

Abstract
Objectives
Duchenne muscular dystrophy is associated with various degrees of cognitive impairment and behavioral disturbances. Emotional and memory deficits also constitute reliable outcome measures to assess efficacy of treatments in the mdx mouse lacking the muscle and neuronal full‐length dystrophins. The present study aimed to evaluate whether these deficits could be alleviated by the restoration of brain dystrophin.
Methods
We performed intracerebroventricular administration of a new potent tricyclo‐DNA antisense oligonucleotide (tcDNA‐ASO) containing a full phosphodiester backbone conjugated to a palmitic acid moiety (tcDNA‐ASO), designed to skip the mutated exon 23 of mdx mice.
Results
We first show that the tcDNA‐ASO rescues expression of brain dystrophin to 10–30% of wild‐type levels and significantly reduces the abnormal unconditioned fear responses in mdx mice in a dose‐dependent manner, 5 weeks post‐injection. Exon skipping efficiency, ASO biodistribution, protein restoration and effect on the fear response were optimal with a dose of 400 μg at 6–7 weeks post‐injection, with synaptic‐like expression in brain tissues such as the hippocampus and amygdala. Furthermore, this dose of tcDNA‐ASO restored long‐term memory retention of mdx mice in an object recognition task, but only had minor effects on fear conditioning.
Interpretation
These results suggest for the first time that postnatal re‐expression of brain dystrophin could reverse or at least alleviate some cognitive deficits associated with Duchenne muscular dystrophy. ANN NEUROL 2022;92:213–229
Introduction
Duchenne muscular dystrophy (DMD) is an X‐linked recessive neuromuscular disorder caused by mutations in the Dmd gene that encodes the dystrophin protein expressed in both muscle and the brain, Dp427. Brain Dp427 is localized at the postsynaptic site of central inhibitory synapses of pyramidal neurons in brain structures involved in cognitive functions, such as the hippocampus, cerebral cortex, cerebellum, and amygdala, where it plays a role in the clustering of gamma‐amino butyric acid‐A (GABAA) receptors. 1 , 2 , 3 , 4 , 5 Several shorter dystrophins with common C‐terminus are also produced in the brain from independent promoters, such as Dp140 and Dp71. 6 Although the position of the mutations might influence the nature and severity of the central alterations, cognitive, neuropsychiatric, and behavioral disturbances, including learning and memory deficits, autism spectrum disorder, attention deficit, and anxiety, have been observed in all subgroups of DMD patients, suggesting a role of full‐length dystrophin (Dp427) in these phenotypes. In contrast, intellectual disability was mostly observed in patients that have a cumulative deficiency on several dystrophins, including the shorter dystrophins Dp140 and Dp71. 7 , 8
The dystrophin‐deficient mdx mouse, one of the most commonly used models of DMD, also shows brain, cognitive, and behavioral alterations. The mdx mouse carries a nonsense mutation in exon 23 of the Dmd gene preventing expression of full‐length Dp427 dystrophin in both muscle and brain, whereas Dp140 and Dp71 expression is preserved. In this model, muscle degeneration is largely compensated by muscle regeneration processes before the age of 6 months, thus allowing behavioral studies without major motor bias. 9 Although learning performance is often preserved in this model, delayed acquisition has been shown in selective conditioning paradigms involving cue–outcome associative learning, as well as deficits in long‐term memory in both spatial and non‐spatial tasks involving recognition memory. 10 , 11 , 12 , 13 The deficits in hippocampal‐dependent and amygdala‐dependent long‐term memories suggested that Dp427 loss could specifically impair memory consolidation processes. In support of this hypothesis, the loss of Dp427 and altered GABAA‐receptor clustering in mdx mice have been associated with alterations in synapse ultrastructural organization and hippocampal synaptic plasticity, which are considered as cellular and neurophysiological features required for learning‐induced remodeling of neuronal networks during memory consolidation. 12 , 14 , 15 Characterization of emotional reactivity in mdx mice showed that despite displaying normal spontaneous locomotor activity in some experimental conditions, 13 , 16 these mice may show enhanced fearfulness (unconditioned fear) characterized by prolonged episodes of tonic immobility (freezing) in response to mild stress. 17 Thus, several robust outcome measures in the mdx mouse model can be used as phenotypic markers to evaluate the effects of specific treatments on central functions in preclinical studies.
In the past few decades, a large number of therapeutic strategies have been explored for DMD. One of the most promising therapeutic strategies is the exon skipping approach using antisense oligonucleotides (ASOs) aiming at restoring the open reading frame. ASO‐based therapies have been evaluated in clinical trials using different chemistries of ASOs, and several phosphorodiamidate morpholino oligomer (PMO) have now been approved by the US Food and Drug Administration despite low clinical benefit. 18 Other antisense molecules are currently being developed to improve the therapeutic potential of ASO and the clinical outcomes in DMD patients. Among these developments, we have previously shown that tricyclo‐DNA (tcDNA) ASOs display unprecedented uptake in many tissues, including cardiac muscle and the central nervous system (CNS), after systemic administration in mouse models of DMD 19 , 20 and SMA. 21 More recently, we showed that conjugation to palmitic acid enhances further the potency of tcDNA‐ASO. 22 The unique ability of tcDNA‐ASO to cross the blood–brain barrier offers the exciting opportunity to treat both the dystrophic muscle phenotype and the central deficits associated with the lack of dystrophin in the brain.
However, in contrast to the muscle and cardiac dystrophic phenotypes, little is known about the reversibility of these central deficits. Only a few therapeutic studies have focused on the brain aspect of DMD. Our groups have previously shown that intra‐hippocampal administration of adeno associated virus vectors encoding a specific U7snRNA targeting the Dmd exon 23 of the mdx mouse induces partial rescue of hippocampal dystrophin (~20%), which was sufficient for a complete recovery of GABAA‐receptor clustering 6 and normalization of hippocampal synaptic plasticity. 23 Furthermore, others reported that intracerebroventricular (ICV) infusion of PMO leads to a partial restoration of brain Dp427 and an improvement of the emotional phenotype in mdx mice. 17 Despite these initial encouraging findings, beneficial effects of ASO treatments on memory deficits have not been tested to date. Furthermore, intracerebral injections of higher doses of ASOs should be considered to test the reversibility of these deficits, as this may likely increase the efficacy of ASOs and help to rule out the possibility that improvements in behavioral performance were in part influenced by unspecific effects at the peripheral level in our previous systemic studies.
In the present study, we investigated the possibility of restoring brain full‐length dystrophin after direct ICV injection of tcDNA‐ASO, and we assessed the extent of emotional and cognitive correction in treated mdx mice. We show that a single bilateral ICV injection of tcDNA‐ASO in mdx mice allows partial restoration of Dp427 in different brain structures, up to 20–30% of WT levels. Treatment effects were characterized by a reduction of mice's stress reactivity, and an unprecedented improvement at the cognitive level, as shown by restored recognition memory retention. However, no beneficial effect was observed in an amygdala‐dependent fear conditioning, suggesting that the ability to alleviate central alterations by postnatal re‐expression of dystrophins might be selective of specific cognitive functions.
Materials and Methods
Animals and Microinjections
Animal procedures were performed in accordance with national and European legislation, approved by ethical committees (CEEA47 and CEEA59) and by the French government (Ministère de l'Enseignement Supérieur et de la Recherche, Autorisation APAFiS #6518 and #1563). C57BL/10ScSc‐Dmd mdx/J dystrophin‐deficient (mdx) and C57BL/10ScSn (WT) mice were bred in our animal facility at the Plateforme 2Care, UFR des Sciences de la santé, Université de Versailles Saint Quentin, and in the animal facility of the Paris‐Saclay Institute of Neuroscience (NeuroPSI, Orsay), where cognitive testing was performed. Male mdx and WT littermate mice were obtained by mating heterozygous mdx females with WT males. Siblings were kept in the same cage from weaning (2–6 per cage) under a 12‐hour light–dark cycle (light on: 7:00 a.m.) with food and water ad libitum. Mouse genotype was verified by polymerase chain reaction (PCR). The tcDNA‐ASO used in this study targeted the donor splice site of exon 23 of the mouse dystrophin pre‐mRNA (sequence M23D 5′‐CCTCGGCTTACCT‐3′ previously described 20 and was synthesized by SQY Therapeutics, Montigny‐le‐Bretonneux, France). Palmitic acid was conjugated at the 5′ end of the tcDNA full phosphodiester via a C6‐amino linker and a phosphorothioate bond.
ICV injections were performed in 8‐week‐old mdx and WT littermate male mice deeply anesthetized by a single intraperitoneal injection of ketamine (95 mg/kg)/medetomidine (1 mg/kg). TcDNA or saline (phosphate‐buffered saline [PBS], 0.1 mol/l) solutions were injected bilaterally into the lateral brain ventricles (−0.5 mm from bregma; 1 mm lateral; −2 mm from dura). A volume of 5 μl was infused in each ventricle at a rate of 0.2 μl/minute. The doses of tcDNA were thus distributed bilaterally to reach a total amount of 100 μg (corresponding to 3.25 mg/kg for a 30 g mouse), 200 μg (6.5 mg/kg), and 400 μg (13 mg/kg). Mice were given at least 3 weeks to recover before being tested in behavioral experiments. Treatment of animals was pseudorandomized in each cage, so that it was balanced within the litters and with comparable distribution among litters. Thus, there was at least one PBS‐WT mouse, one mdx‐treated mouse, and one PBS‐mdx mouse in each litter to avoid bias from litter effects.
Animals were euthanized by cervical dislocation at the end of behavioral experiments to collect brain tissues for molecular and biochemical analyses. Brain regions (hippocampus, cortex, cerebellum) were dissected out and snap‐frozen in liquid nitrogen, then stored at −80°C before further analysis.
Three cohorts of mice were used in this study, as represented in the study design presented in Figures 1A, 3A and 6A, corresponding to a total number of 76 mice for the entire study.
FIGURE 1.
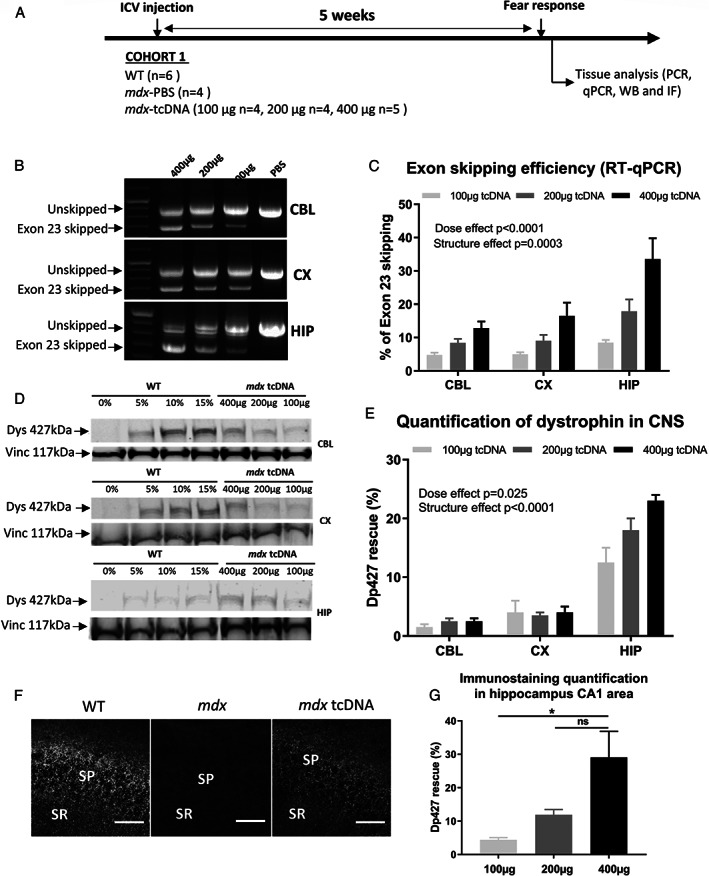
Dose‐dependent effects of tricyclo‐DNA antisense oligonucleotide (tcDNA‐ASO) treatment on exon skipping levels and dystrophin rescue 5 weeks after intracerebroventricular (ICV) injection. (A) Schematic representation of the study design applied to the first cohort of mice (Cohort 1), showing the sample sizes, the doses of tcDNA‐ASO administered by ICV, the post‐injection delay at which fear conditioning was performed (5 weeks) and then the collection of tissues after euthanasia. (B) Detection of exon 23‐skipped dystrophin mRNA by nested RT–PCR in treated mdx mice. (C) Quantification by RT–qPCR of exon 23‐skipping levels in different brain tissues (cerebellum [CBL], cortex [CX], and hippocampus [HIP]) 5 weeks after the ICV injection of different doses of tcDNA‐ASO (100, 200, or 400 μg). Results are expressed as means ± SEM; n = 4 mice/group. (D) Detection and (E) quantification of dystrophin protein restoration by western blot analysis in different brain regions, 5 weeks after the injection of tcDNA‐ASO (100, 200, or 400 μg). The blots show a representative example of dystrophin restoration in CBL, CX, and HIP in one of the four animals analyzed in each group, in which a four‐point standard curve made of 0, 5, 10, and 15% of wild‐type (WT) lysate (mixed with mdx lysate) was loaded for quantification. Results are expressed as means ± SEM; n = 3 mice/group. (F) Detection and (G) quantification of restored dystrophin by immunofluorescence staining using the DYS1 primary antibody, in the stratum pyramidale (SP) and proximal stratum radiatum (SR) of the CA1 hippocampus in WT, mdx mice treated with phosphate‐buffered saline (mdx) and mdx mice treated with 400 μg of tcDNA‐ASO (mdx‐tcDNA). Scale bar, 12 μm. Results in (G) are expressed as means ± SEM; n = 3 mice/group, *p < 0.05 (one‐way ANOVA).
FIGURE 3.
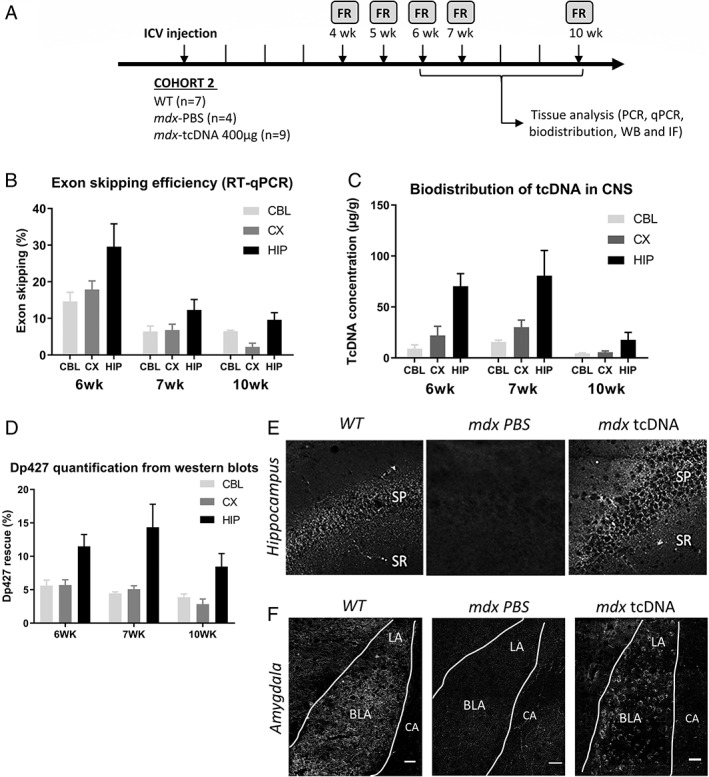
Analysis of exon 23‐skipping levels and protein rescue over time following intracerebroventricular (ICV) treatment with 400 μg of tricyclo‐DNA antisense oligonucleotide (tcDNA‐ASO). (A) Schematic representation of the study design applied to the second cohort of mice (Cohort 2), showing the sample sizes, the ICV injection of 400 μg of tcDNA‐ASO, the post‐injection delays at which the fear response (FR) was performed (4, 5, 6, 7, and 10 weeks) and the tissues collected after euthanasia in subsets of animals at 6, 7 and 10 weeks after the injection. (B) Quantification of exon 23‐skipping levels by RT–qPCR in different brain tissues (cerebellum [CBL], cortex [CX], hippocampus [HIP]) at 6, 7, and 10 weeks post‐injection delays. Results are expressed as means ± SEM; n = 3 mice/group. (C) Quantification by fluorescent hybridization assay of tcDNA‐ASO content in various brain tissues at 6, 7, and 10‐week post‐injection delays. Results are expressed as means ± SEM of ASO concentration (μg/g); n = 3 mice per group. (D) Quantification of dystrophin protein restoration by western blot analysis in different brain regions at 6, 7, and 10‐week post‐injection delays. (E, F) Detection of restored dystrophin protein by immunofluoresecnce in (E) the stratum pyramidale (SP) and proximal stratum radiatum (SR) of the CA1 hippocampus and (F) within the basolateral (BLA) and the lateral (LA) nuclei of amygdala, but not in central amygdala (CA), in wild‐type (WT) and mdx mice injected with phosphate‐buffered saline (PBS; mdx PBS) or tcDNA‐ASO (mdx‐tcDNA) at the 7 week post‐injection delay. Scale bar, 50 μm.
Behavioral Testing
Behavioral experiments were undertaken blind to genotype and treatment using three independent cohorts of mice. Two cohorts were used to assess unconditioned fear 5 weeks after treatment with different doses of ASOs and at different post‐injection delays, respectively. The third cohort was successively submitted to two behavioral tests starting at 6 weeks after injection of 400 μg tcDNA‐ASO, with an interval of 5 days between tests and in the following order: object recognition (at 6 weeks post‐injection) and cued fear conditioning (at 7 weeks post‐injection). Behavioral experiments were performed during the light phase. All experimental procedures were conducted under appropriate biological containment in accordance with European Communities Council Directive (CEE 86/609) for animal care and experimentation, EU Directive 2010/63/EU and French National Committee (87/848), and approved by the ethics committee (Paris Centre et Sud, CEEA59).
Restraint‐Induced Unconditioned Fear
The mouse was restrained by grasping the scruff and back skin between the thumb and index fingers, while securing the tail between the third and little fingers and tilting the animal upside‐down, so that the ventral part of its body faced the experimenter. After 15 seconds, the mouse was released to a novel cage (24 × 19 cm, with 12‐cm high walls) containing clean sawdust, and was then video‐tracked for 5 minutes under dim illumination (80 lx) using the Any‐maze software (Stoelting, Wood Dale, IL). Unconditioned fear responses induced by this short acute stress were typically characterized by periods of tonic immobility (freezing) and quantified during a 5‐minutes recording period. Complete immobilization of the mouse, except for respiration, was regarded as a freezing response. The percentage of time spent freezing was calculated. 11 , 17
Auditory‐Cued Fear Conditioning
The conditioning procedure was carried out using the StartFear system (Panlab S.L., Barcelona, Spain). The conditioning chamber (25 × 25 × 25 cm) had three black methacrylate walls, a transparent front door, a grid floor connected to a shock scrambler to deliver electrical footshocks as unconditioned stimuli (US), and a speaker mounted on the ceiling to deliver audible tones as conditioned stimuli (CS). The conditioning chamber rested on a high sensitivity weight‐transducer system to generate an analogical signal reflecting the animal's movement. The chamber was confined in a ventilated soundproof enclosure (67 × 53 × 55 cm) on an anti‐vibration table with a surrounding 60‐dB white noise. Interchangeable floors and walls (ie, plain floor and white walls) were used to analyze retention of cued fear in a novel context.
On the first day (acquisition), a 2‐minute baseline period was recorded before delivery of five CS–US pairs (Tone: 80 dB, 10 kHz, 30 seconds; footshocks: each at 0.4 mA for 2 seconds) with variable and pseudo‐randomly distributed intervals between pairs of stimuli (60, 120, and 180 seconds). On the next day (retention), the session started by placing the mouse in a different context for 2 minutes (baseline) before delivery of four CS (80 dB, 10 kHz, 30 seconds) separated by intervals of variable durations (60, 90, and 120 seconds). The animal's movements were sampled at 50 Hz for quantitative analysis (FREEZING software; Panlab S.L.). Freezing was analyzed in both acquisition and retention sessions during the delivery of the CS (periods of 30 seconds) to specifically reflect associative learning performance. 9 , 26
Object Recognition
The test box consisted of a square open field (50 × 50 × 50 cm) with black walls and a white floor covered with sawdust. Experiments were undertaken under constant room temperature and homogeneous dim illumination (<50 lx). A set of three objects of different shapes were used, which were either glazed wooden (one set) or plastic objects (two sets). Two objects were simultaneously present in the box during testing, each placed at opposite sides of the box, at 15 cm from the walls. The sequential presentation of the three sets of objects and the spatial arrangement of the objects in the box (left or right sides) were chosen in a pseudorandom order and counterbalanced among individuals.
The testing procedure started with a 4‐day period of habituation consisting of two daily sessions of 10 minutes separated by a 5‐hour delay. On the first day, mouse siblings from a given cage were placed together in the empty apparatus and allowed to move freely for 10 minutes. On the next days, mice were exposed individually to the empty box to let them familiarize with the apparatus and to record spontaneous locomotor activity. On the last pretraining session of day 4, two identical plastic objects were placed in the box and mice were allowed to freely explore the objects for 10 minutes. These two objects were not used for subsequent memory testing. The object recognition test started 48 hours after the end of the habituation period. Mice were first submitted to a single acquisition trial during which they were exposed to two different objects for 10 minutes. Memory retention was tested after a delay of 24 hours. During this test phase, a novel object replaced one of the objects used during acquisition and mice were allowed to explore for 5 minutes.
A video camera above the box was connected to a computer system and mouse behavior was analyzed using the Any‐maze video‐tracking (Stoelting). Behavioral parameters were recorded for 10 minutes (pretraining and acquisition sessions) or 5 minutes (retention session) to quantify locomotor activity, expressed by the distance travelled in the box (horizontal activity), and the number of rearings and leanings (vertical activity). During the acquisition and retention sessions, the latency of the first contact with an object and the time spent by the mouse in contact with it were recorded. A contact was defined as the animal's snout or paws actually touching the object. A recognition index was calculated for each subject (time exploring the novel object × 100 / total object exploration time), and object recognition was evaluated by comparing the relative exploration of the novel object to chance level (50%). 12
In this experiment, five mice were excluded from the analysis (1 WT, 2 mdx‐PBS and 2 mdx‐tcDNA), because they had an exploration of the objects lower than 4 seconds, which is our exclusion criterion, as in our previous characterization study. 12
TcDNA Tissue Quantification by Fluorescent Hybridization Assay
Tissues were homogenized using the Precellys 24 (Bertin Instruments, Montigny le bretonneux, France) in lysis buffer (100 mmol/l Tris–HCl, pH 8.5, 200 mmol/l NaCl, 5 mmol/l EDTA, 0.2% sodium dodecyl sulfate) containing 2 mg/ml of proteinase K (Invitrogen, Waltham, MA, USA; 50 mg tissue/ml of buffer), followed by incubation overnight at 55°C in a hybridization oven. After centrifugation at 12,000 g (Sorval ST 8R centrifuge, 75,005,719 rotor) for 15 minutes, the supernatant was used in the assay. Quantification of tcDNA was performed using a hybridization assay with a molecular beacon probe, as previously described. 36 Briefly, 10 μl of tissue lysates were incubated with a 5′ Cy3‐DNA complementary probe conjugated with HBQ quencher at 3′ in a black non‐binding 96‐well plates (Fischer Scientific, Waltham, MA, USA). PBS was added to a final volume of 100 μl per well, and fluorescence was measured on a spectrophotometer (Ex 544 nm/Em 590 nm using FluoStar Omega). The amount of tcDNA in tissues was determined using a standard curve build on the measurement of known tcDNA quantities dissolved in the respective tissue lysates PBS‐injected animals.
RNA Analyses
Total RNA extracts were isolated using TRIzol reagent according to the manufacturer's instructions (Thermo Fisher Scientific, Waltham, MA, USA). Aliquots of 500 ng of total RNA were used for RT–PCR analysis using the Access RT–PCR System (Promega, Madison, WI, USA) in a 50‐ml reaction using the external primers Ex 20Fo (50‐CAGAATTCTGCCAATTGCTGAG‐30) and Ex 26Ro (50‐TTCTTCAGCTTGTGTCATCC‐30). The cDNA synthesis was carried out at 45°C for 45 minutes, directly followed by the primary PCR of 30 cycles of 95°C (30 seconds), 55°C (1 minute), and 72°C (2 minutes). Two ml of these reactions were then re‐amplified in nested PCRs by 22 cycles of 95°C (30 seconds), 55°C (1 minutes), and 72°C (2 minutes) using the internal primers Ex 20Fi (50‐CCCAGTCTACCACCCTATCAG AGC‐30) and Ex 26Ri (50‐CCTGCCTTTAAGGCTTCCTT‐30). PCR products were analyzed on 2% agarose gels. Exon 23 skipping was also measured by Taqman quantitative PCR as previously described 37 using Taqman assays designed against the exon 23–24 junction (assay Mm.PT.58.43432707: Forward: 5′‐CAGGCCATTCCTCTTTCAGG‐3′; reverse: 5′‐GAAACTTTCCTCCCAGTTGGT‐3′; Probe: 5′‐TCAACTTCAGCCATCCATTTCTGTAAGGT‐3′) and exon 22–24 junction (Forward: 5′‐CTGAATATGAAATAATGGAGGAGAGACTCG‐3′; reverse: 5′‐CTTCAGCCATCCATTTCTGTAAGGT‐3′; Probe: 5′‐ATGTGATTCTGTAATTTCC‐3′; Integrated DNA technology). A total of 150 ng of cDNA was used as input per reaction, and all assays were carried out in triplicate. Assays were performed under fast cycling conditions on a Biorad CFX384 Touch Real‐Time PCR Detection System, and all data were analyzed using the absolute copy number method. For a given sample, the copy number of skipped product (exon 22–24 assay) and unskipped product (exon 23–24 assay) were determined using the standards Ex20‐26 and Ex20‐26Delta23, respectively (gBlocks gene fragments from Integrated DNA technology). Exon 23 skipping was then expressed as a percentage of total dystrophin (calculated by the addition of exon 22–23 and exon 22–24 copy numbers).
Western Blot Analyses
Protein extracts were obtained from dissected brain structures (cortex, hippocampus, cerebellum) treated with RIPA lysis and extraction buffer (Thermo Fisher Scientific) complemented with SDS powder (5% final; Bio‐Rad, Marnes‐la‐Coquette, France), and the total protein concentration was determined with the BCA Protein Assay Kit (Thermo Fisher Scientific). Samples were denatured at 100°C for 3 minutes, and 25 μg of protein were loaded onto NuPAGE 3–8% Tris‐Acetate Protein gels (Invitrogen), following the manufacturer's instructions. Dystrophin protein was detected by probing the membrane with NCL‐DYS1 primary monoclonal antibody (NCL‐DYS1; Novocastra, Newcastle, UK), and vinculin was detected as the internal control with the hVin‐1 primary antibody (Sigma, St Louis, MO, USA), followed by incubation with a goat anti‐mouse secondary antibody (IRDye 800CW Goat anti‐mouse IgG; Li‐Cor, Germany). Bands were visualized using the Odyssey CLx system (Li‐Cor, Lincoln, NE, USA), and quantification was carried out using the Empiria Studio software (Li‐Cor) based on a standard curve specific of each brain structure and made of a mix of WT and mdx control lysates to obtain defined percentages of dystrophin (0, 5, 10, 15%, or 0, 10, 15, 30% of corresponding WT tissues).
Detection of Dp427 by Immunofluorescence in Tissue Sections
Fresh‐frozen brain cryosections of 30 μm thickness were collected onto Superfrost+ slides, thawed for 2 minutes at room temperature, fixed in acetone/methanol (1:1) for 5 minutes at −20°C, washed in PBS, incubated first in a blocking solution for 45 minutes (10% normal goat serum, 0.3% Triton X‐100, and 1% BSA), then overnight at 4°C with a monoclonal anti‐dystrophin primary antibody (DYS1 Leica, Wetzlar, Germany; dilution: neat), and washed and incubated with secondary antibody Alexa 647 (1:400, 1 hour, room temperature). Controls were prepared by omitting the primary antibody showed no specific staining. Images were taken at equivalent locations and exposure times using a laser scanning confocal microscope (Zeiss LSM 700, ×40 objective). Stacks of seven to eight images (1,024 × 1,024 pixels) spaced by 1 μm were recorded at a magnification of 156 nm/pixel. Quantification of Dp427 re‐expression in the CA1 hippocampus was performed in coronal sections from three mice per group at Bregma −2.9 mm (total surface sampled: 25,000 μm2).
Basescope™ Analyses of DMD mRNA
The BaseScope™ Duplex assay from Advanced Cell Diagnostics was used to detect the specific dmd mRNA by in situ hybridization. Fresh frozen sections prepared as above for immunofluorescence were fixed in cold neutral buffered formalin and dehydrated in ethanol bath (50, 70, 100%). The hydrogen peroxide, Protease IV treatment and the RNA in situ hybridization were performed using the BaseScope Duplex reagent kit (323,800; Advanced Cell Diagnostics, Newark, CA, USA) according to the manufacturer's instructions. Specific BaseScope probes were designed against the exon 22–23 junction to detect unskipped mRNA and against exon 22–24 junction to specifically detect exon‐23 skipped mRNA. Control probes targeting peptidylprolyl isomerase B and ubiquitin C were used in parallel as positive controls, and against the bacterial dihydrodipicolinate reductase as a negative control (data not shown). At the end of the in situ hybridization, tissues were counterstained with hematoxyline Gills I (GHS132‐1 L; Sigma) diluted at 50% in water (30‐second staining) and ammonium hydroxide 28‐30WT% (205,840,025; Acros Organics, Geel, Belgium) diluted at 0.02% in water (30‐second staining). Slides were then dried at 60°C for 45 minutes and mounted in Vectamount (321,584; Advanced Cell Diagnostics). Images were obtained using the Aperio AT2 scanner (Leica; zoom ×40) and analyzed with ImageScope software (Leica). Signal quantification was performed in coronal sections from three mice per group at Bregma −2.9 mm, 38 from images taken in various layers of the hippocampus, covering 3,500‐μm length along CA1 subfield and 1,100 μm along CA3, and in all cortical layers above the hippocampal dendate gyrus (primary and secondary visual cortices), as well as along the Purkinje cell layers of cerebellum (4&5Cb and 9Cb lobules) from sagittal sections (lateral 0.60 mm). Discrimination of specific signals was achieved by detection of turquoise staining (corresponding to Ex22/23 junction) with a Hue value of 0.35 and color saturation threshold of 0.2, and specific red staining (corresponding to Ex22/24 junction) with a Hue value of 0.5 and color saturation threshold of 0.14. The positive Pixel Count v9 program of ImageScope software was used for semiquantitative analysis of the relative area covered by each signal (rather than dot counts because of the variability in dot size).
Statistical Analysis
Data expressed as means ± SEM were analyzed with the GraphPad Prism8 software (San Diego, CA, USA). All data passed the normality (Shapiro–Wilk normality test) and equal‐variance tests (F test). Group comparisons were performed using one‐ and two‐way analyses of variance (ANOVA) with repeated measures comparisons when required (effects in distinct brain areas, at different post‐injection delays, or during different sessions/trials in behavioral analyses), followed by Tukey (for one‐way ANOVA) or Holm–Sidak (for 2‐way ANOVA) post‐hoc multiple comparisons. In the object recognition test, comparisons of the recognition index with chance level (50%) were performed using a one‐sample t test. Significant levels were set at *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
Dose‐Dependent Exon‐Skipping in the CNS Following ICV Delivery of tcDNA‐ASO
To evaluate the effect of Dp427 restoration in the brain of mdx mice, we used ICV microinjection of the previously published tcDNA sequence targeting the donor splice site of the mouse Dmd exon 23. 20 For optimal efficacy, palmitic acid conjugated‐tcDNA‐ASOs containing a full phosphodiester backbone were used. 22 To define the optimal conditions for Dp427 restoration in the brain, we first evaluated the effects of different doses of tcDNA‐ASO (100, 200, and 400 μg) administered once to mdx mice by ICV bilateral microinjection (Figure 1A). The maximal feasible dose was 400 μg, a limitation related to the solubility of the tcDNA‐ASOs taken from a 40‐mg/ml stock solution for bilateral injections of a 10‐μl maximal volume. All doses were well tolerated. Brains were analyzed 5 weeks after the injection to determine the extent of exon 23 skipping levels and protein rescue. A dystrophin transcript lacking exon 23 was detected by RT–PCR in cortical, cerebellar, and hippocampal samples from tcDNA‐treated mice at all injected doses (Figure 1B). However, no transcript lacking exon 23 was detected in skeletal muscles (data not shown), indicating no peripheral effect. Quantification by RT–qPCR confirmed efficient exon 23 skipping in all examined tissues. There was an overt dose effect (F[2, 18] = 19.63, p < 0.0001; 100 μg vs 200 μg, p = 0.0695; 100 μg vs 400 μg, p < 0.0001; 200 μg vs 400 μg, p = 0.0034), as the level of exon skipping increased with tcDNA dose increments, reaching levels >30% in the hippocampus with a dose of 400 μg (Figure 1C). Exon skipping levels were variable depending on the brain structure (structure effect: F[2, 18] = 13.05, p = 0.0003) reflecting higher levels in the hippocampus compared with the cerebellum and cortex (CBL vs CX: p = 0.808; CBL vs HIP: p = 0.0005; CX vs HIP: p = 0.0019) at all doses (insignificant dose × structure interaction). ICV injection of up to 400 μg of a control sense tcDNA induced no exon skipping in any of the analyzed regions (data not shown).
We then evaluated restoration of dystrophin expression by western blot (Figure 1D). The amount of dystrophin protein ranged from 3% to 25%, confirming the presence of higher levels of restored protein with a higher dose (dose effect: F[2, 9] = 5.754, p = 0.0246; 100 μg vs 200 μg, p = 0.2337; 100 μg vs 400 μg, p = 0.0197; 200 μg vs 400 μg, p = 0.2860). Alike skipping levels, restoration of dystrophin protein was also higher in the hippocampus compared with other brain structures (structure effect: F[2, 9] = 115.9, p < 0.0001; CBL vs CX, p = 0.3470; CBL vs HIP, p < 0.0001; CX vs HIP, p < 0.0001), with up to 20–25% restoration in mdx mice treated with 400 μg of tcDNA (Figure 1E).
We next performed immunofluorescence (IF) assays in brain sections and analyzed dystrophin expression in the hippocampus, as this structure shows the highest skipped mRNA and protein restoration levels. Dystrophin is normally expressed in inhibitory synapses around neuronal soma in the stratum pyramidale and in the proximal dendritic layer of the stratum radiatum within the CA1‐CA3 hippocampal subfields. As shown in Figure 1F, dystrophin expression was indeed characterized by a punctate immunoreactive signal reflecting synaptic expression in WT mice, as well as in treated mdx mice, whereas it was completely absent in untreated mdx mice (Figure 1F). We used a semiquantitative approach based on confocal image analysis to estimate the level of dystrophin re‐expressed in synapses of hippocampal neurons. 24 Whereas mdx mice showed complete absence of dystrophin immunoreactive signal except for minimal background staining (<2% of WT levels), mdx‐tcDNA mice showed significant levels of dystrophin re‐expression in the CA1 hippocampus, ranging from 5 to approximately 30%, depending of the dose of tcDNA‐ASO (dose effect: F[2, 6] = 7.550, p = 0.0001; 100 μg vs 200 μg, p = 0.052; 100 μg vs 400 μg, p = 0.0212; 200 μg vs 400 μg, p = 0.0857; Figure 1G). We also observed a restoration of dystrophin by IF in the cortex and cerebellum (data not shown).
The loss of dystrophin in the mdx mouse model has been associated with an enhanced defensive behavior in response to mild acute stress, characterized by long periods of tonic immobility (freezing) after a short manual restraint. 17 To evaluate a possible compensation of this behavioral phenotype after treatment, we measured the duration of freezing during 5 minutes after a 15‐second manual restraint. As expected, this short restraint induced a robust fear response in PBS‐treated mdx mice, but not in WT mice. Control mdx mice injected with PBS spent approximately 70% of their time freezing, whereas WT mice only showed approximately 10% immobility (Figure 2A). In line with the detected exon skipping and rescued dystrophin expression in the brain 5 weeks after ICV injection of tcDNA, we measured a significant improvement of the mdx phenotype, characterized by decreased freezing in the treated mice in a dose‐dependent manner (group effect: F[4, 18] = 10.56, p = 0.0001; Figure 2A). Indeed, the percentage of freezing was decreased as compared with PBS‐injected mdx mice, to approximately 60% with a dose of 100 μg (p = 0.99), 40% with 200 μg (p = 0.42), and 30% with 400 μg (p = 0.026; Figure 2A). At the 400‐μg dose, locomotor horizontal activity (Figure 2B) and vertical activity (rearings; Figure 2C) were slightly higher in tcDNA‐treated mdx mice than in PBS controls, but this did not reach the level of significance.
FIGURE 2.
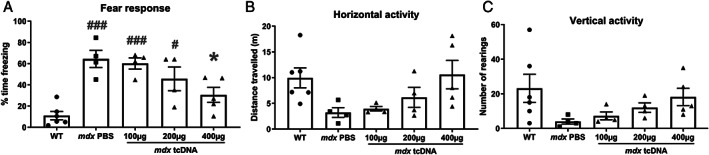
Dose‐dependent effects of tricyclo‐DNA antisense oligonucleotide (tcDNA‐ASO) treatment on emotional response 5 weeks after intracerebroventricular (ICV) injection. (A–C) Behavioral parameters recorded during 5 minutes after a mild stress induced by a short (15 seconds) manual restraint in wild‐type (WT) and mdx mice treated with phosphate‐buffered saline (PBS; mdx PBS) or tcDNA‐ASO (100, 200, or 400 μg) and tested 5 weeks after the ICV treatment, (A) Fear response expressed as the percentage of time spent freezing, (B) horizontal activity expressed as the total distance travelled (m), and (C) vertical activity expressed as the number of rearings. Results are expressed as means ± SEM; n = 5 per treated group at the dose of 400 μg, n = 4 per treated group at 100 μg, 200 μg, and mdx PBS, and n = 6 for WT mice. *p < 0.05 compared with mdx PBS, ### p < 0.001 and # p < 0.05 compared with WT (ANOVA).
Considering the levels of exon skipping, dystrophin rescue and the effects on the emotional response, we selected the dose of 400 μg for subsequent experiments.
Determination of the Optimal Timeframe Following ICV Injection of tcDNA‐ASO
To define the optimal timeframe for further behavioral studies, we then investigated the extent and duration of dystrophin restoration after ICV injection of 400 μg of tcDNA‐ASO. Exon 23 skipping levels and protein restoration were evaluated 6, 7, and 10 weeks after the ICV injection (Figure 3A). As previously shown at 5 weeks post‐injection (Figure 1B,C), exon 23 skipping levels measured by RT–qPCR (Figure 3B) were higher in the hippocampus than in the cortex and cerebellum at all post‐injection delays (structure effect: F[2, 24] = 7.145, p = 0.0037; structure × delay interaction, not significant; CBL vs CX, p = 0.9963; CBL vs HIP, p = 0.0096; CX vs HIP, p = 0.0079). Levels of exon skipping were maximal 6 weeks post‐injection, and then progressively decreased (delay effect: F[2, 24] = 20.59, p < 0.0001). Remarkably, however, the skipped transcript was still detectable 10 weeks after an ICV injection of 400 μg of tcDNA‐ASO. We also quantified the amount of tcDNA‐ASO in the hippocampus, cortex, and cerebellum at the different post‐injection delays (Figure 3C). TcDNA were effectively detected in these three CNS regions, yet with differences in the distribution of ASO among these structures (structure effect: F[2, 24] = 16.06, p < 0.0001). The highest amount of tcDNA was detected in the hippocampus, whereas levels were lower in the cortex and the cerebellum (CBL vs CX, p = 0.5666; CBL vs HIP, p < 0.0001; CX vs HIP, p < 0.0007). Hence, the biodistribution of tcDNA‐ASO after ICV injection was in agreement with the variable levels of exon skipping depending on the brain structure. To further investigate the localization of the skipped and unskipped mRNA in the brain, we performed in situ hybridization using BaseScope duplex probes against exon junction 22–23 (unskipped mRNA) and 22–24 (exon 23 skipped mRNA). We successfully detected the exon 23‐skipped mRNA in the brain of tcDNA‐treated mice (red dots in Figure 4A), whereas only the unskipped mRNA (turquoise dots) was detected in WT and PBS‐treated mdx mice. By quantifying the relative area covered by the red and blue signals, we were able to estimate the level of exon skipping depending on the localization in the brain. Within dorsal CA1 (Figure 4B,C), highest expression was detected close to the subiculum (>80% of exon 23 skipping level), with a progressive decrease along the CA1 layer towards ventral hippocampal regions (Figure 4C,D; F[9, 18] = 55.98, p = 0.0031). Likewise, the skipped mRNA was selectively detected in mdx mice treated with tcDNA‐ASO in cortex (>20% of exon 23 skipping) and cerebellum (>15% of exon 23 skipping), suggesting widespread diffusion of the treatment in a variety of brain structures after ICV injections (not shown).
FIGURE 4.
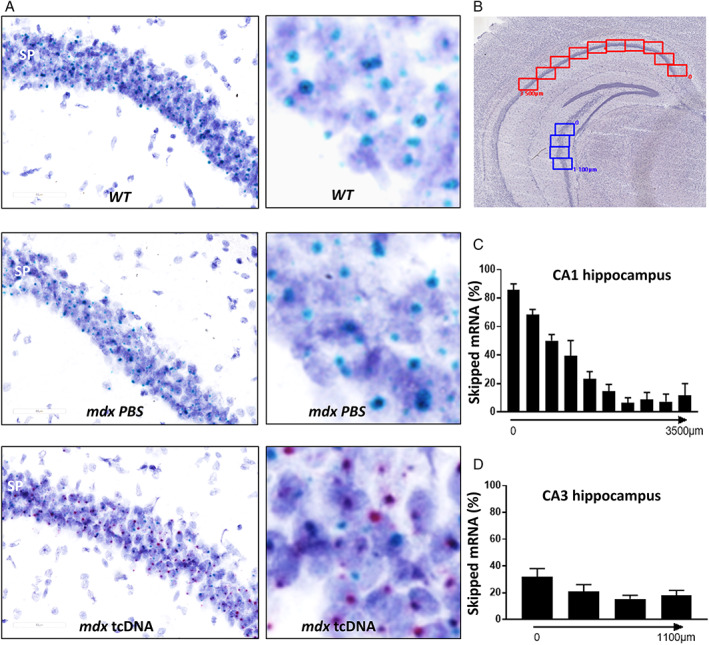
Detection of exon 23‐skipped transcripts after intracerebroventricular (ICV) treatment with tricyclo‐DNA antisense oligonucleotide (tcDNA‐ASO) using BaseScope hybridization technology. (A–D) Detection of skipped dystrophin mRNA with BaseScope hybridization technology in hippocampus at 7‐week post‐injection delay: (A) Visualisation of dmd mRNA in the stratum pyramidale (SP) of the hippocampus of wild‐type (WT), phosphate‐buffered saline (PBS)‐treated and tcDNA‐treated mdx mice detected by BaseScope probes against the exons 22–23 junction for native full mRNA (turquoise dots in the three images) and exons 22–24 junction for exon 23‐skipped mRNA only detected in tcDNA treated mdx mice (red dots in mdx tcDNA, last image on the right). Dark blue elements are counterstained nuclei. Scale bar, 60 μm. The right panels are zoomed parts of each image with higher brightness, allowing better visualization of the turquoise and red dots. (B) Representation of the images used as counting windows along the Ammon's horn of hippocampus for quantification in CA1 (red windows, 0–3,500 μm length, as indicated) and CA3 (blue windows, 0–1,100 μm length, as indicated). (C) Percentage of exon 23‐skipped mRNAs along the 3,500 μm long counting area in CA1 of treated mdx mice, calculated as the red staining (skipped mRNA) normalized to turquoise + red staining (skipped and unskipped mRNA). (D) Percentage of exon 23‐skipped mRNAs along the 1,100‐μm counting area in CA3 of treated mdx mice, calculated as the red staining (skipped mRNA) normalized to turquoise + red staining (skipped and unskipped mRNA). Results are expressed as means ± SEM; n = 3 per group.
In line with the levels of exon skipping, dystrophin protein restoration measured by western blot was the highest in hippocampus (structure effect: F[2, 6] = 40.19, p = 0.0003; CBL vs CX, p = 0.9884; CBL vs HIP, p = 0.0046; CX vs HIP, p = 0.0041), peaking at 7 weeks post‐injection and slightly decreasing at 10 weeks post‐injection (Figure 3D). Levels were slightly higher at 7 weeks, but this was not significant (delay effect: F[2, 6] = 2.582, p = 0.1564; delay × structure interaction, not significant; Figure 3D). IF analyses confirmed synaptic expression of the restored dystrophin protein in tcDNA‐treated mdx mice at 7 weeks post‐injection. As expected, the restored dystrophin protein was expressed in CA1 hippocampal synapses of the stratum pyramidale and proximal stratum radiatum (Figure 3E), and quantitative variations along the CA1‐CA3 cell layer were in line with the localization of skipped mRNA. Dystrophin was also detected in the amygdala, a key structure in the brain fear circuit likely involved in the abnormal emotional responses displayed by mdx mice. Dystrophin was expressed around the soma of neurons in the basolateral and lateral nuclei of amygdala (Figure 3F), but not in the central amygdala, as previously shown by others. 17
We next evaluated the optimal post‐injection delay to observe a maximal effect of the treatment on the fear response (Figure 5). In line with the levels of exon skipping and dystrophin protein re‐expression, the effects of tcDNA treatment on the fear response were optimal 6–7 weeks after the injection of 400 μg tcDNA. Significant improvements of the phenotype were characterized in tcDNA‐treated mdx mice as a decreased amount of freezing (Figure 5A; group effect: F[2, 65] = 63.63, p < 0.0001), and increased horizontal activity (Figure 5B; F[2, 66] = 12.30, p < 0.0001) and vertical activity (Figure 5C; F[2, 68] = 24.35, p < 0.0001). Significant decreases in freezing were detected in tcDNA‐treated mice from 4 to 7 weeks post‐injection compared with control mdx PBS mice (significant post‐hoc tests shown in Figure 5A). The largest improvements were detected at 6 and 7 weeks post‐injection, at which the percent freezing was statistically different from mdx PBS mice (p = 0.0006) and statistically comparable to WT levels (p = 0.2779; Figure 5A). We also found significant improvements in horizontal (Figure 5B) and vertical (Figure 5C) activities between 4 and 7 weeks after treatment. However, 10 weeks post‐injection, tcDNA‐treated mice showed levels similar to that of mdx PBS for all three parameters (fear response, horizontal, and vertical activities), correlating with the decreased levels of skipping and dystrophin restoration observed at these delays.
FIGURE 5.
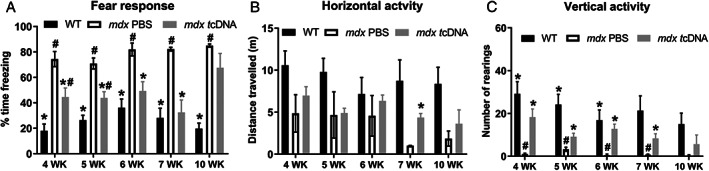
Evolution of the emotional response over time after intracerebroventricular (ICV) injection tricyclo‐DNA antisense oligonucleotide (tcDNA‐ASO). (A‐C) Behavioral parameters recorded during 5 minutes after a mild stress induced by a short (15 seconds) manual restraint in wild‐type (WT) and mdx mice treated with phosphate‐buffered saline (PBS) or 400 μg of tcDNA‐ASO 4, 5, 6, 7, and 10 weeks after the ICV treatment. (A) Restraint‐induced unconditioned fear responses expressed as the percentage of time spent freezing. (B) Horizontal activity expressed as the total distance travelled (m). (C) vertical activity expressed as the number of rearings. Results are expressed as means ± SEM; n = 7 (4 weeks), 6 (5 weeks), 6 (6 weeks), 4 (7 weeks), and 3 (10 weeks) per tcDNA‐treated group with 400 μg, n = 4 for mdx PBS control and n = 7 (4 weeks), 6 (5 weeks), 6 (6 weeks), 4 (7 weeks), and 3 (10 weeks) for WT. *p < 0.05 compared with mdx controls, ### p < 0.001 and # p < 0.05 compared with WT (ANOVA).
Effect of Partial Dp427 Restoration on Cognitive Functions
As the emotional response was most effectively improved between 6 and 7 weeks post‐injection of 400 μg tcDNA, we selected these time points to further evaluate the effects of this treatment on cognitive functions. We successively addressed two different cognitive tasks in which learning and/or memory deficits were previously reported in mdx mice: the novel object recognition test 12 , 25 and cued fear conditioning (Figure 6A). 9 , 26
FIGURE 6.
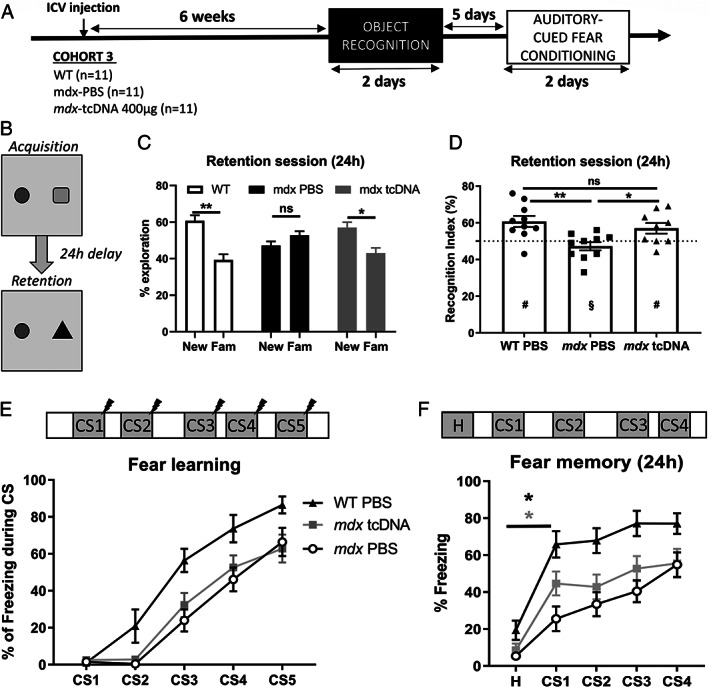
Impact of brain dystrophin restoration on cognition. (A) Schematic representation of the study design applied to the third cohort of mice (Cohort 3), showing the sample sizes, the intracerebroventricular (ICV) injection, the post‐injection delay at which behavioral testing started (6 weeks), which comprised an object recognition test followed by the auditory‐cued fear conditioning with an interval of 5 days between the two tests. (B–D) Object recognition test performed 6 weeks after ICV injection of 400 μg tricyclo‐DNA antisense oligonucleotide (tcDNA‐ASO) or phosphate‐buffered saline (PBS) in mdx mice (n = 9 per group) and of PBS in wild‐type (WT) littermate mice (n = 10). (B) Schematic representation of the object recognition composed of an acquisition session with two different objects, followed 24 hours later by a retention session during which one of the object is replaced by a novel object. (C) Relative time spent (%) exploring the new and familiar (Fam) objects during the retention session, calculated as the time spent exploring one of these objects divided by the total exploration time × 100. (D) Recognition index calculated as the time spent exploring the new object, divided by the total exploration time × 100. The horizontal dotted line represents chance level (50%). Values are means ± SEM. # p < 0.05 significantly different from the 50% chance level, and § p > 0.05 not different from 50%; *p < 0.05, **p < 0.01 significantly different from mdx PBS; ns: no significant difference between mdx tcDNA and WT groups. (E, F) Auditory‐cued fear conditioning performed 7 weeks after the ICV injection of 400 μg of tcDNA‐ASO or PBS in mdx mice (n = 9 per group) and WT littermate mice (n = 10). (E) The top drawing shows the succession of CS‐US pairs of stimuli (30‐second tone followed by electric shock) spaced by variable delays between pairs of stimuli during the acquisition session. The plot shows the fear‐learning performance during the acquisition session, expressed as percentage of freezing during the five 30‐second tones (CS1‐5). (F) Fear memory assessed during the retention session performed 24 hours later in a new context. The top drawing shows the succession of baseline habituation period (H) and CS1 to CS4 delivered alone (30‐second tones, no shock), spaced by variable delays. The plot shows the learning performance expressed as the percentage of time spent freezing during baseline habituation (H) and the presentation of the four conditioned stimulus (CS1–4). Group differences were significant in both sessions (group effects: p < 0.0001; two‐way ANOVAs for repeated measures), *(black star) WT group H versus CS1 p = 0.0032; *(grey star) mdx‐tcDNA treated mice H versus CS1 p = 0.0001.
In the object recognition test, a behavioral paradigm widely studied in humans to probe declarative memory, 27 we first performed a habituation session during which the locomotor activity of the mice, quantified by the distance traveled (m) in the open field, was comparable in the three groups during the four successive habituation sessions (data not shown; F[2, 25] = 0.7057, p = 0.503). During the acquisition session, two different objects were presented to the mice within the open field (Figure 6B, Acquisition). In mice of all three groups, the percentage of time spent exploring each object during this acquisition session was comparable between groups (F[2.25] = 0.00, p > 0.99) and never different from a 50% chance level (all groups, p > 0.05, one‐sample t test). For retention testing 24 hours later, one of the objects was replaced by a novel object (Figure 6B, Retention); the position of the new object was pseudorandomized in each genotype. The absolute time spent exploring objects during this session was not comparable among groups (F[2.26] = 9.56, p < 0.001; WT: 9.4 ± 2 s; mdx‐PBS: 28 ± 2 s; mdx‐TcDNA: 16.2 ± 4.8 s). The exploration time in mdx mice treated with TcDNA was not longer than in mdx‐PBS, indicating that treatment did not increase their motivation to explore. During this retention session, the memory performance was then compared using standard representation of the relative (percent) time spent exploring the objects. As shown in Figure 6C, the relative time spent exploring the novel object compared with the familiar object differed between groups (group × type of object: F[2.26] = 3.88, p < 0.05). Indeed, the percentage of time spent exploring the novel object was significantly longer in the WT group (p = 0.0015) and in the mdx‐tcDNA group (p = 0.0410). In contrast, no difference was found between the time spent exploring the novel and familiar objects in the mdx‐PBS group (p = 0.31). Consequently, both WT mice and tcDNA‐treated mdx mice had a recognition index significantly different from the 50% chance level (WT mice: 60.7%, p = 0.006; mdx‐tcDNA mice: 57%, p = 0.043), indicating preferential exploration of the novel object, whereas PBS treated mdx mice showed impaired memory performance with a recognition index not different from chance level (recognition index: 47.2%; p = 0.24, not significant; Figure 6D). Furthermore, there was a significant group effect on the recognition index (F[2, 26] = 6.666, p = 0.0046), reflecting that the performance of tcDNA‐treated mdx mice was comparable to WT mice (mdx‐tcDNA vs WT, p = 0.61, not significant), and significantly improved as compared with mdx mice injected with PBS (p = 0.048). The present results, therefore, confirm that untreated mdx mice have a long‐term memory deficit (24 hours) in this test, but further show that partial restoration of Dp427 can significantly improve the memory performance of treated mdx mice.
In cued fear conditioning, which involves amygdala‐dependent fear learning and memory processes (Figure 6E), mice first received five CS‐US pairs of stimuli (30‐second tone immediately followed by one 2‐second electric footshock), spaced by variable delays between CS‐US pairs (Figure 6E, top drawing) to evaluate fear learning. Learning performance was evaluated by measuring the percentage of time spent freezing during the presentation of each 30‐second conditioning auditory stimuli (CS1 to CS5). As shown in Figure 6E, all three groups of mice showed a progressive acquisition of associative fear learning, as reflected by incremental increases in the percentage of freezing during presentation of the CS (CS effect over five trials: F[3.581, 107.4] = 104.6, p < 0.0001). Although this indicates that mice progressively learnt that the tone predicts electric shock delivery, there was a significant difference in performance among groups (group effect: F[2, 30] = 10.83, p = 0.003), reflecting that acquisition of this task was delayed in mdx mice compared with WT mice (mdx‐PBS vs WT, p = 0.0086), as previously characterized. 9 , 26 There was no difference between treated and untreated mdx mice mdx‐PBS vs mdx‐tcDNA‐ASO, p = 0.87), as both groups of mdx mice were impaired compared with WT mice (mdx‐tcDNA‐ASO vs WT, p = 0.0284). In contrast, the amount of freezing during inter‐trial intervals (period that follows US delivery) was comparable with WT controls (data not shown), indicating that learning performance was not biased by abnormal reactivity or freezing response to electric footshocks.
The retention session (Figure 6F) was performed on the next day (24 hours later) to evaluate long‐term fear memory, and was characterized by a baseline habituation period (H) followed by the successive delivery of four CS (CS1–4) without US, spaced by variable interstimulus intervals (Figure 6F, top drawing). There was a significant group effect during this session (F[2, 30] = 14.49, p < 0.0001). Post‐hoc analyses showed that the percentage of freezing was comparable in the three groups during the baseline habituation session (H), approximately 15% (WT PBS vs mdx‐PBS, p = 0.25; WT PBS vs mdx‐tcDNA, p = 0.43; mdx‐PBS vs mdx‐tcDNA, p = 0.93). After the presentation of the first CS (CS1), a large and significant increase in freezing was observed in the WT mice (H vs CS1, p = 0.0032) and in the mdx‐tcDNA‐treated mice (H vs CS1, p = 0.0001), whereas presentation of CS1 did not significantly modify freezing in the mdx‐PBS mice (H vs CS1, p = 0.0795). The group effect, significant during the entire retention session, also reflected that both PBS‐ and tcDNA‐treated mdx mice showed a lower rate of freezing induced by delivery of CS1‐4 compared with PBS‐treated WT mice (mdx‐PBS vs WT, p < 0;0001; mdx‐tcDNA‐ASO vs WT, p < 0.0001), whereas the effect of the treatment was only marginally different (mdx‐PBS vs mdx‐tcDNA‐ASO, p = 0.0953).
Overall, tcDNA treatment did not induce significant improvement in the acquisition of cued fear conditioning, and memory retention performance was accordingly impaired at a 24‐hour post‐acquisition delay in both PBS‐ and tcDNA‐treated mdx mice. However, the significant increase in freezing induced by the CS1 in tcDNA‐treated mdx mice suggests that treatment partially improved acquisition and subsequent consolidation of the conditioning task.
Discussion
The emotional and cognitive defects in DMD are progressively being more studied and better characterized. However, in the past few decades, most of the efforts have focused on improving outcomes related to muscle weakness, whereas brain involvement has received less attention. None of the currently approved drugs for DMD or the drugs in clinical development address DMD brain comorbidities. Still, some of the therapeutic approaches, such as antisense mediated exon skipping or gene therapy, could bear such potential if only the tools used had the ability to cross the blood–brain barrier. Recent developments in preclinical research have shown encouraging results with compounds, such as cholesterol‐functionalized DNA/RNA heteroduplexes, 28 ASO conjugated to peptides, 29 or antibodies targeting specific receptors (such as the transferrin receptor 30 ), or tcDNA‐based ASO. 19 , 20 , 22 In our previous studies, 19 , 20 , 22 we showed that systemic treatment of mdx mice with tcDNA‐ASO induced a slight restoration of Dp427 in the CNS leading to the correction of the abnormal restraint‐induced fear response. In the present work, we investigated further the impact of dystrophin restoration on cognitive defects described in the mdx mouse model. For that purpose, we administered tcDNA‐ASO via the ICV route as an experimental model to achieve higher levels of dystrophin protein rescue. We used a tcDNA‐ASO carrying a full phosphodiester backbone and conjugated to palmitic acid, to avoid the presence of phosphorothioate bonds, which have previously been associated with an activation of the innate immune system in the mouse brain. 31 We recently reported that conjugation to palmitic acid enhanced the potency of tcDNA‐ASOs and significantly improved their safety profile. 22
All the tested doses of tcDNA‐ASO (up to the maximum feasible injection dose of 400 μg) were well tolerated. The effect on exon skipping and protein levels were dose‐dependent, and could reach up to 35% of exon 23 skipping in hippocampus, and approximately 15% in the cortex and cerebellum after an ICV injection of the optimal dose of 400 μg. The restoration of the Dp427 protein was up to 20–25% in the hippocampus, and approximately 5% in the cortex and cerebellum, as measured by western blot. Immunohistochemical labeling on hippocampal sections confirmed that the restored Dp427 in the hippocampus was correctly localized in the pyramidal nuclei, and quantifications of IF showed a restoration of approximately 30% of WT levels. This partial restoration of brain dystrophin was sufficient to significantly correct the fear response (30% of freezing for tcDNA‐treated mdx mice vs 70% for mdx‐PBS control mice). We then defined the optimal period for dystrophin restoration after ICV injection to ensure that we would perform behavioral analysis when dystrophin expression reaches its maximum level. Levels of dystrophin rescue were relatively stable between 5 and 7 weeks, but decreased 10 weeks post‐administration, which correlated with lower amount of tcDNA‐ASO in tissues and lower levels of exon skipping.
From a behavioral point of view, 6–7 weeks after treatment also appeared optimal, because the difference in freezing was maximal compared with mdx control mice injected with PBS (30% vs 80%) at these time points, but also because it was no longer statistically different from WT mice (30% vs 15/20%). These results are consistent with the molecular analyses, as behavioral improvement is the most effective when protein restoration is the highest. Similar results were observed in a previous study, in which a PMO was directly administered to the brain. 17 However, in addition to the ASO chemistry being different, the injection method also differed. Sekiguchi et al. administered a total of 1 mg of PMO via ICV infusion using an osmotic infusion pump that persistently delivered the ASO for 1 week, whereas we performed here one single bilateral ICV bolus injection of 400 μg of tcDNA (200 μg in each hemisphere). Despite using more than twice the amount of ASO that we used, the percentage of brain Dp427 restored in both studies was quite similar (~25%), suggesting that tcDNA‐ASO may be more effective than PMO after direct delivery to the CNS. Furthermore, it should also be noted that western blot quantifications were performed on enriched postsynaptic density fractions of the forebrain in the previous study, which may not be directly comparable to western blots performed on total protein extracts. Anyhow, our present results confirm that partial rescue of brain Dp427 leads to significant reduction of the abnormal fear response in mdx mice. We also confirmed that ICV administration of ASO allowed re‐expression of brain Dp427 in various brain structures involved in emotional and cognitive processes, including in the amygdala, which is a key structure of the brain fear circuit involved in the expression of unconditioned fear responses.
Brain comorbidities in DMD are not limited to changes in emotional reactivity, as a range of other cognitive and executive dysfunctions have been reported in DMD patients, 7 , 8 , 13 as well as in mdx mice that show selective learning and memory deficits. 9 , 12 , 13 , 26 We therefore investigated further the impact of partial Dp427 restoration on learning and memory impairment, using a selection of two tests that we previously identified as providing robust outcome measures in mdx mice: An auditory‐cued fear conditioning paradigm in which mdx mice are impaired for both acquisition and long‐term memory retention of the cue–outcome (CS‐US) Pavlovian association, 9 , 26 and a novel object recognition test in which these mice show impaired recognition memory at long retention delays. 12
TcDNA‐treated mice did not show any improvement during acquisition of the cued fear conditioning, as they performed just like the mdx PBS‐control mice, showing a significant acquisition delay compared with WT mice. During the retention session performed 24 hours later, a significant improvement of performance was detected in the mice treated with tcDNA‐ASO in response to the first CS. Hence, the present findings suggest that low restoration of brain Dp427 in various structures including the amygdala was sufficient to compensate for the stress‐induced unconditioned fear responses and slightly improved fear memory, but was insufficient to fully overcome the learning and memory deficits during fear conditioning. Whether higher levels of Dp427 rescue in the amygdala would have an impact on these deficits or whether these are not reversible remains to be elucidated. We may hypothesize that this phenotype is more difficult to correct, because it affects several brain structures that all have undergone abnormal structural neurodevelopment without dystrophin, which could not be reversed postnatally. It would be interesting in the future to develop therapies allowing greater restoration of brain dystrophin to better characterize the therapeutic threshold necessary to fully rescue certain cognitive phenotypes. In addition, restoring brain dystrophin at earlier stages of development (eg, administrating therapies in new born mice or in utero) would shed some light on the reversibility of these deficits and possibly maximize the therapeutic effectiveness. In contrast, other phenotypes related to spontaneous emotional reactivity, such as unconditioned fear responses, might involve a distinct functional role of Dp427 in the mature neuronal networks involved in emotional states.
In the object recognition test, all three groups of mice (WT‐PBS, mdx‐PBS, and tcDNA‐treated mdx) initially explored the two presented objects equally (ie, approximately 50% of exploration of each object) and, therefore, did not show any biased preference for an object or a side of the apparatus during the acquisition session. During the retention phase, however, when one object was replaced by a novel object, the mdx‐PBS mice still explored both objects in the same proportion, unlike the WT and tcDNA‐treated mdx mice that preferentially explored the novel object as compared with the familiar object already explored during acquisition. This efficient discrimination of the novel and familiar objects was statistically different from chance level, thus reflecting unaltered recognition memory, contrary to mdx‐PBS mice who showed no significant recall, as expected from previous studies. This suggests that partial restoration of Dp427 in the CNS of tcDNA‐treated mdx mice compensated the long‐term recognition memory deficit normally shown by this model. This is particularly encouraging, given that Dp427 restoration was only restored by 5–30% of WT levels depending on the brain structure. Object recognition memory expressed at long delays post‐acquisition involves hippocampal functions, 32 , 33 particularly that of the dorsal CA1 hippocampal region, 34 , 35 and it is noteworthy that this structure expressed the highest level of exon skipping and protein restoration following treatment with tcDNA‐ASO. This may have favored the compensation of this phenotype involving this hippocampal subfield.
TcDNA‐ASOs after ICV bolus injection were indeed biodistributed preferentially to the hippocampus, as we observed in biodistribution, RT–qPCR, western blot, and IF, but also using in situ hybridization against the skipped mRNA, which showed higher expression in the dorsal CA1 region. Interestingly, we noted a gradient of skipped mRNA along the CA1 in line with the restored dystrophin expression, which was also at the highest in the dorsal CA1 region. This gradient may be driven by the injection method, as the dorsal hippocampal formation is close to the lateral and third ventricles, and might preferentially capture larger quantities of ASOs after the ICV bolus injection. Nevertheless, tcDNA‐ASOs also distributed widely to the CNS after ICV delivery and effectively reached the cortex and cerebellum. These wide biodistribution properties associated with their particularly well tolerated full phosphodiester backbone make tcDNA‐ASO an attractive chemistry for the treatment of neurological disorders. Yet, the biodistribution of tcDNA may explain that processes in which the hippocampus is predominantly involved are more likely to be restored due to a better protein restoration in this tissue. In this regard, it would be useful in further studies to combine different administration methods, such as ICV and intrathecal delivery, to achieve a restoration of dystrophin as homogenous as possible throughout the brain.
Altogether, these findings show that in addition to emotional features, some cognitive deficits associated with the lack of brain dystrophin, such as long‐term memory deficits, may be reversible or at least improved by partial rescue of Dp427 in the postnatal brain. These results have important implications for the treatment of DMD patients and the development of future therapies. So far, none of the currently approved drugs for DMD have addressed the brain comorbidities, mostly because of their inability to cross the blood–brain barrier. However there is active research developing novel ASO chemistries or conjugates, which may in the near future be able to restore at least some levels of dystrophin in the brain. Demonstrating here that this restoration is likely to have an impact on patient's brain comorbidities represents a major milestone, and should serve as a driving force to further encourage the development of such therapies. These results also imply that brain comorbidities in DMD patients should be assessed in future clinical trials, and highlight the need to develop better outcome measures to assess the potential benefits of these future therapies. Overall, the present study shows promising therapeutic opportunities for DMD, and brings gene‐correction strategies closer to a full compensation of symptoms and comorbidities in DMD patients.
Author Contributions
F.Z., LG., C.V., and A.G. contributed to the conception and design of the study; F.Z., K.R., F.B., T.T., A.G., and C.V. contributed to the acquisition and analysis of data; and F.Z., C.V., and A.G. contributed to drafting the text and figures.
Potential Conflicts of Interest
TT is an employee of SQY Therapeutics which produces tricyclo‐DNA oligomers. LG is co‐founder of SQY Therapeutics.
Acknowledgments
This work was supported by Agence Nationale de la Recherche (ANR, France; initiated and coordinated by CV; grant number ANR‐14‐CE13‐0037‐01 to CV and AG), Centre National de la Recherche Scientifique (CNRS, France), the Institut National de la santé et la recherche médicale (INSERM), Université Paris‐Saclay (France), Paris Ile‐de‐France Region, grants to F.Z. and C.V from Association Monégasque contre les Myopathies (AMM, Monaco), and PhD fellowships from Ministère de l'Enseignement Supérieur et de la Recherche (France) to F.Z. and F.B. We also gratefully acknowledge support to C.V. and A.G. from the European Union's Horizon 2020 research and innovation program “Brain Involvement iN Dystrophinopathies” under grant agreement No 847826. The authors are grateful to the Zootechnic platform of our institutes for mouse breeding, care, and genotyping.
Contributor Information
Cyrille Vaillend, Email: cyrille.vaillend@universite-paris-saclay.fr.
Aurélie Goyenvalle, Email: aurelie.goyenvalle@uvsq.fr.
References
- 1. Brünig I, Suter A, Knuesel I, et al. GABAergic terminals are required for postsynaptic clustering of dystrophin but not of GABA(a) receptors and gephyrin. J Neurosci 2002;22:4805–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim TW, Wu K, Xu JL, Black IB. Detection of dystrophin in the postsynaptic density of rat brain and deficiency in a mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci USA 1992;89:11642–11644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Knuesel I, Mastrocola M, Zuellig RA, et al. Short communication: altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice). Eur J Neurosci 1999;11:4457–4462. [DOI] [PubMed] [Google Scholar]
- 4. Lidov HG, Byers TJ, Kunkel LM. The distribution of dystrophin in the murine central nervous system: an immunocytochemical study. Neuroscience 1993;54:167–187. [DOI] [PubMed] [Google Scholar]
- 5. Lidov HG, Byers TJ, Watkins SC, Kunkel LM. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature 1990;348:725–728. [DOI] [PubMed] [Google Scholar]
- 6. Perronnet C, Vaillend C. Dystrophins, utrophins, and associated scaffolding complexes: role in mammalian brain and implications for therapeutic strategies. J Biomed Biotechnol 2010;2010:849426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ricotti V, Mandy WPL, Scoto M, et al. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol 2016;58:77–84. [DOI] [PubMed] [Google Scholar]
- 8. Snow WM, Anderson JE, Jakobson LS. Neuropsychological and neurobehavioral functioning in Duchenne muscular dystrophy: a review. Neurosci Biobehav Rev 2013;37:743–752. [DOI] [PubMed] [Google Scholar]
- 9. Vaillend C, Chaussenot R. Relationships linking emotional, motor, cognitive and GABAergic dysfunctions in dystrophin‐deficient mdx mice. Hum Mol Genet 2017;26:1041–1055. [DOI] [PubMed] [Google Scholar]
- 10. Muntoni F, Mateddu A, Serra G. Passive avoidance behaviour deficit in the mdx mouse. Neuromuscul Disord 1991;1:121–123. [DOI] [PubMed] [Google Scholar]
- 11. Remmelink E, Aartsma‐Rus A, Smit AB, et al. Cognitive flexibility deficits in a mouse model for the absence of full‐length dystrophin. Genes Brain Behav 2016;15:558–567. [DOI] [PubMed] [Google Scholar]
- 12. Vaillend C, Billard J‐M, Laroche S. Impaired long‐term spatial and recognition memory and enhanced CA1 hippocampal LTP in the dystrophin‐deficient Dmd(mdx) mouse. Neurobiol Dis 2004;17:10–20. [DOI] [PubMed] [Google Scholar]
- 13. Vaillend C, Rendon A, Misslin R, Ungerer A. Influence of dystrophin‐gene mutation on mdx mouse behavior. I. Retention deficits at long delays in spontaneous alternation and bar‐pressing tasks. Behav Genet 1995;25:569–579. [DOI] [PubMed] [Google Scholar]
- 14. Miranda R, Nudel U, Laroche S, Vaillend C. Altered presynaptic ultrastructure in excitatory hippocampal synapses of mice lacking dystrophins Dp427 or Dp71. Neurobiol Dis 2011;43:134–141. [DOI] [PubMed] [Google Scholar]
- 15. Miranda R, Sébrié C, Degrouard J, et al. Reorganization of inhibitory synapses and increased PSD length of perforated excitatory synapses in hippocampal area CA1 of dystrophin‐deficient mdx mice. Cereb Cortex 2009;19:876–888. [DOI] [PubMed] [Google Scholar]
- 16. Manning J, Kulbida R, Rai P, et al. Amitriptyline is efficacious in ameliorating muscle inflammation and depressive symptoms in the mdx mouse model of Duchenne muscular dystrophy. Exp Physiol 2014;99:1370–1386. [DOI] [PubMed] [Google Scholar]
- 17. Sekiguchi M, Zushida K, Yoshida M, et al. A deficit of brain dystrophin impairs specific amygdala GABAergic transmission and enhances defensive behaviour in mice. Brain 2009;132:124–135. [DOI] [PubMed] [Google Scholar]
- 18. Ferlini A, Goyenvalle A, Muntoni F. RNA‐targeted drugs for neuromuscular diseases. Science 2021;371:29–31. [DOI] [PubMed] [Google Scholar]
- 19. Goyenvalle A, Griffith G, Babbs A, et al. Functional correction in mouse models of muscular dystrophy using exon‐skipping tricyclo‐DNA oligomers [internet]. Nat Med 2015;21:270–275. [DOI] [PubMed] [Google Scholar]
- 20. Relizani K, Griffith G, Echevarría L, et al. Efficacy and safety profile of Tricyclo‐DNA antisense oligonucleotides in Duchenne muscular dystrophy mouse model. Mol Ther Nucleic Acids 2017;8:144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robin V, Griffith G, Carter J‐PL, et al. Efficient SMN rescue following subcutaneous Tricyclo‐DNA antisense oligonucleotide treatment. Mol Ther Nucleic Acids 2017;7:81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Relizani K, Echevarría L, Zarrouki F, et al. Palmitic acid conjugation enhances potency of tricyclo‐DNA splice switching oligonucleotides. Nucleic Acids Res 2021;50:17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dallérac G, Perronnet C, Chagneau C, et al. Rescue of a dystrophin‐like protein by exon skipping normalizes synaptic plasticity in the hippocampus of the mdx mouse. Neurobiol Dis 2011;43:635–641. [DOI] [PubMed] [Google Scholar]
- 24. Knuesel I, Zuellig RA, Schaub MC, Fritschy JM. Alterations in dystrophin and utrophin expression parallel the reorganization of GABAergic synapses in a mouse model of temporal lobe epilepsy. Eur J Neurosci 2001;13:1113–1124. [DOI] [PubMed] [Google Scholar]
- 25. Sinadinos A, Young CNJ, Al‐Khalidi R, et al. P2RX7 purinoceptor: a therapeutic target for ameliorating the symptoms of duchenne muscular dystrophy. PLoS Med 2015;12:e1001888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chaussenot R, Edeline J‐M, Le Bec B, et al. Cognitive dysfunction in the dystrophin‐deficient mouse model of Duchenne muscular dystrophy: a reappraisal from sensory to executive processes. Neurobiol Learn Mem 2015;124:111–122. [DOI] [PubMed] [Google Scholar]
- 27. Manns JR, Hopkins RO, Reed JM, et al. Recognition memory and the human hippocampus. Neuron 2003;37:171–180. [DOI] [PubMed] [Google Scholar]
- 28. Nagata T, Dwyer CA, Yoshida‐Tanaka K, et al. Cholesterol‐functionalized DNA/RNA heteroduplexes cross the blood‐brain barrier and knock down genes in the rodent CNS. Nat Biotechnol 2021;39:1529–1536. [DOI] [PubMed] [Google Scholar]
- 29. Hammond SM, Hazell G, Shabanpoor F, et al. Systemic peptide‐mediated oligonucleotide therapy improves long‐term survival in spinal muscular atrophy. Proc Natl Acad Sci USA 2016;113:10962–10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hammond SM, Abendroth F, Goli L, et al. Systemic antibody‐oligonucleotide delivery to the central nervous system ameliorates mouse models of spinal muscular atrophy [internet]. 2021. Available at: https://www.biorxiv.org/content/10.1101/2021.07.29.454272v1 Accessed February 18, 2022.
- 31. Toonen LJA, Casaca‐Carreira J, Pellisé‐Tintoré M, et al. Intracerebroventricular administration of a 2’‐O‐methyl Phosphorothioate antisense oligonucleotide results in activation of the innate immune system in mouse brain. Nucleic Acid Ther 2018;28:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clark RE, Zola SM, Squire LR. Impaired recognition memory in rats after damage to the hippocampus. J Neurosci 2000;20:8853–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Genoux D, Haditsch U, Knobloch M, et al. Protein phosphatase 1 is a molecular constraint on learning and memory. Nature 2002;418:970–975. [DOI] [PubMed] [Google Scholar]
- 34. Pittenger C, Huang YY, Paletzki RF, et al. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus‐dependent spatial memory. Neuron 2002;34:447–462. [DOI] [PubMed] [Google Scholar]
- 35. Rampon C, Tang YP, Goodhouse J, et al. Enrichment induces structural changes and recovery from nonspatial memory deficits in CA1 NMDAR1‐knockout mice. Nat Neurosci 2000;3:238–244. [DOI] [PubMed] [Google Scholar]
- 36. Echevarría L, Aupy P, Relizani K, et al. Evaluating the impact of variable Phosphorothioate content in Tricyclo‐DNA antisense oligonucleotides in a Duchenne muscular dystrophy mouse model. Nucleic Acid Ther 2019;29:148–160. [DOI] [PubMed] [Google Scholar]
- 37. Aupy P, Echevarría L, Relizani K, et al. Identifying and avoiding tcDNA‐ASO sequence‐specific toxicity for the development of DMD exon 51 skipping therapy. Mol Ther Nucleic Acids 2020;19:371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates, second edition. 2nd ed. San Diego: Academic Press, 2001. [Google Scholar]