

Abstract
A population of neutrophils recruited into cystic fibrosis (CF) airways is associated with proteolytic lung damage, exhibiting high expression of primary granule exocytosis marker CD63 and reduced phagocytic receptor CD16. Causative factors for this population are unknown, limiting intervention. Here we present a laboratory model to characterize responses of differentiated airway epithelium and neutrophils following respiratory infection. Pediatric primary airway epithelial cells were cultured at the air–liquid interface, challenged individually or in combination with rhinovirus (RV) and Pseudomonas aeruginosa, then apically washed with medical saline to sample epithelial infection milieus. Cytokine multiplex analysis revealed epithelial antiviral signals, including IP‐10 and RANTES, increased with exclusive RV infection but were diminished if P. aeruginosa was also present. Proinflammatory signals interleukin‐1α and β were dominant in P. aeruginosa infection milieus. Infection washes were also applied to a published model of neutrophil transmigration into the airways. Neutrophils migrating into bacterial and viral–bacterial co‐infection milieus exhibited the in vivo CF phenotype of increased CD63 expression and reduced CD16 expression, while neutrophils migrating into milieus of RV‐infected or uninfected cultures did not. Individually, bacterial products lipopolysaccharide and N‐formylmethionyl‐leucyl‐phenylalanine and isolated cytokine signals only partially activated this phenotype, suggesting that additional soluble factors in the infection microenvironment trigger primary granule release. Findings identify P. aeruginosa as a trigger of acute airway inflammation and neutrophil primary granule exocytosis, underscoring potential roles of airway microbes in prompting this neutrophil subset. Further studies are required to characterize microbes implicated in primary granule release, and identify potential therapeutic targets.
Keywords: Airway epithelium, cystic fibrosis, innate immunity, neutrophils, Pseudomonas, rhinovirus
Neutrophils in cystic fibrosis (CF) airways exhibit high primary granule release and are phagocytosis deficient, with respiratory infection as a possible trigger of this pathological phenotype. To explore this, we challenged differentiated airway epithelial cultures with rhinovirus and/or Pseudomonas aeruginosa, and assessed epithelial cytokine release and neutrophil recruitment to infection milieus. Primary or secondary infection with P. aeruginosa induced pro‐inflammatory cytokine release and activated the CF neutrophil phenotype, suggesting airway bacteria modulate functions of locally recruited neutrophils in CF lung disease.

Introduction
Originally viewed as a functionally rigid, short‐lived cell, neutrophils are increasingly recognized for their functional plasticity and roles in inflammatory disease. 1 Studies continue to identify viable neutrophil subsets that contribute to chronic inflammation following recruitment to mucosal sites, notably through alterations to their traditional functions [phagocytosis, exocytosis or formation of neutrophil extracellular traps (NETosis)]. 1 , 2 In the autosomal recessive disorder cystic fibrosis (CF), neutrophils are a primary cause of airway damage. Mucus obstruction and impaired airway clearance facilitate infection, resulting in large numbers of neutrophils recruited into CF airways via signaling cascades induced by interleukin (IL)‐1 and IL‐6, as well as direct recruitment via IL‐8. 3 Following migration into the lung, neutrophils secrete tissue‐damaging proteases including the serine protease neutrophil elastase (NE), which has been shown to strongly correlate with structural lung disease. 4 , 5 , 6
Release of NE in CF airways was conventionally thought to be caused by the rapid death of recruited neutrophils. However, comprehensive in vivo and in vitro studies demonstrate that viable airway neutrophils are reprogrammed in CF airways into a granule‐releasing, immunoregulatory and metabolically active phenotype (encapsulated by the “GRIM” acronym) with reduced bacterial killing capacity. 7 , 8 , 9 GRIM reprogramming occurs upon neutrophil recruitment into the CF airway lumen and is measurable as changes in surface markers from levels in peripheral blood neutrophils; chiefly, a decrease in phagocytic receptor CD16 with a concomitant increase in CD63, a marker of exocytosis of primary granules (which contain NE). 7 Initially described in adults with CF, 10 we recently established that GRIM neutrophils are present in pediatric CF airways, indicating that mechanisms of neutrophil reprogramming are initiated early in life. 11 Importantly, levels of the exocytosis marker CD63 correlate more robustly with airway damage than NE activity in young children with CF, underscoring the importance of directly assessing neutrophil phenotypes in inflammatory disease. 11
While de novo transcription was recently established as a requirement for GRIM reprogramming, 12 the specific factors in the CF airway milieu that induce this reprograming, and particularly exocytosis, are still unknown. 7 Characterizing the onset of neutrophil reprogramming is key to providing earlier clinical intervention and better outcomes for individuals with CF, as there are currently no available therapies to directly treat the underlying mechanisms. A combined in vivo and in vitro study of neutrophils in non‐CF pediatric patients admitted to hospital for virally induced acute respiratory distress syndrome points to a possible cause, as neutrophils isolated from the airways of patients co‐infected with virus and bacteria, but not viral infection alone, demonstrated increased exocytosis and reduced phagocytic ability. 13 This implies that at least some aspects of GRIM reprogramming are not restricted to CF airway disease and that combined viral–bacterial challenges to the CF airway may be a major contributor to reprogramming of recruited neutrophils.
As a condition characterized by persistent airway infection, epithelial responses to microbes have a major role in driving neutrophil influx into the lungs. 14 Increased nuclear factor‐kappaB activity and IL‐8 secretion are reported in in vitro studies of Pseudomonas aeruginosa infection of CF airway epithelium. 15 , 16 In response to respiratory viral infection, CF airway cells have shown dampened apoptosis and interferon (IFN) responses, as well as elevated inflammatory cytokine production. 17 , 18 However, whether the CF epithelium is intrinsically proinflammatory is debated, as there is also evidence of no difference in inflammation compared with non‐CF tissue. 19 , 20 Pathogens may also dictate airway neutrophil activity independently of epithelial responses. The bacterial peptide N‐formylmethionyl‐leucyl‐phenylalanine (fMLF) is a well‐known powerful neutrophil chemoattractant that can prime neutrophils for degranulation and superoxide release. 21 , 22 Bacterial lipopolysaccharides (LPSs) have also been recognized as neutrophil activators, triggering recruitment, respiratory burst and formation of neutrophil extracellular traps. 23 , 24 , 25
To provide insights into the interplay between infection, the airway epithelium and neutrophils, we recapitulated airway infection microenvironments in a multistep in vitro model. Our aim was to identify which infection scenarios are relevant to neutrophil reprogramming in early CF, with the hypothesis that microenvironments of viral and bacterial co‐infection activate GRIM neutrophils. We performed in vitro pathogen challenges using pediatric CF and non‐CF primary epithelial cell cultures representative of native airway tissue and applied rhinovirus (RV), the most commonly detected respiratory virus in children with CF, 26 and/or P. aeruginosa, which is associated with prolonged NE activity in pediatric CF airways. 27 Saline washes of the apical surface of differentiated cultures, mimicking samples of the in vivo airway lumen, were used to first assess epithelial cytokine release in response to infection, then applied to a laboratory model of airway neutrophil recruitment 7 so that the adaptations by neutrophils to these environments could be assessed by flow cytometry.
Results
Rhinovirus and P. aeruginosa infection in CF and non‐CF epithelium
Primary airway epithelial cells (AECs) were derived from seven children with CF and five non‐CF controls (Table 1). Cells were first expanded as submerged monolayers and then cultured at the air–liquid interface (ALI) on 24‐well permeable inserts, growing into ciliated epithelial layers (Figure 1a, b). Inserts were then challenged with different infection scenarios: exclusive RV infection for 48 h, exclusive P. aeruginosa infection for 24 h or sequential co‐infection with RV for 24 h followed by the addition of P. aeruginosa for an additional 24 h to model respiratory viral infection followed by a secondary bacterial infection (the infection timeline is shown in Supplementary figure 1). A low multiplicity of infection (MOI) of P. aeruginosa (MOI 0.001) was used to model initial bacterial infection with a low starting biomass. Uninfected controls were treated with phosphate‐buffered saline (PBS) and incubated for 48 h. Titers of RV and P. aeruginosa were mostly similar between their respective infection conditions, with only viral load of co‐infected CF cultures reduced 2.6‐fold compared with exclusive CF viral infection, and with no significant differences in titers between non‐CF and CF cultures (Figure 1c). Apical washes from inserts were measured for lactate dehydrogenase activity as a marker of cytotoxicity and results normalized to levels in uninfected washes (0%). When comparing cytotoxicity between non‐CF and CF cultures, exclusive viral and bacterial infection were significantly more cytotoxic to non‐CF cultures (a difference of 83 ± 5% and 15 ± 2%, respectively), while co‐infection elicited similar cytotoxicity in each culture type (Figure 1d). Overall, exclusive RV infection was the most cytotoxic condition to airway cells compared with P. aeruginosa or viral–bacterial co‐infection (211 ± 15% non‐CF, 127 ± 6% CF).
Table 1.
Participant demographics.
| Non‐CF | CF | |
|---|---|---|
| Participants | 5 | 7 |
| Age range (year) | 2.3–6.5 | 0.3–5.8 |
| Male/female | 5/0 | 3/4 |
| Genotype (% F508Del homozygous) | N/A | 100% |
| Respiratory infection at sampling | N/A | 3 |
| Viral | 1 | |
| Bacterial (Pseudomonas aeruginosa) | 2 (0) | |
| Previous respiratory infection | N/A | 4 |
| Viral | 0 | |
| Bacterial (P. aeruginosa) | 4 (1) | |
| Neutrophil elastase positive | N/A | 5 |
| IL‐8 detected | N/A | 7 |
CF, cystic fibrosis; IL, interleukin.
Figure 1.
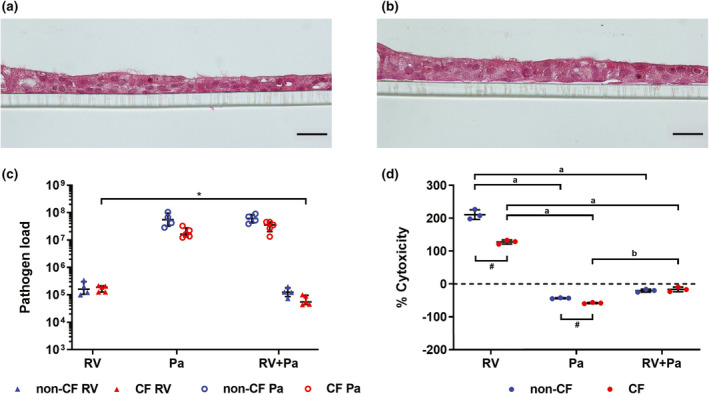
Pathogen load and cytotoxicity following infection. Hematoxylin and eosin staining representative of (a) non‐CF and (b) CF air–liquid interface cultures show formation of a ciliated epithelial layer (scale bar = 20 µm). (c) Titers of rhinovirus (RV) and adherent Pseudomonas aeruginosa calculated as plaque‐forming units and colony‐forming units, relative to RV 5′ untranslated region and P. aeruginosa 16S ribosomal RNA expression, respectively. (*P < 0.05 compared with co‐infection titer, t‐test). (d) Results from lactate dehydrogenase assay, shown as % cytotoxicity of infection conditions normalized to uninfected controls (0% cytotoxicity) (a P < 0.001 compared with P. aeruginosa and RV + P. aeruginosa infection, b P < 0.001 compared with RV + P. aeruginosa infection, one‐way ANOVA; # P < 0.01 non‐CF vs. CF, t‐test). Error bars indicate mean ± standard deviation. CF, cystic fibrosis; Pa, Pseudomonas aeruginosa.
P. aeruginosa elicits inflammatory cytokine release and suppresses antiviral signals
To characterize innate airway epithelial signals in response to infection, washes of infected ALI cultures were tested on a human cytokine multiplex encompassing 48 different analytes (an abbreviation list is provided in Supplementary table 1), complemented by an IL‐8‐specific ELISA to allow for a greater dynamic range for this cytokine. A total of 38 cytokines could be detected (Supplementary table 2; Supplementary figure 2), and out of these 32 cytokines had significant changes in concentration from baseline levels in uninfected washes upon infection (P < 0.05, repeated measures one‐way ANOVA), with cytokine profiles unique to infectious stimuli and having similar expression in non‐CF and CF cultures. Six cytokines had greater than fivefold increases in concentration following infection (Figure 2a). Exclusive infection with RV significantly increased IP‐10 (271‐fold non‐CF; 277‐fold CF), MIG (22‐fold non‐CF; 14‐fold CF) and RANTES (20‐fold non‐CF; 6‐fold CF; Figure 2c–e). However, these responses were strongly reduced upon co‐infection with bacteria, in some instances below baseline expression. P. aeruginosa and co‐infection washes had significant increases in IL‐1α (26–38‐fold non‐CF; 25–40‐fold CF), IL‐1β (52–73‐fold non‐CF; 73–102‐fold CF) as well as IL‐1Ra (6–7‐fold non‐CF; 2‐fold CF), reflecting a strong acute phase reaction in response to bacterial infection (Figure 2f–h). Twenty‐six cytokines had median‐fold changes of five or less (Figure 2b), and also showed distinct patterns of cytokine release for exclusive RV infection and conditions where P. aeruginosa was present. Exclusive RV infection was characterized by significant increases in TRAIL, MIF, IL‐9, SDF‐1α and CTACK in both non‐CF and CF washes (1.3–2.8‐fold; Figure 2b, Supplementary figure 2). With the exception of CTACK, these responses were again diminished with bacterial co‐infection. Both exclusive infection with P. aeruginosa and viral–bacterial co‐infection showed similar patterns of cytokine release, characterized by significant increases in M‐CSF, bFGF, HGF, LIF, IL‐2, IL‐16, IL‐18 and IFNγ (1.4–5.0‐fold; Figure 2b, Supplementary figure 2). P. aeruginosa and co‐infection also significantly increased IFNα2, CTACK and TNFα in CF washes (1.9–4.1‐fold). Cytokines that were significantly reduced from baseline concentrations included GM‐CSF in all infection conditions compared with uninfected controls (0.1–0.4‐fold), as well as SCF, GRO‐α, IL‐6, MIF, TRAIL and TNF‐β in P. aeruginosa and co‐infection washes (0.01–0.7‐fold; Figure 2b, Supplementary figure 2). Of note, the clinically important marker of inflammation IL‐8 did not significantly differ from baseline levels in any infection condition (Supplementary figure 2). Overall, these results highlight inflammatory signals by airway epithelium to P. aeruginosa infection featuring increased release of proinflammatory mediators and dampening of antiviral responses.
Figure 2.
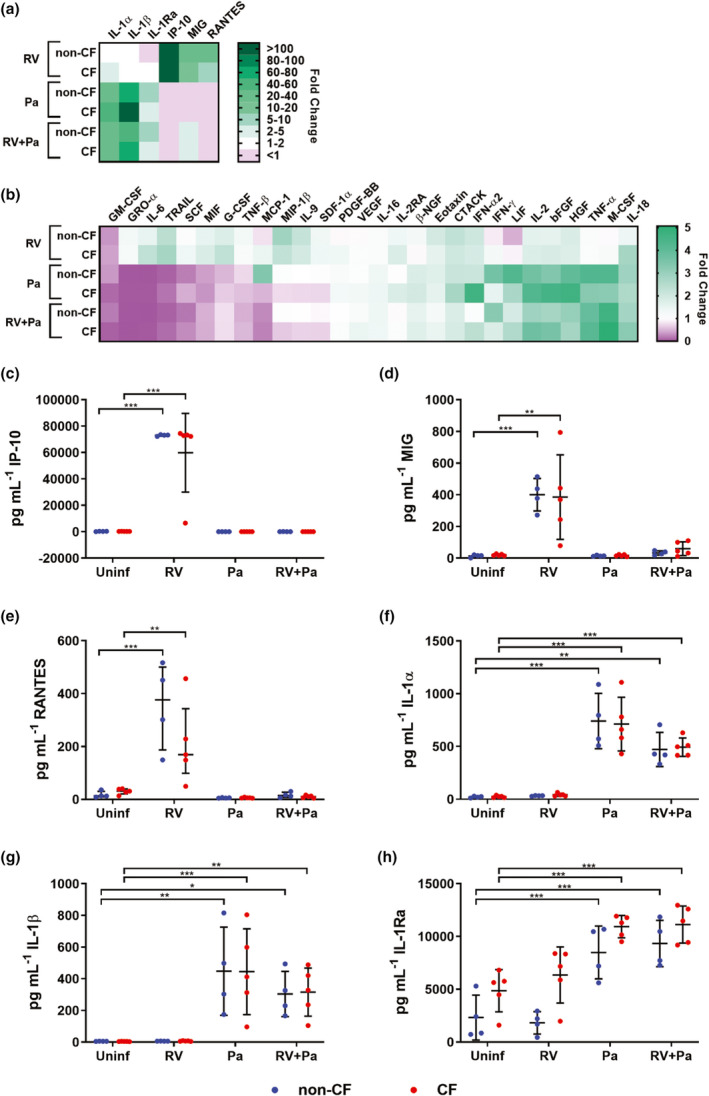
Pseudomonas aeruginosa elicits proinflammatory epithelial signals. Heat maps show median‐fold change (FC) of cytokine concentrations in washes of infected air–liquid interface cultures over washes of uninfected cultures. (a) Cytokines with > 5 FC. (b) Cytokines with ≤ 5 FC. (c) IP‐10, (d) MIG and (e) RANTES increased in response to exclusive rhinovirus (RV) infection and (f) IL‐1α, (g) IL‐1β and (h) IL‐1Ra increased in response to P. aeruginosa and viral–bacterial co‐infection [*P < 0.05, **P < 0.01, ***P < 0.001 compared with uninfected controls, repeated measures (RM) one‐way ANOVA]. Error bars indicate mean ± standard deviation. Data points indicate cytokine concentrations in washes of individual primary cell cultures. CF, cystic fibrosis; IL, interferon gamma‐induced protein 10; interleukin; IP‐10, MIG, monokine induced by gamma interferon; Pa, Pseudomonas aeruginosa; RANTES, regulated on activation, normal T cell expressed and secreted.
Neutrophils reprogram upon encountering bacterial infection milieu
To assess neutrophil recruitment and reprogramming following infection, naïve human peripheral blood neutrophils from non‐CF donors were migrated across an epithelial barrier into filtered (0.22 µm) saline washes of the apical surface of infected and control ALI inserts (Supplementary figure 3), similar to previous studies. 7 , 13 Expression of functional markers in postmigration neutrophils was characterized by flow cytometry using a four‐step gating strategy (Supplementary figure 4). Neutrophil subsets A1 and A2 were also defined according to expression of CD16 and CD63 (Supplementary figure 4), where A1 neutrophils (CD16high CD63low) reflected a baseline unactivated phenotype and A2 neutrophils (CD16low CD63high) indicated GRIM reprogramming. 7 Median fluorescence intensity (MFI) of CD16 was significantly reduced in neutrophils migrating toward exclusive P. aeruginosa and co‐infection washes compared with baseline expression levels in nonmigrated neutrophils (0.1‐fold non‐CF and CF; P < 0.001; Figure 3a). By contrast, MFI of CD63 increased threefold in neutrophils migrating toward P. aeruginosa infected and co‐infection washes (P < 0.001 non‐CF; P < 0.01 CF; Figure 3b). The MFI of CD66b was measured as a marker of secondary granule exocytosis in neutrophils, 7 , 10 with all conditions showing increased surface CD66b to varying degrees from baseline expression. However, exclusive P. aeruginosa and co‐infection scenarios resulted in fourfold increases (P < 0.001 non‐CF and CF; Figure 3c). Total numbers of migrated neutrophils did not differ between non‐CF or CF ALI washes, and while there was a trend of increasing migration with infection, this was only significant for migration into CF P. aeruginosa infection washes compared with uninfected washes (34 593 ± 27 655 uninfected vs. 61 241 ± 48 698 P. aeruginosa; P < 0.05; Figure 3d). However, when assessing subset distribution, we found that the proportion of A1 neutrophils significantly decreased following migration into P. aeruginosa and viral bacterial co‐infection washes (–63 ± 19% non‐CF and –65 ± 16% CF; P < 0.001), and that of A2 neutrophils significantly increased when migrating into P. aeruginosa or viral–bacterial co‐infection washes from non‐CF and CF cultures (58 ± 20% non‐CF and 53 ± 20% CF; P < 0.001; Figure 3e, f), indicative of GRIM neutrophil activation following bacterial infection.
Figure 3.
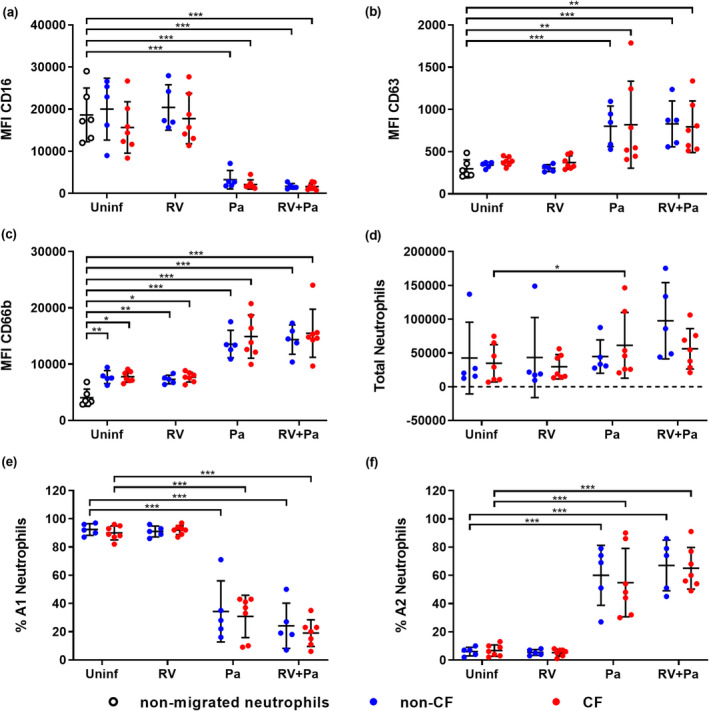
Neutrophil phenotype and populations change following Pseudomonas aeruginosa infection. Median fluorescence intensity (MFI) of stains for neutrophil functional markers (a) CD16, (b) CD63 and (c) CD66b in neutrophils following migration into air–liquid interface washes [*P < 0.05, **P < 0.01, ***P < 0.001 compared with nonmigrated neutrophils, repeated measures (RM) one‐way ANOVA]. (d) Total migrated neutrophils, (e) % A1 (baseline) subset and (f) % A2 (GRIM) subset of neutrophils for each infection condition (*P < 0.05, ***P < 0.001 compared with uninfected controls, RM one‐way ANOVA). Error bars indicate mean ± standard deviation. Data points indicate neutrophil responses to washes of individual primary cell cultures. GRIM, granule‐releasing, immunoregulatory and metabolically active phenotype; Pa, Pseudomonas aeruginosa; RV, rhinovirus.
LPS and fMLF partially influence neutrophil phenotype
Because the A2 neutrophil subset (GRIM phenotype) increased following migration into washes primed by P. aeruginosa, we were curious whether bacterial stimuli could induce this phenotype independently of host epithelial responses. To test this, neutrophils were migrated into 0.9% (v/v) medical saline spiked with LPS and fMLF individually or in combination. LPS was tested at 1500 endotoxin units per mL (EU mL−1), which was the median concentration across all LPS‐positive conditions as measured by a Limulus amebocyte lysate kinetic chromogenic assay (Supplementary figure 5). Control and RV washes were about 0.1 EU mL−1, below the 0.5 EU mL−1 Food and Drug Administration standard for most medical devices. 28 The bacterial chemotactic peptide fMLF was tested at a concentration of 100 ng mL−1 as performed previously. 7 The MFI of CD16 was significantly reduced from baseline expression by all treatments (0.4‐fold; P < 0.001; Figure 4a). LPS, alone or in combination with fMLF, also increased CD63 MFI in recruited neutrophils (2‐fold; P < 0.01; Figure 4b), but this was less than what was observed in neutrophils migrating toward washes from P. aeruginosa‐infected ALI cultures. LPS also elicited a three to fourfold increase in CD66b (Figure 4c). Numbers of total migrated neutrophils were similar between bacterial stimuli. In contrast to results with P. aeruginosa‐primed ALI washes, the proportion of A1 neutrophils were high while A2 neutrophils remained low, indicating few reprogrammed neutrophils with bacterial stimuli alone (Figure 4d–f).
Figure 4.
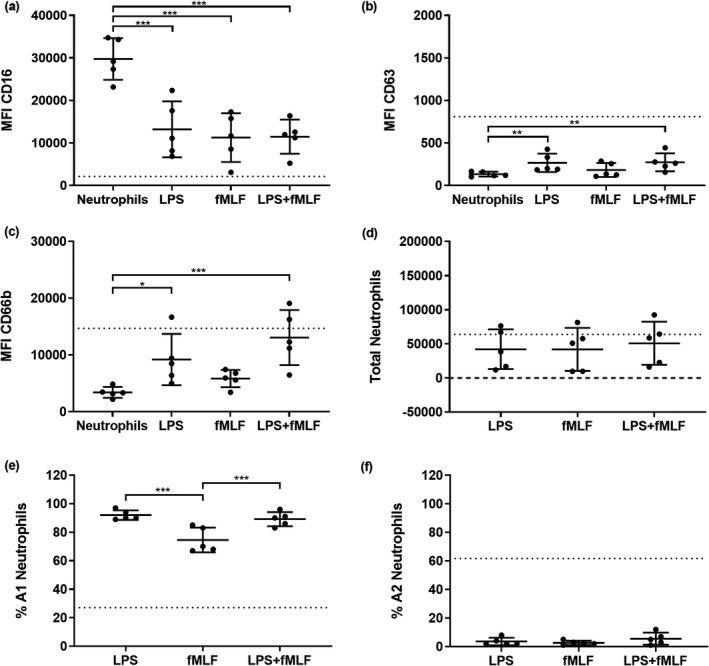
Lipopolysaccharide (LPS) modulates secondary granule mobilization. Median fluorescence intensity (MFI) of stains for neutrophil functional markers (a) CD16, (b) CD63 and (c) CD66b in neutrophils following migration into LPS and/or fMLF [*P < 0.05, **P < 0.01, ***P < 0.001 compared with nonmigrated neutrophils, repeated measures (RM) one‐way ANOVA]. (d) Total migrated neutrophils, (e) % A1 (baseline) population and (f) % A2 (GRIM) population of neutrophils for each treatment (***P < 0.001 compared with other conditions, RM one‐way ANOVA). Error bars indicate mean ± standard deviation. Data points indicate neutrophil responses from five individual neutrophil donors. Dotted lines indicate mean MFI or neutrophil numbers from prior migrations into air–liquid interface washes from Pseudomonas aeruginosa and co‐infected cultures (Figure 3a–f). fMLF, N‐formylmethionyl‐leucyl‐phenylalanine; GRIM, granule‐releasing, immunoregulatory and metabolically active phenotype.
Neutrophil reprogramming is not exclusively triggered by individual cytokine signals
Because of the dominant, inflammatory epithelial responses following bacterial infection (Figure 2), we also tested whether the subset of cytokines upregulated in P. aeruginosa and co‐infection washes could alone trigger reprogramming. Neutrophils were migrated into 0.9% (v/v) medical saline spiked with a mixture of cytokines upregulated by P. aeruginosa and viral–bacterial co‐infection. Each cytokine was suspended according to mean concentrations in apical washes from P. aeruginosa epithelial challenges: IL‐1α (604 pg mL−1), IL‐1β (378 pg mL−1), IL‐1Ra (10,085 pg mL−1), IL‐2 (11 pg mL−1), macrophage colony‐stimulating factor (590 pg mL−1), TNFα (163 pg mL−1), basic fibroblast growth factor (46 pg mL−1), leukemia inhibitory factor (288 pg mL−1) and IFNγ (55 pg mL−1). IL‐1α and IL‐1β, as well as TNFα, were tested separately as some studies suggest they are implicated in neutrophil primary granule exocytosis. 29 , 30 Cytokines were also tested with the addition of LPS and fMLF to assess whether combinations of innate signals and bacterial products had synergistic effects on neutrophil reprogramming. The MFI of CD16 was reduced from baseline levels in nonmigrated neutrophils by all treatments (0.5‐fold; P < 0.001; Figure 5a). However, no changes in CD63 MFI were observed in any conditions (Figure 5b). Migration into cytokines alone did not increase MFI of CD66b from baseline, but the addition of LPS and fMLF did trigger increases of 2.5‐fold (P < 0.001) (Figure 5c). Numbers of total migrated neutrophils were again similar when migrating into cytokine treatments, with high and low proportions of A1 and A2 neutrophils, respectively, suggesting that these cytokines alone cannot induce GRIM neutrophil reprogramming (Figure 5d–f).
Figure 5.
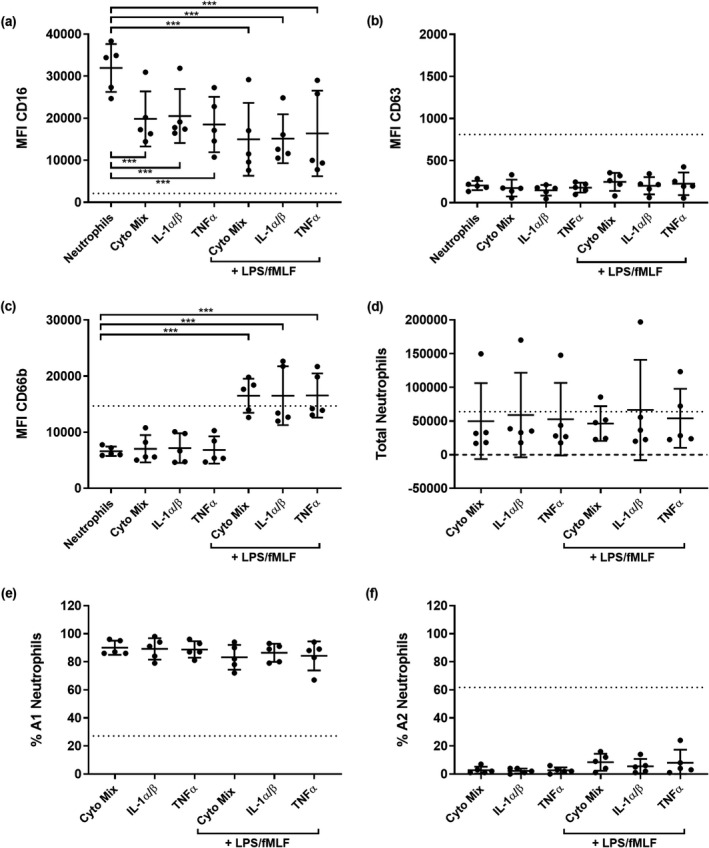
Cytokine signals alone do not induce granule exocytosis. Median fluorescence intensity (MFI) of stains for neutrophil functional markers (a) CD16, (b) CD63 and (c) CD66b in neutrophils following migration into interleukin (IL)‐1α and β, tumor necrosis factor alpha (TNFα) or a cytokine mixture (***P < 0.001 compared with nonmigrated neutrophils, repeated measures one‐way ANOVA). (d) Total migrated neutrophils, (e) % A1 (baseline) population and (f) % A2 (GRIM) population of neutrophils after migration into different cytokine conditions. Error bars indicate mean ± standard deviation. Data points indicate neutrophil responses from five individual neutrophil donors. Dotted lines indicate mean MFI or neutrophil numbers from prior migrations into air–liquid interface washes from Pseudomonas aeruginosa and co‐infected cultures (Figure 3a–f). fMLF, N‐formylmethionyl‐leucyl‐phenylalanine; GRIM, granule‐releasing, immunoregulatory and metabolically active phenotype; LPS, lipopolysaccharide.
Screen of anti‐inflammatory drugs during neutrophil transmigration
In addition to assessing neutrophil phenotype in the presence of individual host and microbial factors, we were interested in determining whether direct inhibition of factors in the P. aeruginosa infection microenvironment itself could have an effect on neutrophil reprogramming. We then proceeded to test a panel of compounds in migration assays for the prevention of GRIM neutrophil activation during recruitment. To generate large volumes of infection washes for drug screens, RV + P. aeruginosa co‐infections were repeated in ALI cultures grown on 6‐well permeable inserts with a larger surface area. Cultures were washed apically with a similar surface area‐to‐volume ratio as infections of 24‐well inserts, with washes pooled by condition and filtered. Neutrophils were then migrated into viral bacterial co‐infection washes in the presence of compounds inhibiting neutrophil recognition of host cytokine signals, LPS and inflammasome activity, including infliximab, high‐dose IL‐1Ra, LPS‐RS, TAK‐242 and MCC950 (Supplementary table 3). In addition, two drugs representative of therapies currently used for treating CF airway inflammation were tested, including S+‐ibuprofen and azithromycin (Supplementary table 3). Washes from uninfected 6‐well ALI cultures were included as negative controls for GRIM reprogramming, and untreated co‐infection washes served as positive controls. Co‐infection washes with added dimethyl sulfoxide and 0.1% bovine serum albumin solution were also used as vehicle controls. MFI of phagocytic receptor CD16 was reduced significantly in neutrophils migrating toward all ALI co‐infection washes compared with nonmigrated neutrophils, with the greatest reduction observed following migration in the presence of S+‐ibuprofen (0.2‐fold; P < 0.001; Figure 6a). MFI of primary granule exocytosis marker CD63 significantly increased in neutrophils migrating into viral bacterial co‐infection washes in the presence of all added compounds (P < 0.001), and again S+‐ibuprofen appeared to intensify marker shift, resulting in the greatest CD63 increase (2.3‐fold; P < 0.001; Figure 6b). Expression of secondary granule exocytosis marker CD66b was similarly increased by co‐infection washes in the presence of most compounds (P < 0.001), except for S+‐ibuprofen, which resulted in the greatest increase in CD66b in recruited neutrophils (4‐fold; P < 0.001; Figure 6c). The number of total migrated neutrophils was mostly similar across conditions, suggesting that drug treatments were not affecting overall neutrophil recruitment (Figure 6d). When organizing neutrophils according to subsets, the proportion of A1 noninflammatory neutrophils decreased upon migration into ALI co‐infection washes compared with uninfected controls, with the greatest reduction of A1 neutrophils observed following migrating into co‐infection washes spiked with S+‐ibuprofen (54 ± 21%; P < 0.001) (Figure 6e). Conversely, the percentage of A2 GRIM neutrophils significantly increased with infection, once again regardless of whether or not compounds were present during migration, and this was highest in the presence of S+‐ibuprofen (40 ± 18%; P < 0.001) (Figure 6f). Altogether, these results indicate that none of the tested compounds had the capacity to reduce infection‐mediated activation of GRIM neutrophils. Curiously, it appeared ibuprofen may be associated with increased GRIM reprogramming.
Figure 6.
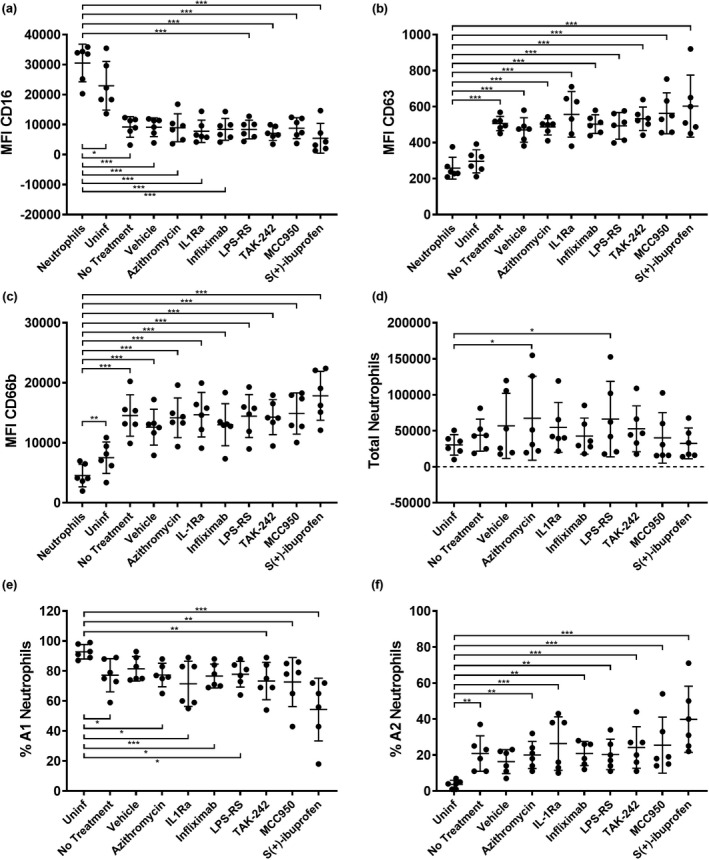
Anti‐GRIM drug screen. Median fluorescence intensity of stains for neutrophil functional markers (a) CD16, (b) CD63 and (c) CD66b in neutrophils following migration into rhinovirus (RV) + Pseudomonas aeruginosa air–liquid interface washes with added compounds [*P < 0.05, **P < 0.01, ***P < 0.001 compared with nonmigrated neutrophils, repeated measures (RM) one‐way ANOVA]. (d) Total migrated neutrophils, (e) % A1 (baseline) subset and (f) % A2 (GRIM) subset of neutrophils for each condition (*P < 0.05, **P < 0.01, ***P < 0.001 compared with uninfected controls, RM one‐way ANOVA). Error bars indicate mean ± standard deviation. Data points indicate neutrophil responses from six individual neutrophil donors. GRIM, granule‐releasing, immunoregulatory and metabolically active phenotype.
Discussion
Currently, it is known that GRIM reprogramming of neutrophils can be observed in CF airway samples, 7 , 11 and reprogramming can be replicated in vitro by migrating naïve blood neutrophils into adult CF sputum. 7 Reprogramming was found to occur independently of LPS and LTB4 signaling, but the specific factors in the CF airway environment triggering changes in functional marker expression remain unidentified. 7 Our study provides further critical insights by demonstrating that RV is unlikely to be a trigger of neutrophil reprogramming in pediatric CF airways, which instead is a response to P. aeruginosa. The observation from our model that even a low starting biomass of P. aeruginosa could induce significant changes in epithelium and neutrophils suggests that subculturable levels of P. aeruginosa could be capable of reprogramming locally recruited neutrophils. Our study also adds to the field of mucosal immunology a new reductive approach to model innate immune responses to airway infection in vitro.
Following infection, we observed that pathogen load was similar for both RV and P. aeruginosa in non‐CF‐ and CF‐differentiated AECs, as were profiles of cytokine signals following infection. Exclusive viral infection elicited canonical antiviral signals, while responses to P. aeruginosa infection reflected an acute inflammatory response and were dominant over antiviral signals in co‐infection with RV, despite high cytotoxicity following exclusive viral infection. This parallels findings where co‐infection of RV with P. aeruginosa dampened viral‐mediated IFN responses, IL‐6 production and increased release of IL‐1β and viral replication in AECs, 31 , 32 , 33 suggesting mechanisms by which P. aeruginosa modulates host responses to RV. Our results show significant reduction of IL‐6, IFN‐associated cytokines and high IL‐1β release upon co‐infection with P. aeruginosa; however, this did not lead to increases in RV titers. Profiles of cytokine signals were highly similar between non‐CF and CF epithelial cultures, analogous to reports finding no differences in inflammation between these tissues. 19 , 20 Why other studies report differences in CF airway cell immune responses could be attributed to a number of factors, including differences in sample collection, experimental design, laboratory culture methods or the viral/bacterial strains used. As an example, antiviral responses mature with airway tissue differentiation, with proliferative basal cells more susceptible to infection and virally induced inflammation. 34 , 35 Our study modeled infection in differentiated ALI cultures, therefore responses are measured in an in vitro system that closely resembles native airway tissue. While CFTR genotype did not result in differences in epithelial cytokine release from non‐CF and CF AECs, intrinsic defects in the CF airway epithelium may still drive in vivo inflammation via mucus accumulation that impairs other innate functions that protect against infection, such as mucociliary clearance.
Following migration into filtered P. aeruginosa and co‐infection washes, neutrophils had reduced MFI of CD16, and increased CD63 and CD66b, resulting in high proportions of the A2 neutrophil (GRIM) subset. These observations are consistent with previously identified signs of GRIM neutrophils. 7 , 9 , 11 , 13 However, contrary to our hypothesis, innate responses to viral–bacterial co‐infection did not exclusively induce the GRIM neutrophil phenotype as P. aeruginosa alone also increased proportions of A2 neutrophils. Furthermore, neutrophil populations and MFI of functional markers did not differ upon migration into washes from non‐CF and CF ALI cultures, suggesting that GRIM neutrophil reprogramming may not be a direct consequence of CFTR dysfunction, and is instead dependent on the microenvironment formed by infectious conditions. The fact that GRIM neutrophils are more commonly observed in pediatric CF airways compared with non‐CF airways may once again be attributed to the mucociliary clearance defect caused by CFTR dysfunction, predisposing young children with CF to bacterial colonization and increased numbers of NE‐releasing GRIM neutrophils. In children without CF, the occurrence of GRIM neutrophils may be limited to instances where bacterial colonization occurs as a result of secondary infection following severe respiratory viral infection, as observed in children with acute respiratory distress syndrome. 13 In our model, we utilized the Gram‐negative bacterium P. aeruginosa which colonizes up to 80% of adults with CF and is associated with increased NE activity. 36 , 37 Although P. aeruginosa infection in young children with CF is less frequent, positive cultures are reported in up to 20% of children in the first 2 years of life. 38 , 39 With increased sensitivity, microbial sequencing of respiratory samples suggests P. aeruginosa prevalence may be as high as 50% in this age group as shown in an Australian pediatric CF cohort 40 ; however, standard administration of amoxicillin as a Staphylococcus prophylactic in recruited participants could have inadvertently selected for increased abundance of P. aeruginosa in this population.
Recently, we have also shown that high NE activity can persist following P. aeruginosa eradication in young children with CF, and persistence is significantly associated with progression of bronchiectasis. 27 In washes from P. aeruginosa and co‐infected cultures, the two cytokines with greatest fold increases were IL‐1 α and IL‐1β, which correlate positively with NE activity and structural lung damage in CF airways. 41 IL‐1α has a role in modulating neutrophil recruitment, 42 while IL‐1β is a proinflammatory mediator and a product of the NLR family pyrin domain containing 3 inflammasome. 43 This inflammasome is implicated in airway diseases featuring neutrophilic inflammation, including chronic obstructive pulmonary disease and neutrophil‐dominant asthma. 44 , 45 In a mouse model of cryopyrin‐associated periodic syndrome, unregulated NLR family pyrin domain containing 3 activity results in increased release of myeloperoxidase, possibly linking the inflammasome and primary granule exocytosis. 29 As such, NLR family pyrin domain containing 3 and IL‐1 are being explored as potential therapeutic targets for CF, with studies in CF mice showing reduced inflammation and improved clearance of P. aeruginosa with pharmaceutical treatment. 46 , 47 A recent study by Forrest and colleagues 48 demonstrated that CF airway fluid is rich in extracellular vesicles containing caspase‐1, an enzyme that activates IL‐1α and β, and these vesicles originate from granule‐releasing neutrophils. Neutrophil extracellular vesicles were observed to induce IL‐1α and β production by primary AECs, and increase CD63 expression in naïve neutrophils, suggesting GRIM neutrophils modulate inflammasome activity in neighboring cells to sustain inflammation and initiate further reprogramming of recruited neutrophils. 48 However, we observed that neutrophil exposure to the NLPR3 inhibitor MCC950 49 during transmigration failed to prevent formation of GRIM neutrophils, as did high‐dose recombinant IL‐1Ra, which is currently approved as an anti‐inflammatory medication for rheumatoid arthritis under the name Anakinra. 50 Similarly, Forrest et al. 48 found that MCC950 did not significantly reduce caspase‐1 activity in neutrophils migrated into CF airway surface fluid over 18 h, suggesting that inflammasome inhibition in GRIM neutrophils may require an extracellular vesicle targeted therapy.
In our experiments P. aeruginosa infection also increased AEC release of TNFα significantly in CF ALI cultures. A study by McLeish et al. 30 found that TNFα‐primed neutrophils stimulated with fMLF were a potent trigger of primary granule exocytosis as measured by release of myeloperoxidase. Similarly in their prior study, Potera et al. 51 observed that secondary fMLF stimulation of TNFα‐primed neutrophils increased NE release. Neutrophil CD63 was also measured, and it was found that TNFα alone did not elicit primary granule surface mobilization, as shown in this study; however, CD63 expression was not assessed following sequential coexposure with TNFα and fMLF. 51 When migrating neutrophils into TNFα and/or fMLF, we did not observe large increases in CD63 as seen following migration into P. aeruginosa‐primed ALI washes. In contrast to the aforementioned studies, this is indicative of low primary granule exocytosis; however, direct measurements of myeloperoxidase and NE in our model may be necessary to verify this. Another factor to consider is that in the studies by McLeish and Potera, the concentration of TNFα was 6–12 times higher than that was tested here (1 and 2 ng mL–1, respectively), as we used a concentration based on what AECs produced in response to P. aeruginosa infection. However, the addition of the Food and Drug Administration‐approved TNFα inhibitor infliximab 52 to RV + P. aeruginosa washes did not prevent GRIM activation in recruited neutrophils, suggesting that epithelial TNFα was not a modulator of this neutrophil response. Overall, cytokines, LPS and fMLF significantly decreased CD16, suggesting shared mechanisms that modulate neutrophil phagocytic ability, but none of these conditions robustly increased CD63, with proportions of A2 subsets remaining low. LPS itself appeared to be an important trigger of secondary granule mobilization, with CD66b MFI increasing in the presence of LPS alone or in combination with other compounds. Neutrophils migrating into RV + P. aeruginosa washes in the presence of LPS‐RS 53 and TAK‐242, 54 inhibitors of LPS detection by Toll‐like receptor 4, still experienced changes consistent with GRIM reprogramming, further suggesting that neutrophil recognition of bacterial endotoxin is not a primary trigger of this phenotype. All together our data indicate cytokine signals, LPS and fMLF are not exclusive activators of GRIM neutrophils and NE release.
Of note was the observation that numbers of GRIM neutrophils appeared to increase following migration into co‐infection washes with added S+‐ibuprofen. While high‐dose ibuprofen has been shown to slow CF lung function decline, 55 a study of inflammatory biomarkers in induced CF sputum showed that ibuprofen treatment reduced IL‐6, but failed to reduce IL‐8, TNF‐α, IL‐1‐β, NE and white cell counts compared with placebo controls. 56 This is interesting considering reports from in vitro and in vivo studies wherein cyclooxygenase inhibition during nonsterile inflammation failed to ameliorate, or even increased, production of NE by unknown mechanisms. 57 , 58 Similarly, a randomized control trial of azithromycin in patients with CF colonized with P. aeruginosa observed improved lung function with treatment, but did not see large differences in IL‐8 or NE in induced sputum between individuals receiving the drug and a placebo group. 59 Our results reflect this, as azithromycin also failed to dampen increases in CD63 in recruited neutrophils. Therefore, despite reduced lung function decline achieved with courses of ibuprofen and azithromycin in vivo, neutrophils recruited during treatment may be prone to NE release and still drive progressive airway disease; however, further studies are required to verify this.
While P. aeruginosa eventually dominates adult CF airways, pediatric CF airways are characterized by a variety of colonizers. Staphylococcus aureus is isolated in 20–30% of children with CF under the age of 2 and is associated with airway inflammation and reduced lung function. 60 , 61 Haemophilus influenzae infection is also frequently identified in this age group and similarly implicated in early lung disease. 39 , 61 Increasingly, the fungal pathogen Aspergillus fumigatus is recognized as a significant airway colonizer linked to early signs of lung damage. 62 , 63 Further investigation is required to assess whether these pathogens contribute to changes in airway neutrophil phenotypes and our model provides a platform to explore this. Because increased inflammatory cytokines, NE and neutrophil CD63 can be identified in early CF in the absence of clinical culture positivity, 11 , 41 a further question is whether nonpathogenic airway microbes may also induce neutrophil reprogramming. Taxa from the oral microbiome are frequently identified in the lower airways of children with CF and may account for the appearance of NE and GRIM neutrophil populations in early CF when priority pathogens such as P. aeruginosa are absent. 40 , 64
Previous studies have relied on ex vivo airway samples to assess GRIM neutrophils. Our study is among the first of its kind in successfully recapitulating this pathological airway neutrophil phenotype, utilizing primary airway cells, low‐dose pathogen challenges and human neutrophils in an in vitro laboratory model. This approach gives us an unprecedented opportunity to characterize the host and microbial factors that drive this process in a controlled experimental platform. One caveat of our method is a lack of signals from tissue‐resident macrophages and cells of the lamina propria, which have important roles in responding to infection and tissue injury. Additionally, we used non‐CF neutrophils in our migration experiments, as we were unable to collect blood from CF donors. While GRIM reprogramming was still observed, CF neutrophils may have additional functional irregularities as a result of CFTR mutations. 65 We are also yet to assess the role of immune signals from neutrophils themselves. As such, our model reflects the initial epithelial response to infection followed by the first wave of neutrophil recruitment to the airways. Finally, our drug screen tested compounds during neutrophil transmigration, with neutrophils exposed to drugs upon recruitment to infection washes. Future studies should consider additional treatment of AEC cultures during microbial challenge to see whether this further modulates the inflammatory capacity of the infection microenvironment.
In summary, our study identified that the P. aeruginosa infection microenvironment induces GRIM reprogramming in recruited airway neutrophils. Additionally, we observed that migration of neutrophils into cytokines, LPS and fMLF, individually or in combination, could not recapitulate this phenotype, suggesting that these are not exclusive activators of reprogramming. Inhibition of certain inflammatory signals and LPS activity in RV + P. aeruginosa‐primed washes also could not prevent GRIM neutrophil activation during recruitment, further highlighting that neutrophil recognition of these factors is likely not involved in reprogramming mechanisms. Finally, two anti‐inflammatory drugs currently used in CF clinical care did not inhibit GRIM neutrophil reprogramming in transmigration experiments. Altogether, these results indicate that there are still unidentified soluble factors in the airway bacterial infection milieu triggering full GRIM reprogramming of neutrophils and underscore a need for development of targeted therapies. Future studies in our model integrating CF peripheral blood neutrophils, proteomics and transcriptomics will provide a robust platform to elucidate the underlying mechanisms and identify potential targets for therapeutic intervention. Beyond CF, we believe our model can be adapted to study other respiratory infections that induce pathological neutrophil activity upon migration to the airway mucosa, including respiratory syncytial virus, 66 a major cause of childhood bronchiolitis; severe acute respiratory syndrome coronavirus 2 67 and bacterial pneumonias. 68
Methods
Participants and primary airway cell culture
This study was conducted using AECs derived from seven young children with CF (Table 1) participating in the Australian Respiratory Early Surveillance Team for CF (AREST CF) early surveillance program. 17 Cells from five children with non‐CF (Table 1) were also obtained from participants recruited thought the Western Australian Epithelial Research Program (WAERP) upon admission to the hospital for elective nonrespiratory surgery. Participant data were collected and managed using REDCap electronic data capture tools. 69 , 70 AECs were collected by brushing of the airway mucosa and primary cultures were established as previously described. 71 After expansion, AECs were seeded onto collagen coated 24‐well or 6‐well transwell inserts (Corning, Corning, NY, USA) and cultured at ALI for 28 days to facilitate differentiation. 71 ALI culture medium was changed basolaterally every 48 h, trans‐epithelial electrical resistance measured weekly and presence of cilia and mucus observed by microscopy. Full differentiation was defined as multilayer formation, the visualization of cilia, production of mucus and a trans‐epithelial electrical resistance > 800 Ω·cm2. 71 Seven days prior to experiments, antibiotics were removed from the ALI culture medium. Twenty‐four hours prior to experiments and for the duration of infection, hydrocortisone and epidermal growth factor were also withdrawn from culture medium to avoid dampening of epithelial immune responses. 72
Virus and Bacteria
Human group A rhinovirus strain 1b (RV) was initially provided by Professor Peter Wark (Hunter Medical Research Institute, Newcastle, NSW, Australia). RV was propagated in HeLa cell suspension culture as previously described, 73 infectious titers determined by plaque assay 74 and aliquots of viral stock prepared and stored at −80°C. A sputum isolate of P. aeruginosa was isolated from a child with CF, cultured and a glycerol stock prepared and stored at −80°C. Prior to use in experiments, subcultures of frozen stocks were prepared on LB agar (BD Difco, Franklin Lakes, NJ, USA) and grown at 37°C for 24 h. A single colony was used to inoculate 5 mL of LB broth (BD Difco) and grown overnight in a shaker at 37°C. The overnight bacterial culture was then diluted to the required working concentration according to optical density at 600 nm, previously determined by diluting overnight cultures to specific optical density at 600 nm values and plated and counted to calculate CFU mL−1.
H441 scaffolds
Human club cell line H441 (ATCC HTB‐174) was expanded as submerged monolayers in Roswell Park Memorial Institute‐1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) heat‐inactivated fetal bovine serum and 0.5% (v/v) penicillin/streptomycin (P/S). To act as epithelial barriers in neutrophil transmigration experiments, H441 cells were cultured at ALI as previously described. 7 H441 cells were seeded onto porous Alvetex 3D Scaffolds (REPROCELL, Beltsville, MD, USA), coated with gelled collagen, at a density of 2.5 × 105 cells per scaffold. The larger void size of scaffolds (33–55 µm) compared with pores on a Transwell membrane (0.4 µm) has been shown to facilitate neutrophil migration and swarming behavior. 7 Scaffolds were initially grown submerged in Dulbecco’s Modified Eagle Medium/F12 (Corning) supplemented with 10% (v/v) heat‐inactivated fetal bovine serum and 0.5% (v/v) P/S for 2 days. Apical media were then removed and scaffolds were cultured at ALI for 2 weeks, fed basolaterally with Dulbecco’s Modified Eagle Medium/F12 (Corning) supplemented with 2% (v/v) Ultroser G (Pall Life Sciences, Port Washington, NY, USA) and 0.5% (v/v) P/S with media changes every 48 h.
Human neutrophils
Whole blood was obtained from donors consented through the Paediatric Epithelial and Airway Research Surrogate Sample (PEARSS) program, and neutrophils were isolated by gradient density. In brief, approximately 15 mL of whole blood was collected into K3EDTA vacutainers (Greiner Bio‐One) via venipuncture from healthy adult volunteers of the PEARSS program. Once transferred to the laboratory, whole blood was immediately layered onto Polymorphprep (Axis‐Shield, Dundee, UK) at a 1:1 ratio by volume. After centrifugation at 400g with minimal brake for 45 min at room temperature, the neutrophil layer was collected and erythrocytes were removed via 30‐s incubation in water to induce hypotonic lysis followed by restoration of tonicity with 1.8% saline solution. Neutrophils were then washed with 1× PBS and resuspended in Roswell Park Memorial Institute‐1640 medium (Thermo Fisher Scientific) for use in migration experiments.
Infection of ALI Cultures
To model different scenarios of acute viral and bacterial respiratory infection, non‐CF and CF ALI cultures were challenged either with RV (MOI 0.5) for 48 h, P. aeruginosa (MOI 0.001) for 24 h or sequentially co‐challenged by permitting infection with RV (MOI 0.5) for 24 h and then inoculating with P. aeruginosa (MOI 0.001) and allowing infection to continue for an additional 24 h. A graphic illustrating our infection timeline is shown in Supplementary figure 1. For addition of pathogens, MOI was calculated according to the number of cells seeded per insert (1.5 × 105 cells per transwell). The required dose of RV or P. aeruginosa was suspended in 1× PBS and loaded onto the apical surface of ALI cultures in a small volume (10 µL) to avoid disruption of the epithelium. Uninfected controls were treated with 1× PBS alone. At the end of the infection period, ALI cultures were washed apically to sample the infected epithelial milieu, analogous to ex vivo bronchoalveolar lavage washes. This was performed by incubating inserts with 100 µL per well of 0.9% (v/v) medical saline for 5 min at 37°C, 5% CO2. Washes from replicate wells were pooled and passed through a 0.22‐µm filter to remove bacteria and cell debris, then stored at −80°C until transmigration assays and cytokine analysis was performed. Inserts were treated to harvest total RNA for assessing pathogen load. For drug screens in transmigration experiments, RV + P. aeruginosa co‐infections were repeated in ALI cultures grown on larger 6‐well transwell inserts to generate large volumes of washes. Infected transwells, and uninfected controls, were washed with 1.2 mL of 0.9% (v/v) medical saline at a similar surface area‐to‐volume ratio as washes of 24‐well inserts, washes pooled according to condition and similarly filtered and stored at −80°C until needed.
Molecular assays
Epithelial cytokines present in apical washes were assessed by multiplex assay using the Bio‐Plex Pro Human Cytokine Screening Panel 48‐Plex (Bio‐Rad, Hercules, CA, USA), with plates read on the Bio‐Plex 200 System (Bio‐Rad) and results calculated on Bio‐Plex Manager Software 6.0 (Bio‐Rad). IL‐8 was assessed by ELISA, using the BD OptEIA Set for human IL‐8 (BD Biosciences, Franklin Lakes, NJ, USA) as previously described. 17 , 41 Endotoxin levels in ALI washes were measured using the KCA‐Endochrome‐K (Charles River, Wilmington, MA, USA) Limulus amoebocyte lysate kinetic chromogenic assay according to manufacturer’s instructions. Lactate dehydrogenase was measured using the CytoTox 96 Non‐Radioactive Cytotoxicity Assay (Promega, Madison, WI, USA).
Model of neutrophil transmigration
Prior to neutrophil transmigration assays, H441 scaffolds were inverted as previously described, 7 such that the apical surface was oriented downward, and the basolateral surface faced the upper compartment. The scaffolds were then placed as standing inserts on top of 200 µL of apical ALI washes from infected wells or uninfected controls (Supplementary figure 3). Isolated neutrophils were counted on a Countess II Automated Cell Counter (Thermo Fisher Scientific), and approximately 4 × 105 neutrophils were directly loaded onto the basolateral side of each scaffold. Neutrophils were permitted to migrate across H441 barriers into washes for 18 h at 37°C, 5%CO2. Migrated neutrophils that settled at the bottom of each well were collected by washing wells with 1 mL of 1× PBS + 2.5 mM ethylenediaminetetraacetic acid. Migrations with bacterial LPS and fMLF were performed similarly, using 200 µL of medical saline per scaffold with 1500 EU mL−1 LPS‐EB Ultrapure (InvivoGen, San Diego, CA, USA) and/or 100 ng mL−1 fMLF (Sigma‐Aldrich, St Louis, MO, USA). Neutrophil migrations into recombinant cytokines were performed using products from the Animal‐Free Cytokine line (PeproTech, Cranbury, NJ, USA). Cytokines were selected based on upregulation in P. aeruginosa and co‐infection washes and availability from the manufacturer, and also suspended in 200 µL medical saline for experiments: IL‐1α (604 pg mL−1), IL‐1β (378 pg mL−1), IL‐1Ra (10,085 pg mL−1), IL‐2 (11 pg mL−1), macrophage colony‐stimulating factor (590 pg mL−1), TNFα (163 pg mL−1), basic fibroblast growth factor (46 pg mL−1), leukemia inhibitory factor (288 pg mL−1) and IFNγ (55 pg mL−1).
Pharmacological compounds
Drugs used in this study included azithromycin (Sigma‐Aldrich), IL‐1Ra (PeproTech), Infliximab (Sigma‐Aldrich), LPS‐RS (InvivoGen), TAK‐242 (Sigma‐Aldrich), MCC950 (Sigma‐Aldrich) and S+‐ibuprofen (Sigma‐Aldrich; Supplementary table 3). Compounds were dissolved in sterile dimethyl sulfoxide, 0.1% solution of bovine serum albumin or ultra‐pure water according to manufacturer’s recommendations (Supplementary table 3). For tests of compounds in neutrophil transmigration assays, a small volume of drug was added to aliquots of ALI co‐infection washes according to concentrations used in prior in vitro studies (Supplementary table 3).
Flow cytometry
Migrated neutrophils, as well as reserved nonmigrated neutrophils, were stained for multiparametric flow cytometry. Initially, single‐cell suspensions were obtained by placing samples on a 35‐µm nylon mesh filter and centrifugation at 400 g for 2 min. Neutrophil suspensions were then incubated with Human TruStain FcX (BioLegend, San Diego, CA, USA) and Zombie Violet as a fixable live/dead measure (BioLegend) for 10 min on ice protected from light. This was followed by incubation with a mixed antibody stain for 20 min on ice away from light. The staining panel included the following antibodies at a 1:16 dilution in 1× PBS + 2.5 mM ethylenediaminetetraacetic acid (all from BioLegend): Brilliant Violet 605 anti‐human CD45 (leukocyte marker), Brilliant Violet 650 anti‐human CD63 (primary granule exocytosis marker), allophycocyanin/Cyanine7 anti‐human CD16 (phagocytosis marker), phycoerythrin anti‐human CD66b (granulocyte and secondary granule exocytosis marker) and phycoerythrin/Cy7 anti‐human CD326/EpCAM (epithelial cell marker). After staining, cells were washed with 3 mL 1× PBS + 2.5 mM ethylenediaminetetraacetic acid, pelleted at 400g for 10 min at 4°C and then fixed in 1 mL of Phosflow Lyse/Fix Buffer (BD Biosciences) at 4°C. Prior to acquisition, Phosflow Lyse/Fix was removed, and fixed cells were resuspended and mixed with CountBright beads (Thermo Fisher Scientific). Acquisition of samples was performed on an LSRFortessa flow cytometer (BD Biosciences) and compensation, gating and analysis were all conducted on FlowJo v10 (FlowJo LLC; Ashland, OR, USA) using single‐stain controls prepared with UltraComp eBeads Compensation Beads (Thermo Fisher Scientific). A four‐step gating strategy was utilized to identify neutrophils consisting of a singlet gate (forward scatter area vs. height), viability gate (EpCAM vs. Zombie UV viability), leukocyte gate (EpCAM vs. CD45) and CD66b+ neutrophils (forward scatter vs. CD66b; Supplementary figure 4). Neutrophil counts were normalized to a standardized number of count beads for all samples, and mean fluorescence intensities of functional markers are reported for CD66b+ neutrophils. The unactivated A1 gate was drawn to contain on average 98% of total neutrophil populations in suspensions of nonmigrated neutrophils used as controls for baseline marker expression. The A2 gate was drawn to include neutrophils that fell outside of the A1 population based on the criteria that both CD16 was reduced and CD63 increased.
RNA extraction and qPCR for pathogen load
Total RNA was extracted from ALI cultures using the Ambion PureLink RNA Mini Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Genomic DNA was removed by treating loaded RNA extraction columns with DNase I (Thermo Fisher Scientific) and the final concentration of eluted RNA measured on a NanoDrop 2000 (Thermo Fisher Scientific). Generation of complementary DNA was performed using Random Hexamers (Thermo Fisher Scientific) with a standardized amount of RNA for all samples. Viral load was quantified relatively using previously described primers complementary to a conserved RV 5′ untranslated region (Supplementary table 4). 75 A standard curve was generated using serial dilutions of RV stock with a known titer. Bacterial load was also relatively quantified by a previously described RNA‐based method using primers specific for P. aeruginosa 16S ribosomal RNA (Supplementary table 4). 76 A standard curve was generated using serial dilutions of P. aeruginosa with a known titer. Reactions were performed using SYBR Green qRT‐PCR (Applied Biosystems, Foster City, CA, USA) on the QuantStudio 7 Flex Real‐Time PCR System (Applied Biosystems). Examples of standard curve plots are shown in Supplementary figure 6.
Statistical analysis
Infections of ALI cultures were performed in four replicate transwells per condition, with postinfection washes pooled. Neutrophil transmigrations were performed in single inserts because of limited sample volume; therefore, individual data points represent biological replicates (n = 7 CF, n = 5 non‐CF). Cytokine data and pathogen load were obtained from five CF cultures and four non‐CF cultures, and assays performed in duplicate wells. Results from cytokine multiplexes were calculated on Bio‐Plex Manager Software 6.0 (Bio‐Rad). lactate dehydrogenase and LPS content were quantified from an apical wash pool and assays performed in triplicate wells. Migrations into saline with added LPS, fMLF or cytokines were performed with neutrophils from five donors. Migrations screening anti‐inflammatory compounds were performed with neutrophils from six donors. Statistical analyses were performed on GraphPad Prism version 9.2.0 (GraphPad Software, San Diego, CA, USA). Normality of data was verified by Shapiro–Wilk normality tests (P > 0.05) and Q–Q plot distributions. Pathogen load between infection conditions was analyzed with a paired t‐test, with comparisons between non‐CF and CF titers performed using a Welch’s unequal variances t‐test. Cytotoxicity results were analyzed using an ordinary one‐way ANOVA. Cytokine data and results from migration experiments were assessed using a repeated measures one‐way ANOVA with a Bonferroni correction for multiple comparisons. Following ANOVAs, comparisons between non‐CF and CF data were performed using multiple unpaired t‐tests, also with a Bonferroni correction. Error bars on all graphs indicate mean ± standard deviation.
Conflict of Interest
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Daniel R Laucirica: Conceptualization; Formal analysis; Investigation; Methodology; Writing – original draft; Writing – review and editing. Craig J Schofield: Investigation; Methodology; Writing – review and editing. Samantha A McLean: Investigation; Methodology; Writing – review and editing. Camilla Margaroli: Methodology; Writing – review and editing. Patricia Agudelo‐Romero: Formal analysis; Methodology; Writing – review and editing. Stephen M Stick: Supervision; Writing – review and editing. Rabindra Tirouvanziam: Methodology; Writing – review and editing. Anthony Kicic: Conceptualization; Supervision; Writing – original draft; Writing – review and editing. Luke W Garratt: Conceptualization; Methodology; Supervision; Writing – original draft; Writing – review and editing.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NHMRC project grant 1142505 and a Telethon Kids Institute Plus 10 Award. SMS is a NHMRC Practitioner Fellow, AK is a Rothwell Family Fellow and LWG is a NHMRC Early Career Fellow (1141479). CM is supported by a Cystic Fibrosis Foundation: Research Development Program Grant ROWE19R0. DRL is funded by a Scholarship for International Research Fees and an Ad Hoc Postgraduate Scholarship through the University of Western Australia, as well as a Stan and Jean Perron Top Up Scholarship through Telethon Kids Institute. This study was authored on behalf of WAERP (walyanrespirato‐ry.telethonkids.org.au/projects/WAERP/) and AREST CF (arestcf.telethonkids.org.au). We thank the families, participants and staff of WAERP, AREST and PEARSS. Open access publishing facilitated by The University of Western Australia, as part of the Wiley–The University of Western Australia agreement via the Council of Australian University Librarians.
References
- 1. Kruger P, Saffarzadeh M, Weber AN, et al. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog 2015; 11: e1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Margaroli C, Tirouvanziam R. Neutrophil plasticity enables the development of pathological microenvironments: implications for cystic fibrosis airway disease. Mol Cell Pediatr 2016; 3: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hartl D, Gaggar A, Bruscia E, et al. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros 2012; 11: 363–382. [DOI] [PubMed] [Google Scholar]
- 4. Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med 2012; 186: 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rosenow T, Mok LC, Turkovic L, et al. The cumulative effect of inflammation and infection on structural lung disease in early cystic fibrosis. Eur Respir J 2019; 54. [DOI] [PubMed] [Google Scholar]
- 6. Sly PD, Gangell CL, Chen L, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med 2013; 368: 1963–1970. [DOI] [PubMed] [Google Scholar]
- 7. Forrest OA, Ingersoll SA, Preininger MK, et al. Frontline Science: Pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J Leukoc Biol 2018; 104: 665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mitchell TC. A GRIM fate for human neutrophils in airway disease. J Leukoc Biol 2018; 104: 657–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tirouvanziam R, Gernez Y, Conrad CK, et al. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc Natl Acad Sci USA 2008; 105: 4335–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Makam M, Diaz D, Laval J, et al. Activation of critical, host‐induced, metabolic and stress pathways marks neutrophil entry into cystic fibrosis lungs. Proc Natl Acad Sci USA 2009; 106: 5779–5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Margaroli C, Garratt LW, Horati H, et al. Elastase exocytosis by airway neutrophils is associated with early lung damage in children with cystic fibrosis. Am J Respir Crit Care Med 2019; 199: 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Margaroli C, Moncada‐Giraldo D, Gulick DA, et al. Transcriptional firing represses bactericidal activity in cystic fibrosis airway neutrophils. Cell Rep Med 2021; 2: 100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grunwell JR, Giacalone VD, Stephenson S, et al. Neutrophil dysfunction in the airways of children with acute respiratory failure due to lower respiratory tract viral and bacterial coinfections. Sci Rep 2019; 9: 2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cabrini G, Rimessi A, Borgatti M, et al. Role of cystic fibrosis bronchial epithelium in neutrophil chemotaxis. Front Immunol 2020; 11: 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joseph T, Look D, Ferkol T. NF‐kappaB activation and sustained IL‐8 gene expression in primary cultures of cystic fibrosis airway epithelial cells stimulated with Pseudomonas aeruginosa . Am J Physiol Lung Cell Mol Physiol 2005; 288: L471–479. [DOI] [PubMed] [Google Scholar]
- 16. Carrabino S, Carpani D, Livraghi A, et al. Dysregulated interleukin‐8 secretion and NF‐kappaB activity in human cystic fibrosis nasal epithelial cells. J Cyst Fibros 2006; 5: 113–119. [DOI] [PubMed] [Google Scholar]
- 17. Sutanto EN, Kicic A, Foo CJ, et al. Innate inflammatory responses of pediatric cystic fibrosis airway epithelial cells: effects of nonviral and viral stimulation. Am J Respir Cell Mol Biol 2011; 44: 761–767. [DOI] [PubMed] [Google Scholar]
- 18. Montgomery ST, Frey DL, Mall MA, Stick SM, Kicic A, Arest CF. Rhinovirus infection is associated with airway epithelial cell necrosis and inflammation via interleukin‐1 in young children with cystic fibrosis. Front Immunol 2020; 11: 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schogler A, Blank F, Brugger M, et al. Characterization of pediatric cystic fibrosis airway epithelial cell cultures at the air‐liquid interface obtained by non‐invasive nasal cytology brush sampling. Respir Res 2017; 18: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Becker MN, Sauer MS, Muhlebach MS, et al. Cytokine secretion by cystic fibrosis airway epithelial cells. Am J Respir Crit Care Med 2004; 169: 645–653. [DOI] [PubMed] [Google Scholar]
- 21. Ebrahimzadeh PR, Hogfors C, Braide M. Neutrophil chemotaxis in moving gradients of fMLP. J Leukoc Biol 2000; 67: 651–661. [DOI] [PubMed] [Google Scholar]
- 22. Sato T, Hongu T, Sakamoto M, Funakoshi Y, Kanaho Y. Molecular mechanisms of N‐formyl‐methionyl‐leucyl‐phenylalanine‐induced superoxide generation and degranulation in mouse neutrophils: phospholipase D is dispensable. Mol Cell Biol 2013; 33: 136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Forehand JR, Pabst MJ, Phillips WA, Johnston RB Jr. Lipopolysaccharide priming of human neutrophils for an enhanced respiratory burst. Role of intracellular free calcium. J Clin Invest 1989; 83: 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reutershan J, Basit A, Galkina EV, Ley K. Sequential recruitment of neutrophils into lung and bronchoalveolar lavage fluid in LPS‐induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 2005; 289: L807–815. [DOI] [PubMed] [Google Scholar]
- 25. Chignard M, Balloy V. Neutrophil recruitment and increased permeability during acute lung injury induced by lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol 2000; 279: L1083–1090. [DOI] [PubMed] [Google Scholar]
- 26. Burns JL, Emerson J, Kuypers J, et al. Respiratory viruses in children with cystic fibrosis: viral detection and clinical findings. Influenza Other Respir Viruses 2012; 6: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garratt LW, Breuer O, Schofield CJ, et al. Changes in airway inflammation with pseudomonas eradication in early cystic fibrosis. J Cyst Fibros 2021; 20: 941–948. [DOI] [PubMed] [Google Scholar]
- 28. FDA . Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers. Maryland: Office of Communications, Division of Drug Information, 2012. [Google Scholar]
- 29. Johnson JL, Ramadass M, Haimovich A, et al. Increased neutrophil secretion induced by NLRP3 mutation links the Inflammasome to Azurophilic granule exocytosis. Front Cell Infect Microbiol 2017; 7: 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McLeish KR, Merchant ML, Creed TM, et al. Frontline Science: Tumor necrosis factor‐α stimulation and priming of human neutrophil granule exocytosis. J Leukoc Biol 2017; 102: 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chattoraj SS, Ganesan S, Faris A, Comstock A, Lee WM, Sajjan US. Pseudomonas aeruginosa suppresses interferon response to rhinovirus infection in cystic fibrosis but not in normal bronchial epithelial cells. Infect Immun 2011; 79: 4131–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Endres A, Schubert R, Braubach P, et al. Pseudomonas aeruginosa influences the inflammatory response of primary bronchial epithelial cells to rhinovirus infection. Eur Respir J 2020; 56: 4343. [Google Scholar]
- 33. Sorensen M, Kantorek J, Byrnes L, et al. Pseudomonas aeruginosa modulates the antiviral response of bronchial epithelial cells. Front Immunol 2020; 11: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jakiela B, Brockman‐Schneider R, Amineva S, Lee WM, Gern JE. Basal cells of differentiated bronchial epithelium are more susceptible to rhinovirus infection. Am J Respir Cell Mol Biol 2008; 38: 517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Warner SM, Wiehler S, Michi AN, Proud D. Rhinovirus replication and innate immunity in highly differentiated human airway epithelial cells. Respir Res 2019; 20: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhagirath AY, Li Y, Somayajula D, Dadashi M, Badr S, Duan K. Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm Med 2016; 16: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oriano M, Gramegna A, Terranova L, et al. Sputum neutrophil elastase associates with microbiota and Pseudomonas aeruginosa in bronchiectasis. Eur Respir J 2020; 56: 2000769. [DOI] [PubMed] [Google Scholar]
- 38. Douglas TA, Brennan S, Gard S, et al. Acquisition and eradication of P. aeruginosa in young children with cystic fibrosis. Eur Respir J 2009; 33: 305–311. [DOI] [PubMed] [Google Scholar]
- 39. Salsgiver EL, Fink AK, Knapp EA, et al. Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis. Chest 2016; 149: 390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muhlebach MS, Zorn BT, Esther CR, et al. Initial acquisition and succession of the cystic fibrosis lung microbiome is associated with disease progression in infants and preschool children. PLoS Pathog 2018; 14: e1006798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Montgomery ST, Dittrich AS, Garratt LW, et al. Interleukin‐1 is associated with inflammation and structural lung disease in young children with cystic fibrosis. J Cyst Fibros 2018; 17: 715–722. [DOI] [PubMed] [Google Scholar]
- 42. Rider P, Carmi Y, Guttman O, et al. IL‐1α and IL‐1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol 2011; 187: 4835–4843. [DOI] [PubMed] [Google Scholar]
- 43. Dinarello CA. Overview of the IL‐1 family in innate inflammation and acquired immunity. Immunol Rev 2018; 281: 8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang H, Lv C, Wang S, Ying H, Weng Y, Yu W. NLRP3 inflammasome involves in the acute exacerbation of patients with chronic obstructive pulmonary disease. Inflammation 2018; 41: 1321–1333. [DOI] [PubMed] [Google Scholar]
- 45. Kim RY, Pinkerton JW, Essilfie AT, et al. Role for NLRP3 inflammasome‐mediated, IL‐1β‐dependent responses in severe, steroid‐resistant asthma. Am J Respir Crit Care Med 2017; 196: 283–297. [DOI] [PubMed] [Google Scholar]
- 46. McElvaney OJ, Zaslona Z, Becker‐Flegler K, et al. Specific inhibition of the NLRP3 inflammasome as an antiinflammatory strategy in cystic fibrosis. Am J Respir Crit Care Med 2019; 200: 1381–1391. [DOI] [PubMed] [Google Scholar]
- 47. Palomo J, Marchiol T, Piotet J, et al. Role of IL‐1β in experimental cystic fibrosis upon P. aeruginosa infection. PLoS One 2014; 9: e114884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Forrest OA, Dobosh B, Ingersoll SA, et al. Neutrophil‐derived extracellular vesicles promote feed‐forward inflammasome signaling in cystic fibrosis airways. J Leukoc Biol 2022. 10.1002/JLB.3AB0321-149R [DOI] [PubMed] [Google Scholar]
- 49. Coll RC, Robertson AA, Chae JJ, et al. A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015; 21: 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fleischmann RM, Schechtman J, Bennett R, et al. Anakinra, a recombinant human interleukin‐1 receptor antagonist (r‐metHuIL‐1ra), in patients with rheumatoid arthritis: A large, international, multicenter, placebo‐controlled trial. Arthritis Rheum 2003; 48: 927–934. [DOI] [PubMed] [Google Scholar]
- 51. Potera RM, Jensen MJ, Hilkin BM, et al. Neutrophil azurophilic granule exocytosis is primed by TNF‐α and partially regulated by NADPH oxidase. Innate Immun 2016; 22: 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang C, Shu W, Zhou G, et al. Anti‐TNF‐α therapy suppresses proinflammatory activities of mucosal neutrophils in inflammatory bowel disease. Mediators Inflamm 2018; 2018: 3021863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abdelmageed ME, El‐Awady MS, Abdelrahim M, Suddek GM. LPS‐RS attenuation of lipopolysaccharide‐induced acute lung injury involves NF‐kappaB inhibition. Can J Physiol Pharmacol 2016; 94: 140–146. [DOI] [PubMed] [Google Scholar]
- 54. Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. TAK‐242 (resatorvid), a small‐molecule inhibitor of Toll‐like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol 2011; 79: 34–41. [DOI] [PubMed] [Google Scholar]
- 55. Lands LC, Stanojevic S. Oral non‐steroidal anti‐inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst Rev 2019; 9: CD001505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chmiel JF, Konstan MW, Accurso FJ, et al. Use of ibuprofen to assess inflammatory biomarkers in induced sputum: Implications for clinical trials in cystic fibrosis. J Cyst Fibros 2015; 14: 720–726. [DOI] [PubMed] [Google Scholar]
- 57. Spinas GA, Bloesch D, Keller U, Zimmerli W, Cammisuli S. Pretreatment with ibuprofen augments circulating tumor necrosis factor‐α, interleukin‐6, and elastase during acute endotoxinemia. J Infect Dis 1991; 163: 89–95. [DOI] [PubMed] [Google Scholar]
- 58. Kimura T, Iwase M, Kondo G, et al. Suppressive effect of selective cyclooxygenase‐2 inhibitor on cytokine release in human neutrophils. Int Immunopharmacol 2003; 3: 1519–1528. [DOI] [PubMed] [Google Scholar]
- 59. Saiman L, Marshall BC, Mayer‐Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2003; 290: 1749–1756. [DOI] [PubMed] [Google Scholar]
- 60. Sagel SD, Gibson RL, Emerson J, et al. Impact of Pseudomonas and Staphylococcus infection on inflammation and clinical status in young children with cystic fibrosis. J Pediatr 2009; 154: 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ramsey KA, Ranganathan S, Park J, et al. Early respiratory infection is associated with reduced spirometry in children with cystic fibrosis. Am J Respir Crit Care Med 2014; 190: 1111–1116. [DOI] [PubMed] [Google Scholar]
- 62. Breuer O, Schultz A, Turkovic L, et al. The changing prevalence of lower airway infections in young children with cystic fibrosis. Am J Respir Crit Care Med 2019; 200: 590–599. [DOI] [PubMed] [Google Scholar]
- 63. Breuer O, Schultz A, Garratt LW, et al. Aspergillus infections and progression of structural lung disease in children with cystic fibrosis. Am J Respir Crit Care Med 2020; 201: 688–696. [DOI] [PubMed] [Google Scholar]
- 64. Zemanick ET, Wagner BD, Robertson CE, et al. Airway microbiota across age and disease spectrum in cystic fibrosis. Eur Respir J 2017; 50: 1700832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Laucirica DR, Garratt LW, Kicic A. Progress in model systems of cystic fibrosis mucosal inflammation to understand aberrant neutrophil activity. Front Immunol 2020; 11: 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sebina I, Phipps S. The contribution of neutrophils to the pathogenesis of RSV bronchiolitis. Viruses 2020; 12: 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Arcanjo A, Logullo J, Menezes CCB, et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID‐19). Sci Rep 2020; 10: 19630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pechous RD. With friends like these: The complex role of neutrophils in the progression of severe pneumonia. Front Cell Infect Microbiol 2017; 7: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Harris PA, Taylor R, Minor BL, et al. The REDCap consortium: Building an international community of software platform partners. J Biomed Inform 2019; 95: 103208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009; 42: 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Martinovich KM, Iosifidis T, Buckley AG, et al. Conditionally reprogrammed primary airway epithelial cells maintain morphology, lineage and disease specific functional characteristics. Sci Rep 2017; 7: 17971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kalinowski A, Ueki I, Min‐Oo G, et al. EGFR activation suppresses respiratory virus‐induced IRF1‐dependent CXCL10 production. Am J Physiol Lung Cell Mol Physiol 2014; 307: L186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lee WM, Chen Y, Wang W, Mosser A. Growth of human rhinovirus in H1‐HeLa cell suspension culture and purification of virions. Methods Mol Biol 2015; 1221: 49–61. [DOI] [PubMed] [Google Scholar]
- 74. Lee WM, Chen Y, Wang W, Mosser A. Infectivity assays of human rhinovirus‐A and ‐B serotypes. Methods Mol Biol 2015; 1221: 71–81. [DOI] [PubMed] [Google Scholar]
- 75. Bochkov YA, Palmenberg AC, Lee WM, et al. Molecular modeling, organ culture and reverse genetics for a newly identified human rhinovirus C. Nat Med 2011; 17: 627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Magalhaes AP, Franca A, Pereira MO, Cerca N. RNA‐based qPCR as a tool to quantify and to characterize dual‐species biofilms. Sci Rep 2019; 9: 13639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material