

Summary
Background
Increased collagen remodelling is a key pathophysiological component underlying intestinal stricture and fistula development in Crohn's disease (CD).
Aims
To investigate associations between serological biomarkers of collagen turnover and disease behaviour according to the Montreal classification in patients with CD.
Methods
Serological biomarkers of type III/IV collagen formation (PRO‐C3, PRO‐C4) and matrix metalloproteinase (MMP) or granzyme‐B (GrzB)‐mediated type I, III, IV and VI collagen degradation (C1M, C3M, C4M, C4G, C6Ma3) were measured using neo‐epitope protein fingerprint assays in 101 patients with CD (Montreal B1: n = 37; B2: n = 27; B3: n = 37) and 96 controls. Patients were followed up until their last outpatient visit to monitor stricturing/penetrating disease progression and recurrence and the occurrence of surgical interventions.
Results
C1M, C3M and C4M were significantly reduced in patients with stricturing disease (Montreal B2) and accurately differentiated them from patients with either non‐stricturing, non‐penetrating (B1) or penetrating (B3) disease (all p < 0.001, multivariable analysis). Similarly, the type IV collagen formation/degradation (PRO‐C4/C4M) ratio demonstrated high discriminative capacity (B1/B2: AUC = 0.90; B1/B3: AUC = 0.87, both p < 0.001, multivariable analysis). Prospectively, higher baseline levels of C1M and C4G were associated with an increased risk of penetrating disease progression (C4G: hazard ratio [HR] 1.71 [1.05–2.81], p < 0.05).
Conclusions
Elevated degradation of type I, III and IV collagen and excessive (relative) formation of type IV collagen strongly associates with stricturing CD. Type I and IV collagen fragments show predictive potential for the risk of penetrating disease progression. These biomarkers may become valuable tools for detection and prediction of stricturing and penetrating CD.
Serological biomarkers of collagen type I, type III and type IV turnover are associated with the presence and future progression of stricturing and penetrating Crohn's disease.

1. INTRODUCTION
Crohn's disease (CD) is a chronic ulcerative inflammatory disease mainly affecting the gastrointestinal (GI) tract and is characterised by an inappropriate and uncontrolled immune response that is putatively triggered by the gut microbiome in genetically susceptible individuals. 1 Longstanding (often subclinical) disease activity may progress to disease complications, including stricturing (i.e. intestinal stenosis) and penetrating (i.e. intestinal fistulae, abscesses or perforations) disease phenotypes, which are already present in 30%–50% of patients at the time of diagnosis and eventually occur in more than 70% of patients. 2 , 3 Stricturing and/or penetrating disease complications are often classified according to the Montreal classification as disease behaviour subtypes, which consist of non‐stricturing, non‐penetrating disease (B1), stricturing (B2) and penetrating (B3) disease. 4
The intestinal (sub)mucosa is rich in extracellular matrix (ECM) proteins, including collagens, which are very important to maintain epithelial integrity and structure and tensile strength of the intestinal tissue. The ECM can be divided into two layers, the basement membrane (BM) and interstitial matrix (IM). The most abundant collagen of the BM is type IV collagen, whereas type I and III collagens are the most abundant collagens of the IM, which are directly associated with the intestinal epithelium (Figure 1). At the interface between the BM and IM, type VI collagen is highly expressed and acts as an anchor of the BM, by interacting directly with type IV collagen and perlecan. 5 Fibrosis is considered the primary pathophysiological mechanism underlying these disease complications, which is a result of excessive ECM deposition, mainly collagens, and abnormal remodelling due to chronic inflammation and impaired wound healing. 6 This process is mediated by increased proliferation and differentiation of intestinal (myo) fibroblasts, and is considered to drive the development and progression of intestinal stricture formation. 5 , 7 On the other hand, chronic intestinal inflammation may lead to ECM breakdown and remodelling, as many local cells secrete proteases and structural proteins. 7 , 8 Inflammatory cells, for example, macrophages and neutrophilic granulocytes, produce matrix metalloproteinases (MMPs), which are collagenases able to destroy ECM components. Moreover, T‐lymphocytes express granzyme‐B, resulting in epithelial barrier disruption, mucosal damage and giving rise to intestinal fistula formation. 5 , 9 Fistulae may occur when healing of chronic ulcers is impaired due to increased activity of granulocytes and T‐cells leading to increased protease activity, including MMPs, resulting in chronic tracts of granulation tissue between two epithelial‐lined surfaces after re‐epithelialisation of penetrating ulcers. 5 , 10 , 11 , 12
FIGURE 1.
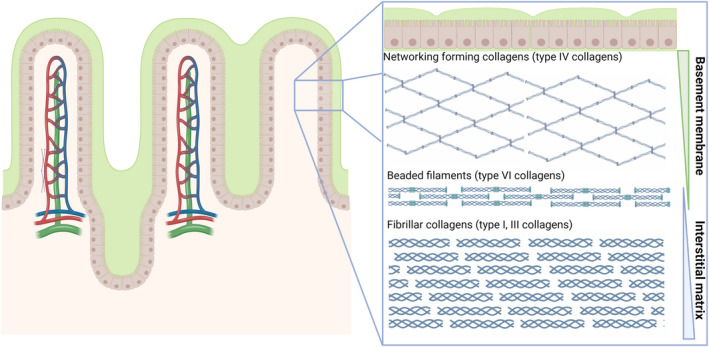
Schematic representation of the extracellular matrix (ECM) composition of the intestinal mucosa. The ECM can roughly be divided into two layers: the basement membrane (BM) and interstitial matrix (IM). The BM is mainly composed of type IV collagen, which are networking forming collagens. The IM primarily consists of type I and type III collagens, which are fibrillar collagens, as well as type VI collagens, which consists of beaded filaments.
As fragments of intestinal collagens derived from an increased proteolytic activity are released into the systemic circulation, they can be measured in blood and potentially serve as serological biomarkers for stricturing and/or penetrating disease in patients with CD. 5 For that purpose, protein fingerprint assays that quantify specific neo‐epitope fragments of collagen formation and degradation, including MMP‐ and granzyme‐B‐derived fragments of type I, III, IV and VI collagens, have recently been developed and validated in a variety of (fibrotic) diseases. 13 , 14 , 15 , 16 , 17 , 18 , 19 Previously, specific biomarkers of collagen formation and degradation were demonstrated to be strongly associated with CD, as well as with CD disease activity, disease behaviour and response to biological therapy. 20 , 21 , 22 , 23 For instance, an imbalance in type III collagen formation and degradation showed potential as a biomarker for penetrating CD and as a monitoring tool for the dynamics of mucosal damage and healing. 22 , 23 , 24 Although these results are promising, further validation studies are warranted, together with an assessment of these biomarkers for their ability to predict future disease course. Importantly, the latter goal may help to early detect and monitor disease complications in CD, thereby enabling prompt therapeutic intervention and prevention of severe disease.
In this study, we aimed to evaluate the potential of circulating collagen formation and degradation fragments as discriminative biomarkers for Montreal disease behaviour subclasses in patients with CD. Furthermore, we aimed to determine associations of these biomarkers with inflammatory disease activity, and to assess their predictive value in relation to the risk of progression of stricturing and penetrating disease and the risk of future surgical interventions.
2. MATERIALS AND METHODS
2.1. Study design and study population
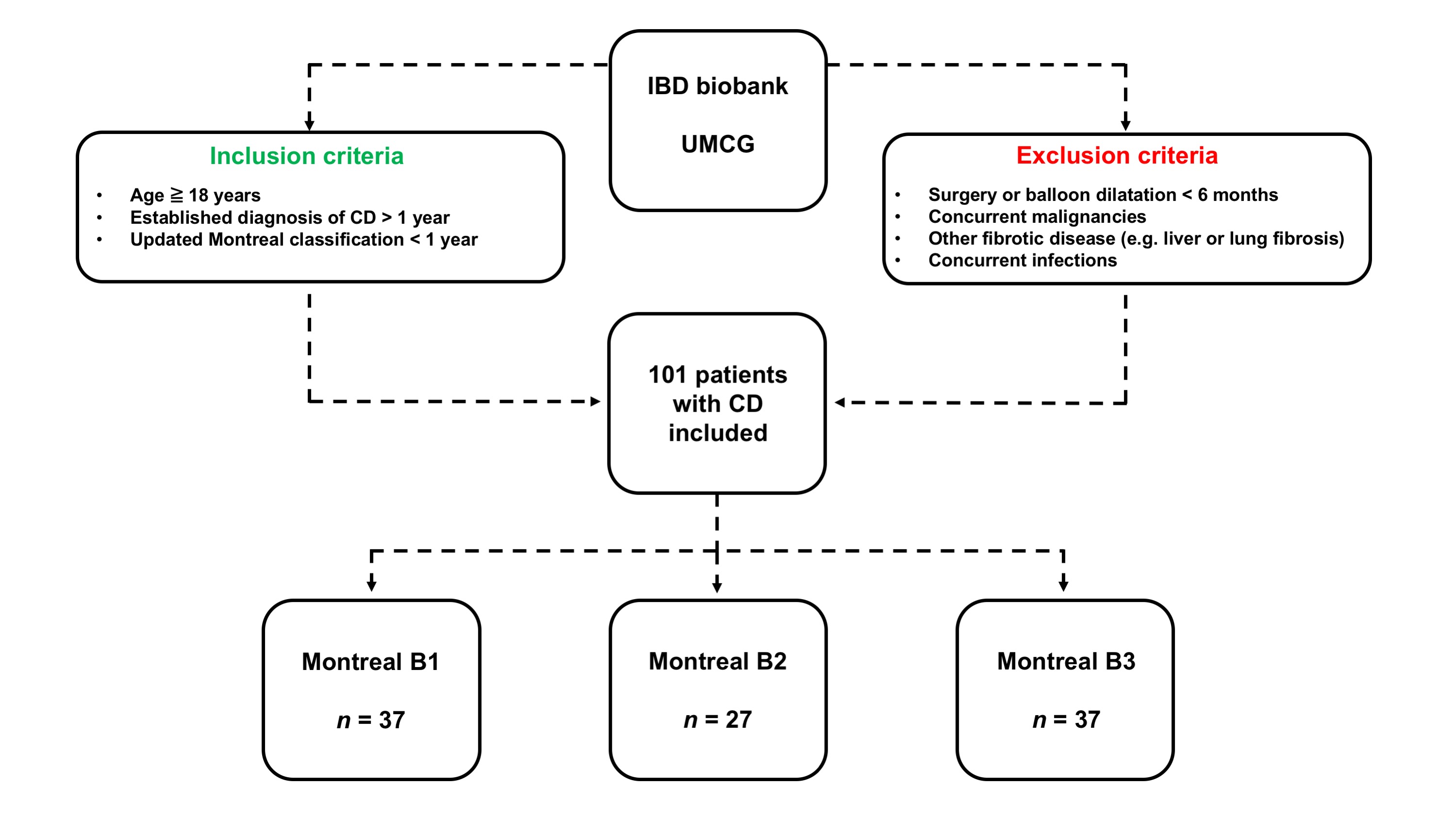
Patients were included from the IBD center database and biobank of the University Medical Center Groningen (UMCG), Groningen, the Netherlands. In total, serum samples from 101 patients with CD were collected. Samples were collected in the period from February 2011 to December 2018 and were stored at −80°C until further analysis. CD diagnosis was based on clinical, endoscopic and histological criteria. Inclusion criteria for this study were as follows: an established diagnosis of CD existing for at least 1 year, age ≥18 years, and having an updated Montreal disease classification within 1 year of serum sampling that remained stable during follow‐up. Exclusion criteria consisted of patients undergoing any surgery or endoscopic balloon dilatation <6 months before sampling, patients with concurrent malignancies (except for skin cancer and haematological malignancies), other fibrotic diseases (e.g. liver fibrosis/cirrhosis, lung fibrosis) or concurrent infections. In addition, serum samples from 96 healthy controls (HCs) were collected, which were obtained from BioIVT (Westbury, NY, USA). The study was approved by the Institutional Review Board (IRB) of the UMCG (IRB no. 08/338). All patients and HCs provided written informed consent for the use of their data and serum. The study was conducted according to the principles of the Declaration of Helsinki (2013).
2.2. Data collection
Detailed phenotypic data were collected for all patients, including age, sex, body‐mass index (BMI), smoking status, Montreal disease classification, medication use, history of bowel surgery, disease activity and standard laboratory parameters, all of which were assessed at the time of serum sampling. Clinical disease activity was established using the Harvey–Bradshaw Index (HBI). 25 The Montreal disease classification was recorded from the closest visit to the outpatient clinic at the time of sampling. In addition to clinical data, routine diagnostic laboratory parameters were collected that were measured as part of routine clinical care, including haemoglobin (Hb), C‐reactive protein (CRP), white blood cell count, platelet counts, creatinine and the estimated glomerular filtration rate (eGFR). Faecal calprotectin (FC) levels were collected from a subset of included patients for which these were available (≤90 days of sampling, n = 38) as part of routine clinical care, which were quantified by enzyme‐linked immunosorbent assays (ELISAs) (Bühlmann Laboratories AG).
2.3. Study outcomes and definitions
2.3.1. Classification of disease behaviour
Disease behaviour according to the Montreal classification was considered the primary study outcome and was classified as follows: B1: non‐stricturing, non‐penetrating disease, representing inflammatory CD without any prior stricturing or penetrating disease complications; B2: stricturing disease, representing the presence of stenosis, either asymptomatic or symptomatic, and either previous or current, as well as previous surgery because of stenosis, or postoperative stenosis in anastomosis; B3: penetrating disease, representing the history or current presence of fistulising disease, including intra‐abdominal fistulae, perforations and abscesses. Here, at baseline, the presence of perianal (P) disease was classified as a separate entity from the Montreal disease behaviour classification. Classifications were based on clinical data and objectively confirmed by retrospectively available endoscopies (images, reports), histopathological reports (e.g. from bowel resections), and radiologic information (e.g. MRI or CT images). Disease behaviour was recorded as the patients' most severe phenotype (where B1 < B2 < B3) at the time of sampling.
2.3.2. Prospective (follow‐up) outcomes and definitions
Patients were followed up from baseline until the most recent date of contact (either outpatient visit, clinical visit, endoscopic investigation or telephone appointment) with their treating gastroenterologist (as of May 4, 2021). During follow‐up, the progression or recurrence of stricturing disease, penetrating disease and the occurrence of surgical interventions was recorded. Stricturing disease by radiographic assessment was defined as an intestinal wall thickness >4 mm, or the presence of luminal narrowing or a pre‐stenotic dilation. 26 , 27 Penetrating disease was defined as a chronic tract of granulation tissue between two epithelial‐lined surfaces, which was evidently described in endoscopic, radiologic or physical examination reports. 10 No differentiation was made between fistula type (i.e. perianal, enterocutaneous, or rectovaginal). Surgical interventions consisted of all CD‐related surgeries, which were classified by indication as described in surgical reports. Indications consisted of stenosis‐ or fistula‐related surgeries or intestinal resections due to therapeutic failure.
Progression or recurrence of stricturing and/or penetrating disease was defined as either the development of new stricturing/penetrating disease (progression) or as the recurrence of active stricturing (recurring symptomatic stenosis after a period of remission or after intervention, i.e. balloon dilation or surgery) or penetrating disease (recurring fistulizing disease after a period of remission or after intervention). These definitions were based on either endoscopic evidence for progression or recurrence (based on physician's assessment, retrieved from endoscopy and physical examination reports) or radiologic evidence (i.e. by MRI or rectal endosonography).
2.4. Biomarker assays
All biomarkers analysed in this study are listed in Table 1. Neo‐epitope fragments of ECM synthesis and degradation were measured using protein fingerprint assays with solid‐phase competitive ELISAs. Assays were based on either colorimetry or chemiluminescence. Ninety‐six well plates pre‐coated with streptavidin (Roche Diagnostics, CAT no. 119‐40‐279) were coated with biotinylated peptides corresponding to each biomarker for 30 min at 20°C. Samples were diluted in assay buffer (50 mM PBS‐BTB 8 g/L NaCl, pH 7.4). Subsequently, samples were incubated with horseradish peroxidase‐conjugated target‐specific monoclonal antibodies for 1 h at 20°C or for 20 h at 4°C, depending on the specific assay, and shaken at 300 rpm. Each incubation step was followed by washing of the plates with washing buffer (25 mM TRIZMA, 50 mM NaCl, 0.036% Bronidox L5, 0.1% Tween‐20) using a standardised ELISA plate washing machine (BioTek Instruments, Microplate washer, ELx405 Select CW). For colorimetric assays, tetramethylbenzidine (TMB) (Kem‐En‐Tec, CAT no. 438 OH) was added as 100 μl per well; the plates were incubated for 15 min at room temperature and shaken again at 300 rpm. After this, 1% H2SO4 stopping buffer was added to stop the TMB reaction. An ELISA reader (VersaMAX; Molecular Devices) was applied to read optical densities at 450 and 650 nm. For chemiluminescence assays, BM Chemiluminescence ELISA Substrate (Merck, CAT no. 11582950001) was added as 100 μl per well. The plates were then shaken at 300 rpm while incubating for 3 min at 20°C. A fluorescence plate reader (Fluoroskan FL, Thermo Fisher) was applied to read light emission at 1000 ms with no filter. Finally, standard curves were created using 4‐parameter logistic models. Detection limits and detection rates of biomarkers can be found in Table S1.
TABLE 1.
Serological biomarkers of extracellular matrix turnover and intestinal inflammation
| Protein | Biomarker of degradation | Biomarker of formation | BM/IM | References |
|---|---|---|---|---|
| Type I collagen | C1M: Specific fragment of MMP‐2, ‐9, ‐13‐mediated degradation of type I collagen | IM | 13 | |
| Type III collagen | C3M: Specific fragment of MMP‐9‐mediated degradation of type III collagen | PRO‐C3: Released N‐terminal pro‐peptide of type III collagen | IM | 14, 15 |
| Type IV collagen |
C4M: Neo‐epitope generated by MMP‐2, ‐9, ‐12‐mediated degradation of type IV collagen C4G: Neo‐epitope generated by T‐cell granzyme‐B‐mediated degradation of type IV collagen |
PRO‐C4: Internal epitope in 7s domain of type IV collagen | BM | 16, 17, 18 |
| Type VI collagen | C6Ma3: MMP‐2 and ‐9‐degraded type VI collagen (alpha chain) | BM/IM | 19 |
Abbreviations: BM, basement membrane; IM, interstitial matrix; MMP, matrix metalloproteinase.
2.5. Statistical analysis
2.5.1. Descriptive and general inferential statistics
Baseline characteristics of the study population were presented as means ± standard deviations (SD), medians with interquartile ranges (IQRs) or as proportions n with corresponding percentages (%). Assessment of normality of continuous variables was performed by visual inspection of normal probability (Q–Q) plots and histograms. Differences in demographic and clinical data were compared using independent sample t‐tests, Mann–Whitney U‐tests or chi‐squared tests, depending on normality and type of variable. Serum biomarker levels were presented as median [IQR], and differences between groups were tested non‐parametrically using Kruskal–Wallis tests and Mann–Whitney U‐tests with post‐hoc Bonferroni correction for multiple comparisons, as appropriate. Correlations between different biomarkers were calculated using Spearman's rank correlation coefficients. Statistical analyses were performed using the Python programming language (v.3.8.5, Python Software Foundation, https://www.python.org), using the pandas (v.1.2.3) and numpy (v.1.20.0) packages and SPSS Statistics software package (v.25.0) (SPSS Inc.). Data visualisation was performed using seaborn (v. 0.11.1) and matplotlib (v. 3.4.1) packages in Python and GraphPad Prism (v.9.1). p‐values ≤0.05 were considered statistically significant.
2.5.2. Discrimination analyses of Montreal Behaviour subclasses
Univariable logistic regression analyses (method: enter) were performed to assess the discriminative ability of the biomarkers regarding Montreal disease classifications. Receiver operating characteristic (ROC) statistics with the area under the curve (AUC) as an overall measure of fit and corresponding 95% confidence intervals (CI) were used to assess the discriminative ability of biomarkers with regard to the outcomes. ROC curves and AUCs were computed using the non‐parametric, tie‐corrected trapezoidal approximation method. Significant results (pre‐selection threshold: nominal p ≤ 0.05) from univariable analyses were incorporated into multivariable logistic regression analyses. First, biomarker levels were adjusted for demographic or clinical characteristics by performing multivariable backward logistic regression analyses, taking into account variables that were significantly associated with the outcome of interest (derived from univariable logistic regression analyses). Second, biomarker levels were adjusted for their unstandardized residual values by performing multivariable logistic regression analysis in order to determine the predictive value of solely the biomarker. Unstandardized residual values were derived from linear regression using the same confounding factors from the first multivariate analysis in relation to the specific biomarker. The discriminative performance of adjusted models was determined by ROC estimation of combined predicted probabilities from the models with bootstrap inference (n = 500 iterations). Additionally, multivariable models were internally validated using k‐fold cross‐validation (k = 10). In this procedure, the dataset was randomly divided into k equally sized folds, where each fold was then left out (10% of cases) while the model was fitted to the remaining k‐1 folds (90% of cases, ‘training set’) and predictions were obtained for the left‐out part (‘test set’). This procedure was repeated 10 times where AUCs from each fold were averaged and bootstrapped to achieve statistical inference, resulting in a cross‐validated AUC (cv‐AUC).
2.5.3. Prospective (follow‐up) analyses
Biomarkers were analysed for associations with the risk of future progression or recurrence of stricturing or penetrating disease or the risk of surgical interventions using Kaplan–Meier survival analysis. Survival distributions were assessed for tertiles of biomarker levels, which were pairwise compared using log‐rank tests. Survival time was defined from the time of sampling (baseline) until the first date that progression was evident (by endoscopy and/or radiography) or CD‐related surgical intervention was performed, or until the last contact date with their treating gastroenterologist (end of follow‐up). Cox proportional hazards regression analyses were performed to assess the prospective associations between biomarker levels and the risk of progression or occurrence of surgical interventions. Results were expressed as hazard ratios (HRs) with corresponding 95% CIs, where biomarker levels were 2log‐transformed before entry to facilitate results interpretation (per doubling). The proportionality of hazards assumption was checked for all biomarkers to confirm the absence of violation. Multivariable Cox regression models were intended to be performed to adjust for potential confounding factors, but omitted due to a low number of occurring events. Cox regression analyses were repeated using restricted cubic splines (RCS) with three knots to evaluate non‐linearity of associations between biomarkers and progression of stricturing or penetrating disease, or the risk of surgical interventions. RCS is a flexible method to model and visualise relationships between continuous predictor variables and prospective outcomes as entered in Cox proportional hazards regression analysis. Furthermore, RCS can be used to test the hypothesis for absence of linear relationships, or summarising relationships that seem to be too non‐linear to be adequately summarised in a linear relationship. Non‐linearity in RCS was evaluated using likelihood ratio tests, where nested models were compared using linear or linear and cubic spline terms.
3. RESULTS
3.1. Characteristics of the study population
Demographic and clinical characteristics of the study population, both for the total cohort (n = 101) and separated by disease behaviour according to the Montreal classification, are presented in Table 2. A flow diagram of the study patient inclusion can be found in Figure S1. The mean age of the total cohort was 40.5 ± 14.7 years, and proportions of males and females were 37.6% and 62.4%, respectively. Patients with penetrating CD (Montreal B3) more often had perianal disease activity, which frequently co‐occurs with this type of disease behaviour (p < 0.001). Patients with stricturing CD (Montreal B2) more often underwent an ileocaecal resection compared with patients with non‐stricturing, non‐penetrating (B1) or penetrating (B3) CD (p < 0.001), and had lower CRP (p < 0.01), platelet counts (p < 0.05) and FC levels (the latter only in a subset of patients, n = 38) compared with patients categorised as Montreal B1 or B3. Finally, patients with stricturing and penetrating disease more often used immunosuppressive drugs compared with non‐stricturing, non‐penetrating CD (p < 0.05). The remaining demographic and clinical characteristics were not significantly different between the Montreal classification subgroups.
TABLE 2.
Cohort demographic and clinical characteristics, separated by disease behaviour according to the Montreal classification
| Total (n = 101) | Montreal B1 (n = 37) | Montreal B2 (n = 27) | Montreal B3 (n = 37) | HC (n = 96) | p‐value | |
|---|---|---|---|---|---|---|
| Age (years) | 40.5 ± 14.7 | 38.5 ± 14.1 | 45.8 ± 18.1 | 38.6 ± 11.7 | 48.9 ± 12.2 | 0.090 |
| Sex, n (%) | ||||||
| Male | 38 (37.6) | 9 (24.3) | 13 (48.1) | 16 (43.2) | 56 (58.3) | 0.102 |
| Female | 63 (62.4) | 28 (75.7) | 14 (51.9) | 21 (56.8) | 40 (41.7) | |
| BMI (kg/m2) | 25.1 ± 5.3 | 26.6 ± 6.8 | 23.4 ± 4.7 | 24.9 ± 3.4 | — | 0.055 |
| Smoking, n (%) | ||||||
| No | 35 (34.7) | 16 (43.2) | 3 (11.1) | 16 (43.2) | 0.055 | |
| Previous | 31 (30.7) | 9 (24.3) | 12 (44.4) | 10 (27.0) | ||
| Current | 35 (34.7) | 12 (32.4) | 12 (44.4) | 11 (29.7) | ||
| Montreal classification | ||||||
| Montreal age (A) | ||||||
| A1 (≤16 years) | 16 (15.8) | 7 (18.9) | 6 (22.2) | 3 (8.1) | 0.164 | |
| A2 (17–40 years) | 67 (66.3) | 23 (62.2) | 14 (51.9) | 30 (81.1) | ||
| A3 (>40 years) | 18 (17.8) | 7 (18.9) | 7 (25.9) | 4 (10.8) | ||
| Montreal location (L), CD | ||||||
| L1 (ileal disease) | 31 (30.7) | 9 (24.3) | 14 (51.9) | 8 (21.6) | 0.221 | |
| L2 (colonic disease) | 15 (14.9) | 6 (16.2) | 2 (7.4) | 7 (18.9) | ||
| L3 (ileocolonic disease) | 54 (53.5) | 21 (56.8) | 11 (40.7) | 22 (59.5) | ||
| L4 (upper GI disease) | 7 (6.9) | 5 (13.5) | 0 (0.0) | 2 (5.4) | ||
| Montreal perianal disease (P), CD | 31 (30.7) | 3 (8.1) | 4 (14.8) | 24 (64.9) | — | <0.001 |
| Medicationuse, n (%) | Total | Montreal B1 | Montreal B2 | Montreal B3 | HC | p‐value |
|---|---|---|---|---|---|---|
| Aminosalicylates | 7 (6.9) | 4 (10.8) | 2 (7.4) | 1 (2.7) | 0.387 | |
| Steroids | 37 (36.6) | 17 (45.9) | 10 (37.0) | 10 (27.0) | 0.240 | |
| Immunosuppressives | 65 (64.4) | 18 (48.6) | 20 (74.1) | 27 (73.0) | 0.043 | |
| Prior anti‐TNF‐α | 43 (42.6) | 15 (40.5) | 15 (55.6) | 13 (35.1) | 0.346 | |
| Surgical history | ||||||
| Ileocaecal resection, n (%) | 38 (37.6) | 4 (10.8) | 19 (70.4) | 15 (40.5) | <0.001 | |
| Colon resection (or partial), n (%) | 7 (6.9) | 1 (2.7) | 1 (3.7) | 5 (13.5) | 0.139 | |
| Clinical disease activity score a | Total (n = 68) | Montreal B1 (n = 28) | Montreal B2 (n = 22) | Montreal B3 (n = 18) | HC | p‐value |
| HBI | ||||||
| Remission (<5) | 24 (35.3) | 12 (42.9) | 6 (27.3) | 6 (33.3) | 0.638 | |
| Mild disease (5–7) | 17 (25.0) | 5 (17.9) | 8 (36.4) | 4 (22.2 | ||
| Moderate disease (8–16) | 24 (35.3) | 9 (32.1) | 8 (36.4) | 7 (38.9) | ||
| Severe disease (>16) | 3 (4.4) | 2 (7.1) | 0 (0.0) | 1 (5.6) | ||
| Laboratory parameters | ||||||
| Haemoglobin (mmol/L) | 7.7 ± 0.9 | 7.8 ± 1.0 | 7.9 ± 0.9 | 7.6 ± 0.9 | 0.349 | |
| CRP (mg/L) | 5.0 [2.1;14.8] | 9.6 [4.2;17.8] | 2.0 [1.0;5.7] | 6.2 [3.2;15.5] | 0.003 | |
| WBC (×109/L) | 7.1 [6.9;9.8] | 8.5 [6.5;11.3] | 6.4 [4.9;9.2] | 6.8 [6.0;9.1] | 0.085 | |
| Platelets (×109/L) | 334 ± 102 | 366 ± 107 | 289 ± 82 | 337 ± 99 | 0.010 | |
| eGFR (ml/min/1.73m2) | 106 ± 23 | 109 ± 20 | 108 ± 30 | 102 ± 20 | 0.400 | |
| Creatinine (μmol/L) | 66.2 ± 15.1 | 63.1 ± 13.7 | 66.4 ± 18.1 | 69.1 ± 13.8 | 0.238 | |
| Faecal calprotectin (μg/g) b | 1105 [553; 2185] | 1895 [1170; 2473] | 625 [425; 805] | 865 [400; 1490] | 0.010 | |
Note: Data are presented as proportions n with corresponding percentages (%), means ± standard deviation (SD) or medians [interquartile range, IQR] in case of continuous variables. p‐values <0.05 were considered statistically significant and are indicated in bold.
Abbreviations: BMI, body mass index; CD, Crohn's disease; CRP, C‐reactive protein; eGFR, estimated glomerular filtration rate; HBI, Harvey–Bradshaw Index; HC, healthy control; TNF‐α, tumour necrosis factor alpha; WBC, white blood cell count.
Clinical disease activity scores (HBI) were available for n = 68 patients.
Faecal calprotectin levels at baseline were available for n = 38 patients (B1: n = 16; B2: n = 11; B3: n = 11).
3.2. CD is characterised by distinct biomarker signatures of type I, III, IV and VI collagen formation and degradation
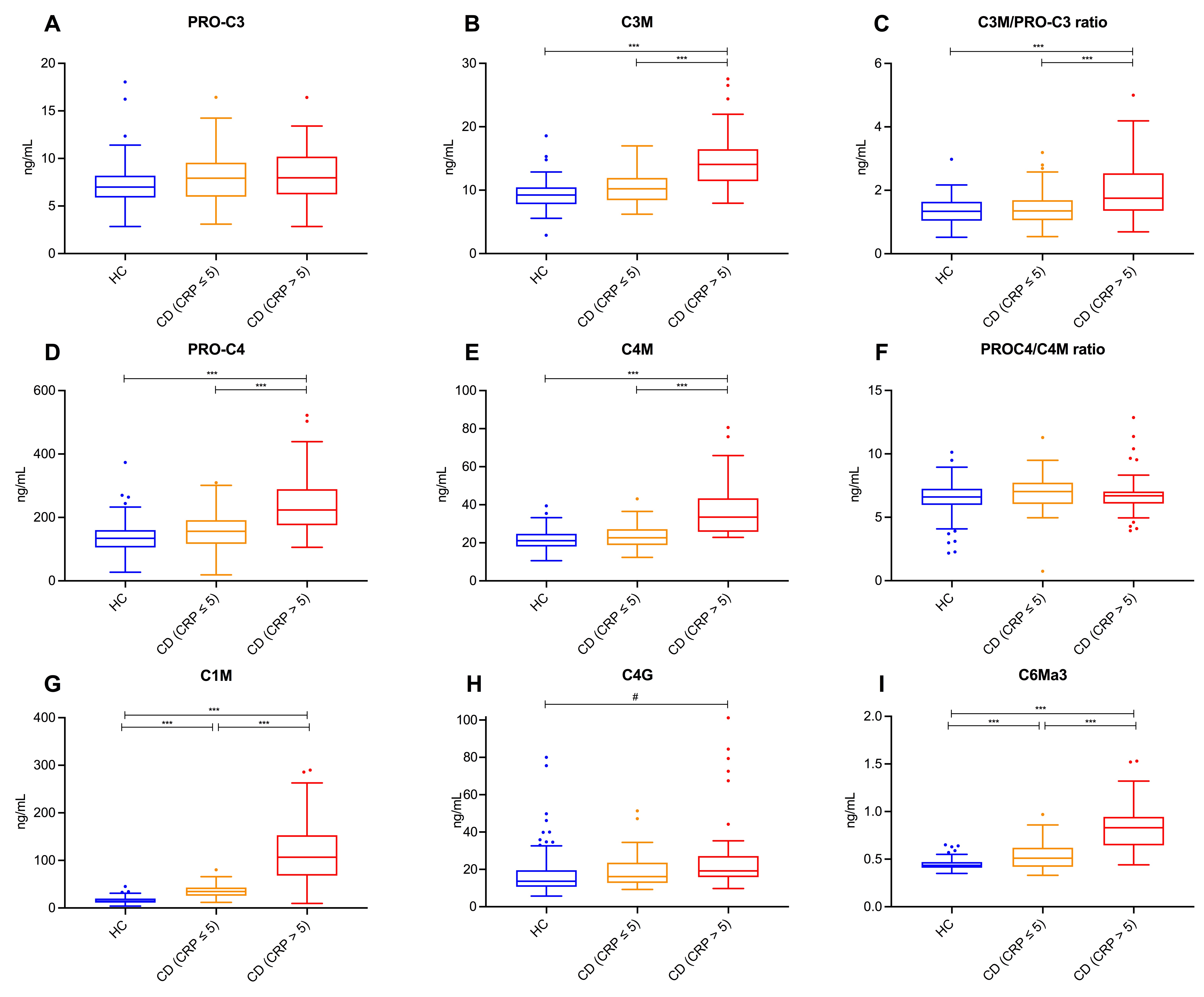
Serum concentrations of type I, III, IV and VI collagen formation and degradation biomarkers in patients with CD and in HCs are presented in Table S2 and separated out according to Montreal classification in Figure 2. Serum levels of C1M and C6Ma3 were substantially elevated in patients with CD compared with HCs (C1M: 50.6 [33.9;105.7] vs 14.3 [11.4;19.9] ng/ml; C6Ma3: 0.62[0.49;0.84] vs 0.44[0.41;0.47] ng/ml, both p < 0.001, Table S2). No significant differences in serum C1M (after adjustment for multiple comparisons) and C6Ma3 levels were observed between disease behaviour subclasses (i.e., without considering HC), although patients with stricturing disease showed a trend towards lower levels than the remaining patients. Serum C3M levels were significantly elevated in patients with CD compared with HCs (median [IQR] 11.8 [9.7;14.6] vs 9.2 [7.8;10.4] ng/ml, p < 0.001), indicating relatively increased degradation of type III collagen, whereas PRO‐C3 levels were not significantly different between groups (nominal p‐value: 0.029) (Table S2). Taken together, the balance between type III collagen formation and degradation (C3M/PRO‐C3 ratio) was higher in patients with non‐stricturing, non‐penetrating CD (Montreal B1) compared with HCs (albeit only nominally significant, p = 0.010; Table S2). Serum PRO‐C4 and C4M levels were higher in patients with CD than in HCs (183.3 [144.7; 239.1] vs 134.2 [105.2160.1] ng/ml and 26.3[22.6, 34.5] vs 21.1[18.1, 24.7] ng/ml, both p < 0.001) (Table S2). However, the balance between type IV collagen formation and degradation (PRO‐C4/C4M ratio) was not significantly elevated in patients with stricturing CD (Montreal B2), compared with HCs or patients with CD having non‐stricturing, non‐penetrating disease (B1) and penetrating disease (B3) (p = 0.153 and p = 0.009, respectively). Serum C4G levels were higher in patients with penetrating CD compared with HCs (18.0 [14.0, 26.3] vs 13.7[10.7, 19.6] ng/ml, respectively, overall p = 0.006), indicating net increased T‐cell granzyme‐B‐mediated degradation of type IV collagen, although there was no statistically significant difference between disease behaviour phenotypes.
FIGURE 2.
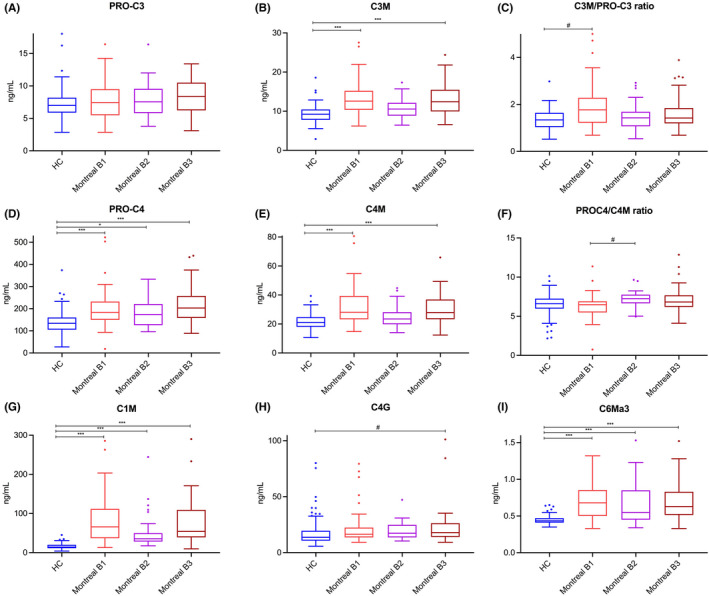
(A–I) Serum concentrations and ratios of type I (C1M), III (C3M, PRO‐C3, C3M/PRO‐C3), IV (C4M, PRO‐C4, C4G, PRO‐C4/C4M) and VI (C6Ma3) collagen formation and degradation biomarkers in patients with CD (n = 101), stratified by disease behaviour, and in healthy controls (HC, n = 96). (A–C) Serum C3M levels were significantly elevated in patients with CD compared with HC, indicating relatively increased degradation of type III collagen, whereas PRO‐C3 levels were equal among groups, resulting in a moderately elevated C3M/PRO‐C3 ratio in patients with CD compared with controls (especially in patients with non‐stricturing, non‐penetrating disease), which was however only nominally statistically significant. (D–F) Serum PRO‐C4 and C4M levels were elevated in patients with CD compared with HC, but the PRO‐C4/C4M ratio was nominally significantly elevated in patients with stricturing disease, compared with both HC and patients with non‐stricturing, non‐penetrating disease. (G) A specific fragment of MMP‐2, 9, 13‐mediated type I collagen degradation (C1M) was markedly elevated in patients with CD compared with HC. (H) Serum C4G levels were nominally significantly elevated in patients with CD, particularly in patients with penetrating CD, compared with HC. (I) Serum C6Ma3 levels were elevated in patients with CD compared with HC. Boxplots were drawn according to the Tukey method, with inner fences defined as 25th/75th percentile ±1.5 IQR. Significances were calculated from Kruskal‐Wallis tests with post‐hoc Bonferroni correction for multiple comparisons. *p < 0.05; **p < 0.01; ***p < 0.001. #Only nominally significant, but not statistically significant after Bonferroni correction for multiple comparisons.
3.3. Type I, III and IV collagen degradation fragments are decreased in patients with stricturing (Montreal B2) CD and accurately differentiate them from both non‐stricturing, non‐penetrating (Montreal B1) and penetrating (Montreal B3) Crohn’s disease
Subsequently, the ability of serum biomarkers to differentiate between Montreal disease behaviour subclasses was determined using ROC statistics and logistic regression modelling (Table 3, Figures 3 and 4, Tables S3 and S4). Unadjusted analyses revealed that serum C1M, C3M and C4M levels accurately discriminated between patients with non‐stricturing, non‐penetrating disease and stricturing disease (AUC with 95% CI: C1M 0.69 [0.56–0.83], p < 0.01; C3M 0.66 [0.53–0.80], p < 0.05; C4M 0.68 [0.55–0.82], p < 0.05) (Figure 3a–c, Table S3). Using multivariable (backwards) logistic regression analyses, allowing adjustment for confounders (history of ileocaecal resection, concurrent use of immunosuppressive drugs, and platelet counts), all biomarkers retained their ability to differentiate between non‐stricturing, non‐penetrating and stricturing CD (Figure 3e–g,i–k). In these analyses, CRP dropped out as a non‐significant confounding factor, but to double‐check whether these results remained robust while adjusting for CRP, analyses were repeated accordingly, which demonstrated comparable results (Table S5). When combining the biomarkers with their residual values (derived from linear regression, corrected for significant confounders), the discriminative ability of C3M was superior in differentiating between non‐stricturing, non‐penetrating and stricturing CD (AUC 0.91[0.83–0.98], p < 0.001), followed by serum C4M levels (AUC 0.87 [0.79–0.96], p < 0.001) and C1M levels (AUC 0.78[0.67–0.98], p < 0.001) (Table S4).
TABLE 3.
The discriminative ability of serological biomarkers with regard to disease behaviour subtypes in patients with Crohn's disease
| Biomarker | Unadjusted | Adjusted (full model) a | Adjusted (residual marker value) b | |||
|---|---|---|---|---|---|---|
| Non‐penetrating, non‐stricturing CD (Montreal B1) versus stricturing CD (Montreal B2) | ||||||
| AUC (95% CI) | Nominal p‐value | AUC (95% CI) | p‐value | AUC (95% CI) | p‐value | |
| C1M | 0.69 (0.56–0.83) | 0.009 | 0.91 (0.84–0.98) | <0.001 | 0.78 (0.67–0.89) | <0.001 |
| C3M | 0.66 (0.53–0.80) | 0.028 | 0.92 (0.84–0.99) | <0.001 | 0.91 (0.83–0.98) | <0.001 |
| PRO‐C3 | 0.52 (0.38–0.67) | 0.760 | ||||
| C3M/PRO‐C3 | 0.61 (0.47–0.75) | 0.126 | ||||
| C4M | 0.68 (0.55–0.82) | 0.013 | 0.92 (0.85–0.99) | <0.001 | 0.87 (0.79–0.96) | <0.001 |
| PRO‐C4 | 0.57 (0.42–0.71) | 0.359 | ||||
| PRO‐C4/C4M | 0.71 (0.57–0.84) | 0.005 | 0.92 (0.85–0.99) | <0.001 | 0.90 (0.82–0.98) | <0.001 |
| C4G | 0.50 (0.36–0.65) | 0.962 | ||||
| C6Ma3 | 0.59 (0.44–0.73) | 0.234 | ||||
| Biomarker | Unadjusted | Adjusted (full model) b | Adjusted (residual marker value) b | |||
|---|---|---|---|---|---|---|
| Non‐penetrating, non‐stricturing CD (Montreal B1) versus penetrating CD (Montreal B3) | ||||||
| AUC (95% CI) | Nominal p‐value | AUC (95% CI) | p‐value | AUC (95% CI) | p‐value | |
| C1M | 0.51 (0.38–0.64) | 0.877 | ||||
| C3M | 0.52 (0.39–0.65) | 0.775 | ||||
| PRO‐C3 | 0.58 (0.45–0.71) | 0.232 | ||||
| C3M/PRO‐C3 | 0.58 (0.45–0.71) | 0.236 | ||||
| C4M | 0.52 (0.39–0.65) | 0.791 | ||||
| PRO‐C4 | 0.56 (0.42–0.69) | 0.414 | ||||
| PRO‐C4/C4M | 0.64 (0.51–0.76) | 0.043 | 0.87 (0.79–0.96) | <0.001 | 0.87 (0.79–0.96) | <0.001 |
| C4G | 0.55 (0.42–0.69) | 0.433 | ||||
| C6Ma3 | 0.51 (0.37–0.64) | 0.935 | ||||
| Biomarker | Unadjusted | Adjusted (full model) d | Adjusted (residual marker value) b | |||
|---|---|---|---|---|---|---|
| Stricturing CD (Montreal B2) versus penetrating CD (Montreal B3) | ||||||
| AUC (95% CI) | Nominal p‐value | AUC (95% CI) | p‐value | AUC (95% CI) | p‐value | |
| C1M | 0.70 (0.56–0.83) | 0.008 | 0.81 (0.70–0.92) | <0.001 | 0.81 (0.70–0.92) | <0.001 |
| C3M | 0.65 (0.51–0.78) | 0.048 | 0.80 (0.68–0.91) | <0.001 | 0.80 (0.68–0.91) | <0.001 |
| PRO‐C3 | 0.58 (0.43–0.72) | 0.289 | ||||
| C3M/PRO‐C3 | 0.54 (0.39–0.68) | 0.610 | ||||
| C4M | 0.67 (0.53–0.80) | 0.023 | 0.81 (0.70–0.92) | <0.001 | 0.81 (0.70–0.92) | <0.001 |
| PRO‐C4 | 0.61 (0.47–0.75) | 0.137 | ||||
| PRO‐C4/C4M | 0.59 (0.44–0.73) | 0.251 | ||||
| C4G | 0.56 (0.42–0.70) | 0.411 | ||||
| C6Ma3 | 0.59 (0.45–0.74) | 0.204 | ||||
Adjusted for history of ileocecal resection, concurrent use of immunosuppressives and platelet counts (derived from multivariable backwards logistic regression analysis containing these confounding factors).
Adjusted for unstandardized residual value of the biomarker (derived from linear regression analysis, adjusted for the same confounders) to the determine the prognostic value of the biomarker only.
Adjusted for perianal disease and history of ileocaecal resection.
Adjusted for perianal disease.
FIGURE 3.
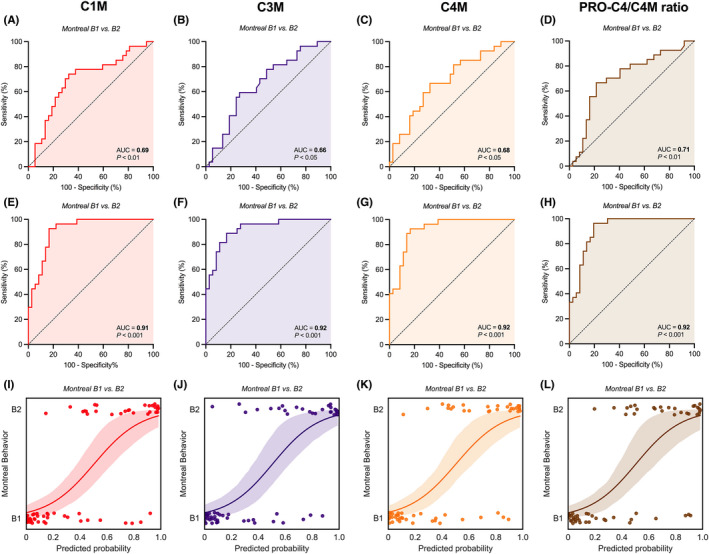
(A–L) Capacity of serological biomarkers of type I, III and IV collagen degradation (C1M, C3M and C4M, respectively) and the type IV collagen formation/degradation ratio (PRO‐C4/C4M ratio) to discriminate between non‐stricturing, non‐penetrating (Montreal B1) and stricturing (Montreal B2) CD. Unadjusted (A–D) and adjusted (E–H) ROC curves demonstrate significant discriminative capacity of serum C1M, C3M and C4M levels and the PRO‐C4/C4M ratio with regard to non‐stricturing, non‐penetrating disease (B1) versus stricturing disease (B2). Predicted probabilities (I–L) derived from the multivariable logistic regression models, representing the odds of having stricturing (Montreal B2) CD and determining the course of the ROC curves as shown in panels (E–H), are substantially separated between both disease behaviour subtypes. The lines with associated 95% confidence intervals (colour shade) represent the fitted logistic regression lines. Abbreviation: AUC, area under the ROC curve.
FIGURE 4.
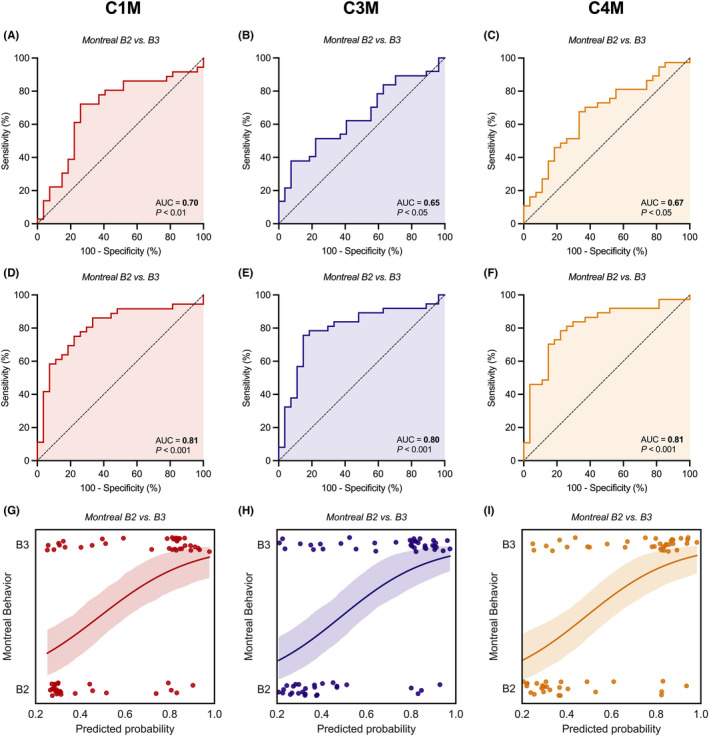
(A–I) Capacity of serological biomarkers of type I, III and IV collagen degradation (C1M, C3M and C4M, respectively) to discriminate between stricturing (Montreal B2) and penetrating (Montreal B3) CD. Unadjusted (A–C) and adjusted (D–F) ROC curves demonstrate significant discriminative capacity of serum C1M, C3M, and C4M levels with regard to stricturing (B2) vs penetrating disease (B3). Predicted probabilities (G–I) derived from the multivariable logistic regression models, representing the odds of having penetrating (Montreal B3) CD and determining the course of ROC curves as shown in panels D‐F are well separated between both disease behaviour subtypes. The lines with associated 95% confidence intervals (colour shade) represent the fitted logistic regression lines. Abbreviation: AUC, area under the ROC curve.
Similarly, serum C1M, C3M and C4M levels demonstrated moderately accurate discriminative ability between stricturing and penetrating CD (Figure 4, Tables S3 and S4). Unadjusted analyses showed that C1M levels were best to discriminate between stricturing and penetrating disease (AUC 0.70 [0.56–0.83, p < 0.01), followed by C4M (AUC: 0.67 [0.53–0.80], p < 0.05), and C3M (AUC 0.65 [0.51–0.78], p < 0.05) (Figure 4a–c). Multivariable (backwards) logistic regression analyses, with adjustment for the presence of perianal disease as a relevant confounder, demonstrated preserved discriminative capacity of these biomarkers (Figure 4d–i). Residual‐corrected biomarker values revealed that C1M was superior in differentiating stricturing from penetrating CD (AUC 0.81 [0.70–0.92], p < 0.001), closely followed by C4M (AUC 0.81 [0.70–0.92], p < 0.001), and C3M (AUC 0.80 [0.68–0.91], p < 0.001) (Table S4).
3.4. Type IV collagen formation/degradation (PRO‐C4/C4M) ratios are lower in patients with non‐stricturing, non‐penetrating (Montreal B1) Crohn’s disease and accurately differentiate them from stricturing (B2) and penetrating (B3) disease
The type IV collagen formation/degradation (PRO‐C4/C4M) ratio moderately accurately discriminated patients with non‐stricturing, non‐penetrating (B1) CD from stricturing CD (AUC, 95% CI: 0.71[0.57–0.84], p < 0.01, Figure 3d) and penetrating CD (AUC, 95% CI: 0.64 [0.51;0.76], p < 0.05) (Table S3). Multivariable analyses showed preserved discriminative accuracy (Figure 3h,l), and residual‐corrected values demonstrated high discriminative accuracy (B1 vs B2: AUC 0.90[0.82–0.98], p < 0.001; B1 vs B3: AUC 0.87 [0.79–0.96], p < 0.001) (Table S4).
3.5. Active inflammation associates with elevated serum levels of fragments of type I, III, IV and VI collagen formation and degradation
As a next step, relationships between biomarker levels and disease activity were explored by examining associations with CRP and faecal calprotectin (FCal) levels (Figure 5, Tables S6 and S7, Figure S2, Results S1). Most biomarkers strongly associated with serum CRP levels, including C1M (ρ = 0.89, p < 0.001), C6Ma3 (ρ = 0.70, p < 0.001), C4M (ρ = 0.67, p < 0.001), C3M (ρ = 0.59, p < 0.001) and PRO‐C4 (ρ = 0.52, p < 0.001) (Figure 5a). In a subset of patients of whom FC data were available (n = 38), FC levels correlated with C6Ma3 levels (ρ = 0.48, p < 0.01), followed by C1M (ρ = 0.39, p < 0.05), C4M (ρ = 0.36, p < 0.05) and C3M levels (ρ = 0.34, p < 0.05). Importantly, the strong positive correlations between serum C1M, C3M and C4M levels and serum CRP levels were independent of disease behaviour (Figure 5b–d). Conversely, differences in biomarker levels between the Montreal behaviour subclasses were independent of active inflammation because CRP did not correlate with disease behaviour according to the Montreal classification (Spearman's ρ = −0.04, p = 0.666). Although patients with stricturing (Montreal B2) CD did demonstrate relatively lower CRP levels (Table 2), this difference was not statistically relevant as CRP did not emerge as a significant confounder in multivariable logistic regression analyses discriminating patients with non‐stricturing, non‐penetrating CD (B1) from patients with stricturing CD (B2) (Table 3).
FIGURE 5.
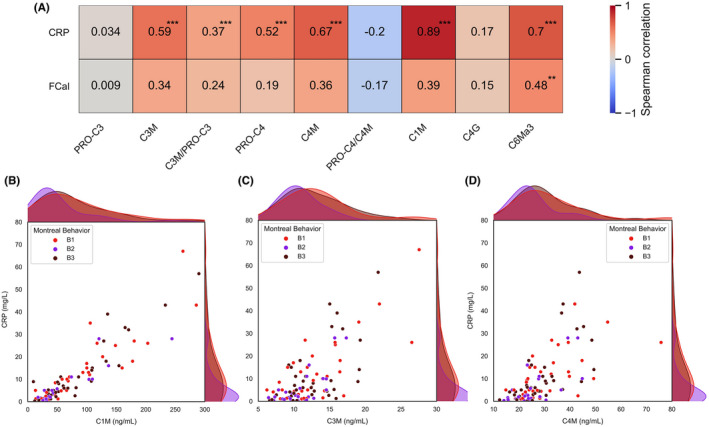
(A–D) Associations between biomarkers and biochemical disease activity (CRP and FCal). (A) Heatmap demonstrating strength and significance of associations between biomarkers and disease activity measures. (B–D) Scatterplots with marginal distributions plotted as kernel density estimates demonstrating associations between serum CRP levels and serum C1M, C3M and C4M levels, respectively, labelled by disease behaviour subtype. Abbreviations: CRP, C‐reactive protein; FCal, faecal calprotectin. **p < 0.01; ***p < 0.001.
3.6. Type I and IV collagen fragments modestly associate with the risk of progression of penetrating Crohn’s disease
Mean follow‐up of patients was 4.5 ± 2.2 years, during which 23 (22.8%) patients showed either progression or recurrence of stricturing disease, 22 (21.8%) patients showed either progression or recurrence of penetrating disease and 22 (21.8%) patients underwent CD‐related surgical interventions (Table S8). Cox proportional hazards regression analyses did not demonstrate statistically significant associations between the biomarkers and all outcomes, but solely demonstrated nominally significant univariable associations between serum C1M, PRO‐C4, C4G and the PRO‐C4/C4M ratio and the risk of progression or recurrence of penetrating disease, either as (2log‐transformed) continuous predictor (PRO‐C4, PRO‐C4/C4M ratio), as a categorical predictor (by tertile division) (C1M) or both (C4G) (Table 4). No significant associations were observed between biomarkers and the risk of progression or recurrence of stricturing disease or the risk of future surgical interventions. Multivariable Cox proportional hazards regression analyses were not performed due to the small number of events, resulting in limited statistical power. Kaplan–Meier survival analyses demonstrated only nominally significantly different survival distributions for tertiles of serum C1M and C4G levels among patients with and without progression or recurrence of penetrating disease during follow‐up (p = 0.035 and p = 0.008, respectively, log‐rank test) (Figure 6a,b). In contrast, tertiles of serum PRO‐C4 levels and the PRO‐C4/C4M ratio did not show differential survival distributions (Figure 6c,d). To assess the shape and (non‐)linearity of these associations, RCS with three knots were fitted, which showed no significant deviances from linear associations with the risk of progression or recurrence of penetrating disease (C1M: χ2 = 3.07, p = 0.08; C4G: χ2 = 0.59, p = 0.44; PRO‐C4: χ2 = 0.65, p = 0.42; PRO‐C4/C4M ratio: χ2 = 2.00, p = 0.16) (Figure S3).
TABLE 4.
Cox proportional hazards regression analyses of associations between (2log‐transformed) serum biomarker levels and the risk of (A) progression or recurrence of stricturing disease, (B) progression or recurrence of penetrating disease and (C) CD‐related surgical interventions
| HR per doubling a (95% CI) | Tertiles per biomarker b | |||
|---|---|---|---|---|
| Tertile 1 (low) | Tertile 2 (mid) | Tertile 3 (high) | ||
| A. Stricturing disease | ||||
| C1M | 1.17 (0.79–1.73), p = 0.424 | 0.77 (0.30–1.99), p = 0.768 | 0.51 (0.18–1.44), p = 0.511 | 1.00 (reference) |
| C3M | 0.76 (0.29–1.95), p = 0.563 | 1.06 (0.40–2.85), p = 0.906 | 0.96 (0.34–2.75), p = 0.944 | 1.00 (reference) |
| PRO‐C3 | 0.66 (0.30–1.45), p = 0.301 | 1.43 (0.53–3.85), p = 0.478 | 0.90 (0.31–2.56), p = 0.836 | 1.00 (reference) |
| C3M/PRO‐C3 | 1.16 (0.59–2.30), p = 0.669 | 0.77 (0.30–2.00), p = 0.769 | 0.54 (0.19–1.53), p = 0.542 | 1.00 (reference) |
| C4M | 0.85 (0.36–1.97), p = 0.699 | 1.32 (0.51–3.42), p = 0.562 | 0.63 (0.20–1.99), p = 0.630 | 1.00 (reference) |
| PRO‐C4 | 1.11 (0.56–2.18), p = 0.764 | 0.53 (0.20–1.38), p = 0.193 | 0.54 (0.20–1.49), p = 0.236 | 1.00 (reference) |
| PRO‐C4/C4M | 1.78 (0.53–5.99), p = 0.352 | 0.37 (0.13–1.06), p = 0.063 | 0.54 (0.21–1.40), p = 0.207 | 1.00 (reference) |
| C4G | 1.08 (0.61–1.89), p = 0.800 | 0.86 (0.36–2.07), p = 0.739 | 0.26 (0.07–1.03), p = 0.065 | 1.00 (reference) |
| C6Ma3 | 2.01 (0.89–4.57), p = 0.095 | 0.61 (0.21–1.76), p = 0.360 | 1.19 (0.46–3.09), p = 0.720 | 1.00 (reference) |
| B. Penetrating disease | ||||
| C1M | 1.33 (0.90–1.97), p = 0.154 | 1.00 (reference) | 5.91 (1.31–26.7), p = 0.021 | 4.73 (1.00–22.3), p = 0.049 |
| C3M | 2.18 (0.87–5.47), p = 0.098 | 1.00 (reference) | 0.51 (0.15–1.68), p = 0.265 | 1.48 (0.58–3.77), p = 0.411 |
| PRO‐C3 | 1.16 (0.50–2.69), p = 0.723 | 1.00 (reference) | 0.75 (0.25–2.25), p = 0.609 | 1.21 (0.45–3.26), p = 0.707 |
| C3M/PRO‐C3 | 1.33 (0.69–2.53), p = 0.393 | 1.00 (reference) | 1.17 (0.39–3.49), p = 0.777 | 1.74 (0.62–4.93), p = 0.296 |
| C4M | 1.53 (0.69–3.39), p = 0.297 | 1.00 (reference) | 1.52 (0.53–4.38), p = 0.440 | 1.64 (0.57–4.74), p = 0.365 |
| PRO‐C4 | 2.24 (1.07–4.71), p = 0.033 | 1.00 (reference) | 1.91 (0.62–5.89), p = 0.258 | 2.10 (0.70–6.28), p = 0.184 |
| PRO‐C4/C4M | 4.81 (1.13–20.4), p = 0.033 | 1.00 (reference) | 1.58 (0.52–4.85), p = 0.420 | 1.86 (0.62–5.55), p = 0.266 |
| C4G | 1.71 (1.05–2.81), p = 0.032 | 1.00 (reference) | 0.53 (0.13–2.11), p = 0.366 | 2.66 (1.01–7.01), p = 0.048 |
| C6Ma3 | 2.00 (0.87–4.62), p = 0.104 | 1.00 (reference) | 3.03 (0.95–9.67), p = 0.062 | 2.30 (0.69–7.64), p = 0.175 |
| C. Surgical interventionsc | ||||
| C1M | 1.39 (0.94–2.06), p = 0.099 | 1.00 (reference) | 2.14 (0.66–6.98), p = 0.208 | 3.13 (0.96–10.2), p = 0.059 |
| C3M | 1.24 (0.49–3.12), p = 0.648 | 1.00 (reference) | 0.48 (0.15–1.57), p = 0.226 | 1.19 (0.47–3.01), p = 0.714 |
| PRO‐C3 | 1.01 (0.42–2.44), p = 0.975 | 1.00 (reference) | 0.50 (0.16–1.53), p = 0.223 | 1.05 (0.40–2.72), p = 0.922 |
| C3M/PRO‐C3 | 1.12 (0.56–2.25), p = 0.752 | 1.00 (reference) | 0.70 (0.24–2.01), p = 0.504 | 1.13 (0.42–3.04), p = 0.806 |
| C4M | 1.07 (0.47–2.39), p = 0.879 | 1.00 (reference) | 1.01 (0.36–2.78), p = 0.993 | 1.10 (0.40–3.06), p = 0.854 |
| PRO‐C4 | 1.41 (0.70–2.87), p = 0.340 | 1.00 (reference) | 1.67 (0.58–4.86), p = 0.345 | 1.75 (0.60–5.08), p = 0.304 |
| PRO‐C4/C4M | 2.76 (0.64–11.9), p = 0.173 | 1.00 (reference) | 1.18 (0.40–3.51), p = 0.767 | 1.64 (0.58–4.61), p = 0.352 |
| C4G | 1.00 (0.55–1.80), p = 0.990 | 1.00 (reference) | 0.23 (0.05–1.05), p = 0.058 | 1.46 (0.61–3.54), p = 0.398 |
| C6Ma3 | 2.08 (0.87–5.00), p = 0.101 | 1.00 (reference) | 2.61 (0.88–7.67), p = 0.082 | 1.80 (0.57–5.71), p = 0.320 |
Abbreviations: CI, confidence interval; HR, hazard ratio; MV, multivariable analysis; UV, univariable analysis.
Biomarker levels were 2log‐transformed before entry into the model as a continuous predictor, facilitating results interpretation (per doubling).
Per biomarker, levels were divided into tertiles with the lowest tertile (Tertile 1) set as a reference standard in the model.
FIGURE 6.
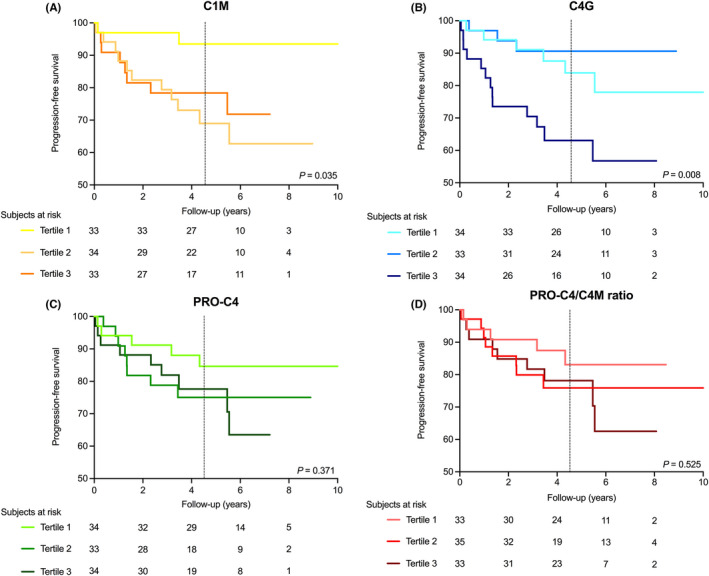
(A–D) Kaplan–Meier survival distributions for tertiles of the biomarkers that showed significant associations with the risk of progression or recurrence of penetrating disease in Cox proportional hazards regression analyses, either continuously or by tertile division. (A) Kaplan–Meier curves representing progression‐free survival for tertiles of serum C1M levels, with the lowest rate of progression occurring in the lowest tertile (p < 0.05, log‐rank test). (B) Kaplan–Meier curves for tertiles of serum C4G levels, with the highest rate of progression occurring in the highest tertile (p < 0.01, log‐rank test). (C, D) Kaplan–Meier curves for tertiles of serum PRO‐C4 levels and the PRO‐C4/C4M ratio, showing no significant curve deviations (p = 0.37 and p = 0.53, respectively). The black dashed vertical lines indicate the median survival time (4.5 years, IQR: [3.0, 6.1]).
4. DISCUSSION
This study demonstrates that CD is characterised by distinct biomarker signatures of collagen turnover, as represented by altered serum levels of fragments of type I (C1M), III (PRO‐C3, C3M), IV (PRO‐C4, C4M, C4G) and VI (C6Ma3) collagen formation and degradation in comparison to healthy individuals. The collagen degradation fragments C1M, C3M, C4M, and the type IV collagen formation/degradation ratio (PRO‐C4/C4M ratio) were able to accurately distinguish between CD disease behaviour subtypes as classified by the Montreal disease classification. Notably, baseline levels of type I and particularly type IV collagen fragments were prospectively associated with an increased risk of penetrating disease progression, as evidenced by endoscopy and/or imaging during follow‐up. Collectively, these results demonstrate that circulating fragments of collagen turnover hold promise as serological biomarkers for detection of disease behaviour as well as prediction of future progression of penetrating CD.
In the present study, we observed markedly elevated levels of C1M and especially C6Ma3 in patients with CD compared with healthy individuals. C6Ma3 reflects the MMP‐2 and MMP‐9‐mediated degradation of the α3 chain of type VI collagen, which is involved in intestinal tissue remodelling in CD. Type VI collagen is mainly located at the BM across the intestinal crypt‐villus axis and regulates the formation of fibronectin fibrils and the morphology and behaviour of intestinal epithelial cells. 28 Recently, C6Ma3 was shown to be strongly associated with endoscopic disease activity in CD. 29 Similarly, in our study, C6Ma3 levels were significantly associated with active inflammation as reflected by serum CRP and FCal levels. Furthermore, we observed lower levels of C1M, C3M and C4M in patients with stricturing CD, representing the dynamics between deposition and degradation of collagens, favouring that of collagen deposition as these markers are reflective of tissue degradation. Indeed, intestinal stricture formation in CD occurs through a net accumulation of several types of collagens, including interstitial fibrillar collagens (e.g. types I and III) and non‐fibrillar BM collagens, the latter being mainly represented by type IV collagen. 30 , 31 Net increased collagen formation may lead to thickening and stiffening of the intestinal wall, culminating in intestinal stenosis. 32 Previous studies have also shown that expression and activity of MMPs, especially MMP‐2, ‐9, ‐12 and ‐13, which mediate degradation of type I, III and IV collagens, were decreased in mucosae overlying stenotic intestinal tissue. In contrast, the expression of tissue inhibitors of MMPs (TIMPs) was increased. 33 , 34 , 35 In our previous study, increased type I and III collagen degradation was mainly present in patients with penetrating disease. This is in line with the current data, especially when adjusting for confounders, as we demonstrated that increased degradation of type I, III and IV collagen can discriminate between patients with stricturing and penetrating CD. In addition, our study is the first to show the accuracy of type IV collagen biomarkers in differentiating between non‐stricturing, non‐penetrating and stricturing or penetrating CD. In fact, the type IV collagen formation/degradation ratio (PRO‐C4/C4M) most accurately discriminated between non‐stricturing, non‐penetrating versus stricturing or penetrating disease. Taken together, the observed reduction in MMP‐mediated collagen degradation fragments together with an increased type IV collagen formation/degradation ratio may be indicative of a relative (net) increase in collagen synthesis/fibrogenesis versus collagen degradation/fibrolysis in patients with stricturing disease.
The frequency of disease flares has been associated with an increased risk of early development of a stricturing or penetrating disease pattern. 36 Importantly, in the present study, associations between biomarker levels and disease behaviour subclasses were independent of active inflammation, as CRP was not significantly associated with the Montreal behaviour classification. Although CRP was initially included as a covariate in multivariable analyses, it eventually dropped out as a non‐significant confounder. Instead, platelet counts remained a significant confounder in some models, which is a less sensitive biomarker of active inflammation, albeit reactive thrombocytosis is a typical laboratory finding in CD. 37 Nevertheless, these observations support the accumulating evidence that the development of stricturing or penetrating disease is not solely driven by inflammation, which may only be part of the initiation and progression of disturbed ECM remodelling in IBD. 38
C4G (a T‐cell activity marker derived from granzyme‐B‐mediated degradation of type IV collagen) was particularly increased in patients with penetrating CD. In prospective analyses, it showed the strongest association with the risk of penetrating CD progression, where patients having the highest serum C4G levels (belonging to the third tertile) had the lowest progression‐free survival until the last follow‐up. C4G is a recently developed biomarker of granzyme B‐mediated degradation of type IV collagen and T‐lymphocyte activity. 18 T‐lymphocytes express the serine protease granzyme B that induces type IV collagen breakdown, enabling migration through BMs towards the intestinal mucosa and facilitating their cytotoxic effector mechanisms. 18 , 39 Granzyme B is typically associated with active CD and is mainly expressed by activated CD4+‐ and CD8+‐Th1‐differentiated lymphocytes in the lamina propria of the intestinal mucosa. 39 , 40 Furthermore, type IV collagen degradation is preferentially mediated by MMP‐2, ‐3, and ‐9, which are markedly upregulated in CD, especially highly active in the case of penetrating disease, and predominantly expressed and secreted by Th1‐lymphocytes. 11 , 41 , 42 , 43 , 44 As penetrating CD is associated with Th1‐lymphocyte infiltration, this may be a plausible mechanistic explanation as to why patients with higher levels of (granzyme B‐mediated) type IV collagen degradation could be at increased risk of penetrating disease progression.
Pharmacological interventions targeting intestinal fibrosis do not yet exist, whereas they recently have become available for other fibrotic indications, such as idiopathic pulmonary fibrosis. 45 Current treatment modalities for intestinal fibrosis consist of endoscopic treatment (e.g. pneumatic dilation) and surgery, both of which are accompanied by high rates of side‐effects and recurrences. 2 The ultimate aim of anti‐fibrotic therapy for fibrostenotic CD would be to reverse fibrosis back to physiological tissue regeneration and ECM remodelling. However, the potential reversibility of intestinal fibrosis will largely depend on the stage of fibrosis: although early stages may be sensitive to at least partial therapeutic amelioration, more advanced fibrosis may show complete therapeutic resistance. 46 In clinical practice, early identification of the development of stricturing and/or penetrating disease is, therefore, paramount to enable prompt therapeutic intervention, to prevent progression of stricturing and/or penetrating disease and thus ultimately prevent the need for surgical procedures. Serological biomarkers of collagen turnover may help to facilitate early detection of stricturing and penetrating disease and even resolution of fibrosis, even before it becomes clinically apparent. Furthermore, appropriate biomarkers may help to predict the progression of stricturing and/or penetrating disease and may even show prognostic value with regard to response to future anti‐fibrotic therapy. To achieve these goals, however, prospective, longitudinal clinical trials focusing on intestinal fibrosis or penetrating disease and extended follow‐up would be required in order to ultimately define the value of such biomarkers. In addition, the quest for accurate fibrosis biomarkers should continue to explore the potential involvement of other relevant ECM proteins, including fibronectins, laminins, and other types of collagens, as well as proteins enhancing matrix stiffness by increasing collagen cross‐linkage within the ECM, which further contributes to stricture formation. 8 , 23
This study was strengthened by the extensive characterisation of the patient cohort, including balanced groups of patients with different disease behaviours and the relatively long follow‐up, which enabled us to prospectively evaluate the predictive capacity of biomarkers with respect to disease course. Furthermore, biomarkers analysed were measured using an innovative neo‐epitope protein fingerprint assay, in which separate quantification of post‐translational modification protein features (e.g., formation, degradation, or signalling domains) provides an additional layer of biomarker specificity compared with standard ELISAs that often provide rather crude protein measurements. 47 Several limitations of this study, however, also warrant recognition. First, our study was of retrospective nature, in which disease behaviour and follow‐up data were registered based on available (endoscopic and/or radiologic) imaging and pathology reports of intestinal resections. This may have introduced information bias, as we had to rely on others for accurate recordkeeping. Similarly, we used the Montreal disease classification as the main study outcome, which is subject to interobserver variability and instability due to the dynamic nature of CD. To partially overcome this, we only included patients who had an updated Montreal disease classification registered at least within 1 year of sampling, and whose disease behaviour classification remained stable during follow‐up. In addition, this was a single‐center study, which ensured a rather homogeneous assessment of disease behaviour subclasses. In this respect, more objective, homogeneous classifications systems for intestinal fibrosis solely based on cross‐sectional imaging or histopathology would have been preferred. Unfortunately, however, there is still a lack of standardised criteria for defining stricturing and penetrating disease (progression) in CD, which are usually of low‐quality evidence and limited to consensus‐based recommendations. 48 , 49 Similarly, there is a lack of validated scoring systems to reliably determine the severity of stricturing/penetrating disease in CD. Another limitation pertaining to the Montreal classification is the fact that for patients with penetrating (B3) disease, it does not distinguish between those who do or do not have concurrent stricturing disease. Therefore, we verified whether patients with and without stricturing disease within the B3 subclass showed differences in biomarker levels, which appeared not to be the case. Moreover, limited FC values were available to evaluate mucosal disease activity in our study cohort, which may be due to low patient compliance in collecting stools. This, however, also emphasises the utility of serological biomarkers instead of faecal biomarkers for disease assessment. Finally, due to the limited sample size of patient subgroups, we could not reliably perform stratification for other relevant covariates, such as age, medication use, disease duration or subgroup analyses for progression and recurrence of stricturing or penetrating disease. For the same reason, analysis of the predictive value of combinations of biomarkers was omitted due to the risk of model overfitting. Future studies should focus on the integrative analysis of these biomarkers as input for clinical algorithms for predictive (machine learning‐based) modelling in larger patient cohorts. In addition, prospective longitudinal studies would be required to study the temporal dynamics of the studied collagen biomarkers and to validate their discriminative accuracy with regard to CD behaviour subtypes. External validation in an independent replication cohort of patients with CD would be crucial to validate the utility and behaviour of the investigated biomarkers, preferably in relation to outcomes derived from modalities such as intestinal imaging and histopathology.
In conclusion, we show that serological biomarkers of type I, III and IV collagen turnover strongly associate with disease behaviour subclasses as defined by the Montreal disease classification in patients with CD. Our results highlight the potential significance and clinical applicability of serological biomarkers of collagen turnover to improve detection of stricturing and/or penetrating disease and predict future progression of penetrating CD. Future studies are warranted to further validate these biomarkers in relation to early detection and monitoring of stricturing/penetrating complications in patients with CD and to assess their value for guiding therapeutic decision‐making to prevent (progression of) these complications.
AUTHOR CONTRIBUTIONS
Arno R. Bourgonje: Conceptualization (lead); data curation (lead); formal analysis (lead); funding acquisition (supporting); investigation (lead); methodology (lead); project administration (equal); resources (supporting); software (lead); validation (lead); visualization (lead); writing – original draft (lead); writing – review and editing (lead). Marta S. Alexdottir: Conceptualization (equal); data curation (supporting); formal analysis (supporting); investigation (equal); methodology (equal); resources (supporting); software (supporting); validation (equal); writing – review and editing (equal). Antonius Timotheus Otten: Data curation (supporting); investigation (supporting); methodology (supporting); validation (supporting); writing – review and editing (supporting). Roberta Loveikyte: Data curation (supporting); investigation (supporting); methodology (supporting); validation (supporting); writing – review and editing (supporting). Anne‐Christine Bay‐Jensen: Funding acquisition (supporting); investigation (supporting); methodology (supporting); resources (supporting); validation (supporting); writing – review and editing (supporting). Martin Pehrsson: Funding acquisition (supporting); investigation (supporting); methodology (supporting); resources (supporting); validation (supporting). Hendrik M van Dullemen: Data curation (supporting); investigation (supporting); resources (supporting); validation (supporting). Marijn C. Visschedijk: Data curation (supporting); investigation (supporting); resources (supporting); validation (supporting); writing – review and editing (supporting). Eleonora A.M. Festen: Data curation (supporting); investigation (supporting); resources (supporting); validation (supporting). Rinse K Weersma: Data curation (supporting); investigation (supporting); methodology (supporting); resources (supporting); supervision (supporting); validation (supporting); writing – review and editing (supporting). Morten Karsdal: Conceptualization (supporting); funding acquisition (equal); investigation (supporting); methodology (supporting); project administration (supporting); resources (equal); supervision (supporting); validation (supporting); writing – review and editing (supporting). Klaas Nico Faber: Data curation (supporting); investigation (supporting); methodology (supporting); project administration (supporting); supervision (supporting); validation (supporting); writing – review and editing (supporting). Joachim Hg Mortensen: Conceptualization (lead); data curation (supporting); formal analysis (supporting); funding acquisition (equal); investigation (lead); methodology (lead); project administration (lead); resources (lead); supervision (lead); validation (lead); visualization (equal); writing – review and editing (lead). Gerard Dijkstra: Conceptualization (lead); data curation (lead); funding acquisition (equal); investigation (lead); methodology (supporting); project administration (equal); resources (lead); supervision (lead); validation (lead); writing – review and editing (equal). All authors approved the final version of the manuscript to be submitted for publication.
AUTHORSHIP
Guarantors of the article: Arno R. Bourgonje and Gerard Dijkstra.
Supporting information
Figure S1
{kind=link}
Figure S2
{kind=link}
Figure S3
Tables S1‐S8
ACKNOWLEDGEMENT
The authors would like to thank all patients who participated in this study. In addition, the authors thank the Dutch Initiative of Crohn's and Colitis (ICC) and the Parelsnoer Institute for providing the data‐ and biobank infrastructure.
Declaration of personal interests: MA, ACBJ, MAK and JHM are employees of Nordic Bioscience. ACBJ and MAK own stocks in Nordic Bioscience. GD received research grants from Royal DSM and speaker's fees from Janssen Pharmaceuticals, Takeda, Pfizer and Abbvie. RKW acted as consultant for Takeda, received unrestricted research grants from Takeda, Johnson & Johnson, Tramedico, and Ferring, and received speaker fees from MSD, Abbvie, and Janssen Pharmaceuticals. All other authors have no conflicts of interest to declare.
Bourgonje AR, Alexdottir MS, Otten AT, Loveikyte R, Bay‐Jensen A‐C, Pehrsson M, et al. Serological biomarkers of type I, III and IV collagen turnover are associated with the presence and future progression of stricturing and penetrating Crohnʼs disease. Aliment Pharmacol Ther. 2022;56:675–693. 10.1111/apt.17063
Arno R. Bourgonje and Marta S. Alexdottir are joint first authors.
Joachim H. Mortensen and Gerard Dijkstra are joint senior authors
The Handling Editor for this article was Dr Colin Howden, and it was accepted for publication after full peer‐review.
Funding information
This work was supported by the Junior Scientific Masterclass (JSM) of the University of Groningen, the Netherlands (grant no. 17‐57, to A.R.B.)
REFERENCES
- 1. Chang JT. Pathophysiology of inflammatory bowel diseases. N Engl J Med. 2021;383(27):2652–64. [DOI] [PubMed] [Google Scholar]
- 2. Rieder F, Latella G, Magro F, Yuksel ES, Higgins PD, Di Sabatino A, et al. European Crohn's and Colitis Organisation topical review on prediction, diagnosis and management of fibrostenosing Crohn's disease. J Crohns Colitis. 2016;10(8):873–5. [DOI] [PubMed] [Google Scholar]
- 3. Siegmund B, Feakins RM, Barmias G, Ludvig JC, Teixeira FV, Rogler G, et al. Results of the Fifth Scientific Workshop of the ECCO (II): pathophysiology of perianal fistulizing disease. J Crohns Colitis. 2016;10(4):377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Silverberg MS, Satsangi J, Ahmad T, Arnott IDR, Bernstein CN, Brant SR, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19 Suppl A:5A–36A. [DOI] [PubMed] [Google Scholar]
- 5. Mortensen JH, Lindholm M, Langholm LL, Kjeldsen J, Bay‐Jensen AC, Karsdal MA, et al. The intestinal tissue homeostasis – the role of extracellular matrix remodeling in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. 2019;13(10):977–93. [DOI] [PubMed] [Google Scholar]
- 6. Shimshoni E, Yablecovitch D, Baram L, Dotan I, Sagi I. ECM remodelling in IBD: innocent bystander or partner in crime? The emerging role of extracellular molecular events in sustaining intestinal inflammation. Gut. 2015;64(3):367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rieder F, Fiocchi C. Intestinal fibrosis in IBD—a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. 2009;6(4):228–35. [DOI] [PubMed] [Google Scholar]
- 8. Ravi A, Garg P, Sitaraman SV. Matrix metalloproteinases in inflammatory bowel disease: boon or a bane? Inflamm Bowel Dis. 2007;13(1):97–107. [DOI] [PubMed] [Google Scholar]
- 9. Warnaar N, Hofker HS, Maathuis MHJ, Niesing J, Bruggink AH, Dijkstra G, et al. Matrix metalloproteinases as profibrotic factors in terminal ileum in Crohn's disease. Inflamm Bowel Dis. 2006;12(9):863–9. [DOI] [PubMed] [Google Scholar]
- 10. Gecse K, Khanna R, Stoker J, Jenkins JT, Gabe S, Hahnloser D, et al. Fistulizing Crohn's disease: diagnosis and management. United European Gastroenterol J. 2013;1(3):206–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kirkegaard T, Hansen A, Bruun E, Brynskov J. Expression and localisation of matrix metalloproteinases and their natural inhibitors in fistulae of patients with Crohn's disease. Gut. 2004;53(5):701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sandborn WJ, Fazio VW, Feagan BG, Hanauer SB. AGA technical review on perianal Crohn's disease. Gastroenterology. 2003;125(5):1508–30. [DOI] [PubMed] [Google Scholar]
- 13. Leeming DJ, He Y, Veidal S, Nguyen Q, Larsen D, Koizumi M, et al. A novel marker for assessment of liver matrix remodeling: an enzyme‐linked immunosorbent assay (ELISA) detecting a MMP generated type I collagen neo‐epitope (C1M). Biomarkers. 2011;16(7):616–28. [DOI] [PubMed] [Google Scholar]
- 14. Barascuk N, Veidal SS, Larsen L, Larsen DV, Larsen MR, Wang J, et al. A novel assay for extracellular matrix remodeling associated with liver fibrosis: an enzyme‐linked immunosorbent assay (ELISA) for a MMP‐9 proteolytically revealed neo‐epitope of type III collagen. Clin Biochem. 2010;43(10–11):899–904. [DOI] [PubMed] [Google Scholar]
- 15. Nielsen MJ, Nedergaard AF, Sun S, Veidal SS, Larsen L, Zheng Q, et al. The neo‐epitope specific PRO‐C3 ELISA measures true formation of type III collagen associated with liver and muscle parameters. Am J Transl Res. 2013;5(3):303–15. [PMC free article] [PubMed] [Google Scholar]
- 16. Leeming DJ, Nielsen MJ, Dai Y, Veidal SS, Vassiliadis E, Zhang C, et al. Enzyme‐linked immunosorbent serum assay specific for the 7S domain of Collagen Type IV (P4NP 7S): a marker related to the extracellular matrix remodeling during liver fibrogenesis. Hepatol Res. 2012;42(5):482–93. [DOI] [PubMed] [Google Scholar]
- 17. Sand JM, Larsen L, Hogaboam C, Martinez F, Han M, Larsen MR, et al. MMP mediated degradation of type IV collagen alpha 1 and alpha 3 chains reflects basement membrane remodeling in experimental and clinical fibrosis—validation of two novel biomarker assays. PLoS One. 2013;8(12):e84934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jensen C, Sinkeviciute D, Madsen DH, Önnerfjord P, Hansen M, Schmidt H, et al. Granzyme B degraded type IV collagen products in serum identify melanoma patients responding to immune checkpoint inhibitors. Cancers (Basel). 2020;12(10):E2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Veidal SS, Karsdal MA, Vassiliadis E, Nawrocki A, Larsen MR, Nguyen QH, et al. MMP mediated degradation of type VI collagen is highly associated with liver fibrosis—identification and validation of a novel biochemical marker assay. PLoS One. 2011;6(9):e24753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mortensen JH, Godskesen LE, Jensen MD, van Haaften WT, Klinge LG, Olinga P, et al. Fragments of citrullinated and MMP‐degraded vimentin and MMP‐degraded Type III Collagen are novel serological biomarkers to differentiate Crohn's disease from ulcerative colitis. J Crohns Colitis. 2015;9(10):863–72. [DOI] [PubMed] [Google Scholar]
- 21. Mortensen JH, Manon‐Jensen T, Jensen MD, Hägglund P, Klinge LG, Kjeldsen J, et al. Ulcerative colitis, Crohn's disease, and irritable bowel syndrome have different profiles of extracellular matrix turnover, which also reflects disease activity in Crohn's disease. PLoS One. 2017;12(10):e0185855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Haaften WT, Mortensen JH, Karsdal MA, Bay‐Jensen AC, Dijkstra G, Olinga P. Misbalance in type III collagen formation/degradation as a novel serological biomarker for penetrating (Montreal B3) Crohn's disease. Aliment Pharmacol Ther. 2017;46(1):26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Haaften WT, Mortensen JH, Dige AK, Grønbæk H, Hvas CL, Bay‐Jensen AC, et al. Serological biomarkers of tissue turnover identify responders to anti‐TNF therapy in Crohn's disease: a pilot study. Clin Transl Gastroenterol. 2020;11(9):e00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lindholm M, Manon‐Jensen T, Madsen GI, Krag A, Karsdal MA, Kjeldsen J, et al. Extracellular matrix fragments of the basement membrane and the interstitial matrix are serological markers of intestinal tissue remodeling and disease activity in dextran sulfate sodium colitis. Dig Dis Sci. 2019;64(11):3134–42. [DOI] [PubMed] [Google Scholar]
- 25. Harvey RF, Bradshaw JM. A simple index of Crohn's disease‐activity. Lancet. 1980;1(8167):514. [DOI] [PubMed] [Google Scholar]
- 26. Bettenworth D, Nowacki TM, Cordes F, Buerke B, Lenze F. Assessment of stricturing Crohn's disease: current clinical practice and future avenues. World J Gastroenterol. 2016;22(3):1008–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rimola J, Ordás I, Rodriguez S, García‐Bosch O, Aceituno M, Llach J, et al. Magnetic resonance imaging for evaluation of Crohn's disease: validation of parameters of severity and quantitative index of activity. Inflamm Bowel Dis. 2011;17(8):1759–68. [DOI] [PubMed] [Google Scholar]
- 28. Groulx J, Gagné D, Benoit YD, Martel D, Basora N, Beaulieu JF. Collagen VI is a basement membrane component that regulates epithelial cell‐fibronectin interactions. Matrix Biol. 2011;30(3):195–206. [DOI] [PubMed] [Google Scholar]
- 29. Lindholm M, Godskesen LE, Manon‐Jensen T, Kjeldsen J, Krag A, Karsdal MA, et al. Endotrophin and C6Ma3, serological biomarkers of type VI collagen remodeling, reflect endoscopic and clinical disease activity in IBD. Sci Rep. 2021;11(1):14713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Graham MF, Diegelmann RF, Elson CO, Lindblad WJ, Gotschalk N, Gay S, et al. Collagen content and types in the intestinal strictures of Crohn's disease. Gastroenterology. 1988;94(2):257–65. [DOI] [PubMed] [Google Scholar]
- 31. Koutroubakis IE, Petinaki E, Dimoulios P, Vardas E, Roussomoustakaki M, Maniatis AN, et al. Serum laminin and collagen IV in inflammatory bowel disease. J Clin Pathol. 2003;56(11):817–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Haaften WT, Blokzijl T, Hofker HS, Olinga P, Dijkstra G, Bank RA, Boersema M Intestinal stenosis in Crohn's disease shows a generalized upregulation of genes involved in collagen metabolism and recognition that could serve as novel anti‐fibrotic drug targets. Therap Adv Gastroenterol 2020;13:1756284820952578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Di Sabatino A, Jackson CL, Pickard KM, Buckley M, Rovedatti L, Leakey NAB, et al. Transforming growth factor beta signaling and matrix metalloproteinases in the mucosa overlying Crohn's disease strictures. Gut. 2009;58(6):777–89. [DOI] [PubMed] [Google Scholar]
- 34. McKaig BC, McWilliams D, Watson SA, Mahida YR. Expression and regulation of tissue inhibitor of metalloproteinase‐1 and matrix metalloproteinases by intestinal myofibroblasts in inflammatory bowel disease. Am J Pathol. 2003;162(4):1355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Bruyn JR, van den Brink GR, Steenkamer J, Buskens CJ, Bemelman WA, Meisner S, et al. Fibrostenotic phenotype of myofibroblasts in Crohn's disease is dependent on tissue stiffness and reversed by LOX inhibition. J Crohns Colitis. 2018;12(7):849–59. [DOI] [PubMed] [Google Scholar]
- 36. Louis E, Michel V, Hugot JP, Reenaers C, Fontaine F, Delforge M, et al. Early development of stricturing or penetrating pattern in Crohn's disease is influenced by disease location, number of flares, and smoking but not by NOD2/CARD15 genotype. Gut. 2003;52(4):552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morowitz DA, Allen LW, Kirsner JB. Thrombocytosis in chronic inflammatory bowel disease. Ann Intern Med. 1968;68(5):1013–21. [DOI] [PubMed] [Google Scholar]
- 38. Latella G, Di Gregorio J, Flati V, Rieder F, Lawrance IC. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand J Gastroenterol. 2015;50(1):53–65. [DOI] [PubMed] [Google Scholar]
- 39. Jenkins D, Seth R, Kummer JA, Scott BB, Hawkey CJ, Robins RA. Differential levels of granzyme B, regulatory cytokines, and apoptosis in Crohn's disease and ulcerative colitis at first presentation. J Pathol. 2000;190(2):184–9. [DOI] [PubMed] [Google Scholar]
- 40. Riaz T, Sollid LM, Olsen I, de Souza GA. Quantitative proteomics of gut‐derived Th1 and Th1/Th17 clones reveal the presence of CD28+ NKG2D‐Th1 cytotoxic CD4+ T cells. Mol Cell Proteomics. 2016;15(3):1007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Efsen E, Saermark T, Hansen A, Bruun E, Brynskov J. Ramiprilate inhibits functional matrix metalloproteinase activity in Crohn's disease fistulas. Basic Clin Pharmacol Toxicol. 2011;109(3):208–16. [DOI] [PubMed] [Google Scholar]
- 42. Goetzl EJ, Banda MJ, Leppert D. Matrix metalloproteinases in immunity. J Immunol. 1996;156(1):1–4. [PubMed] [Google Scholar]
- 43. Oviedo‐Orta E, Bermudez‐Fajardo A, Karanam S, Benbow U, Newby AC. Comparison of MMP‐2 and MMP‐9 secretion from T helper 0, 1 and 2 lymphocytes alone and in coculture with macrophages. Immunology. 2008;124(1):42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin L, Couturier J, Yu X, Medina MA, Kozinetz CA, Lewis DE. Granzyme B secretion by human memory CD4 T cells is less strictly regulated compared to memory CD8 T cells. BMC Immunol. 2014;15:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074. [DOI] [PubMed] [Google Scholar]
- 46. Bettenworth D, Rieder F. Reversibility of stricturing Crohn's disease‐fact or fiction? Inflamm Bowel Dis. 2016;22(1):241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karsdal MA, Henriksen K, Leeming DJ, Woodworth T, Vassiliadis E, Bay‐Jensen AC. Novel combinations of post‐translational modification (PTM) neo‐epitopes provide tissue‐specific biochemical markers—are they the cause or the consequence of the disease? Clin Biochem. 2010;43(10–11):793–804. [DOI] [PubMed] [Google Scholar]
- 48. Rieder F, Bettenworth D, Ma C, Parker CE, Williamson LA, Nelson SA, et al. An expert consensus to standardise definitions, diagnosis and treatment targets for anti‐fibrotic stricture therapies in Crohn's disease. Aliment Pharmacol Ther. 2018;48(3):347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lamb CA, Kennedy NA, Raine T, Hendy PA, Smith PJ, Limdi JK, et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut. 2019;68(Suppl 3):s1–s106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Tables S1‐S8