMed] [Google Scholar]
- 129. Gao X, Thomsen H, Zhang Y, Breitling LP, Brenner H. The impact of methylation quantitative trait loci (mQTLs) on active smoking‐related DNA methylation changes. Clin Epigenetics. 2017;9:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zeilinger S, Kühnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, et al. Tobacco smoking leads to extensive genome‐wide changes in DNA methylation. PLoS One. 2013;8:e63812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Shenker NS, Ueland PM, Polidoro S, Van Veldhoven K, Ricceri F, Brown R, et al. DNA methylation as a long‐term biomarker of exposure to tobacco smoke. Epidemiology. 2013;24:712–6. [DOI] [PubMed] [Google Scholar]
- 132. Philibert R, Hollenbeck N, Andersen E, Osborn T, Gerrard M, Gibbons FX, et al. A quantitative epigenetic approach for the assessment of cigarette consumption. Front Psychol. 2015;6:656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Philibert R, Hollenbeck N, Andersen E, Mcelroy S, Wilson S, Vercande K, et al. Reversion of AHRR demethylation is a quantitative biomarker of smoking cessation. Front Psychiatry. 2016;7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhang Y, Yang R, Burwinkel B, Breitling LP, Brenner H. F2RL3 methylation as a biomarker of current and lifetime smoking exposures. Environ Heal Perspect. 2014;122:131–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour‐promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. [DOI] [PubMed] [Google Scholar]
- 136. Ringh MV, Hagemann‐Jensen M, Needhamsen M, Kular L, Breeze CE, Sjöholm LK, et al. Tobacco smoking induces changes in true DNA methylation, hydroxymethylation and gene expression in bronchoalveolar lavage cells. EBioMedicine. 2019;46:290–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Christiansen C, Castillo‐Fernandez JE, Domingo‐Relloso A, Zhao W, El‐Sayed Moustafa JS, Tsai P‐C, et al. Novel DNA methylation signatures of tobacco smoking with trans‐ethnic effects. Clin Epigenetics. 2021;13:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Guida F, Sandanger TM, Castagné R, Campanella G, Polidoro S, Palli D, et al. Dynamics of smoking‐induced genome‐wide methylation changes with time since smoking cessation. Hum Mol Genet. 2015;24:2349–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. 2012;21:3073–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Dugué P‐A, Jung C‐H, Joo JE, Wang X, Wong EM, Makalic E, et al. Smoking and blood DNA methylation: an epigenome‐wide association study and assessment of reversibility. Epigenetics. 2020;15:358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Teschendorff AE, Yang Z, Wong A, Pipinikas CP, Jiao Y, Jones A, et al. Correlation of smoking‐associated DNA methylation changes in buccal cells with DNA methylation changes in epithelial cancer. JAMA Oncol. 2015;1:476–85. [DOI] [PubMed] [Google Scholar]
- 142. Bell CC. DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders. JAMA. 1994;272:828–9. [Google Scholar]
- 143. Heatherton TF, Kozlowski LT, Frecker RC, Fagerstrom K‐O. The Fagerström Test for Nicotine Dependence: a revision of the Fagerström Tolerance Questionnaire. Br J Addict. 1991;86:1119–27. [DOI] [PubMed] [Google Scholar]
- 144. Centers for Disease Control and Prevention (CDC) . Quitting smoking among adults—United States, 2001–2010. MMWR Morb Mortal Wkly Rep. 2011;60:1513–9. [PubMed] [Google Scholar]
- 145. Benowitz NL, Schultz KE, Haller CA, Wu AHB, Dains KM, Jacob P. Prevalence of smoking assessed biochemically in an urban public hospital: a rationale for routine cotinine screening. Am J Epidemiol. 2009;170:885–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hsieh SJ, Ware LB, Eisner MD, Yu L, Jacob P, Havel C, et al. Biomarkers increase detection of active smoking and secondhand smoke exposure in critically ill patients. Crit Care Med. 2011;39:40–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Sandberg A, Sköld CM, Grunewald J, Eklund A, Wheelock ÅM. Assessing recent smoking status by measuring exhaled carbon monoxide levels. PLoS One. 2011;6:e28864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Tantoh DM, Wu M‐C, Chuang C‐C, Chen P‐H, Tyan YS, Nfor ON, et al. AHRR cg05575921 methylation in relation to smoking and PM2.5 exposure among Taiwanese men and women. Clin Epigenetics. 2020;12:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Yan Z, Ben S, Ines F, Stock C, Butterbach K, Holleczek B, et al. Smoking‐associated DNA methylation biomarkers and their predictive value for all‐cause and cardiovascular mortality. Environ Health Perspect. 2016;124:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zhang Y, Elgizouli M, Schöttker B, Holleczek B, Nieters A, Brenner H. Smoking‐associated DNA methylation markers predict lung cancer incidence. Clin Epigenetics. 2016;8:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Andersen AM, Philibert RA, Gibbons FX, Simons RL, Long J. Accuracy and utility of an epigenetic biomarker for smoking in populations with varying rates of false self‐report. Am J Med Genet Part B Neuropsychiatr Genet. 2017;174:641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Dawes K, Andersen A, Reimer R, Mills JA, Hoffman E, Long JD, et al. The relationship of smoking to cg05575921 methylation in blood and saliva DNA samples from several studies. Sci Rep. 2021;11:21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Langdon RJ, Yousefi P, Relton CL, Suderman MJ. Epigenetic modelling of former, current and never smokers. Clin Epigenetics. 2021;13:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Sugden K, Hannon EJ, Arseneault L, Belsky DW, Broadbent JM, Corcoran DL, et al. Establishing a generalized polyepigenetic biomarker for tobacco smoking. Transl Psychiatry. 2019;9:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hulls PM, De Vocht F, Bao Y, Relton CL, Martin RM, Richmond RC. DNA methylation signature of passive smoke exposure is less pronounced than active smoking: the Understanding Society study. Environ Res. 2020;190:109971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Richmond RC, Sillero‐Rejon C, Khouja JN, Prince C, Board A, Sharp G, et al. Investigating the DNA methylation profile of e‐cigarette use. Clin Epigenetics. 2021;13:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Andersen A, Reimer R, Dawes K, Becker A, Hutchens N, Miller S, et al. DNA methylation differentiates smoking from vaping and non‐combustible tobacco use. Epigenetics. 2022;17:178–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Avşar TS, Mcleod H, Jackson L. Health outcomes of smoking during pregnancy and the postpartum period: an umbrella review. BMC Pregnancy Childbirth. 2021;21:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Richmond RC, Suderman M, Langdon R, Relton CL, Davey Smith G. DNA methylation as a marker for prenatal smoke exposure in adults. Int J Epidemiol. 2018;47:1120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Reese SE, Zhao S, Wu MC, Joubert BR, Parr CL, Håberg SE, et al. DNA methylation score as a biomarker in newborns for sustained maternal smoking during pregnancy. Environ Health Perspect. 2017;125:760–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Sebastian R, Melton PE, Anni H, Karhunen V, Burdge G, Craig JM, et al. Machine learning‐based DNA methylation score for fetal exposure to maternal smoking: development and validation in samples collected from adolescents and adults. Environ Health Perspect. 2022;128:97003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. He C, Zhang M, Li J, Wang Y, Chen L, Qi B, et al. Novel insights into the consequences of obesity: a phenotype‐wide Mendelian randomization study. Eur J Hum Genet. 2022. 10.1038/s41431-021-00978-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Singh GM, Danaei G, Farzadfar F, Stevens GA, Woodward M, Wormser D, et al. The age‐specific quantitative effects of metabolic risk factors on cardiovascular diseases and diabetes: a pooled analysis. PLoS One. 2013;8:e65174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Di Angelantonio E, Bhupathiraju SN, Wormser D, Gao P, Kaptoge S, de Gonzalez AB, et al. Body‐mass index and all‐cause mortality: individual‐participant‐data meta‐analysis of 239 prospective studies in four continents. Lancet. 2016;388:776–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Peters SAE, Bots SH, Woodward M. Sex differences in the association between measures of general and central adiposity and the risk of myocardial infarction: results from the UK Biobank. J Am Heart Assoc. 2022;7:e008507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Pischon T, Boeing H, Hoffmann K, Bergmann M, Schulze MB, Overvad K, et al. General and abdominal adiposity and risk of death in Europe. N Engl J Med. 2008;359:2105–20. [DOI] [PubMed] [Google Scholar]
- 167. Battineni G, Sagaro GG, Chintalapudi N, Amenta F, Tomassoni D, Tayebati SK. Impact of obesity‐induced inflammation on cardiovascular diseases (CVD). Int J Mol Sci. 2021;22:4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Li S, Shin HJ, Ding EL, Van Dam RM. Adiponectin levels and risk of type 2 diabetes: a systematic review and meta‐analysis. JAMA. 2009;302:179–88. [DOI] [PubMed] [Google Scholar]
- 169. Xun P, Wu Y, He Q, He K. Fasting insulin concentrations and incidence of hypertension, stroke, and coronary heart disease: a meta‐analysis of prospective cohort studies. Am J Clin Nutr. 2013;98:1543–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. National Institute of Diabetes and Digestive and Kidney Diseases . Diabetes tests & diagnosis. https://www.niddk.nih.gov/health‐information/diabetes/overview/tests‐diagnosis (accessed 1 February 2022) (2016).
- 171. Mortensen MB, Nordestgaard BG. Elevated LDL cholesterol and increased risk of myocardial infarction and atherosclerotic cardiovascular disease in individuals aged 70–100 years: a contemporary primary prevention cohort. Lancet. 2020;396:1644–52. [DOI] [PubMed] [Google Scholar]
- 172. Johannesen CDL, Langsted A, Mortensen MB, Nordestgaard BG. Association between low density lipoprotein and all cause and cause specific mortality in Denmark: prospective cohort study. BMJ. 2020;371:m4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. The Emerging Risk Factors Collaboration . C‐reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta‐analysis. Lancet. 2010;375:132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Meisner A, Kundu P, Zhang YD, Lan LV, Kim S, Ghandwani D, et al. Combined utility of 25 disease and risk factor polygenic risk scores for stratifying risk of all‐cause mortality. Am J Hum Genet. 2020;107:418–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Khera AV, Chaffin M, Wade KH, Zahid S, Brancale J, Xia R, et al. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell. 2019;177:587–96.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome‐wide consortium meta‐analysis. Am J Hum Genet. 2016;98:680–96. [DOI] [PMC free article] [PubMed] [Google Scholar]