

Abstract
Clonal hematopoiesis (CH) is an aging-associated condition characterized by the clonal outgrowth of mutated pre-leukemic cells. Individuals with CH are at an increased risk of developing hematopoietic malignancies. Here, we describe a novel animal model carrying a recurrent TET2 missense mutation, frequently found in CH and leukemic patients. In a fashion similar to CH, animals show signs of disease late in life when they develop a wide range of myeloid neoplasms, including acute myeloid leukemia (AML). Using single cell transcriptomic profiling of the bone marrow, we show that disease progression in aged animals correlates with an enhanced inflammatory response and the emergence of an aberrant inflammatory monocytic cell population. The gene signature characteristic of this inflammatory population is associated to poor prognosis in AML patients. Our study illustrates an example of collaboration between a genetic lesion found in CH and inflammation, leading to transformation and the establishment of blood neoplasms.
INTRODUCTION
Hematopoiesis is a tightly-regulated process where hematopoietic stem cells (HSC) differentiate into functional and mature subsets evenly, with the progressive plasticity to meet the needs of various physiological demands such as regeneration and infections(1). Through aging, the acquisition of somatic mutations in hematologic malignancy-associated genes (leads to the clonal expansion of progenitors, resulting in an indolent condition referred to as Clonal Hematopoiesis of Indeterminate Potential (CHIP), also known as Age-Related Clonal Hematopoiesis (ARCH)(2–4). As a consequence, the mutant clone gains selective advantage leading to an increased contribution of cell populations within the normal range (defined with a variant of allele frequency > 2%)(5). Interestingly, the rate of progression to a bona fide myeloid neoplasm in individuals with CHIP is only ~1% per year(2), suggesting that additional contributing factors collaborate to promote neoplasia. Still, the specific events preventing or causing the progression to malignancy are mainly unknown.
The epigenetic regulator ten-eleven-translocation enzyme 2 (TET2) is among the most commonly mutated genes in both CHIP and myeloid malignancies. TET2 mutations occur in ~30%−50% of myelodysplastic syndromes (MDS), ~13% of myeloproliferative neoplasms (MPN), and ~10%−30% of acute myeloid leukemia (AML) cases(6),(7). Together with TET1 and TET3, TET2 belongs to a family of proteins which comprise of 2-oxoglutarate- and FE2(II) dependent dioxygenase activity and catalyze the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and its iterative products(8). Murine modeling has shown that deletion of Tet2 is associated with an increase in HSC self-renewal, arrest in differentiation, myelomonocytic bias, and myeloid transformation(9–12). However, the precise function of TET2 in suppressing clonal hematopoiesis and leukemia remains mainly unknown.
To better characterize the molecular basis underlying the phenomenon of TET2-driven clonal evolution leading to aberrancies in hematopoiesis, we have generated a genetically engineered mouse strain, carrying one of the most frequent TET2 missense mutations found in human myeloid neoplasms (p.H1881R) and individuals with CHIP. We leveraged single-cell technologies to delineate the transcriptional landscape of an aging-induced progression between an indolent state (similar to human CHIP) and bona fide myeloid malignancy. Surprisingly, our data demonstrate that an enhanced inflammatory response is sufficient to collaborate with Tet2 deficiency in promoting myeloid neoplasia. Specifically, our results reveal that inflammation transcriptionally rewires Tet2 mutant to differentiate into cells with the characteristics of inflammatory monocytes that egress the bone marrow to infiltrate peripheral tissues. Interestingly, our data suggest that the inflammatory transcriptional program expressed by these aberrant monocytes has importance beyond TET2 associated leukemogenesis, as its presence strongly associates with poor prognosis in AML patients. Together, our work provides novel insights on the mechanisms of inflammation-induced myeloid neoplasia, potentially relevant for the generation of early prognostic markers that may predict the progression to myeloid malignancy from a precursor CHIP state and for the development of target therapies.
RESULTS
Biological function of TET2H1881R, a recurring mutation in CH and myeloid malignancy
In addition to nonsense/frameshift mutations, CH/MDS/AML patients frequently present with a broad spectrum of TET2 missense mutations, most commonly within the two highly conserved regions comprising the catalytic domain (AA1140-1478 and AA1845–2002, Fig. 1A). The impact of these diverse mutations in the catalytic and non-catalytic functions of Tet2 and malignant transformation is currently unclear as the vast majority of studies utilize Tet2 loss of function models(9–11,13). One of the most frequent missense mutations impacts the Histidine 1881, essential for coordinating the active site iron, a critical cofactor for Tet2 catalytic activity(6). Previous and our own in vitro studies have suggested that the overexpression of Tet2 H1881R in 293T cells leads to a decreased 5-hmC compared to the wild-type, suggesting that this mutant is catalytic dead(8). (Fig. 1B–C). To better characterize the effects of the H1881R mutation on the structure and function of TET2, molecular dynamics (MD) simulations(14) were carried out on wild-type (WT) TET2 and the TET2H1881R mutant with three possible oxidized substrates: 5mC, 5hmC, and 5fC(15). Root mean squared fluctuations (RMSF), sugar puckering, correlated motions, and energy decomposition analyses (EDA) were calculated from the resulting trajectories. EDA provides a qualitative approach to investigate the change in average interaction energy over the calculated ensemble. This analysis showed a reduction in the total interaction (destabilization) of 10.1 (32.2, 10.5) kcal/mol for 5mC (5hmC, 5fC) for the H1881R mutant compared with WT TET2. The largest destabilization was observed for 5hmC, in which the substrate is stabilized overall in WT TET2 by 32.2 kcal/mol compared with the TET2 H1881R mutant. Several residues around the active site and beyond exhibit significant changes in the interaction between WT and H1881R TET2 (Fig. 1D). The addition of a positive charge drives the decrease in stability of 5hmC upon mutation at position 1881 and promotes differences in how the substrate base is oriented in the active site. By contrast, the H1881 residue becomes more stable upon mutation to Arg by 24.4 kcal/mol, owing to the favorable interaction between the positive charge on the residue and the DNA substrate, even in the presence of the divalent cation in the active site. The disruption of the active site alignment is reflected in the changes to the backbone puckering and delta and chi angles of the ribose on the substrate base (Fig. 1E–G), and in changes in RMSF and correlated motions of TET2 (SI), where less correlated motions are observed for the cancer variant compared to WT. Further, the intermolecular interaction energy differences per residue of WT-5hmC and H1881R-5hmC (ΔE) presented in Figure 1D show that R1261 significantly stabilizes WT by ~18 kcal/mol, in agreement with our previous studies on TET2 oxidation using QM/MM modeling(16,17). Additionally, LC/MS/MS analysis of replicate transfections using the H1881R mutant and controls (WT TET2, catalytically inactive TET2 and an empty vector) demonstrated that the H1881R mutant is inactive, with no detectable 5hmC/5fC/5caC generated upon expression (Fig. 1H). Together, these results underscore the importance of H1881 in the coordination of the divalent cation, as well as in the stability and orientation of the DNA substrate in the active site, suggesting that the TET2H1881R mutant is unlikely to be able to catalyze the oxidation reaction on any of the oxidation states.
Figure 1. TET2 p.H1881R mutations impacts structure and induces a catalytic dead enzyme.
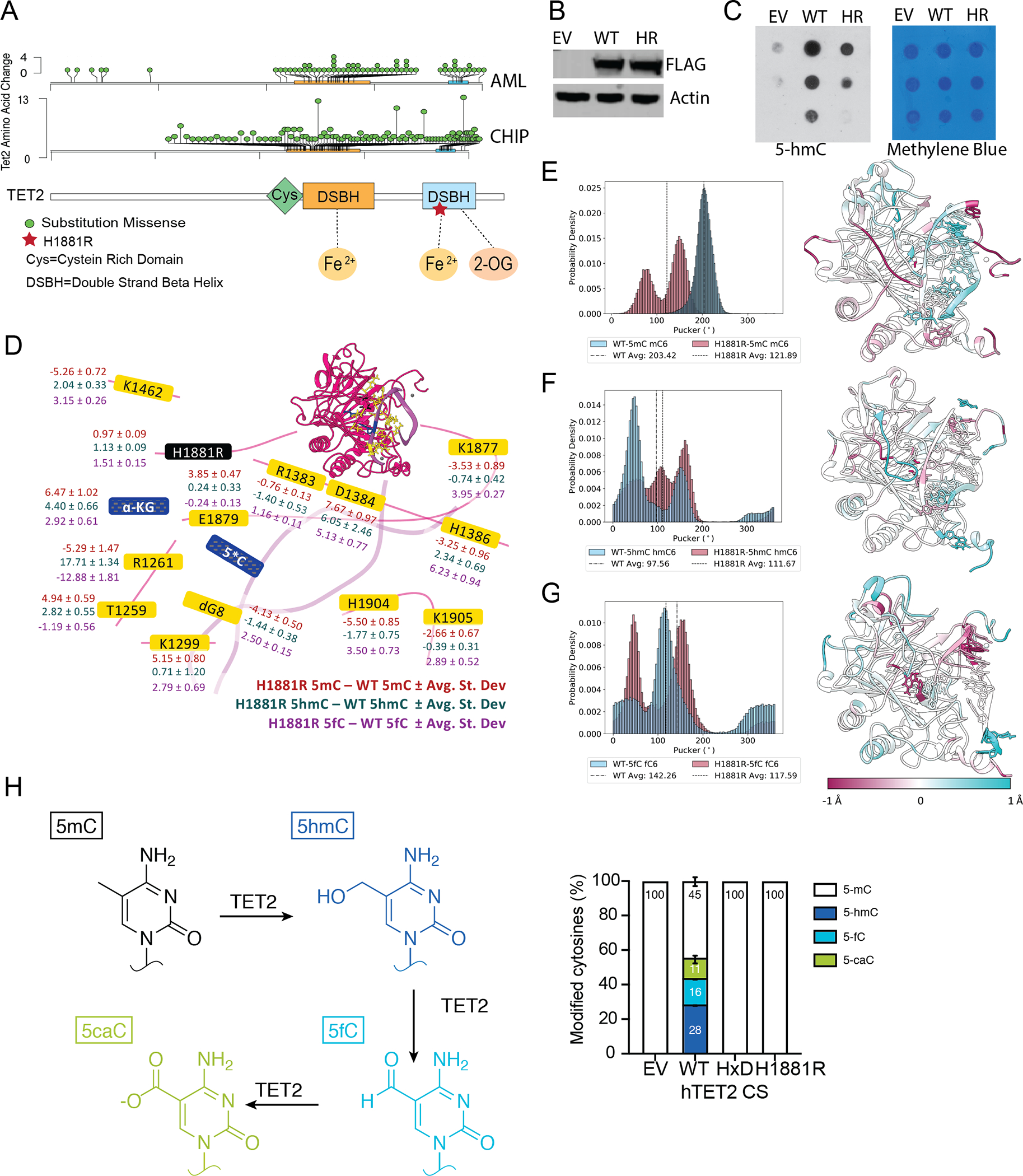
A, Lolliplot representation of TET2 missense mutations found in AML and CHIP samples acquired from previously published datasets39,64 and TCGA.
B, Expression levels of TET2-Flag fusion protein in 293T cells transfected with Empty-Vector (EV), TET2-FLAG (WT) and TET2-H1881R-FLAG (HR).
C, 5hmC DNA dot blot of samples collected from 293T cells transfected with Empty-Vector (EV), TET2-FLAG (WT) and TET2-H1881R-FLAG (HR) (left). Methylene Blue staining of the DNA is showed as a loading control (Right).
D, Differences in total interaction energy (Coulomb and van der Waals) of H1881R – WT for each of the modeled oxidation states (5-mC, 5-hmC, and 5-fC). These residues changed by at least |1| kcal/mol in their interactions with at least 1 oxidation state.
E-G, Sugar puckering distribution and averages for the modified cytosine base (left), and differences in root mean square fluctuation (RMSF) for H1881R – WT (right) mapped onto the protein structure in the (E) 5-mC (F) 5-hmC and (G) 5-fC systems. The WT is shown in blue and the H1881R mutant is shown in pink.
H, Schematic of TET2-catalyzed sequential oxidations of 5mC to 5hmC and to 5fC and 5caC (top). Genomic levels of 5mC, 5hmC, 5fC, and 5caC modifications produced by each TET2 construct overexpressed in HEK293T cells shown as the percent of total modified cytosine bases. EV, empty vector; WT, wild-type; HxD, catalytically inactive mutant; H1881R, mutant. Percentages were derived from 3 independent experiments, with mean and associated standard error shown.
An animal model carrying a patient-derived TET2 p.H1881R missense mutation
To better understand how such missense mutations of TET2 promote clonal hematopoiesis and delineate the molecular mechanisms inducing progression to malignancies, we generated a mouse model that expresses HuTET2 p.H1881R (Mm Tet2 H1794R) from the endogenous locus (referred as HR, Fig. S1A). RNA expression levels of the wild-type and HR allele were comparable in stem and progenitor Tet2HR/+ (here referred to as Tet2HR) cells (HSPC: Lineage c-Kit+ cells, Fig. 2A). Likewise, wild-type and Tet2HR showed similar Tet2 protein levels, indicating that the mutation doesn’t affect the protein stability (Fig. 2B). In contrast, the endogenous expression of Tet2HR mutant in one allele was sufficient to decrease the levels of 5-hydroxy-methylation of the DNA in these progenitor cells (Fig. 2C), supporting the molecular dynamics predictions illustrated in Figure 1. Tet2HR and Tet2HR/HR mice were born in the expected mendelian ratios and show normal development. Similar to Tet2KO cells(9), Tet2HR HSPCs collected from mice 8–16 weeks old showed increased colony-forming re-plating capacity in vitro (Fig. S1B) and increased competitive reconstitution capacity in vivo (Fig. S1C). However, we observed that young Tet2HR mice showed normal hematopoiesis. In contrast, upon aging, approximately 90% of the Tet2HR mutant cohorts developed a spectrum of myeloid neoplasms (with latencies between 1.2–2 years) and increased mortality (Fig. 2D). To evaluate the potential role of Tet2 missense mutations in leukemic transformation, we assessed for cooperativity between Tet2HR mutation and genetic lesions that have previously been shown to co-occur in human AML patients(18–20). We observed that the Tet2HR mutation cooperates with both Flt3-ITD, AML-ETO9a translocations as well as cohesin (Stag2) mutations leading to acute myeloid leukemias with significantly faster kinetics (Fig. S1D–K). Surprisingly, whole exome sequencing of Tet2HR mice tumor cells (from 8 different Tet2HR mice) showed that not all tumors carry additional somatic mutations in genes frequently mutated in hematologic malignancies (Fig. S1L.) Such findings suggested that progression to transformation can be in some cases independent of acquisition of secondary mutations in agreement with emerging notions of epigenetic and environmental triggers in human cancer.
Figure 2. Tet2 H1881R mutation results in a spectrum of myeloid malignancies.
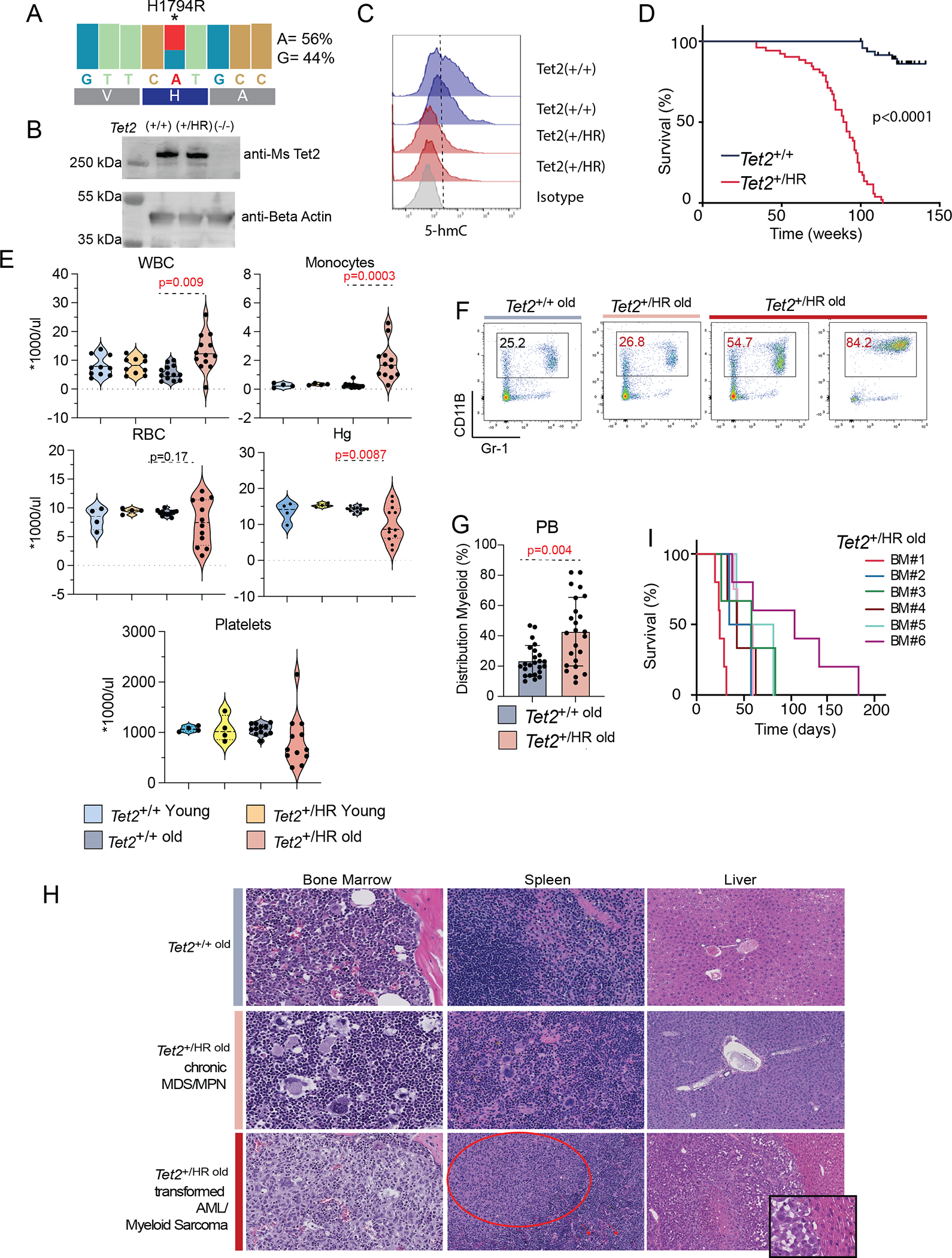
A, Expression levels (measured by RNA-seq) of Tet2 wild-type and H1794R alleles in Tet2+/HR HSPCs (Lineageneg Kit+ cells).
B, Western blot analysis of Tet2 protein expression from Tet2+/+, Tet2+/HR, and Tet2−/− mice bone marrow cells.
C, Flow cytometry analysis of 5hmC expression levels in Tet2+/+ and Tet2+/HR HSPCs (Lineageneg Kit+ cells).
D, Kaplan Meier survival curves of Tet2WT and Tet2HR
E, Levels of White Blood Counts (WBC), Monocytes, Red Blood Cells (RBC), Hemoglobin (Hg) and platelets from peripheral blood samples collected from young and old Tet2HR and control samples
F, Representative flow cytometry analysis plots showing myeloid cell population percentages (Cd11B and GR-1) in peripheral blood of Tet2WT and Tet2HR old mice.
G, Percentage of myeloid cells (Cd11B and Gr-1+) in the peripheral blood of Tet2WT and Tet2HR mice.
H, Top panel: Tet2WT old BM shows orderly trilineage hematopoiesis, spleen and liver. Middle panel: In Tet2HR old, the chronic (mild) phase of disease shows MDS/MPN-like features with proliferation of atypical megakaryocytes and increased myeloid:erythroid ratio; myeloid maturation is preserved. The splenic red pulp is infiltrated by atypical megakaryocytes. The liver was uninvolved. Bottom panel: “transformed” or “severe” disease shows BM involvement by acute myeloid leukemia with sheets of blasts. The splenic red pulp is infiltrated by abnormal megakaryocytes (right half; arrow); in additional nodular aggregate of mononuclear cells is also present, largely replacing normal splenic tissue (myeloid sarcoma); and the liver is also involved by myeloid sarcoma. I, Kaplan Meier curve of sub-lethally irradiated mice transplanted with total bone marrow cells collected from Tet2HR mice with transformed disease.
The myeloid neoplasms developing in these animals with long latency showed a broad spectrum of aggressiveness and disease manifestation. Overall, Tet2HR-induced myeloid malignancies were associated with leukocytosis, monocytosis, frequent anemia, and thrombocytopenia (Fig. 2E). Peripheral blood (PB) immunophenotyping of the Tet2HR mice older than 14 months revealed a significant expansion of myeloid cells (CD11b+, Gr-1+, Fig. 2F–G). Additionally, in the bone marrow, the HSPC compartment (LSK, Lineageneg c-Kit+ Sca-1+ cells) was significantly increased in old Tet2HR mice, (Fig. S2A), correlating with an expansion of myeloid multipotent progenitors (MPP3) and a loss of lymphoid multipotent progenitors (MPP4, Fig. S2B–D). Compared with Tet2WT and Tet2HR young mice, old Tet2HR showed significantly larger spleen size (Fig. S2E) driven by extramedullary hematopoiesis (Fig. S2F–H). Morphologic evaluation of the bone marrow and extramedullary tissues of the Tet2HR mice (spleen, liver, lymph node) showed a spectrum of disease ranging from a more chronic phase of the disease with features of myelodysplastic/myeloproliferative neoplasm (characterized by atypical megakaryocytic hyperplasia and increased myeloid: erythroid ratio with preserved maturation of the myeloid lineage in the BM, often with various degrees of extramedullary involvement predominantly involving the spleen and liver, Fig. 2H middle panel), to a fully “transformed” AML with involvement of extramedullary tissues manifesting frequently as myeloid sarcoma (Fig. 2H, bottom panel). Finally, sub-lethally irradiated mice transplanted with bone marrow cells from aged mice with AML phenotype developed myeloid neoplasms more and the mice more rapidly succumbed to the disease (Fig. 2I), confirming that Tet2HR leukemic cells were fully transformed. Together, our data indicate that although the loss of Tet2 catalytic activity increases the self-renewal potential of the progenitor cells, it is not sufficient to promote transformation in Tet2HR young mice and suggest that secondary events, acquired through aging, collaborate with the loss of Tet2 catalytic activity to induce myeloid neoplasms, including AML.
Inflammatory signals correlate with the acquisition of malignancy in Tet2HR mice
To delineate transcriptional events contributing to the progression to AML in the Tet2HR mutant animal model, we performed single-cell RNA-sequencing (scRNA-Seq) on total bone marrow cells collected from young animals (8–10 weeks old) with no signs of disease and older animals (>60 weeks old) showing both chronic (MDS/MPN-like) and transformed myeloid disease (AML-like). Cells with more than 10% of mitochondrial reads and cells expressing less than 400 genes or more than 6000 genes were removed from the analysis. Following this quality control, we collected 37,355 transcriptomes from young and old wild-type and Tet2HR mice (Fig. 3A). Seurat anchor-based integration taking the 2,000 most variable genes as input to account for batch effect(21). The resulting scaled data was used to calculate PCA, followed by Uniform Manifold Approximation and Projection visualization (UMAP)(22) using the first 20 principal components. Next, cells were assigned to their specific cell-type identity in an iterative fashion. First, unbiased clusters were given their respective hematopoietic lineage based on the expression of previously established lineage markers: hematopoietic stem and progenitor cells (HSPCs, Meis1, Kit), monocytes and dendritic cells (Irf8, Ly6c), neutrophils (Cebpe, Ly6G) B-cells (Cd79, Vpreb3, Bach2), T-cells (Cd3, Cd8, Cd4, Cd28) and erythroid (Klf1, Hba-a2, Rhd) Fig. S3A–C). Additionally, each cell type was subset, re-clustered, and assigned to a more specific cell type based on the expression of well-known markers (Fig. S3D and Supp.Table 1 and Supp.Table 2). This analysis resulted in 19 different cell states, including HSPCs, common lymphoid progenitors, B-cells, plasma cells, T-cells, NK-cells, erythroid cells, myeloid progenitors, monoblasts, classical monocytes, non-classical macrophages, conventional dendritic cells, plasmacytoid dendritic cells, basophils, pro-neutrophils, pre-neutrophils and neutrophils (Fig. 3B). Both wild-type and Tet2HR young transcriptomes appeared intermingling through the different cell types showing no significant differences in their distribution in the UMAP. Moreover, differential expression analysis of young animal transcriptomes showed no statistical significance (Fig. 3C). Cell cycle assignments per cell were determined, but no differences between conditions were found (Fig S3A). In contrast, our analysis revealed significant transcriptional differences later in life in animals that develop transformed disease (AML-like phenotype) compared to aged-matched wild-type counterparts (Fig. 3C–D). In agreement with the immunophenotype analysis, loss-of-Tet2 catalytic activity in old mice resulted in an increased percentage of monocytes and a decreased percentage of granulocytes and B-cells, especially in mice with the transformed phenotype (Fig. 3C–D and Fig. S3E). Furthermore, the mutant granulocytes and monocytes from old mice accumulated in occupied distinct regions of the UMAP, suggesting that these cells shared expression profiles distinct from the wild-type controls. These results indicate that aging-mediated acquisition of aberrant transcriptional programs promotes transformation in TET2 mutant-associated myeloid neoplasms.
Figure 3. Inflammation correlates with Tet2HR-induced transformation.
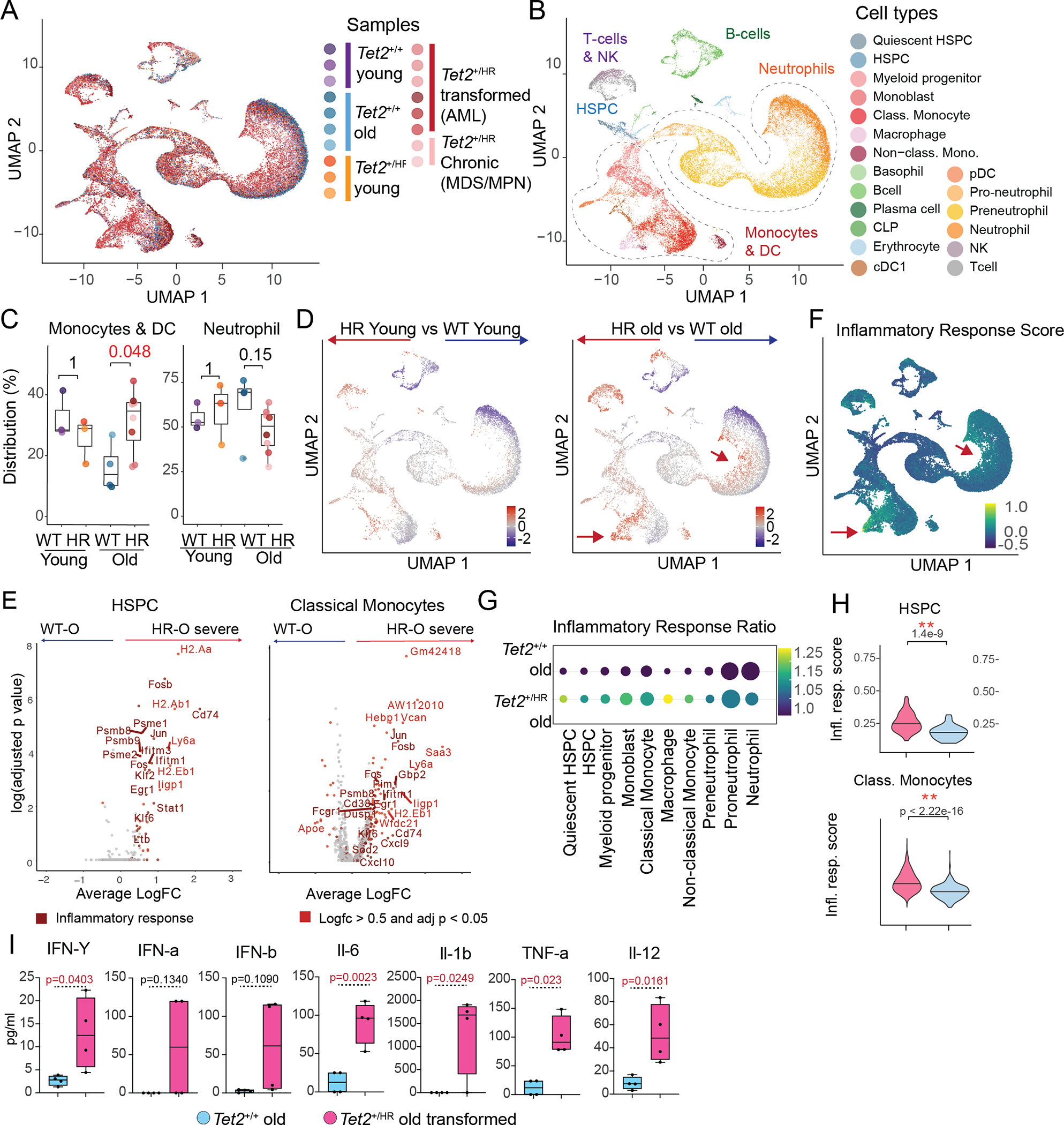
A, Uniform manifold approximation and projection (UMAP) representation of 37,355 cells collected from whole bone marrow of 18 mice, including Tet2WT young (n=3), Tet2HR young (n=3), Tet2WT old (n = 4), and Tet2HR old (transformed n = 6, chronic n = 2).
B, Uniform manifold approximation and projection (UMAP) representation of 37,355 cells collected from whole bone marrow from Tet2WT young (n=3), Tet2HR young (n=3), Tet2WT old (n = 4), and Tet2HR old (transformed n = 6, chronic n = 2) color code by cell type.
C, Percentage of Monocyte and Neutrophils, relative to the total bone marrow cells, separated by mutation status and age.
D, Heatmap of the density ratio of Tet2HR vs Tet2WT cells from young and old mice projected into the UMAP.
E, Volcano plots of differentially expressed genes between Tet2WT old and Tet2HR old in HSPCs and Classical Monocytes. Genes with a log.fc > 0.5 and and adj.pval < 0.05 are highlighted in bright red and genes involved in Interferon response are highlighted in dark red.
F, Interferon Response score calculated from the Hallmarks of Interferon Response projected on the UMAP.
G, Dot plot detailing the difference in interferon score between Tet2WT old and Tet2HR old in the indicated populations. The size of the dot corresponds to the relative number of cells in each cell type, and the color corresponds to the fold change in mean IFN score between Tet2WT old and Tet2HR.
H, Violin plots of Interferon response score in HSPCs and Classical Monocytes.
I, Box plots showing levels of key pro-inflammatory cytokines in the bone marrow fluids of Tet2WT old and Tet2HR old with transformed disease.
We next focused our analysis in the myelomonocytic compartment. To identify potential oncogenic pathways in Tet2HR cells, we performed differential expression analysis within each cell population between age-matched mutant and wild-type HSPCs and the different myeloid cell states. Tet2HR cells from young mice showed no significant differences in gene expression compared to young wild-type cells (data not shown). In contrast, Tet2HR cells from aged mice showed significantly differentially expressed genes in multiple cell states. Specifically, we identified 31 differentially expressed genes (DEG) in HSPCs and 262 in classical CD14+ monocytes (Fig. 3E). We did not observe upregulation of established oncogenic pathways. However, gene-set enrichment analysis identified inflammation-related signatures such as interferon gamma, interferon alpha and TNF alpha hallmark signatures as the primary upregulated signatures in Tet2HR with transformed disease. We created an overarching inflammatory response gene signature combining several inflammatory response signatures from the hallmark database. We found this inflammatory response pathway upregulated in different cell populations, from the HSPCs to the differentiated lineages of the myeloid compartment (Fig. 3F–H). Furthermore, we plot the inflammatory response score on the UMAP representation and we observed that the highest values correlated with the regions with increased density of Tet2HR myeloid cells (Fig. 3F) representing the major transcriptional differences in the transformed samples. To gain deeper transcriptional coverage, we expanded the transcriptional profile of the inflammatory monocytes. We sorted total monocytes from Tet2WT and Tet2HR mice and performed bulk-RNA sequencing. This analysis helped us confirm our finding of increased inflammation in the monocyte compartment of the Tet2HR mice, this time using bulk RNA sequencing of the monocyte compartment of Tet2WT and Tet2HR mice. Indeed, genes related to interferon response, inflammatory response and JAK/STAT signaling were enriched in Tet2HR monocytes (Fig S4 A–B).
Finally, and due to this induction of inflammatory gene signatures, we quantified the levels of proinflammatory cytokines in the bone marrow fluid of aged wild-type and Tet2HR animals with aggressive neoplasms. We found an accumulation of a number of inflammatory cytokines, including IFN-γ, IL-6, IL-1b, TNF-α, and IL-12, suggesting the establishment of a pro-inflammatory micro-environment in the bone marrow of these animals (Fig. 3I). Overall, all these data indicate that the progression to myeloid transformation correlates with an increased inflammatory response signaling in the myeloid compartment of the diseased mice.
Inflammation induces the emergence of an aberrant monocyte population in Tet2HR mice
We next subset the transcriptomes progressing from the HSPCs to the differentiated monocytes and generated a new UMAP (Fig. 4A). Unbiased clustering coupled with cell calling revealed 7 states: HSPCs, myeloid progenitors, monoblast/promonocyte, classical monocytes, non-classical monocytes, macrophages and a distinct “monocytic-like” population present predominantly in Tet2HR mice with transformed disease (Fig. 4A–C, Fig. S4A–B and Supp.Table 3). This separate cluster of monocytes (referred henceforth as MHC IIhigh monocytes due to its high levels of expression for MHC-II) expressed transcriptional markers similar to the classical monocytes and different from non-classical monocytes and dendritic cells (Fig. 4B–C and Fig. S4.C–E). In addition, MHC IIhigh monocytes were characterized by a unique inflammatory signature including genes related to the interferon response pathway (Ly6a, Irf1, Irf3, Ifi204, Ifi209, Ifitm3, Isg15, Cxcl9, Cxl10), components of the Major Histocompatibility Complex (MCH) Class II (Cd74, H2-Aa, H2-Ab1…) as well as crucial inflammatory transcription factors, including Stat1, Jun, Junb, Fos, and Fosb. Furthermore, we observed that this unique inflammatory response signature was also imprinted at earlier stages of monocyte development (in a subset of HSPCs, myeloid progenitors, and monoblast cells) in Tet2HR mice (Fig. 4B), suggesting that emergence of inflammation is also apparent at earlier differentiation stages and could shape their differentiation potential. To assess how inflammatory response affects the differentiation of the myeloid lineage, we used Monocle3 to calculate a pseudotime ordering along monocytic differentiation and to project differentiation trajectories onto the UMAP(23). We found that Tet2HR mice with histologically AML/myeloid sarcoma disease showed a breakpoint in the transition of the monoblasts leading to the Tet2 mutant MHC IIhigh monocytes (Fig. 4D–E). We applied local regression (loess) to model variations of gene expression through pseudotime. Interestingly, the expression levels of myeloid markers through pseudotime show no differences between the different groups (Fig. 4F). In contrast, inflammatory genes were progressively upregulated through the different transcriptional states until reaching MHC IIhigh monocytic subset in the Tet2HR mice with severe transformed disease (Fig. 4G). These results suggest that aging and inflammatory signals dramatically alter the developmental trajectory of the Tet2HR mutant myeloid cells which are imprinted early in the differentiation stages and promote the emergence of the MHC IIhigh monocytes.
Figure 4. Emergence of an inflammatory monocytic population in leukemic Tet2HR mice.
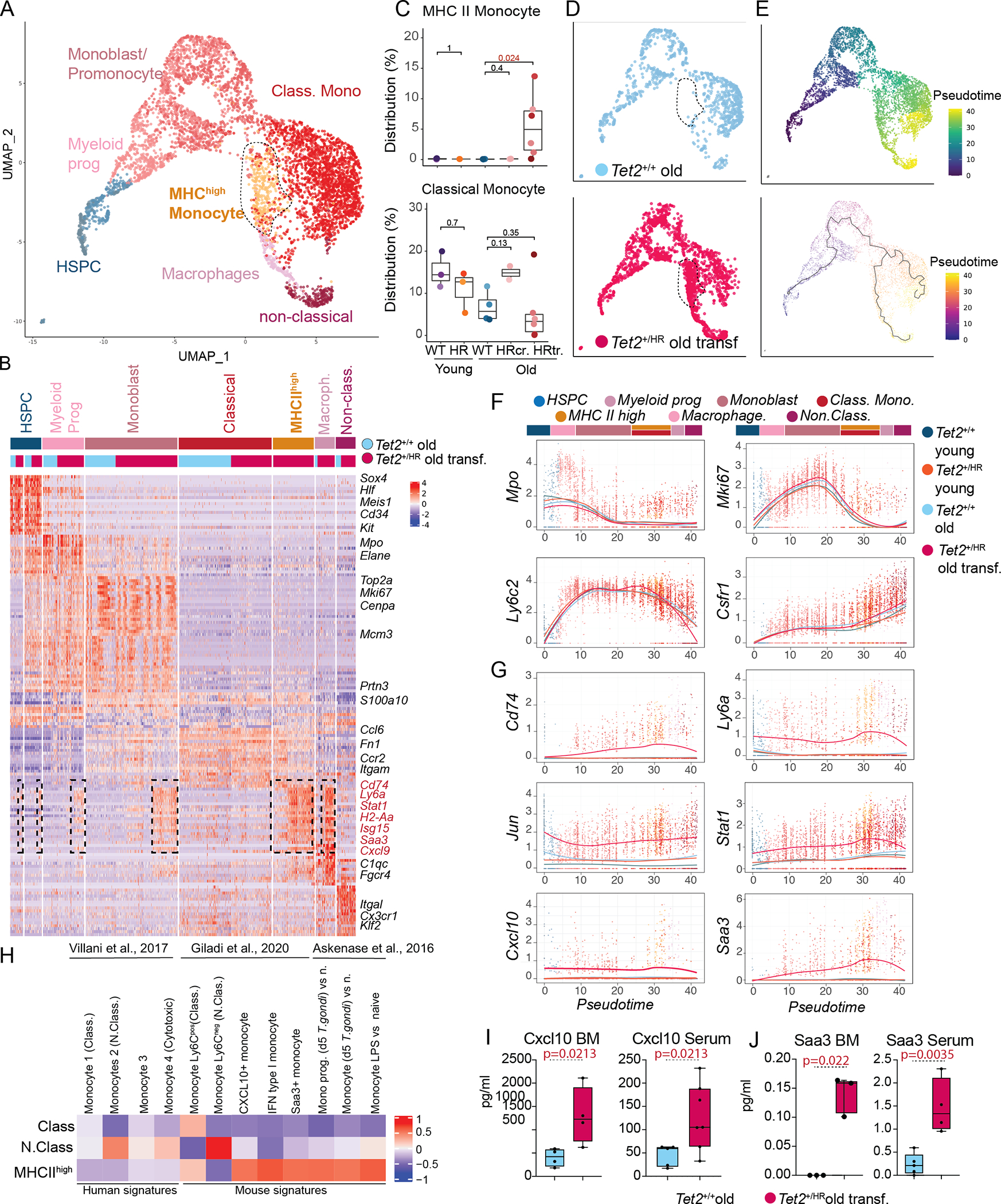
A, UMAP representation of the indicated myeloid populations (n=5597 cells).
B, Heatmap representing the top 50 genes expressed in each of the indicated populations.
C, Percentage of the indicated cell types in the total bone marrow of Tet2WT young, Tet2HR young, Tet2WT old, and Tet2HR.
D, UMAP representation showing only transcriptomes from Tet2WT old mice (blue) and Tet2HR with transformed disease (pink).
E, UMAP including Tet2WT old and Tet2HR old with transformed disease, color coded by pseudotime (top), and UMAP representation with a principal graph representing differentiation overlaid (low).
F, Gene expression of myeloid differentiation and cell cycle genes along pseudotime. Gene expression for each condition was modelled using local regression (loess) and overlaid.
G, Gene expression of genes high in the MHCII high population along pseudotime.
H, Heatmap GSVA analysis showing enrichment of signatures from published monocytic populations in the classical monocyte, non-classical monocyte and MHCII high monocyte populations.
I, Levels of Cxcl10 measured in the bone marrow fluids and Serum of Tet2WT old and Tet2HR old with transformed disease.
J, Levels of Saa3 measured in the bone marrow fluids and Serum of Tet2WT old and Tet2HR old with transformed disease.
Recent studies have described that the bone marrow is the source of an IFN gamma-mediated subset of inflammatory monocytes (characterized by the high expression of MCH II and Sca-1 surface markers) in response to gastrointestinal infections(24). Furthermore, in a model of multiple sclerosis, another study has identified bone marrow-mediated generation of pathologic monocytes expressing inflammatory mediators like the chemokine Cxcl10 and the inflammatory acute phase reactant serum amyloid A3 gene (Saa3)(25,26). Interestingly, the Tet2HR MCH IIhigh monocytes showed transcriptional programs very similar to these subsets of inflammatory and pathological monocytes previously described (Fig. 4H)(24,25,27). Given that Tet2HR MCH IIhigh monocytes overexpressed Cxcl10 and Saa3, we interrogated the levels of these ligands in the bone marrow and the serum of aged mice, and found that both inflammatory mediators were significantly increased in Tet2HR mice with transformed disease (Fig. 4I–J). Taken together, our results indicate that enhanced inflammatory signaling in Tet2 mutant mice leads to the generation of a distinct subset of inflammatory-primed monocytic population with pathological potential.
MHC IIhigh monocyte transcriptional signature correlates with worse prognosis in AML
To gain further insights into the biological and potential clinical significance of these findings and to assess the impact of the MCH IIhigh monocytes in AML patients, we generated an MHC IIhigh monocyte-specific gene signature composed of genes specific to the MHC IIhigh population from the single cell analysis, genes upregulated in Tet2HR monocytes from the bulk RNA sequencing (Fig. S4F–G), and 10 monocytic genes, that define the cell type (CTSS, KLF4, ZEB2, IRF5, F13A1, CSF1R, CD14, FCGR3A, ITGAM, CCR2) (Supp.Table 4, 5, and 6). Next, we compared the survival outcomes of TET2-mutated AML patients with high or low expression of the MHC IIhigh signature in a younger (< 60 years old n=60) and older (≥ 60 years old n=186) cohort using the Alliance study protocols(27). We found that the MHC IIhigh monocyte gene signature is significantly associated with survival in the younger cohort of TET2 mutated patients (3-year OS High vs Low; <60 years 24% vs 58% p <0.001; >=60 years, 0% vs 7% p < 0.22; Fig. 5A and Supp Table 7). This result gives additional credence to our model, in which leukemia progression is supported not only by mutations and aberrant cytogenetics(28), but by induction of inflammation and a population of inflammatory monocytes. Notably, the adverse prognostic impact of the MHC IIhigh monocyte signature was also applicable in the setting of AML in general, in both <60 years old (n=686) and ≥ 60 years old (n=186) de novo AML patients. Similarly, in this cohort, the MHC IIhigh monocyte signature was associated with significantly shorter survival (3-year OS High vs Low; < 60 years 32% vs 58% p <0.001; >=60 years, 4% vs 21% p < 0.001; Fig. 5B and Supp Table 8). When considered with ELN, the MHC IIhigh monocyte signature was an independent predictor of OS in younger (<60y) and older (≥60y) adult AML patients, demonstrating a “biomarker potential” for the presence of the MHC IIhigh population (Supp Table 9).
Figure 5. MHC IIhigh monocytic signature correlates with poor prognosis in AML patients.
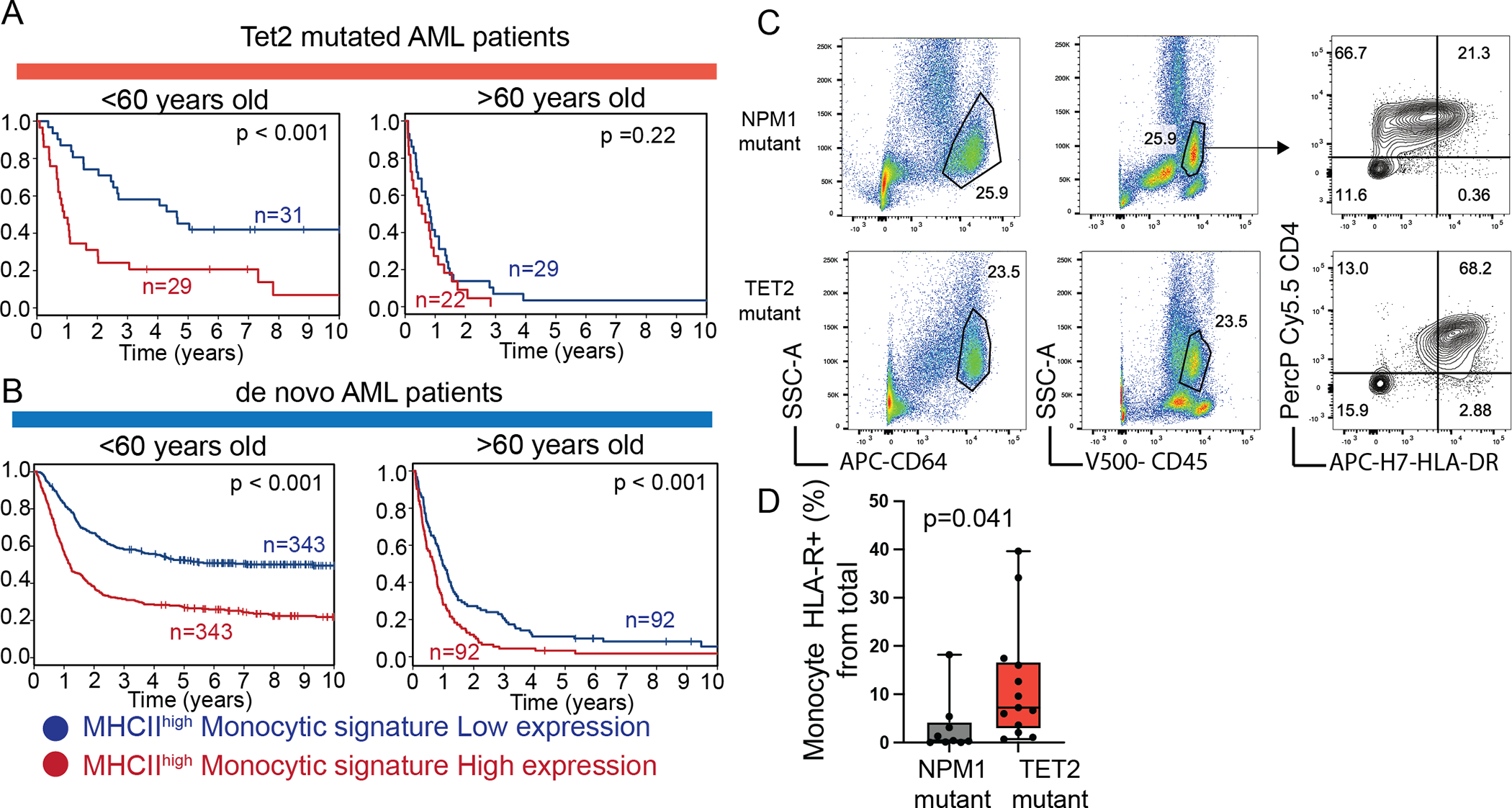
A, Survival plots of TET2 mutated AML patients stratified MHC IIhigh monocytic signature.
B, Survival plots of AML patients stratified by MHC IIhigh monocytic signature.
C, Flow Cytometry representative plots of HLA-DR+ monocytes found in AML patient samples. Monocytes were gated based on their CD64high and CD4intermediate expression levels.
D, Quantification of Monocyte HLA-DR+ cells from NPM1 mutant (n=11) and TET2HR mutant human samples (n=13).
Emerging studies are connecting inflammation to myeloid neoplasms, supporting our Tet2HR animal findings (29). This notion and prompted us to search for a monocytic population similar to the MHC IIhigh monocytes described here in human samples. We thus analyzed several (n=36) bone marrow samples with mutations in the catalytic domain of TET2 including 11 samples with a TET2H1881 mutation using flow cytometry. As a control, we used a cohort of human AML samples with wild-type TET2 and mutated NPM1. We found that cases with TET2 mutation had an increased percentage of CD64+CD4+ monocytes with increased expression of HLA-DR (Fig. 5C–D) compared with TET2 wild-type NPM1 mutated samples (p= 0.041). Future studies are required to test the biological role of such monocytes in human AML.
MHC IIhigh inflammatory monocytes are part of the leukemic infiltrate but they lack leukemia initiating capacity
Given the high levels of expression of the components of the MHC IIhigh cluster and inflammatory response genes like Sca-1, both encoding for proteins expressed on the cell surface, we next assessed whether these inflammatory monocytes can be detectable in mice by flow cytometry. By immunophenotyping the blood and bone marrow myeloid compartment of aged animals (>16 months old), we observed a progressive expansion of an MHC IIhigh, Sca-1high monocyte (MHC IIhigh) population in the Tet2HR mice with AML (Fig. 6A–B). Interestingly, we found that MHC IIhigh monocytes can also be detected in aged Tet2KO animals (Fig. S5A)(29), suggesting that this inflammatory population is not exclusive of the Tet2HR mutation and correlates with Tet2 deficiency. Moreover, MHC IIhigh monocytes were also increased in the peripheral blood and the spleen of leukemic Tet2HR mice (Fig. 6C and Fig. S5B). In agreement with such animal studies, as described above (Fig. 5C–D), one could detect such monocytes in the bone marrow of patients carrying TET2 H1881 mutations using a combination of antibodies against CD45, CD4, CD64 and HLA-DR. However, we believe that emergence of this MHC IIhigh monocytic population is not a universal phenomenon in murine myeloid leukemia as we failed to detect such cells in carrying a number of additional mutations (Npm1/Flt3-ITD and Jak2V617F) (Fig. S5C).
Figure 6. Inflammatory Tet2HR monocytes are detectable by immunophenotyping in mouse samples.
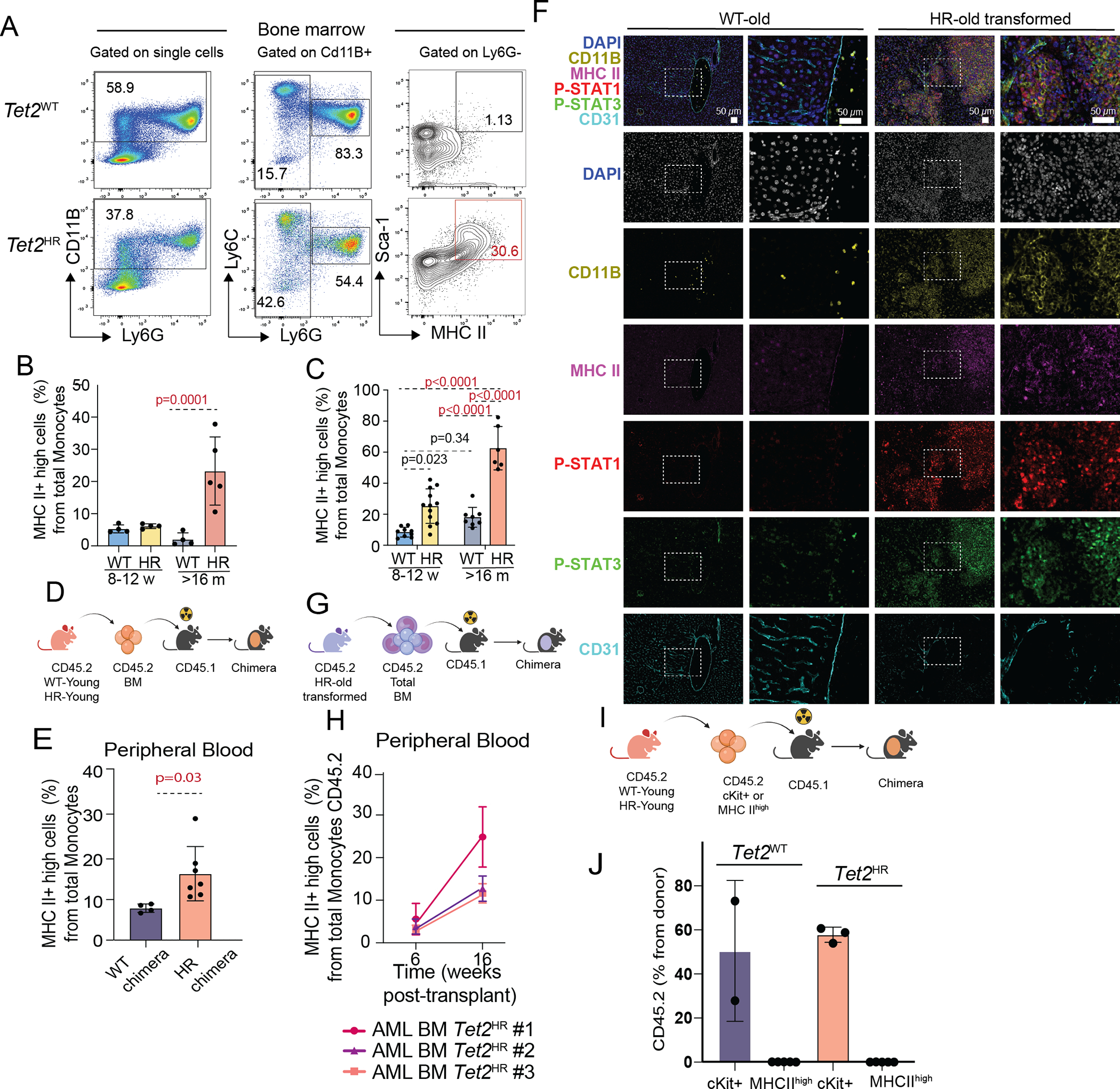
A, Representative flow cytometry gating strategy and quantification of the percentage of MHC IIhigh monocytes in Tet2WT and Tet2HR bone marrow cells collected from 16-weeks-old mice.
B, Percentage of MHC IIhigh monocytes from total monocytes in the bone marrow of Tet2WT and Tet2HR mice.
C, Percentage of MHC IIhigh monocytes from total monocytes in the peripheral blood of Tet2WT and Tet2HR peripheral blood cells.
D, Experimental design of transplantation experiment of young CD45.2 Tet2WT and CD45.2 Tet2HR bone marrow into lethally irradiated CD45.1 young mice.
E, Quantification of MHC IIhigh monocytes from total monocytes in the peripheral blood of CD45.2 Tet2WT and CD45.2 Tet2HR chimera (described in D) one year post-transplantation.
F, Multiplex Immunohistochemistry/Immunofluorescence for the simultaneous detection of the indicated antibodies in Tet2WT and Tet2HR liver samples.
G, Experimental design of transplantation experiment of total bone marrow collected from CD45.2 Tet2HR mice with transformed disease into CD45.1 young mice.
H, Percentage of MHC IIhigh monocytes in the peripheral blood of the indicated chimeras (described in G) 16 weeks following transplantation.
I, Experimental design of transplantation of cKit enriched CD45.2 Tet2WT and CD45.2 Tet2HR and MHC II high sorted CD45.2 Tet2WT and CD45.2 Tet2HR monocytes into CD45.1 young mice.
J, Percentage of CD45.2 engraftment in the peripheral blood of the indicated chimeras (described in J) 2 months following transplantation.
To define whether the emergence of inflammatory MHC IIhigh monocytes is a cell-intrinsic Tet2-driven phenomenon, we generated hematopoietic chimeras by transplanting CD45.2+ bone marrow cells from young (8 weeks) Tet2WT or Tet2HR mice into lethally irradiated CD45.1+ recipient mice. At early time-points after reconstitution, both Tet2WT and Tet2HR chimeras showed emergence of lymphoid and myeloid reconstitution and no presence of inflammatory monocytes in the peripheral blood of any of the groups. However, one year after transplantation, we were able to see that MHC IIhigh monocytes emerged and expanded in Tet2HR chimeras (Fig. 6D–E), demonstrating that the emergence of this population is driven by the mutation of Tet2 in the hematopoietic compartment.
Interestingly, morphologic assessment of the liver and spleen of a mouse with the histologically aggressive disease had shown marked infiltration by leukemic immature mononuclear cells, predominantly with an intra-sinusoidal pattern of distribution, and some areas with morphologic features suggestive of monocytic differentiation (characterized by ample eosinophilic cytoplasm and highly convoluted nuclear contours, best appreciated in the spleen in Fig. S5D–E). Therefore, we wondered whether this monocytic component of the leukemic infiltrates corresponded to the MHC IIhigh monocytes identified in our transcriptional analysis and also by flow cytometry. To test this, we applied a multiplex immunofluorescence platform using Cd11b (a mature myeloid cell marker), MHC II antigen, and antibodies recognizing active modifications (phosphorylation) of the inflammatory transcription factors Stat1 and Stat3 (Fig. 6F and Fig. S5E). This analysis showed that monocytic cells from the leukemic infiltrate (Cd11b+ cells) also expressed high levels of MHCII, p-Stat1, and p-Stat3, indicating that the inflammatory monocytes identified in our transcriptional analysis egress the bone marrow and infiltrate peripheral tissues. To evaluate if the MHC IIhigh monocytes are part of the tumor, we transplanted bone marrow cells from three Tet2HR old mice with signs of myeloid leukemia into lethally irradiated CD45.1 recipient animals. Six weeks after transplantation, we confirmed that the MHC IIhigh monocytes expanded in the recipient mice (Fig. 6G–H). To test whether the MHC IIhigh monocytes are transformed cells, we transplanted c-Kit+ enriched bone marrow cells as well as MHC IIhigh sorted cells from CD45.2 Tet2WT and Tet2HR mice into lethally irradiated CD45.1 recipients. Six weeks after transplantation, we confirmed that the c-Kit+ HSPC cells expanded in the recipient mice, however, MHC IIhigh monocytes had no ability to transplant (Fig. 6I–J). Overall, our results demonstrate that inflammation in conjunction with Tet2 mutations promotes the emergence of a monocytic population (MHC IIhigh), which acquires the ability to infiltrate peripheral tissues.
Inflammatory stimuli induce MHC IIhigh monocytes and accelerate disease onset
To further prove that inflammation can induce the emergence of this MHC IIhigh population, we induced a chronically inflammatory environment by administering consecutive doses of lipopolysaccharide (LPS) by an intraperitoneal injection into young mice (8-week-old) (Fig. 7A). Interestingly, we found that LPS promoted the expansion of MHC IIhigh monocytes in Tet2HR mice but not in wild-type controls (Fig. 7B). Furthermore, the expansion of this inflammatory population remained sustained for at least eight months after the last LPS injection (Fig. 7B). At the end point (8 months following the first injection), we observed overt splenomegaly in Tet2HR mice treated with LPS (Fig. 7C–D). Morphological assessment of spleen tissue of these mice showed extramedullary hematopoiesis, whereas no involvement of the spleen was observed in the Tet2WT mice treated with LPS nor Tet2HR mice treated with PBS (Fig. 7E). The peripheral blood analysis at 8 months, confirmed the increased levels of monocytes in Tet2HR mice treated with LPS together with the decreased of RBC and WBC (Fig. 7F,G and Fig. S6A). Immunophenotyping of bone marrow and spleen samples showed significant expansion of the LSK compartment (Fig. 7H and Fig. S6B). Moreover, the expansion of MHC IIhigh monocytes correlated with an increased level of Saa3 in the peripheral blood of Tet2HR mice injected with LPS (Fig. S6C). Interestingly, we observed increased colony formation capacity, consistent over four consecutive passages in the Tet2HR LPS treated mice compared to the controls (Fig. 7I). This finding demonstrates that inflammatory signals such as LPS can accelerate the production of MHC IIhigh monocytes in Tet2HR mice, suggesting that an enhanced inflammatory response cooperates with Tet2 loss-of-function genetic events in promoting malignant transformation.
Figure 7. Inflammatory stimulation accelerates emergence of MHC IIhigh monocytes and leukemic transformation.

A, Experimental design of chronic LPS stimulation experiment. Tet2WT and Tet2HR young mice (8-weeks-old) were injected with 16 doses of 35 ug of LPS (every three days).
B, Percentage of MHC IIhigh Monocytes in the peripheral blood of the indicated mice during the LPS treatment described in A.
C, Representative spleens of Tet2WT and Tet2HR treated with PBS or LPS with LPS treated Tet2HR showing overt splenomegaly.
Experimental design. Bone marrow cells from Tet2HR mice with transformed disease were transplanted into recipients sub-lethally irradiated CD45.1.
D, Spleen weights of of Tet2WT and Tet2HR treated with PBS and LPS
E, Hematoxylin & Eosin of spleen sections of the indicated mice. No to minimal involvement observed in Tet2WT PBS and LPS treated samples. Chronic phase disease and signs of extramedullary MPN/MDS-MPN observed in Tet2HR LPS treated samples.
F, Hemavet results showing red blood count (RBC) in Tet2WT and Tet2HR PBS and LPS treated mice
G, Percentage of MHC IIhigh Monocytes in the peripheral blood of the indicated mice 8 months following LPS treatment.
H, Percentage of LSK in bone marrow of old Tet2 +/+ or old Tet2+/HR with transformed disease phenotype quantified by flow cytometry immunophenotyping.
I, Colony formation assay. Quantification of the number of colonies derived from serial plating of 10,000 unfractionated bone marrow cells collected from PBS and LPS treated Tet2 WT and Tet2 +/HR (n=3).
J, Tet2 mutant progenitors maintain a tight regulation through their hematopoietic differentiation during steady-state. However, inflammatory conditions such as aging and infections triggers the proliferation of the progenitor cells and their myeloid-bias differentiation into inflammatory monocytes. The uncontrolled inflammatory response, in part maintained by the Tet2HR inflammatory monocyte contributes to the transformation of the progenitor cells and the sustained generation of leukemic inflammatory monocyte
LPS triggers the TLR signaling pathway, and previous studies have shown that targeting factors in this signaling pathway such as IRAK1/4 suppresses inflammatory activation in MDS and AML(30). Additionally, studies have shown that infections drive pre-leukemic myeloproliferation (PMP) and that antibiotic treatment prevented and reversed PMP(31). To investigate if antibiotic and IRAK1/4 inhibitor treatments can prevent the emergence of the MHC IIhigh monocytes, we transplanted CD45.2 Tet2HR mice into lethally irradiated CD45.1 Tet2WT mice and allowed them to recover for 4 weeks post-transplantation. Mice were treated with either PBS, antibiotic cocktail, or IRAK1/4 inhibitor (Fig. S6D). Antibiotic cocktail was provided by oral gavage for 1 week (given at a 1:50 concentration in drinking water) (31). IRAK1/4 inhibitor was injected intraperitoneally 5 days a week for 4 weeks(32). We observed that antibiotic and IRAK1/4 inhibitor treatment prevented the expansion of MHC IIhigh monocytes in transplanted mice but not in the PBS treated controls (Fig. S6 E–F). Taken together, our results demonstrate that blocking inflammatory signals disrupts the generation of MHC IIhigh monocytes in leukemic mice.
DISCUSSION
During the last decade, a number of studies have suggested a relationship between dysregulation of inflammatory- and immune-response related pathways and the development of myeloid neoplasms (MDS, MPN, and AML)(33). However, the cellular and molecular mechanisms by which inflammation interacts with or even induces carcinogenesis in blood malignancies are under investigation. It is known that in the bone marrow, inflammatory signals activate HSPCs, inducing their proliferation and rapid differentiation to increase the myeloid output under stress conditions(34). Therefore, several studies have postulated that inflammation is detrimental to wild-type progenitor cells but could preferentially promote the emergence of clones that carry CHIP mutations, endowing cells with enhanced self-renewal(34). In agreement with this notion, we show here that a TET2 mutation found in CHIP and myeloid neoplasms can lead to myeloid malignancy and that this process of cellular transformation is coupled to organismal aging as well as the induction of inflammatory gene expression programs and an inflamed microenvironment.
A number of elegant studies have recently challenged the notion that monocyte function is only modulated by local signals at the infiltrating tissues(24,25,35–38). They have proposed that inflammation, particularly IFN-signaling, increases the myeloid output in the BM and rewires stem and progenitor cells (HSPC), resulting in the generation of inflammatory-primed monocytes, which respond faster to the exogenous stimuli. For instance, a subset of bone marrow inflammatory monocytes, characterized by their elevated levels of the MHC II and Sca-1 surface antigens, show increased regulatory and effector responses during T.gondi intestinal infection(24). Work presented here reveals that there could be parallels between infection and early stages of transformation in the bone marrow. Specifically, our single-cell transcriptomic profiling suggests that inflammation reshapes the differentiation trajectory of pre-leukemic HSPC, giving rise to a novel subset of inflammatory MHC IIhigh monocytes. Although they lack leukemia initiating capacity (LIC), Tet2HR inflammatory monocytes acquire the ability to infiltrate peripheral tissues such as the liver and the spleen. They express inflammatory response genes, proteins of the MHC class II antigen presentation cluster and a number of secreted factors. Such gene expression profile would suggest that these inflammatory monocytes could impact on the microenvironment and support disease progression (Fig. 7J). Indeed, it was recently suggested by van Galen and colleagues that myeloid cells in human AML could play an immunomodulatory role35. Further supporting the notion that inflammatory monocytes also occur in human AML, we were able to show that a gene expression signature composed by genes highly expressed in mouse inflammatory Tet2HR monocytes is tightly associated with poor prognosis in AML patients.
It is well established that clonal hematopoiesis (including CHIP carrying TET2 mutations) is associated with inflammatory pathologies like cardiovascular disease, pulmonary disease, and type 2 diabetes(39–42). Previous studies have shown that Tet2-deficient macrophages promote inflammation, aggravating atherosclerosis (43–45). Specifically, myeloid Tet2 deficient cells showed enhanced production of IL-6 following an inflammatory trigger due to the direct effect of Tet2 in repressing the IL-6 promoter by recruiting HDAC2(46). Likewise, in response to bacterial infection-induced IL-6, Tet2 deficient HSPCs show increased proliferation and myeloid differentiation45. Additionally, Tet2 deficient cells have a strong proliferative and intrinsic anti-apoptotic advantage in response to TNF-a and IL-6 activation(47,48). These studies suggest an enhanced cell-extrinsic and cell-intrinsic component of the Tet2 response during inflammation and transformation. Our transcriptional analysis reveals that the inflammatory response pathway is aberrantly upregulated in Tet2 mutant leukemic cells, suggesting that Tet2 may regulate a large inflammatory program and not only IL-6. Interestingly, inflammatory mediators (like Cxcl10 and Saa3), are expressed by Tet2 mutant monocytes, and they are also increased in the bone marrow fluid and plasma of the Tet2 mutant mice with AML. Previous studies have shown that Saa3 has a pro-tumorigenic role in pancreatic cancer, colon cancer, as well as promoting metastasis in breast cancer models (49)(26,50), suggesting similar roles also in blood malignancy. Future studies on the crosstalk between inflammatory monocytes and their microenvironment will be crucial to delineate potential roles in supporting the malignant self-renewal progenitors and modulating the immune response, facilitating tumor progression (51).
METHODS
Generation of a TetHR mouse model
To construct the targeting vector with the mutation H1794R, we digested the plasmid K427, previously generated by Oliver Bernard to target exon 12 of mouse Tet2, with Kpn I and Nhe I enzymes(52). Next, we subcloned the targeting sequence into a pCDNA 3.1 plasmid and generated the mutation H1794R by using the Quick Change Stratagene kit. The mutated targeting sequence was then cloned back to the plasmid K427. The targeting vector K427-H1794R was electroporated to mESC by Applied Stem Cells. We PCR-screened for the clones carrying the proper homologous recombination by using the primers GTAGAAGGTGGCGCGAAGGGGC and CACCCATAATTTTGTGTCTGAAGC, placed in the Neo cassette and outside the 3’ homology arm. The Rodent Genetic Engineering Laboratory at NYU Langone generated the Tet2HR-Frt mice by tetraploid complementation of two different targeted clones. Finally, Flp cassette was removed by crossing the Tet2HR-Frt mice with Flp mice (JAX stock 003946). The primers CACCCATAATTTTGTGTCTGAAGC and GAGGCATGTTGAATGACTCTGG were used for PCR-genotyping the mutant allele, discriminating a wild-type allele of 111 bp and HR allele of 250 bp. All mice were housed at New York University School of Medicine under pathogen-free conditions. All procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees at New York University School of Medicine.
Blood Analysis (Hemavet)
Blood was collected by either retro-orbital or a transterminal intracardiac bleed into EDTA-coated tubes. Blood samples were analyzed on an element HT5 Hemavet (Heska) according to the manufacturer’s instructions.
Bone Marrow Transplantation
Total bone marrow cells were collected from flushing the femur and tibia of CD45.2+ mice. The cell suspension was treated with lysate using 2 mL of ACK lysing buffer (Quality Biological). Donor cells (0.5 × 10^6 per genotype per mouse) were transplanted via retro-orbital injection into lethally irradiated (2 × 450 rad) congenic CD45.1+ recipients (B6.SJL-Ptprca). Reconstitution was monitored 4 weeks after transplantation.
Colony- Forming Cell Assays
Mouse bone marrow cells were enriched using a CD117 microbeads kit (Miltenyi Biotec) and separated using a MACS (Miltenyi Biotec) cell separator. After enrichment of CD117 cells, 2000 cells/well were seeded in Methocult (M3434) medium (STEMCELL Technologies). The methoCult medium-cell mixture was plated into 12-well plates with water supply in the inter-well chamber. After 7 days (1 passage) of incubation at 37C in 5% CO2, colonies were counted in 3 replicates per sample. The process was repeated for a total of 5 passages.
Flow Cytometry
Peripheral blood was collected via retroorbital bleeding and lysed using 2ml ACK Lysing buffer. Bone marrow cells were collected from the femur and tibia of mice, and red blood cells were lysed using 2mL ACK Lysing buffer (Quality Biological). For obtaining splenocytes hepatocytes for flow cytometry, spleens and livers were harvested from mice and were physically dissociated by pressing through a 0.40 μm cell strainer (Falcon) and lysed with 5mL ACK Lysing buffer (Quality Biological). Single-cell suspensions were re-suspended in PBS with 2% FCS and blocking solutions. All samples were gated on viable cells using forward scatter (FSC) and sideward scatter (SSC) size discrimination, followed by exclusion of doublets. Surface staining of mouse HSC cells was performed in FACS blocking buffer using monoclonal antibodies against APC-Cy7-conjugated Gr-1, Cd11b, Cd4, Cd8, Ter119, Cd45r, and Nk1.1 (Biolegend), APC-conjugated Cd117 (Biolegend), PE-Cy7-conjugated Sca-1 (Biolegend), FITC-conjugated Cd150, Pacific Blue-conjugated Cd48. Surface staining of mouse progenitor cells was performed in FACS blocking buffer using monoclonal antibodies against APC-Cy7-conjugated Gr-1, Cd11b, Cd4, Cd8, Ter119, Cd45r, and Nk1.1 (Biolegend), APC-conjugated Cd117 (Biolegend), PE-Cy7-conjugated Sca-1 (Biolegend), FITC-conjugated Cd34 (Invitrogen), Pacific Blue-conjugated Cd16/32 (Invitrogen). Surface staining of mouse lineage cells was performed in FACS blocking buffer using monoclonal antibodies against FITC-conjugated Cd4 (Pharmigen) and Cd8 (Biolegend), PE-conjugated Cd71 (Biolegend), PECy7 conjugated Cd11b, and Pacific Blue conjugated CD45r (Biolegend). Surface staining of mouse MHC II high cells was performed in FACS blocking buffer using monoclonal antibodies against APC-Cy7 conjugated Ly6c (Biolegend), BV605 conjugated Ly6g (Biolegend), Pacific Blue conjugated Cd11b (Biolegend), PE conjugated I-A/I-E (Biolegend), PE-Cy7 conjugated Ly6a (Biolegend). Samples were incubated with antibodies for 1 hour in the dark at 4°C. Samples were acquired using an LSRFortessa (BD Biosciences), and data were analyzed with FlowJo. Cell sorting was performed on SY3200 Cell Sorter (Sony).
Hematoxylin and Eosin Staining
Tissue samples were fixed in 4% paraformaldehyde for 48 hours in 4°C. Samples were washed three times with PBS (Corning) and were placed in 70% Ethanol solution. Hematoxylin and eosin staining were performed using standard methods.
Western Blot
Cell lysate was generated from wild type and Tet2 mutant mouse bone marrow and were collected from spine, hips, femur and tibia. A single-cell suspension was generated after red blood cell lysis ACK buffer and was treated with 2x Laemmli buffer and β-mercapetoethanol and quantified using BCA protein assay (Pierce). Equal amount of proteins was applied to a 4–12% Bis-Tris Gel (Invitrogen) and SDS-PAGE was performed. Wet transfer was performed following gel electrophoresis, followed by 1 hour blocking with 5% non-fat milk and TBST and overnight primary antibody incubation. Primary antibody was incubated at 1:1000 concentration. After washing the excess primary antibody, secondary antibody incubation was performed for 1 hour at room temperature before developing. Secondary antibody was incubated at 1:2000 concentration.
Cytokine quantification
For collecting bone marrow fluids, the four long bones (two femurs and two tibiae) of each mouse were flushed with the same 200 μl of PBS. Cells were spun down by centrifugation at 1,000 RPM for 5 min. Next, supernatants were clarified by a second centrifugation at 12,000 RPM for 5 min. IL-1α and IL-1β were measured by ELISA (mouse IL-1β and mouse IL-1α ELISA kit; R&D). IFN-α, IL-1β and other cytokines were measured using the Legendplex Anti-virus response panel (BioLegend).
Saa3 quantification
Blood was collected by either retro-orbital bleed into BD Microtainer Serum Separator Tube. Blood was allowed to clot for 30 minutes at room temperature. After incubation, samples were centrifuged at 15,000 g for 3 minutes. Serum was collected for the Mouse SAA ELISA Kit (Abcam) following the direction of the manufacturer.
Antibiotic treatment
Mice were treated with an antibiotic cocktail as previously described(53). All antibiotics used are listed as follows (concentration used, company, catalogue number). In detail, in the first week mice received a daily intragastric gavage with 100μl of a mixture of kanamycin (4mg/ml, Sigma-Aldrich, 60615), gentamicin (0.35mg/ml, Sigma-Aldrich, G1914), colistin (8500U/ml, Sigma-Aldrich, C4461), metronidazole (2.15mg/ml, Sigma-Aldrich, M3761), and vancomycin (0.45mg/ml, Sigma-Aldrich, V2002). For the following three weeks antibiotics were administered in the autoclaved (non-acidified) drinking water at 50-fold dilution except for vancomycin, which was maintained at 0.5mg/ml. Autoclaved (non-acidified) water was used to dissolve antibiotics. Antibiotic water was prepared fresh and replaced weekly.
IRAK1 /4 Inhibitor Treatment
Mice were treated with an IRAK 1 /4 inhibitor as previously described(30,54). The IRAK1 inhibitor (IRAK1/4 inhibitor; Amgen Inc.) was purchased from Sigma-Aldrich (I5409). Animals were intraperitoneally injected with NCGC-1481 (30 mg/kg).
Single-cell RNA-seq Library Preparation
Bone marrow cells were isolated from the spine, hips, femur and tibia of mice. A single-cell suspension was generated after red blood cell lysis ACK buffer. Cells were tagged with cell hashing(55) oligo-tagged antibodies (BioLegend) according to manufacturer’s instructions and pooled on a 10x Chromium instrument (10x Genomics) to generate barcoded single-cell beads-in emulsion (GEM) according to the manufacturer’s protocol. scRNA-seq libraries were prepared using the Chromium Single-Cell’ v3 Reagent Kit (10x Genomics) following the direction of the manufacturer. The libraries were quantified using Qubit fluorometric quantification (Thermo Fisher Scientific). The quality was assessed on an Agilent Bioanalyzer 2000, obtaining an average library size of 455 bp. Libraries were pooled and normalized to 2 nmol/L and clustered on a HiSeq4000 high output mode on a paired-end read flow cell and sequenced for 28 cycles on R1 (10x barcode and the UMIs). Followed by 8 cycles of i7 index (sample index), and 98 bases on R2 (transcript), with a coverage around 130M reads per sample.
scRNA-Seq data pre-processing
Single-cell RNA sequencing data were demultiplexed and converted to FASTQ format using Illumina bcl2fastq software and then processed using Cell Ranger for barcode processing and gene counting using the mm10 reference. Quality control of the data was done using the R package Seurat19. Low quality cells were excluded with a cutoff of a minimum of 400 genes and a maximum of 6000 genes expressed per cell, as well as a cutoff of a maximum of 10% of the reads coming from mitochondrial reads. Samples that were multiplexed using hashtag oligos were demultiplexed using Seurat function HTODemux using a positive quantile of 0.99. Cells determined to be doublets or singlets after demultiplexing were removed. After filtering we obtained 37,355 cells. Data were then normalized by total expression per cell, multiplied by 10,000 and then log transformed.
Integrated analysis of single cell datasets
Samples split between several libraries and run at the same time were combined using the Seurat function merge. The Seurat anchor-based integration method was used to account for technical differences between libraries run on different days, using 2,000 most variable genes. The resulting scaled data was used to calculate PCA followed by UMAP using 20 neighbors and 20 principal components. The resulting principal components were also used as input to Smart Local Moving clustering algorithm used to partition the dataset. A range of resolutions (0.1–10) were used to explore clusters that corresponded to cell populations and biological processes.
UMAP plots colored by density ratios were determined by calculating densities for each condition, by splitting the UMAP plane into 2D bins and counting the percentage of points in each bin. The log2FC between the values for each condition are calculated and used to color each cell.
Pseudotime analysis was conducted using Monocle322 on the monocyte subset of the integrated and scaled matrix. Loess curves were fit to gene expression values for each condition along the pseudotime axis.
Cells were assigned cell-type identity in an iterative fashion. First, all cells were clustered. Clusters were assigned known broad cell-types based on the expression of established markers. Specifically, the cluster-average expression values were filtered for genes expressed in any clusters based on a detection threshold of 10 counts per million (~3 transcripts per cell), the values for each gene were normalized relative to the maximum level across all clusters, and the rescaled values were averaged across all markers for each population, with each cell assigned to the population with the highest value. Following separation into the broad cell-types, each cell-type was subset into its own dataset and re-clustered. Clusters within each cell-type were then assigned granular cell-type labels based on expression of established markers using the average expression levels of every signature and subtracting the aggregated expression of randomly selected control genes. Cell cycle analysis was performed per-cell, based on expression of established markers using the average expression levels of each signature and subtracting the aggregated expression of all genes.
Boxplots of cell-type abundances were created by calculating per mouse, the percent of cells in each cell-type, relative to the total number of cells per mouse. Statistical analysis was conducted using the ggpubr package to calculate the wilcox test statistic between conditions, and ggplot was used to create the plots(56).
Differential expression between conditions or between cell-types was performed using wilcox test statistic, using the FindMarkers function from the package Seurat. The MHC II monocyte gene signature was determined by comparing, within the monocytes, genes significantly expressed in the MHCIIhigh monocytes.
Inflammatory response genes were defined as genes included in either the Hallmark Interferon Alpha Response, Hallmark Interferon Gamma Response, Hallmark Inflammatory response, Hallmark IL6 JAK STAT3 Signaling, Hallmark IL2 STAT5 signaling, or Hallmark TNFA signaling via NFKB genesets from MSIGDB. Inflammatory response score was calculated by taking the difference between the mean expression of genes within the gene list, compared to mean expression of all genes.
Bulk RNA Sequencing
Analysis of bulk RNA sequencing was performed using the pipelines provided in the Seq-N-Slide pipeline(57) https://github.com/igordot/sns. Briefly, after trimming using Trimmomatic(57), reads were aligned to the mm10 reference genome using STAR(58), and count tables were generated using featureCounts(59). DESeq2(60) was then used to identify differentially expressed genes between the Tet2HR and Tet2WT condition.
Whole Exome Sequencing
Analysis of whole exome sequencing data was performed using the Seq-N-Slide pipeline(57) https://github.com/igordot/sns. In short, adapters were trimmed using Trimmomatic(57), and aligned to reference genome mm10 using BWA-MEM(61). Duplicate reads were removed using Sambamba(62). Somatic point mutations and small indels were identified using Strelka(63).
Lolliplot Representation of AML and ChIP samples
CHIP data were acquired from the Bick at al., 2021, Zink et al., 2017, and Jaiswal et al., 2017 papers(45,64,65). AML data was acquired from the Tet2 Cosmic database (66). Tet2 missense mutations were identified and plotted using the trackViewer package (67). Tet2 domains were defined according to Hu et al. Cell, 2013(14).
Comparison of MHC IIhigh monocytes to published monocyte signatures
Average gene expression of classical monocytes, non-classical monocytes, and MHC IIhigh monocytes were calculated and used in an enrichment analysis for genes signatures characterizing inflammatory and pathological monocytes from Villani et al., 2017, Giladi et al., 2020, and Askenase et al., 2016(24,25,27). The GSVA package was used to calculate enrichment (68).
Computational Modeling
The H1881R mutation was incorporated into the 4NM6 crystal structure using the LEaP program of AMBER (14). The structure was prepared and compared to WT as previously described with Mg(II) in place of Fe(II) using the published 5-position modified cytosine parameters(15). Three systems, with either 5mC, 5hmC, or 5fC as the everted base, were simulated in triplicate for two-hundred nanoseconds of production dynamics each with TIP3P water and the ff99SB force field58. Production dynamics were performed with the Langevin thermostat in the NPT ensemble using the pmemd.cuda AMBER18 code(16). The H1881R results were compared to our previous WT simulations extended out to 500 ns (15). Cpptraj was used for structural analysis, including calculating root mean square fluctuations and backbone angles (69). An open-source program was used to perform the by-residue energy decomposition analysis with respect to the 5-position cytosine (https://zenodo.org/record/4469899#.YSGFmx0pBZo). Data were further processed using R v4.0.3 with the abind, data.table, and tidyverse packages and Python v3.6.7 with the NumPy, pandas, snakemake, and matplotlib modules(56,70–72). UCSF Chimera was used to create images (73).
Characterization of the TET2 H1881R mutation
The human TET2-CS variant (1129–1936 Δ1481–1843) with an N-terminal FLAG-tag was expressed using a pLEXm expression vector as previously described(10,15). The H1881R mutation or mutation of the catalytic H1382Y and D1384A (HxD mutant) were generated by standard means. HEK293T cells were cultured in DMEM with GlutaMAX (Thermo Fisher Scientific) and 10% FBS (Sigma). Cells were transfected with wild-type (WT) hTET2-CS, mutant hTET2-CS, or an empty pLEXm vector control using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. Medium was changed 24 h after transfection, and cells were collected by trypsinization 48 h after transfection and resuspended in phosphate-buffered saline. Genomic DNA was isolated from the cells using the DNeasy Blood and Tissue Kit (Qiagen).
For liquid chromatography tandem-mass spectrometry (LC-MS/MS), 1 μg of genomic DNA from each sample was degraded to component nucleosides with Nucleoside Digestion Mix (New England Biolabs) at 37°C overnight. The nucleoside mixture was diluted ten-fold into 0.1% formic acid, and injected onto an Agilent 1200 Series HPLC with a 5 μm, 2.1 × 250 mm Supelcosil LC-18-S analytical column (Sigma) equilibrated to 45°C in Buffer A (0.1% formic acid). The nucleosides were separated by column affinity over 10 min in Buffer A at a flow rate of 0.5 mL/minute, and then the column was washed in 0 to 100% Buffer B (0.1% formic acid, 30% (v/v) acetonitrile). Tandem MS/MS was performed by positive ion mode ESI on a 6460 triple-quadrupole mass spectrometer (Agilent) with a gas temperature of 225°C, a gas flow of 12 L/minute, a nebulizer pressure of 35 psi, a sheath gas temperature of 300°C, a sheath gas flow of 11 L/minute, a capillary voltage of 3,500 V, a fragmentor voltage of 70 V, and a delta EMV of +1,000 V. Collision energies were optimized to 10 V for 5mC and 5fC; 15 V for 5caC; and 25 V for 5hmC. Multiple reaction monitoring (MRM) mass transitions were 5mC 242.11→126.066 m/z; 5hmC 258.11→124.051; 5fC 256.09→140.046; 5caC 272.09→156.041; and T 243.10→127.050. Standard curves were generated using standard nucleosides (Berry & Associates, Inc.) ranging from 1.85 μM to 113 pM (9.25 pmol to 0.565 fmol total). The sample peak areas were fit to the standard curve to determine the amount of each modified cytosine in the genomic DNA sample. Amounts are expressed as the percentage of total cytosine modifications.
Patient sample analysis and treatment
We investigated 872 adult patients with de novo acute myeloid leukemia (AML) who were enrolled on CALGB/Alliance study protocols and received treatment regimens, as detailed below. Patients were excluded from outcome analyses if they received an allogeneic hematopoietic stem cell transplantation in first complete remission (CR).
All patients gave written informed consent for participation in the studies. All study protocols were in accordance with the Declaration of Helsinki and approved by Institutional Review Boards at each treatment center. All patients were enrolled on CALGB 8461 (cytogenetic studies), CALGB 9665 (leukemia tissue bank) and CALGB 20202 (molecular studies) companion protocols. Patients were treated on CALGB/Alliance protocols (74–83). CALGB 8525 (n=28), 8621 (n=1), 8721 (n=1), 8821 (n=3), 8923 (n=10), 9022 (n=3), 9120 (n=1), 9222 (n=53), 9420 (n=9), 9621 (n=101), 9720 (n=66), 10201 (n=62), 10502 (n=19), 10503 (n=227), 10603 (n=54), 11001 (n=5), 11002 (n=7), and 19808 (n=232).
Definition of clinical endpoints and statistics for Alliance data set
Clinical endpoints were defined according to generally accepted criteria. A CR was defined as recovery of morphologically normal bone marrow and blood counts (i.e., neutrophils ≥1.5 × 109/L and platelets >100 × 109/L), and no circulating leukemic blasts or evidence of extramedullary leukemia, all of which had to persist for ≥4 weeks. DFS was measured from the date of achievement of a CR until the date of relapse or death from any cause; patients not known to have relapsed or died at last follow-up were censored on the date they were last examined. OS was measured from the date of diagnosis to the date of death from any cause; patients not known to have died at last follow-up are censored on the date they were last known to be alive. Event-free survival (EFS) was measured from the date of study entry until the date of failure to achieve CR, relapse or death. Patients alive and in CR at last follow-up were censored.
Low and high monocyte signature groups were compared using the Fisher’s exact test for categorical variables. Estimated probabilities of DFS, OS and EFS were calculated using the Kaplan-Meier method,(23) and the log-rank test evaluated differences between survival distributions. Cox proportional hazard models were used to calculate hazard ratios (HR) for DFS, OS and EFS. All statistical analyses on Alliance patients were performed by the Alliance Statistics and Data Center.
The MHChigh monocyte signature for the survival analysis in the Alliance patients was calculated using 204 genes identified either as marker genes of the MHCIIhigh monocyte population in the single-cell data, or as differentially expressed in the Tet2 monocytes profiled by bulk RNA sequencing. Additionally, 10 known monocytes genes were added to the list (CTSS, KLF4, ZEB2, IRF5, F13A1, CSF1R, CD14, FCGR3A, ITGAM, CCR2). The signature was derived as a linear combination of the expression of the 214 genes. The monocyte signature for patient i was ci = Σ wj xij, where xij was the expression value for gene j in patient i, and wj was the weight assigned to gene j. The univariable Cox regression coefficients for OS for each of the 214 genes included in the signature were used as the weights (wj) in the monocyte signature.
Mutational Profiling of Alliance Patient Set
Viable cryopreserved BM or blood cells of patients enrolled onto the CALGB 9665 tissue bank protocol were stored for future analyses prior to starting treatment. Mononuclear cells were enriched through Ficoll-Hypaque gradient centrifugation and cryopreserved until use. Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (QIAGEN, Hilden, Germany). The mutational status of 80 protein-coding genes was determined centrally at The Ohio State University by targeted amplicon sequencing using the MiSeq platform (Illumina, San Diego, CA). Furthermore, testing for CEBPA mutations was performed with the Sanger sequencing method, thus adding up to a total of 81 genes analyzed.
Data and Code Availability
All data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE182615, GSE201659 and SRA with BioProject ID PRJNA833651. Patient data used in survival analysis was obtained from Alliance Statistics and Data Center and is available upon reasonable request; generated data are deposited in GEO GSE137851, GSE63646. Code used for the single cell data analysis is available at https://github.com/igordot/scooter.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Progression from a pre-leukemic state to transformation, in the presence of TET2 mutations, is coupled to emergence of inflammation and a novel population of inflammatory monocytes. Genes characteristic of this inflammatory population are associated to worst prognosis in AML patients. These studies connect inflammation to progression to leukemia.
ACKNOWLEDGEMENTS
We thank all members of the Aifantis laboratory for discussions throughout this project; A. Heguy and the NYU Genome Technology Center (GTC) for expert in sequencing, the Applied Bioinformatics Laboratories (ABL) for providing bioinformatics support and helping with the analysis and interpretation of the data; GTC and ABL are shared resources partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center; the NYU Histology Core (5P30CA16087-31) for assistance; C. Loomis for immune-multiplex experiments; The Rodent Genetic Engineering Laboratory at NYU Langone for electroporated the ESC clones and generated the Tet2 mutant mice; the NYU Flow Cytometry facility for expert cell sorting; Olivier Bernard (Gustave Roussy & Université Paris-Saclay) for the Tet2 targeting vector; Kristian Helin (MSKCC) for the mouse Tet2 antibody; The results in this work here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. This work has used computing resources at the NYU School of Medicine High Performance Computing Facility. A.Y. is supported by a Translational Research TL1 Grant. G.A.C. is supported by the National Institutes of Health (grant no. R01GM108583). A.T. is supported by the NCI/NIH R01CA260028-01, NCI/NIH P01CA229086 and NCI/NIH R01CA252239-01 and is a St. Baldrick Scholar. A.K.E is supported by Pelotonia, the American Society of Hematology, Leukemia and Lymphoma Society (LLS), the Leukemia Research Foundation. V.P is an EMBO postdoctoral fellow (ALTF 742–2020) M.G. is supported by the Ramon Areces Foundation, the American Society of Hematology (ASH Restart Research Award) and is a Gilead Research Fellow. I.A. is supported by the NIH/NCI (RO1CA216421, RO1CA173636, RO1CA228135, RO1CA242020, RO1HL159175 and RO1CA271455), The EvansMDS Foundation, and the Leukemia and Lymphoma Society (LLS). U10CA180821, U10CA180882, and U24CA196171 have supported the Alliance for Clinical Trials in Oncology.
Footnotes
Conflict of Interest Disclosure Statement
I.A. is a consultant for Foresite Labs, LLC
M.G. is an employee of Loxo Oncology/ Eli Lilly since January 2022.
REFERENCES
- 1.Laurenti E, Gottgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018;553(7689):418–26 doi 10.1038/nature25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitchell SR, Gopakumar J, Jaiswal S. Insights into clonal hematopoiesis and its relation to cancer risk. Curr Opin Genet Dev 2021;66:63–9 doi 10.1016/j.gde.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer 2017;17(1):5–19 doi 10.1038/nrc.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corces MR, Chang HY, Majeti R. Preleukemic Hematopoietic Stem Cells in Human Acute Myeloid Leukemia. Front Oncol 2017;7:263 doi 10.3389/fonc.2017.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steensma DP. Clinical consequences of clonal hematopoiesis of indeterminate potential. Hematology Am Soc Hematol Educ Program 2018;2018(1):264–9 doi 10.1182/asheducation-2018.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009;114(1):144–7 doi 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrone CK, Blydt-Hansen M, Rauh MJ. Age-Associated TET2 Mutations: Common Drivers of Myeloid Dysfunction, Cancer and Cardiovascular Disease. Int J Mol Sci 2020;21(2) doi 10.3390/ijms21020626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A 2011;108(35):14566–71 doi 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011;20(1):11–24 doi 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011;20(1):25–38 doi 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011;118(17):4509–18 doi 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cimmino L, Neel BG, Aifantis I. Vitamin C in Stem Cell Reprogramming and Cancer. Trends Cell Biol 2018;28(9):698–708 doi 10.1016/j.tcb.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito K, Lee J, Chrysanthou S, Zhao Y, Josephs K, Sato H, et al. Non-catalytic Roles of Tet2 Are Essential to Regulate Hematopoietic Stem and Progenitor Cell Homeostasis. Cell Rep 2019;28(10):2480–90 e4 doi 10.1016/j.celrep.2019.07.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, et al. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell 2013;155(7):1545–55 doi 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 15.Liu MY, Torabifard H, Crawford DJ, DeNizio JE, Cao XJ, Garcia BA, et al. Mutations along a TET2 active site scaffold stall oxidation at 5-hydroxymethylcytosine. Nat Chem Biol 2017;13(2):181–7 doi 10.1038/nchembio.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput 2013;9(9):3878–88 doi 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 17.Hix MA, Leddin EM, Cisneros GA. Combining Evolutionary Conservation and Quantum Topological Analyses To Determine Quantum Mechanics Subsystems for Biomolecular Quantum Mechanics/Molecular Mechanics Simulations. Journal of Chemical Theory and Computation 2021;17(7):4524–37 doi 10.1021/acs.jctc.1c00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasmussen KD, Jia G, Johansen JV, Pedersen MT, Rapin N, Bagger FO, et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev 2015;29(9):910–22 doi 10.1101/gad.260174.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shih AH, Meydan C, Shank K, Garrett-Bakelman FE, Ward PS, Intlekofer AM, et al. Combination Targeted Therapy to Disrupt Aberrant Oncogenic Signaling and Reverse Epigenetic Dysfunction in IDH2- and TET2-Mutant Acute Myeloid Leukemia. Cancer Discov 2017;7(5):494–505 doi 10.1158/2159-8290.CD-16-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tothova Z, Krill-Burger JM, Popova KD, Landers CC, Sievers QL, Yudovich D, et al. Multiplex CRISPR/Cas9-Based Genome Editing in Human Hematopoietic Stem Cells Models Clonal Hematopoiesis and Myeloid Neoplasia. Cell Stem Cell 2017;21(4):547–55 e8 doi 10.1016/j.stem.2017.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36(5):411–20 doi 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 2019; 37, 38–44 doi 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 23.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019;566(7745):496–502 doi 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Askenase MH, Han SJ, Byrd AL, Morais da Fonseca D, Bouladoux N, Wilhelm C, et al. Bone-Marrow-Resident NK Cells Prime Monocytes for Regulatory Function during Infection. Immunity 2015;42(6):1130–42 doi 10.1016/j.immuni.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giladi A, Wagner LK, Li H, Dorr D, Medaglia C, Paul F, et al. Cxcl10(+) monocytes define a pathogenic subset in the central nervous system during autoimmune neuroinflammation. Nat Immunol 2020;21(5):525–34 doi 10.1038/s41590-020-0661-1. [DOI] [PubMed] [Google Scholar]
- 26.Lee JY, Hall JA, Kroehling L, Wu L, Najar T, Nguyen HH, et al. Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease. Cell 2020;180(1):79–91 e16 doi 10.1016/j.cell.2019.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017;356(6335) doi 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao AV, Valk PJ, Metzeler KH, Acharya CR, Tuchman SA, Stevenson MM, et al. Age-specific differences in oncogenic pathway dysregulation and anthracycline sensitivity in patients with acute myeloid leukemia. J Clin Oncol 2009;27(33):5580–6 doi 10.1200/jco.2009.22.2547. [DOI] [PubMed] [Google Scholar]
- 29.Ellegast JM, Alexe G, Hamze A, Lin S, Pimkin M, Ross L, et al. Unleashing Cell-Intrinsic Inflammation As a Strategy to Kill AML Blasts. Blood 2021;138:3305 doi 10.1182/blood-2021-151511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rhyasen GW, Bolanos L, Fang J, Jerez A, Wunderlich M, Rigolino C, et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell 2013;24(1):90–104 doi 10.1016/j.ccr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018;557(7706):580–4 doi 10.1038/s41586-018-0125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melgar K, Walker MM, Jones LM, Bolanos LC, Hueneman K, Wunderlich M, et al. Overcoming adaptive therapy resistance in AML by targeting immune response pathways. Science Translational Medicine 2019;11(508):eaaw8828 doi doi: 10.1126/scitranslmed.aaw8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trowbridge JJ, Starczynowski DT. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med 2021;218(7) doi 10.1084/jem.20201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caiado F, Pietras EM, Manz MG. Inflammation as a regulator of hematopoietic stem cell function in disease, aging, and clonal selection. J Exp Med 2021;218(7) doi 10.1084/jem.20201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2018;172(1–2):176–90 e19 doi 10.1016/j.cell.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 36.Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018;172(1–2):147–61 e12 doi 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018;172(1–2):162–75 e14 doi 10.1016/j.cell.2017.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jordan S, Tung N, Casanova-Acebes M, Chang C, Cantoni C, Zhang D, et al. Dietary Intake Regulates the Circulating Inflammatory Monocyte Pool. Cell 2019;178(5):1102–14 e17 doi 10.1016/j.cell.2019.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaiswal S Clonal hematopoiesis and nonhematologic disorders. Blood 2020;136(14):1606–14 doi 10.1182/blood.2019000989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuster JJ, Zuriaga MA, Zorita V, MacLauchlan S, Polackal MN, Viana-Huete V, et al. TET2-Loss-of-Function-Driven Clonal Hematopoiesis Exacerbates Experimental Insulin Resistance in Aging and Obesity. Cell Rep 2020;33(4):108326 doi 10.1016/j.celrep.2020.108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371(26):2488–98 doi 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sidlow R, Lin AE, Gupta D, Bolton KL, Steensma DP, Levine RL, et al. The Clinical Challenge of Clonal Hematopoiesis, a Newly Recognized Cardiovascular Risk Factor. JAMA Cardiol 2020;5(8):958–61 doi 10.1001/jamacardio.2020.1271. [DOI] [PubMed] [Google Scholar]
- 43.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017;355(6327):842–7 doi 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol 2017;55:56–70 e13 doi 10.1016/j.exphem.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 45.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 2017;377(2):111–21 doi 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015;525(7569):389–93 doi 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abegunde SO, Buckstein R, Wells RA, Rauh MJ. An inflammatory environment containing TNFalpha favors Tet2-mutant clonal hematopoiesis. Exp Hematol 2018;59:60–5 doi 10.1016/j.exphem.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 48.Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018;23(6):833–49 e5 doi 10.1016/j.stem.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Djurec M, Graña O, Lee A, Troulé K, Espinet E, Cabras L, et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proceedings of the National Academy of Sciences 2018;115(6):E1147–E56 doi doi: 10.1073/pnas.1717802115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang G, Liu J, Wu L, Fan Y, Sun L, Qian F, et al. Elevated Expression of Serum Amyloid A 3 Protects Colon Epithelium Against Acute Injury Through TLR2-Dependent Induction of Neutrophil IL-22 Expression in a Mouse Model of Colitis. Frontiers in Immunology 2018;9 doi 10.3389/fimmu.2018.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood 2011;118(9):2551–5 doi 10.1182/blood-2010-12-324707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Damm F, Mylonas E, Cosson A, Yoshida K, Della Valle V, Mouly E, et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov 2014;4(9):1088–101 doi 10.1158/2159-8290.Cd-14-0104. [DOI] [PubMed] [Google Scholar]
- 53.Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018;557(7706):580–4 doi 10.1038/s41586-018-0125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Melgar K, Walker MM, Jones LM, Bolanos LC, Hueneman K, Wunderlich M, et al. Overcoming adaptive therapy resistance in AML by targeting immune response pathways. Sci Transl Med 2019;11(508) doi 10.1126/scitranslmed.aaw8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 2018;19(1):224 doi 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, et al. Welcome to the Tidyverse. Journal of Open Source Software 2019;4:1686 doi 10.21105/joss.01686. [DOI] [Google Scholar]
- 57.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30(15):2114–20 doi 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30(7):923–30 doi 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 60.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 2015;31(12):2032–4 doi 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28(14):1811–7 doi 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 64.Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020;586(7831):763–8 doi 10.1038/s41586-020-2819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017;130(6):742–52 doi 10.1182/blood-2017-02-769869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019;47(D1):D941–d7 doi 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ou J, Zhu LJ. trackViewer: a Bioconductor package for interactive and integrative visualization of multi-omics data. Nat Methods 2019;16(6):453–4 doi 10.1038/s41592-019-0430-y. [DOI] [PubMed] [Google Scholar]
- 68.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 2013;14(1):7 doi 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olson WK, Bansal M, Burley SK, Dickerson RE, Gerstein M, Harvey SC, et al. A standard reference frame for the description of nucleic acid base-pair geometry. J Mol Biol 2001;313(1):229–37 doi 10.1006/jmbi.2001.4987. [DOI] [PubMed] [Google Scholar]
- 70.Hunter JD. Matplotlib: A 2D Graphics Environment. Computing in Science & Engineering 2007;9(3):90–5 doi 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 71.Mˆlder F, Jablonski K, Letcher B, Hall M, Tomkins-Tinch C, Sochat V, et al. Sustainable data analysis with Snakemake [version 2; peer review: 2 approved]. F1000Research 2021;10(33) doi 10.12688/f1000research.29032.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McKinney W Data Structures for Statistical Computing in Python. PROC. OF THE 9th PYTHON IN SCIENCE CONF2010. DOI: 10.25080/Majora-92bf1922-00a [DOI] [Google Scholar]
- 73.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004;25(13):1605–12 doi 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 74.Roboz GJ, Mandrekar SJ, Desai P, Laumann K, Walker AR, Wang ES, et al. Randomized trial of 10 days of decitabine +/− bortezomib in untreated older patients with AML: CALGB 11002 (Alliance). Blood Adv 2018;2(24):3608–17 doi 10.1182/bloodadvances.2018023689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mayer RJ, Davis RB, Schiffer CA, Berg DT, Powell BL, Schulman P, et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med 1994;331(14):896–903 doi 10.1056/NEJM199410063311402. [DOI] [PubMed] [Google Scholar]
- 76.Moore JO, Dodge RK, Amrein PC, Kolitz J, Lee EJ, Powell B, et al. Granulocyte-colony stimulating factor (filgrastim) accelerates granulocyte recovery after intensive postremission chemotherapy for acute myeloid leukemia with aziridinyl benzoquinone and mitoxantrone: Cancer and Leukemia Group B study 9022. Blood 1997;89(3):780–8. [PubMed] [Google Scholar]
- 77.Stone RM, Berg DT, George SL, Dodge RK, Paciucci PA, Schulman P, et al. Granulocyte-macrophage colony-stimulating factor after initial chemotherapy for elderly patients with primary acute myelogenous leukemia. Cancer and Leukemia Group B. N Engl J Med 1995;332(25):1671–7 doi 10.1056/NEJM199506223322503. [DOI] [PubMed] [Google Scholar]
- 78.Attar EC, Johnson JL, Amrein PC, Lozanski G, Wadleigh M, DeAngelo DJ, et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J Clin Oncol 2013;31(7):923–9 doi 10.1200/JCO.2012.45.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blum W, Sanford BL, Klisovic R, DeAngelo DJ, Uy G, Powell BL, et al. Maintenance therapy with decitabine in younger adults with acute myeloid leukemia in first remission: a phase 2 Cancer and Leukemia Group B Study (CALGB 10503). Leukemia 2017;31(1):34–9 doi 10.1038/leu.2016.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kolitz JE, George SL, Marcucci G, Vij R, Powell BL, Allen SL, et al. P-glycoprotein inhibition using valspodar (PSC-833) does not improve outcomes for patients younger than age 60 years with newly diagnosed acute myeloid leukemia: Cancer and Leukemia Group B study 19808. Blood 2010;116(9):1413–21 doi 10.1182/blood-2009-07-229492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moore JO, George SL, Dodge RK, Amrein PC, Powell BL, Kolitz JE, et al. Sequential multiagent chemotherapy is not superior to high-dose cytarabine alone as postremission intensification therapy for acute myeloid leukemia in adults under 60 years of age: Cancer and Leukemia Group B Study 9222. Blood 2005;105(9):3420–7 doi 10.1182/blood-2004-08-2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Uy GL, Mandrekar SJ, Laumann K, Marcucci G, Zhao W, Levis MJ, et al. A phase 2 study incorporating sorafenib into the chemotherapy for older adults with FLT3-mutated acute myeloid leukemia: CALGB 11001. Blood Adv 2017;1(5):331–40 doi 10.1182/bloodadvances.2016003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cassileth PA, Harrington DP, Appelbaum FR, Lazarus HM, Rowe JM, Paietta E, et al. Chemotherapy compared with autologous or allogeneic bone marrow transplantation in the management of acute myeloid leukemia in first remission. N Engl J Med 1998;339(23):1649–56 doi 10.1056/NEJM199812033392301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE182615, GSE201659 and SRA with BioProject ID PRJNA833651. Patient data used in survival analysis was obtained from Alliance Statistics and Data Center and is available upon reasonable request; generated data are deposited in GEO GSE137851, GSE63646. Code used for the single cell data analysis is available at https://github.com/igordot/scooter.