

Abstract
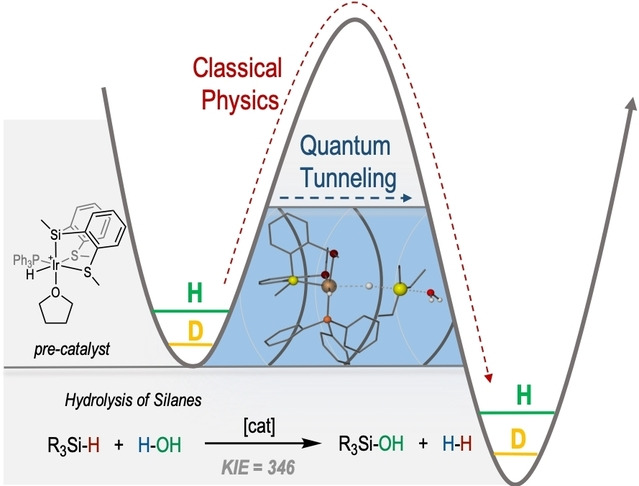
An unprecedented quantum tunneling effect has been observed in catalytic Si−H bond activations at room temperature. The cationic hydrido‐silyl‐iridium(III) complex, {Ir[SiMe(o‐C6H4SMe)2](H)(PPh3)(THF)}[BArF 4], has proven to be a highly efficient catalyst for the hydrolysis and the alcoholysis of organosilanes. When triethylsilane was used as a substrate, the system revealed the largest kinetic isotopic effect (KIESi−H/Si−D=346±4) ever reported for this type of reaction. This unexpectedly high KIE, measured at room temperature, together with the calculated Arrhenius preexponential factor ratio (AH/AD=0.0004) and difference in the observed activation energy [(E −E )=34.07 kJ mol−1] are consistent with the participation of quantum tunneling in the catalytic process. DFT calculations have been used to unravel the reaction pathway and identify the rate‐determining step. Aditionally, isotopic effects were considered by different methods, and tunneling effects have been calculated to be crucial in the process.
Keywords: Homogeneous Catalysis, Iridium, Quantum Tunneling, Silanes
A “giant” kinetic isotope effect (KIE=346) has been observed for the generation of hydrogen from the hydrolysis of silanes catalyzed by a silyl‐iridium(III) complex. This and other experimental evidence together with theoretical calculations have been used to demonstrate the participation of hydrogen quantum tunneling in this catalytic process.
Introduction
The metal‐catalysed hydrolysis and alcoholysis of hydrosilanes are well‐known processes. Early studies focused on the formed silanols/siloxides, considered versatile reaction intermediates in organic transformations.[ 1 , 2 ] More recently, the attention drove towards the equimolecular H2 evolved through this reaction, which converted hydrosilanes into promising candidates as hydrogen source materials. [3] The hydrolysis of the Si−H bonds is a thermodynamically favourable process, but it suffers from slow kinetics. [4] For this reason, many catalytic systems based on transition metals such as gold,[ 5 , 6 ] iron, [7] ruthenium,[ 8 , 9 ] rhenium,[ 10 , 11 ] rhodium[ 12 , 13 ] and iridium[ 14 , 15 , 16 , 17 , 18 , 19 , 20 ] have been developed. Among them, the iridium‐based catalysts stand out due to their high activities (Figure 1). In 1989, Luo and Crabtree reported on the high catalytic activity of a cationic dihydride‐IrIII complex in the methanolysis of triethylsilane (Figure 1). [15] In 2014, Oro et al. reported on a neutral hydrido‐silyl‐IrIII complex, which behaves as an excellent catalyst in the hydrolysis of Et3SiH (Figure 1). [17] Among the different IrIII catalytic systems reported to date, cationic complexes seem to outperform neutral derivatives in both hydrolysis and alcoholysis of silanes (Figure 1).[ 15 , 19 ] The efficiency of the cationic iridium systems is attributed to the electrophilic character of the metal centre.[ 15 , 19 ] With these systems the hydrosilane activation should occur by coordination to the electrophilic metal centre, as σ‐H‐SiR3 ligand, rather than through oxidative addition.
Figure 1.
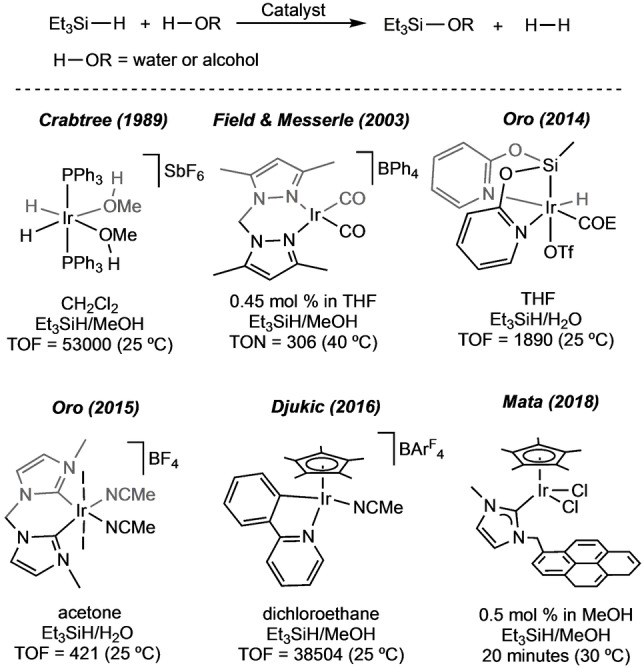
Schematic hydrolysis/alcoholysis of triethylsilane and iridium precatalysts reported for this reaction (TOFs in h−1).[ 15 , 16 , 17 , 18 , 19 , 20 ]
The catalytic cycle has been described as an outer‐sphere nucleophilic attack of alcohol or water on the activated silicon center, generating an iridium‐hydride and a solvent‐stabilized silylium cation, which immediately reacts to form an iridium‐dihydrogen complex and the corresponding silanol or silylether. Liberation of hydrogen from the metal center and coordination of a new hydrosilane close the catalytic cycle, requiring only one vacant coordination site on the metal to proceed (Scheme 1).
Scheme 1.
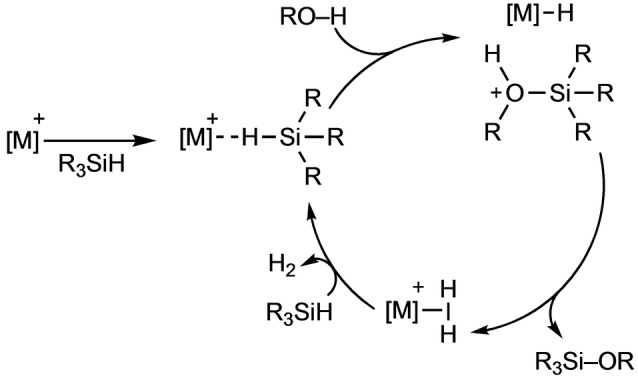
Proposed electrophilic mechanism for the hydrolysis/alcoholysis of silanes.
This mechanism, initially proposed by Luo and Crabtree in 1989, [15] has been accepted for the hydrolysis and alcoholysis of hydrosilanes involving different electrophilic catalysts.[ 7 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 ] While there are examples of electrophilic mechanism for the hydrolysis/alcoholysis of silanes with other transition metals,[ 7 , 21 , 22 , 23 , 24 , 25 , 26 ] mechanistic studies involving cationic iridium complexes are still very attractive due to its high capability to transfer electrophilicity inducing Si−H activation.[ 27 , 28 ] Considering iridium(III) complexes, this kind of mechanism has been well studied theoretically by Oro et al. in 2014 [17] and Mata et al. in 2018. [20] Moreover, in 2016, the group of Djukic has extensively studied the mechanism of the alcoholysis of triethylsilane experimentally. The work reported by Djukic et al. includes the full characterization of a cationic iridium η1‐silane complex and other possible intermediates of this reaction, as for example hydride species. [19]
In the last years, our research group has been working on the synthesis and applications of rhodium and iridium complexes with silyl‐thioether multidentate ligands. This kind of ligands proved useful to stabilize unsaturated rhodium and iridium cationic complexes leaving a coordinative vacant site in trans with respect to the coordinated silicon center (often occupied by a solvent molecule).[ 29 , 30 ] Assuming an electrophilic mechanism for the hydrolysis/alcoholysis of silanes (Scheme 1), unsaturated iridium(III) compounds are perfectly suited to activate hydrosilane through coordination prompting the subsequent nucleophilic attack of water/alcohol.
Herein we present combined experimental and theoretical studies on the catalytic activity of a cationic iridium species {Ir[SiMe(o‐C6H4SMe)2](H)(PPh3)(THF)}[BArF 4] (1 in Figure 2) in the hydrolysis and alcoholysis of silanes. The involvement of hydrogen tunneling in the rate‐determining step (RDS) of the process permitted us to explain the high catalytic activity and the unexpectedly large KIE observed at room temperature.[ 31 , 32 , 33 , 34 ]
Figure 2.
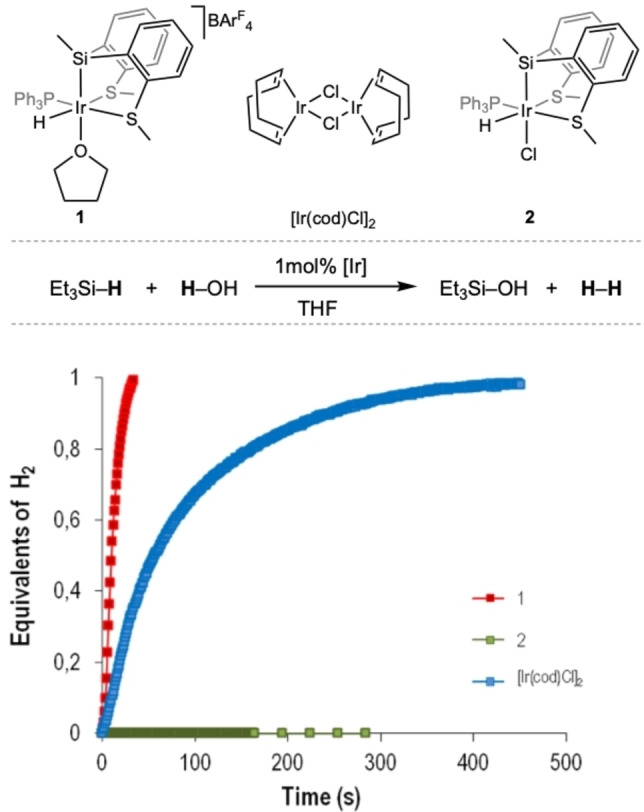
Iridium precatalysts used in this work, hydrolysis of Et3SiH catalyzed by 1, 2 and [Ir(cod)Cl]2 and reaction profiles (equiv. of H2 generated vs time). Reaction conditions: Et3SiH (0.25 mmol), H2O (2.5 mmol), catalyst (0.0025 mmol based on iridium) in THF (1 mL) at 25 °C. H2 production calculated by continuous monitoring of the pressure evolution using a pressure transducer (Man on the Moon X102 kit).
Results and Discussion
Iridium(III) compounds 1 and 2 [30] have been studied as precatalysts for the hydrolysis of Et3SiH in THF under standard reaction conditions (1 mol % catalyst, [Et3SiH]=0.25 M, 10 equiv H2O). The parent iridium dimer [Ir(cod)Cl]2 has also been included in the initial screening for comparative purposes. [14] The reaction profiles obtained (equiv. of H2 vs time) are shown in Figure 2. The hydrogen evolution during the reaction was continuously monitored through a pressure transduced implemented in the Man on the Moon X102 kit (see Supporting Information for details).
The results obtained showed that no hydrogen was liberated when the saturated complex 2 was used as precatalyst. This result indicates that a vacant coordination site on the catalyst is required for the reaction to proceed. In contrast, compound 1, containing a labile solvent molecule, was a very effective catalyst for the hydrolysis of Et3SiH, liberating 1 equiv of H2 in only 35 seconds (Table 1, entry 1). The activity observed (turnover frequency calculated at 50 % of conversion, TOF1/2) was 2.8 times larger than that observed for the dimeric iridium precursor [Ir(cod)Cl]2 under identical reaction conditions. 1H and 1H‐29Si HMBC NMR analysis of evaporated samples at the end of the reaction permitted us to identify triethylsilanol as the only silane‐containing product (Figure S1, Supporting Information). The robustness of the catalytic system derived from 1 was confirmed through successive additions of Et3SiH. The sequential reaction profiles obtained showed that the catalyst maintained its activity for at least 10 successive cycles (Figure 3).
Table 1.
Catalytic hydrolysis/alcoholysis of hydrosilanes.[a]
|
Entry |
Catalyst |
Silane |
Nucleophile |
TOF1/2 (h−1)[b] |
|---|---|---|---|---|
|
1 |
1 |
Et3SiH |
H2O |
20 385(2850) |
|
2 |
2 |
Et3SiH |
H2O |
0 |
|
3 |
[Ir(cod)Cl]2 |
Et3SiH |
H2O |
6276[c] |
|
4 |
1 |
Me2PhSiH |
H2O |
13 5678(20 509) |
|
5 |
1 |
MePh2SiH |
H2O |
44 776(297) |
|
6 |
1 |
Ph3SiH |
H2O |
950(29) |
|
7 |
1 |
Et3SiH |
MeOH |
13 107(671) |
|
8 |
1 |
Et3SiH |
EtOH |
11 952(289) |
|
9 |
1 |
Et3SiH |
iPrOH |
1301(47) |
[a] Reaction conditions: Silane (0.25 mmol), H2O or alcohol (2.5 mmol), 1 mol % of catalyst (based on iridium) in 1 mL of THF at 25 °C. [b] TOF1/2: turnover frequency calculated at a reaction time corresponding to 50 % of conversion, values averaged from 3 runs, errors in parenthesis. [c] Only one run.
Figure 3.

Reaction profiles obtained by successive additions of 0.25 mmol of Et3SiH to a THF solution containing 0.0025 mmol of compound 1 and 5.5 mmol of H2O. H2 production calculated by continuous monitoring of the pressure evolution using a pressure transducer (Man on the Moon X102 kit).
The hydrolytic reaction with precatalyst 1 was also studied using a range of hydrosilanes under standard conditions (see Table 1). In all cases, the corresponding silanol was obtained as the sole reaction product, as confirmed by 1H and 1H‐29Si HMBC NMR analysis of evaporated reaction mixtures (see Figures S2–S4, Supporting Information). The results obtained showed a clear electronic influence of the silane substituents on the reaction rate (entries 4, 5 and 6, Table 1), being faster when increasing the electron‐donating nature of the substituents except for Et3SiH. The low activity of Et3SiH compared with Me2PhSiH and MePh2SiH could be attributed to the larger buried volume of the latter which would hinder it from approaching the metal center (see Figure S8, Supporting Information). [35]
The catalytic activity of compound 1 towards the alcoholysis of Et3SiH was also explored (Table 1, entries 7–9, and Figure S10, Supporting Information). The results obtained showed that in all cases one equivalent of hydrogen was liberated, and the corresponding silylethers were the only silicon‐containing reaction products, as confirmed by 1H and 1H‐29Si HMBC NMR analysis of evaporated reaction mixtures (Figures S5–S7, Supporting Information). The reaction proceeded at similar rates for the primary alcohols assayed (entries 7 and 8, Table 1). It is worth noticing that, even if at a much‐reduced rate, compound 1 was also an effective catalyst when a secondary alcohol (iPrOH) was used as a nucleophile.
These results positioned the cationic iridium complex 1 among the most active catalytic systems for the hydrolysis and alcoholysis of organosilanes.[ 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 ]
Induction periods were observed in most of the catalytic reactions. The addition of excess Hg during the hydrolysis of triethylsilane led to similar results, and no darkening of the solution was observed, suggesting the homogeneous nature of the catalytic process. [36] Therefore, the induction period was, at this point, tentatively attributed to decoordination of THF being required to obtain an active species to start the catalytic reaction.
A series of NMR experiments were performed to obtain some information about the reaction mechanism. Initially, the reactivity of 1 toward H2O was studied. 1 was reacted with 100 equivalents of H2O in THF‐d8 for 12 hours. 1H NMR spectrum of the starting compound 1 remained apparently unaltered apart from a small displacement (0.03 ppm) and broadening of the signal attributed to the coordinated hydride (Figure S20, Supporting Information). This can be explained by Ir−H/H2O exchange via short‐lived dihydrogen complexes [37] or because an Ir−THF⇌Ir−OH2 equilibrium is taking place. Both possibilities would involve a H2O coordination to the metal center. When the same reaction was performed in CD2Cl2, displacements of the Ir−H signal (0.82 ppm) and Si‐CH3 signal (0.1 ppm) were observed (Figure S21). This fact reinforces the idea of an Ir−THF⇌Ir−OH2 equilibrium. No hydrogen evolution was observed during these experiments, therefore, a mechanism operating through IrIII‐OH intermediates, as proposed by Luo et al., [38] can be excluded. To study the ability of complex 1 to activate Et3Si−H, as proposed within the electrophilic mechanism, compound 1 was reacted with 10 equivalents of Et3Si−H in THF‐d8. 1H NMR spectrum shows a 1.1 ppm displacement on the Ir−H signal, which could indicate the formation of a cationic iridium η1‐silane complex. Unfortunately, sigma interaction between Et3SiH and 1 was not observed in a static 1H NMR (−90 °C) in this solvent. Triethylsilyl iridium complex is not observed, which would exclude a mechanism via oxidative addition/reductive elimination steps. The same reaction (1 and 10 equivalents of Et3Si−H) was studied in CD2Cl2 at −90 °C. The hydride region of the 1H NMR spectrum showed the transformation of the signal attributed to the hydride of 1 (δ=−12.13, J H‐P=18.5 Hz), into two new signals at −3.88 and −13.27 ppm, respectively (Figure 4 and Figures S22 and S23 in Supporting Information). The former signal was a singlet accompanied by the characteristic 29Si satellites (J H‐Si=98 Hz) (Figure S23, Supporting Information). It was assigned to an Ir−H−Si hydride, by comparison with a cationic iridium η1‐silane complex previously reported by Brookhart. [39] The resonance at higher field resolved into a doublet (J H‐P=16.3 Hz), and resembled that of the parent compound 1. Therefore, it was assigned to a terminal hydride trans to a sulfur atom and cis to PPh3. Additionally, the 31P{1H} NMR spectrum showed the transformation of the original singlet at 19.7 ppm into a new singlet at 16.0 ppm (Figure S24, Supporting Information).
Figure 4.
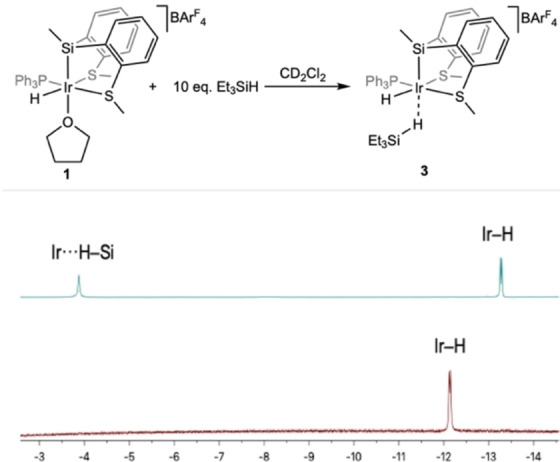
Scheme of the reaction of 1 with Et3SiH in CD2Cl2 and hydride region of the 1H NMR spectra of 1 (bottom) and after addition of 10 equivalents of Et3SiH (top), at −90 °C.
These observations are consistent with the formation of compound 3 (Figure 4) containing a η1‐coordinated hydrosilane,[ 19 , 39 ] which would demonstrate the capacity of 1 to promote the electrophilic activation of hydrogen‐silicon bonds as proposed within the electrophilic mechanism.
In an effort to obtain evidence of the origin of the formed hydrogen, several catalytic reactions using Et3SiH/H2O, Et3SiH/D2O, and Et3SiD/H2O (ratio silane/water of 1/10 in all cases) were performed in a sealed NMR tube. As expected, using Et3SiH/H2O, only H2 was observed in the 1H NMR spectrum (Figure S19, Supporting Information). Therefore, the relative integration of the H2 signals of the spectra compared to that of triethoxysilanol (used as internal reference) were used to calculate the total amount of gas dissolved in a fully converted sample. Under identical experimental conditions, the hydrolysis of Et3SiD with H2O led to the formation of H2 mainly (Figure S19, Supporting Information). Finally, when Et3SiH/D2O are used as reactants, a H2/HD mixture in a ratio of 14/86 was observed by 1H NMR (Figure S19, Supporting Information). According to our calculations, this mixture corresponds to approximately 10 % of the total gas evolved, being the main component 1H NMR‐blind D2 (amount of D2 estimated of 90 %, see Supporting Information for more details). The formation of H2 (≈1.4 %) would be due to traces of H2O. These results indicate that, eventually, the isotope composition of the hydrogen released is determined by the water isotope used, which may be due to quick exchange between Ir−H(D) and the water protons as previously reported by Himeda et al. for iridium‐catalyzed formic acid dehydrogenation reactions. [40]
The results obtained using 1 as precatalyst showed a first‐order dependence of the reaction rate on Et3SiH and catalyst concentration, but a zero‐order dependence on H2O (v=k [1] [Et3SiH]) (Figures S11–S12, Supporting Information). Kinetic isotopic effect (KIE) was analyzed, using compound 1, on both the hydrolysis and the methanolysis of Et3SiH. The results obtained showed no effect on the reaction rate when a deuterated nucleophile was used (KIE =KIEMeOH/MeOD=1), which is consistent with the zero‐order dependence of the reaction rate on H2O concentration (see above). Surprisingly, extraordinarily high KIEs (25 °C) were calculated when the reaction rate using Et3SiD was compared to that of Et3SiH (KIE (25 °C)=346±4 (hydrolysis); KIE (25 °C)=143±1 (methanolysis), see Figures S14 and S16, Supporting Information. These results clearly point to H−Si bond activation being involved in the rate‐determining step (RDS) of the catalytic reaction, and suggest the possible participation of hydrogen quantum tunneling in this step of the process.[ 31 , 32 , 33 , 34 ] Rate constants and KIEs were also calculated for the catalytic hydrolysis of other hydrosilanes (KIE =4.3±0.5; KIE =16.8±0.2 (Figure S17, Supporting Information). These values show that the KIE (and probability of quantum tunneling) strongly depends on the hydrosilane. Considering that with deuterated derivatives the probability of hydrogen tunneling is very low, the rate constants (k =50.6±1.5 s−1 M−1; k =0.10±0.01 s−1 M−1) would be indicative of a much lower classic barrier for the former. Therefore, the relatively low KIE observed for PhMe2SiH(D) could be explained by an increase of the classical component to the overall rate that outruns tunneling. These observations would be in agreement with our mechanistic proposal (see below) revealing the importance of the Ir⋅⋅⋅H⋅⋅⋅Si distances in the active species for the reaction to proceed through quantum tunneling. When [Ir(cod)Cl]2 was used as precatalyst, KIE =1.10±0.03 and KIE =2.26±0.06 (Figure S18, Supporting Information) were measured. These results also pointed to a related mechanism, with H−Si bond activation being involved in the RDS, but hydrogen quantum tunneling could not be claimed in this case. In fact, a comparison of rate constants between precatalysts 1 and [Ir(cod)Cl]2 using Et3SiD as a nucleophile (k(1)=0.101 s−1 M−1; k([Ir(cod)Cl]2)=8.45 s−1 M−1) reinforces the idea that the high activities observed for our system are caused by a quantum tunneling mechanism, strongly inhibited when deuterosilanes were used as nucleophiles. No tunneling has been observed for the methanolysis of Et3SiH(D) using the cationic iridium(III) complex [IrCp*(phenylpiridine)(MeCN)]+. [19] To reinforce the hypothesis that quantum tunneling participates in the RDS of this reaction, we looked for additional experimental pieces of evidence. As generally accepted, the observation of an unusually low Arrhenius preexponential factor ratio (AH/AD<0.7) and a difference in the observed activation energy larger than the ones expected from zero‐point energy (E −E >5.02 kJ mol−1) would support this idea.[ 41 , 42 ] Therefore, the hydrolysis of Et3SiH and Et3SiD were studied at different temperatures (5–30 °C) (Figures S13 and S15, Supporting Information). Comparative Eyring and Arrhenius analyses of the data obtained showed linear but non‐parallel correlations (Figure 5). From these data a AH/AD ratio of 0.0004±0.0003 and E −E =34.1±0.2 kJ mol−1 were calculated. These results are consistent with an important contribution of hydrogen tunneling to the process and most probably being responsible for the extraordinarily high activities observed when non‐deuterated nucleophiles were used. Moreover, using the Eyring analysis, the activation barriers were determined: Et3SiH (ΔH ≠=27±2 KJ mol−1, ΔS ≠=−125±7 J mol−1 K−1 and ΔG(298)≠=64±4 KJ mol−1); Et3SiD (ΔH ≠=60±2 KJ mol−1, ΔS ≠=−60±6 J mol−1 K−1 and ΔG(298)≠=79±4 KJ mol−1). The significant negative activation entropy suggests a transition state considerably more ordered than the reactants, which agrees with a SN2‐type mechanism in the RDS, as has been previously reported for an iron catalyst. [7] Moreover, negative entropies are known to be an indication for tunneling. [43]
Figure 5.
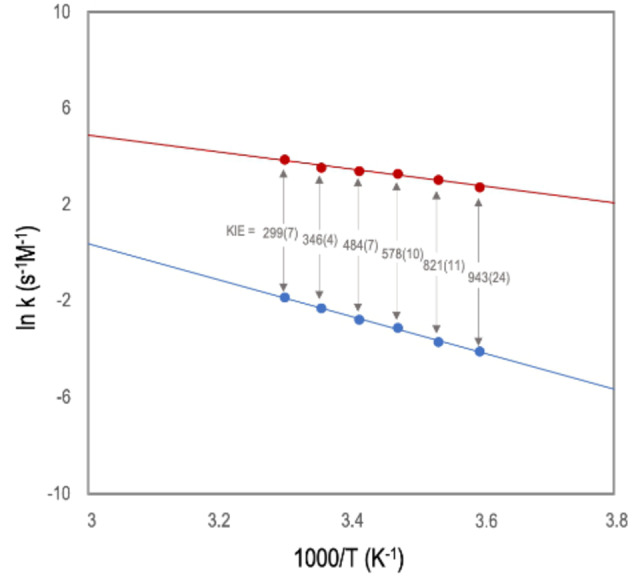
Arrhenius plots for the hydrolysis of Et3Si−H (red) and Et3Si−D (blue) catalyzed by 1, leading to values of A (from the intercept) and E a (from the slope). From the differences of the plots the KIE at each temperature is calculated, errors in parenthesis.
The high activities observed in the hydrolysis and alcoholysis of hydrosilanes, together with the experimental pieces of evidence pointing to the involvement of hydrogen tunneling in the process, prompted us to study the reaction from a theoretical point of view. DFT calculations[ 44 , 45 ] have been carried out using the M06 [46] functional and considering the implicit solvent effect by means of IEFPCM [47] method for both geometry optimizations and frequency calculations, as implemented in Gaussian 16 program package. [48] According to our calculations, described in the Supporting Information, the reactant complex would be the C0 complex (Figure 6).
Figure 6.
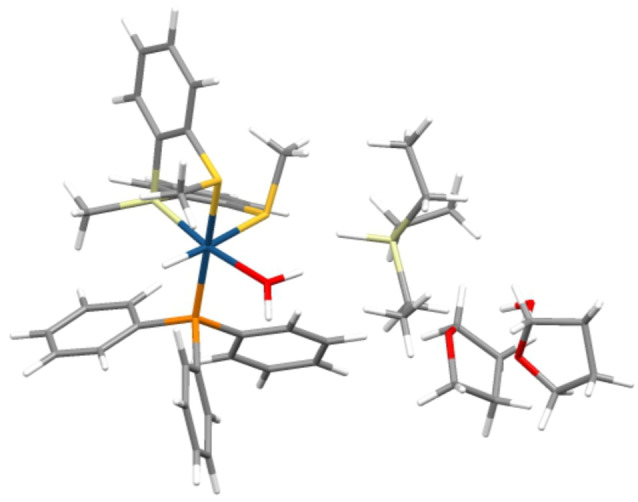
Calculated structure of the C0 reactant complex.
This reactant complex is an iridium‐aquo complex, formed by ligand exchange from the starting iridium‐THF compound. A triethylsilane molecule (not located in the first coordination sphere; d Ir⋅⋅⋅Si=5.14 Å), which contains a back‐side H2O molecule solvated with two THF solvent molecules, is also part of C0 . Starting from this reactant complex, the reaction may take place, and the calculated full reaction mechanism is depicted in Figure 7. [49] The optimized geometry for C0 , showed that the hydridic hydrogen atom of the hydrosilane is oriented towards one of the protic hydrogens of the coordinated H2O (d H⋅⋅⋅H=1.80 Å), which may indicate a kind of H∂+−H∂− interaction. Indeed, calculated Natural Bond Orbital charges (NBO charges)[ 50 , 51 , 52 ] show values of 0.541 e and −0.271 e, for the H−OH and Si−H, respectively. These values show that there is in fact a stabilizing H∂+−H∂− interaction that attracts the triethylsilane moiety towards the catalyst. In order to check for the importance of this interaction along the reaction mechanism, we have calculated the partial charges and interatomic distances all along the reaction steps, and the results are collected in Table 2.
Figure 7.

Full Gibbs Free Energy Surface for the studied reaction mechanism, in kJ mol−1. Red values correspond to deuterated species.
Table 2.
Calculated Natural Bond Orbital charges (NBO)[ 49 , 50 , 51 ] for the protic and hydridic hydrogens, and the interatomic distances between them.
|
|
H∂+ (e) |
H∂− (e) |
d (H∂+–H∂−) [Å] |
|---|---|---|---|
|
C1′ |
0.518 |
−0.290 |
4.49 |
|
C0 |
0.541 |
−0.271 |
1.80 |
|
TS1 |
0.511 |
−0.372 |
1.68 |
|
C1 |
0.518 |
−0.124 |
1.71 |
|
C2 |
0.547 |
−0.118 |
1.57 |
|
TS2 |
0.379 |
−0.139 |
0.99 |
|
C3 |
0.147 |
0.115 |
0.83 |
|
C4 |
0.001 |
−0.001 |
0.74 |
One of the possible paths starting from C0 is the complete substitution of the coordinated H2O molecule by the Et3SiH, leading to the formation of C1′ complex. In this vein, the H∂+−H∂− interaction found in C0 is broken, being the interatomic distance too long for such interaction (4.49 Å). This structure can be described as an iridium η1‐silane complex where the Si⋅⋅⋅Ir distance is 3.53 Å, which proved to be a stable structure. The consequence of such stability is that all attempts to calculate a nucleophilic attack to the Si atom eventually ended on the same structure, which shows that this is a non‐reactive path for these species. It is worth noting that analogue species have been commonly proposed to be the reactant complex in related processes.[ 20 , 28 ]
An alternative pathway was considered starting from C0 species. According to our calculations, the first step of this mechanism would be the substitution of the H2O molecule from the coordination sphere of the metal center by the H atom of the triethylsilane, leading to the C1 intermediate, where an Ir−H bond is formed. This step occurs through the transition state TS1 , where the originally coordinated H2O molecule is already displaced from the Ir coordination sphere (d Ir⋅⋅⋅O=3.98 Å), leaving a vacant position for the H transfer. This H transfer needs a nucleophilic attack of the back‐side water to happen, helped by the released front‐side water, which is oriented towards the triethylsilane moiety (d H⋅⋅⋅H=1.69 Å). Notice that this short H−H distance suggests that the H∂+−H∂− interaction observed in C0 is present in the TS1 as well. The H transfer occurs at Ir⋅⋅⋅Si distance of 4.44 Å, being the hydrogen atom aligned midway the Ir and Si atoms, at 2.54 Å and 2.05 Å, respectively. Accordingly, in TS1 , the Si atom is in a tbp coordination environment, as expected for the TS of a SN2 nucleophilic attack.
C1 could be described as the adduct formed by the neutral dihydrido‐Ir compound and the tetracoordinated silylium cation formed, stabilized by the labile front‐side H2O molecule previously coordinated to the iridium center. This H2O molecule is H‐oriented towards the Ir−H moiety (d HO−H⋅⋅⋅H−Ir=1.71 Å). The partial atomic charges are calculated to be 0.518 e and −0.124 e. Both bond lengths and atomic charges are clear indications of the previously mentioned H∂+−H∂− interaction.
The next step would be the formation of a pentacoordinated silylium cation [Et3Si(OH2)2]+, [53] which is the consequence of the coordination of the front‐side H2O molecule to the silylium cation. This front side H2O is oriented like in C1 , keeping the silylium cation close to the dihydrido‐iridium complex. The so‐formed intermediate is labelled as C2 . In this complex, the electrostatic (H∂+−H∂−) interaction remains, as it may be concluded from the distance between the protic H−OH the hydride H−Ir, 1.57 Å, and the partial charges 0.547 and −0.118 e, respectively. From this intermediate, the protic H atom is ready to be transferred from the front‐side H2O to the dihydrido‐iridium complex through TS2 , leading to the formation of an iridium‐H2 complex (C3 ) and Et3Si−OH species after the decomplexation of the back‐side H2O.
Finally, the decomplexation of dihydrogen would result in the formation of an iridium complex with a coordinative vacancy (intermediate C4 ), which would be occupied by a H2O molecule, leading the reactant complex C0 , and closing the cycle. According to the calculated mechanism, the RDS would be the hydrogen transfer from the silicon to the iridium, through TS1 , which is in good agreement with the experimental observations that suggest that Si−H activation is involved in the RDS.
In an effort to demonstrate theoretically the importance of hydrogen tunneling in the hydrolysis of triethylsilane catalyzed by 1, theoretical KIEs have been calculated using Eyring and Bigelesein theories[ 54 , 55 , 56 ] (Table S.2, Supporting Information). These data (KIEEyring=3.44; KIEBigelesein=3.32), which does not include tunneling corrections, are very different from the experimental data (KIEexp=346). The difference between both values would be caused to the lack of tunneling corrections in the theoretical values, and are indicative of the importance of tunneling in these processes. [57] Hence, the inclusion of tunneling effects in the theoretical calculations should increase the calculated KIE values. In this vein and in a simple way, one‐dimensional tunneling approaches, such as Wigner[ 31 , 58 , 59 ] and Bell's inverse parabola [60] corrections to the KIE calculated by the Bigeleisen method have been implemented (Table S.2, Supporting Information). [56] As expected, these two approaches underestimate the tunneling corrections,[ 56 , 61 ] but still are able to increase slightly the calculated KIE.
Hence, more sophisticated approaches are mandatory to account for the expected tunneling effects to explain the large observed experimental KIE. In this vein, tunneling transmission probability for both hydrogen and deuterium were calculated via the one‐dimensional Wentzel‐Kramers‐Brillouin (WKB) semiclassical approach, [62] which with a reasonable accuracy/time‐consuming balance provides a qualitatively accurate picture of the tunneling nature of the process. Thus, WKB method has been applied systematically, considering tunneling from all vibrational levels of H and D and their occupation probability using the Boltzmann distribution function (all the details are given in the Supporting Information), and the obtained results are collected in Table 3. At 298 K, the KIE including the tunneling effect is calculated to be 26.7. Now, the calculated KIE is one order of magnitude larger than the calculated with Wigner and/or Bell's tunneling corrections, being a clear indication of the importance of tunneling. Notice that even with this one‐dimensional approach, the importance of tunneling appears to be clear. In addition, the temperature effect in the calculated KIEs has been also calculated (Table 3). As may be observed, the predicted KIE values decrease as the temperature increases, as experimental ones. In addition, notice that the transmission probabilities calculated for both H and D increase with temperature, as expected, but those of D increase more rapidly, leading to smaller KIE values (see Supporting Information for a full discussion). Moreover, from predicted tunneling rate constants a AH/AD ratio of 0.27 and E −E =11.4 kJ mol−1 were calculated, which are also indicative of tunnel effects.[ 41 , 42 ] In this way, we have been able to demonstrate the participation of quantum tunneling by means of a one‐dimensional simple model.
Table 3.
Calculated transmission probabilities for H, τ(H), and D, τ(D), and calculated theoretical and experimental KIEs at different temperatures, in K, errors in parenthesis.
|
T [K] |
τ(H) |
τ(D) |
KIEtheo |
KIEexp |
|---|---|---|---|---|
|
278 |
3.98×10−9 |
1.47×10−10 |
37.5 |
943(24) |
|
283 |
4.81×10−9 |
1.95×10−10 |
34.3 |
821(11) |
|
288 |
5.78×10−9 |
2.55×10−10 |
31.4 |
578(10) |
|
293 |
6.92×10−9 |
3.32×10−10 |
28.9 |
484(7) |
|
298 |
8.23×10−9 |
4.27×10−10 |
26.7 |
346(4) |
|
303 |
9.73×10−9 |
5.45×10−10 |
24.7 |
299(7) |
Summarizing the computational results, they confirm the key role of hydrogen tunneling in the reaction mechanism, and provide a detailed mechanism for this reaction (Scheme 2). Slight differences with the accepted electrophilic mechanism for the hydrolysis of silanes have been found: i) the η1‐silane complex (C1′ in Scheme 2) is not involved in the catalytic cycle; ii) the silylium cation is a pentacoordinated species ([Et3Si(OH2)2]+, C2 in scheme 2). Scheme 2 shows the new alternative proposed mechanism for the hydrolysis of triethylsilane and the green dashed line represents the shortcut in the reaction pathway due to the quantum tunneling.
Scheme 2.
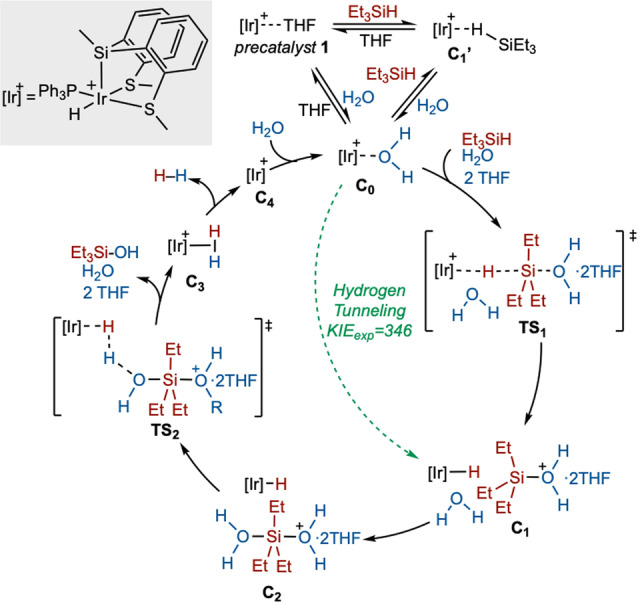
Catalytic cycle proposed for the hydrolysis of Et3SiH when 1 is used as catalyst.
Conclusion
The cationic hydrido‐silyl‐IrIII complex 1, behaved as an efficient precatalyst for the hydrolysis/alcoholysis of tertiary silanes leading to the release of one equivalent of dihydrogen and the corresponding silanols/silylethers. Kinetic studies on the process revealed k Si‐H/k Si‐D=346, which represents the largest one involving Si−H/D bonds observed up to date. This huge KIE in addition to the calculated Arrhenius factor and the difference in the observed activation energy are experimental indications of the participation of hydrogen quantum tunneling in the RDS of the process at room temperature. Detailed DFT calculation on the reaction mechanism allowed us to propose a slightly modified electrophilic mechanism for the reaction compared to the previously reported ones. In addition to this, theoretically calculated KIEs at different temperatures using the WKB one‐dimensional tunneling correction, support the importance of quantum tunneling. To the best of our knowledge, this is the first reported case of hydrosilane activation with the participation of quantum tunneling.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This research was supported by the Universidad del País Vasco (UPV/EHU) (GIU13/06), Ministerio de Economía y Competitividad (PID2019‐111281GB‐00), Gobierno Vasco (IT1880‐19 and IT1254‐19). Technical and human support provided by IZO‐SGI, SGIKER (UPV/EHU, MICINN, GV/EJERDF and ESF), is gratefully acknowledged for assistance and generous allocation of computational resources. N.A. is grateful to Diputacion Foral de Gipuzkoa (OF215/2016), and M.A.H. and Z.F. to IKERBASQUE for funding. We would like to thank Dr. Eugene E. Kwan for his support and fruitful discussion using PyQuiver program.
N. Almenara, M. A. Garralda, X. Lopez, J. M. Matxain, Z. Freixa, M. A. Huertos, Angew. Chem. Int. Ed. 2022, 61, e202204558; Angew. Chem. 2022, 134, e202204558.
Contributor Information
Dr. Jon M. Matxain, Email: jonmattin.matxain@ehu.es.
Dr. Zoraida Freixa, Email: zoraida_freixa@ehu.es.
Dr. Miguel A. Huertos, Email: miguelangel.huertos@ehu.es.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Murugavel R., Voigt A., Walawalkar M. G., Roesky H. W., Chem. Rev. 1996, 96, 2205–2236. [DOI] [PubMed] [Google Scholar]
- 2. Denmark S. E., Regens C. S., Acc. Chem. Res. 2008, 41, 1486–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eberle U., Felderhoff M., Schüth F., Angew. Chem. Int. Ed. 2009, 48, 6608–6630; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6732–6757. [Google Scholar]
- 4. Walsh R., Acc. Chem. Res. 1981, 14, 246–252. [Google Scholar]
- 5. Asao N., Ihikawa Y., Hatakeyama N., Menggencateer, Tamamoto Y., Chen M., Zhang W., Inoue A., Angew. Chem. Int. Ed. 2010, 49, 10093–10095; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 10291–10293. [Google Scholar]
- 6. John J., Gravel E., Hagege A., Li H., Gacoin T., Doris D., Angew. Chem. Int. Ed. 2011, 50, 7533–7536; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7675–7678. [Google Scholar]
- 7. Teo A. K. L., Fan W. Y., Chem. Commun. 2014, 50, 7191–7194. [DOI] [PubMed] [Google Scholar]
- 8. Lee M., Ko S., Chang S., J. Am. Chem. Soc. 2000, 122, 12011–12012. [Google Scholar]
- 9. Tan S. T., Kee J. W., Fan W. Y., Organometallics 2011, 30, 4008–4013. [Google Scholar]
- 10. Ison E. A., Corbin R. A., Abu-Omar M. M., J. Am. Chem. Soc. 2005, 127, 11938–11939. [DOI] [PubMed] [Google Scholar]
- 11. Corbin R. A., Ison E. A., Abu-Omar M. M., Dalton Trans. 2009, 2850–2855. [DOI] [PubMed] [Google Scholar]
- 12. Krüger A., Albrecht M., Chem. Eur. J. 2012, 18, 652–658. [DOI] [PubMed] [Google Scholar]
- 13. Yu M., Jing M., Fu X., Inorg. Chem. 2013, 52, 10741–10743. [DOI] [PubMed] [Google Scholar]
- 14.The catalytic activity of [Ir(cod)Cl]2 has been already reported under other reactions conditions: Lee Y., Seomoon D., Kim S., Han H., Chang S., Lee P. H., J. Org. Chem. 2004, 69, 1741–1743. [DOI] [PubMed] [Google Scholar]
- 15. Luo X.-L., Crabtree R. H., J. Am. Chem. Soc. 1989, 111, 2527–2535. [Google Scholar]
- 16. Field L. D., Messerle B. A., Rehr M., Soler L. P., Hambley T. W., Organometallics 2003, 22, 2387–2395. [Google Scholar]
- 17. Garcés K., Fernández-Alvarez F. J., Polo V., Lalrempuia R., Pérez-Torrente J. J., Oro L. A., ChemCatChem 2014, 6, 1691–1697. [Google Scholar]
- 18. Aliaga-Lavrijsen M., Iglesias M., Cebollada A., Garces K., Garcia N., Sanz Miguel P. J., Fernandez-Alvarez F. J., Perez-Torrente J. J., Oro L. A., Organometallics 2015, 34, 2378–2385. [Google Scholar]
- 19. Hamdaoui M., Ney M., Sarda V., Karmazin L., Bailly C., Sieffert N., Dohm S., Hansen A., Grimme S., Djukic J.-P., Organometallics 2016, 35, 2207–2223. [Google Scholar]
- 20. Ventura-Espinosa D., Sabater S., Carretero-Cerdan A., Baya M., Mata J. A., ACS Catal. 2018, 8, 2558–2566. [Google Scholar]
- 21. Barber D. E., Lu Z., Richardson T., Crabtree R. H., Inorg. Chem. 1992, 31, 4709–7411. [Google Scholar]
- 22. Matthews S. L., Pons V., Heinekey D. M., Inorg. Chem. 2006, 45, 6453–6459. [DOI] [PubMed] [Google Scholar]
- 23. Doyle M. P., High K. G., Bagheri V., Pieters R. J., Lewis P. J., Pearson M. P., J. Org. Chem. 1990, 55, 6082–6086. [Google Scholar]
- 24. Chang S., Scharrer E., Brookhart M., J. Mol. Catal. A 1998, 130, 107–119. [Google Scholar]
- 25. Bühl M., Mauschick F. T., Organometallics 2003, 22, 1422–1431. [Google Scholar]
- 26. Wang W., Wang J., Huang L., Wei H., Catal. Sci. Technol. 2015, 5, 2157–2166. [Google Scholar]
- 27. Robert T., Ostreich M., Angew. Chem. Int. Ed. 2013, 52, 5216–5218; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5324–5326. [Google Scholar]
- 28. Iglesias M., Fernandez-Alvarez F. J., Oro L. A., ChemCatChem 2014, 6, 2486–2489. [Google Scholar]
- 29. Azpeitia S., Garralda M. A., Huertos M. A., ChemCatChem 2017, 9, 1901–1905. [Google Scholar]
- 30. Azpeitia S., Rodriguez-Dieguez A., Garralda M. A., Huertos M. A., ChemCatChem 2018, 10, 2210–2213. [Google Scholar]
- 31. Bell R. P. in The Tunnel Effect in Chemistry, Chapman and Hall, London, 1980. [Google Scholar]
- 32. Isotope Effects in Chemistry and Biology (Eds.: Kohen A., Limbach H.-H.), Taylor and Francis, Boca Raton, 2006. [Google Scholar]
- 33. Meisner J., Kästner J., Angew. Chem. Int. Ed. 2016, 55, 5400–5413; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5488–5502. [Google Scholar]
- 34. Schreiner P. R., Trends Chem. 2020, 2, 980–989. [Google Scholar]
- 35. Falivene L., Credendino R., Poater A., Petta A., Serra L., Oliva R., Scarano V., Cavallo L., Organometallics 2016, 35, 2286–2293. [Google Scholar]
- 36. Adams G. M., Ryan D. E., Beattie N. A., McKay A. I., Lloyd-Jones G. C., Weller A. S., ACS Catal. 2019, 9, 3657–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Friedrich A., Drees M., Schmedt auf der Günne J., Schneider S., J. Am. Chem. Soc. 2009, 131, 17552–17553. [DOI] [PubMed] [Google Scholar]
- 38. Luo N., Liao J., Ouyang L., Wen H., Zhong Y., Liu J., Tang W., Luo R., Organometallics 2020, 39, 165–171. [Google Scholar]
- 39. Yang J., White P. S., Schauer C. K., Brookhart M., Angew. Chem. Int. Ed. 2008, 47, 4141–4143; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4209–4211. [Google Scholar]
- 40. Wang W.-H., Hull J. F., Muckerman J. T., Fujita E., Hirose T., Himeda Y., Chem. Eur. J. 2012, 18, 9397–9404. [DOI] [PubMed] [Google Scholar]
- 41. Cong Z., Kinemuchi H., Kurahashi T., Fujii H., Inorg. Chem. 2014, 53, 10632–10641. [DOI] [PubMed] [Google Scholar]
- 42. Kwart H., Acc. Chem. Res. 1982, 15, 401–408. [Google Scholar]
- 43. Dix E. J., Herman M. S., Goodman J. L., J. Am. Chem. Soc. 1993, 115, 10424–10425. [Google Scholar]
- 44. Hohenberg P., Kohn W., Phys. Rev. [Sect.] B 1964, 136, B864–B871. [Google Scholar]
- 45. Kohn W., Sham L. J., Phys. Rev. [Sect.] A 1965, 140, A1133–A1138. [Google Scholar]
- 46. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]
- 47.Gaussian 16, Rev. B.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, F. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Wallingford, CT, 2016.
- 48. Scalmani G., Frisch M. J., J. Chem. Phys. 2010, 132, 114110. [DOI] [PubMed] [Google Scholar]
- 49.The cartesian coordinates of this structure, along with the cartesian coordinates of all other species discussed hereafter are given in the Supporting Information.
- 50. Reed A. E., Weinstock R. B., Weinhold F., J. Chem. Phys. 1985, 83, 735–746. [Google Scholar]
- 51. Reed A. E., Weinhold F., J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar]
- 52. Reed A. E., Curtiss L. A., Weinhold F., Chem. Rev. 1988, 88, 899–926. [Google Scholar]
- 53. Olsson L., Ottoson C.-H., Cremer D., J. Am. Chem. Soc. 1995, 117, 7460–7479. [Google Scholar]
- 54. Bigeleisen J., Mayer M. G., J. Chem. Phys. 1947, 15, 261–267. [Google Scholar]
- 55. Wolfsberg M., Acc. Chem. Res. 1972, 5, 225–233. [Google Scholar]
- 56.T. L. Anderson, E. E. Kwan, PyQuiver 2020, www.github.com/ekwan/PyQuiver.
- 57. Bercaw J. E., Chen G. S., Labinger J. A., Lin B.-L., Organometallics 2010, 29, 4354–4359. [Google Scholar]
- 58. Wigner E. P., Phys. Chem. 1932, 19B, 203. [Google Scholar]
- 59. Garrett B. C., Truhlar D. G., J. Phys. Chem. 1979, 83, 200–203. [Google Scholar]
- 60. Bell R. P., Chem. Soc. Rev. 1974, 3, 513–544. [Google Scholar]
- 61. Karandashev K., Xu Z.-H., Meuwly M., Vaníček J., Richardson J. O., Struct. Dyn. 2017, 4, 061501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Froman N., Froman P. O., Wkb approximation, contributions to the theory, North Holland Publishing Company, Amsterdam, 1965. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.