

Summary
Endoplasmic reticulum (ER) homeostasis has been implicated in the pathogenesis of various forms of cancer; however, our understanding of the role of ER quality control mechanisms in tumorigenesis remains incomplete. Here, we show that the SEL1L-HRD1 complex of ER-associated degradation (ERAD) suppresses hepatocyte proliferation and tumorigenesis in mice. Hepatocyte-specific deletion of Sel1L or Hrd1 predisposed mice to diet/chemical-induced tumors. Proteomics screen from SEL1L-deficient livers revealed WNT5A, a tumor suppressor, as an ERAD substrate. Indeed, nascent WNT5A was misfolding prone and degraded by SEL1L-HRD1 ERAD in a quality control capacity. In the absence of ERAD, WNT5A misfolds is largely retained in the ER and forms high-molecular weight aggregates, thereby depicting a loss-of-function effect and attenuating WNT5A-mediated suppression of hepatocyte proliferation. In humans, SEL1L-HRD1 ERAD expression correlated positively with survival time for patients with liver cancer. Overall, our data reveal a key role of SEL1L-HRD1 ERAD in suppressing hepatocyte proliferation and liver cancer.
Subject areas: Molecular biology, Cell biology, Cancer
Graphical abstract
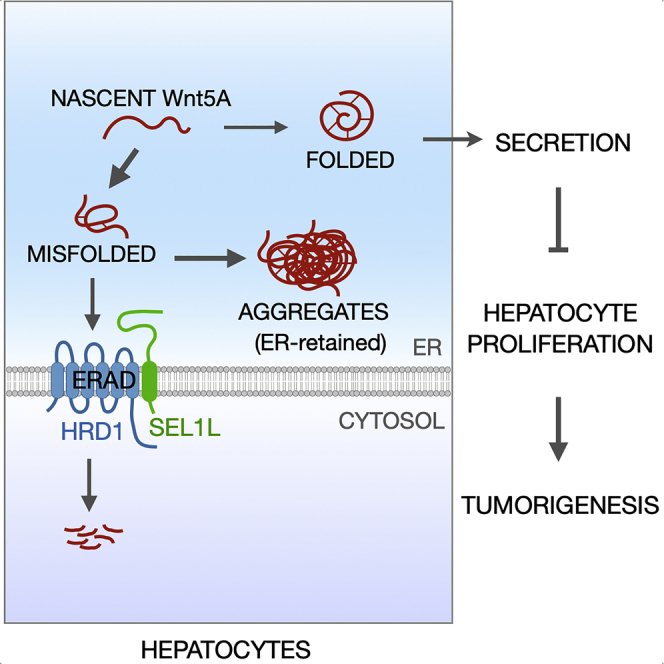
Highlights
-
•
Loss of hepatic SEL1L-HRD1 ERAD causes hyperproliferation and propensity to cancer
-
•
Proteomics of SEL1L-deficient hepatic microsomes reveal WNT5A as a potential link
-
•
WNT5A is misfolding prone and an endogenous SEL1L-HRD1 ERAD substrate in the liver
-
•
Hepatic SEL1L-HRD1 levels correlate with better prognosis in patients with liver cancer
Molecular biology; Cell biology; Cancer.
Introduction
Endoplasmic reticulum (ER) homeostasis has been implicated in the pathogenesis of various forms of cancer (Kim et al., 2015). Assorted ER processes, such as ER folding chaperones and unfolded protein response (UPR), have been investigated for their role in cancer progression. For example, ER chaperones such as BIP and CALRETICULIN have been suggested to affect cell viability and oncogenic signaling in a wide range of solid as well blood cancers (Luo and Lee, 2013). Moreover, the inositol-requiring enzyme 1α (IRE1α) – X-box binding protein 1 (XBP1) pathway of UPR has been reported to promote triple-negative breast cancer (Chen et al., 2014; Zhao et al., 2018), Burkitt’s lymphoma (Xie et al., 2018), multiple myeloma (Harnoss et al., 2019), and prostate cancer (Sheng et al., 2019), while suppressing colon cancer (Niederreiter et al., 2013; Xie et al., 2018) and managing Ras oncogenicity in epidermal tumors (Blazanin et al., 2017). In addition, the PKR-like ER kinase (PERK) – Eukaryotic translation initiation factor 2α (eIF2α) pathway of UPR is known to sensitize metastatic cells to ER stress (Feng et al., 2014), while playing opposing roles in melanoma (Pytel et al., 2016) and breast cancer (Bobrovnikova-Marjon et al., 2010; Sequeira et al., 2007). However, the role of ER-associated degradation (ERAD)—the principal degradative machinery for ER proteins, in cancer remains unexplored.
ERAD recognizes and retro-translocates specific ER proteins to the cytosol for proteasomal degradation (Bhattacharya and Qi, 2019; Guerriero and Brodsky, 2012; Qi et al., 2017). The protein complex composed of E3 ligase HMG-CoA reductase degradation protein 1 (HRD1) and its cofactor suppressor/enhancer of lin-12-like (SEL1L) represents the most conserved branch of mammalian ERAD complex (Mehnert et al., 2015; Mueller et al., 2008; Sun et al., 2014; Vashistha et al., 2016). Global deletion of either gene in mice causes embryonic lethality (Francisco et al., 2010; Yagishita et al., 2005). Previous work from our laboratories and others have shown, using tissue-specific knockout mice, that SEL1L-HRD1 ERAD functions to safeguard against various pathophysiological processes in a substrate-specific manner (Bhattacharya and Qi, 2019; Kim et al., 2018; Qi et al., 2017; Shi et al., 2017; Shrestha et al., 2020; Sun et al., 2015; Wei et al., 2018; Yang et al., 2018; Zhou et al., 2020). In the liver, we recently showed that hepatocyte-specific ERAD regulates the transcription of fibroblast growth factor 21 (FGF21) and systemic energy metabolism in young mice by mediating the turnover of an ER-resident transcription factor, cAMP-responsive element-binding protein, hepatocyte specific (CREBH) (Bhattacharya et al., 2018; Wei et al., 2018).
SEL1L and HRD1, both independently and collectively, have been previously associated with various cancer pathogeneses in humans (Ashktorab et al., 2012; Cardano et al., 2011; Cattaneo et al., 2005; Granelli et al., 2004; Kim et al., 2015; Liu et al., 2012, 2020; Orlandi et al., 2002; Yamasaki et al., 2007); however, the causality and molecular mechanism underlying the role of ERAD in cancer remain unknown. Using different liver cancer and injury models as well as human samples, here we demonstrated a critical role of SEL1L-HRD1 ERAD in hepatocyte proliferation and tumorigenesis. Using non-biased proteomics and cDNA profiling assays, we identified WNT5A, a known tumor suppressor belonging to the WNT family of cysteine-rich, secreted glycoproteins, as a bona fide endogenous substrate of ERAD-mediated quality control in the liver. WNT5A has widely been reported to suppress proliferation and exhibit tumor suppressor activity in hepatocytes (Fan et al., 2017; Wang et al., 2019; Yang et al., 2015), B cells (Liang et al., 2003), skin cells (Wang et al., 2018; Zhang et al., 2013), mammary tissue (Roarty and Serra, 2007), ovarian cancer (Bitler et al., 2011), β cells (Wu et al., 2017), cortical neurons (Zhou et al., 2017a), developing lung (Li et al., 2002), thyroid carcinoma (Kremenevskaja et al., 2005), and colonic epithelium (Cheng et al., 2014; Miyoshi et al., 2012; Ying et al., 2008).
In this study, we describe how ERAD regulates proper folding of WNT5A, thereby indirectly guarding against excess proliferation. In absence of ERAD quality control, misfolded WNT5A aggregates and fails to suppress hepatocyte proliferation, thereby allowing for tumor growth. Indeed, we found ERAD expression levels to be elevated in human tumors, which could be an adaptive response to the excessive proliferation. Overall, this study uncovers a novel regulatory mechanism linking SEL1L-HRD1 ERAD to liver cancer, and also characterizes an unprecedented quality control mechanism overseeing nascent WNT5A maturation in the ER.
Results
Sel1LAlb mice are prone to developing chemically induced liver tumors
To examine the role of SEL1L-HRD1 ERAD in hepatocytes, we recently generated hepatocyte-specific SEL1L-deficient (Sel1LAlb) (Bhattacharya et al., 2018) and Hrd1Alb mice (Wei et al., 2018) by using the albumin promoter-driven Cre. These mice showed modest growth retardation due to elevated FGF21 production at postnatal 4–12 weeks of age on normal chow diet (Bhattacharya et al., 2018; Wei et al., 2018). However, upon reaching adult stage, they appeared normal and healthy, and grew comparably to wild-type Sel1Lf/f littermates (Figure 1A).
Figure 1.

Loss of hepatic SEL1L predisposes mice to chemically induced tumors
(A) Body weight curve for male mice over 22 months (n = 8 per group).
(B–J) Male mice were injected i.p. with 25 mg/kg diethylnitrosamine (DEN) at 2 weeks of age, placed on high-fat diet (HFD) at 4 weeks of age, and sacrificed at 6–7 months of age: (B) Schematic outline; (C and D) Representative images of liver (C, arrows: visible tumors), and external visible tumor count (D) (n = 5–7 per group); (E and F) Western blot analysis of tumor and normal liver tissue with quantitation shown below the blot after normalization to the loading control HSP90 (n = 2 per group, 2 independent repeats); (G–J) Representative images of H&E of normal liver, adenoma, and carcinoma (G and H, arrows: expanded hepatic parenchyma (1) with compressed adjacent tissue (2), asterisks: dense cellular packing), cyclin D1 (I), and KI67 (J) staining at the end of experiment, with quantitation shown in Figures S1A and S1B (n = 5–7 per group). Values, mean ± SEM; ∗∗, p < 0.01; ∗∗∗, p < 0.001 by Student’s t test.
To explore the role of hepatocyte ERAD in liver cancer, we i.p. injected Sel1Lf/f and Sel1LAlb mice once with a potent carcinogen diethylnitrosamine (DEN) (25 mg/kg body weight) at 2 weeks of age, followed by high-fat diet (HFD) feeding starting at 4 weeks of age for a total of 24 weeks (day 196) (Figure 1B). On day 196, there were none to very few tumors visible in Sel1Lf/f livers. By contrast, all Sel1LAlb mice exhibited high hepatic tumor load (arrows, Figures 1C and 1D). Cyclin D1 is classically known to be a primary regulator and marker of hepatocyte proliferation (Nelsen et al., 2003; Núñez et al., 2017; Rickheim et al., 2002). Some of Sel1LAlb liver tumors were adenomas with moderately elevated cyclin D1 and E-cadherin levels (Figure 1E), while others were advanced carcinomas with high cyclin D1 and low E-cadherin levels (Figure 1F). Histological analysis showed normal fatty liver morphology for Sel1Lf/f mice, with the accumulation of lipid droplets (Figure 1G). However, Sel1LAlb livers showed multiple adenomas with abnormal regenerative hyperplasia, hepatic parenchyma expansion (arrows, area 1), and compression of adjacent tissue (area 2, Figure 1H). In some, carcinomas with trabecular patterning and dense cellular packing were visible (asterisks, Figure 1H). Immunostaining of cyclin D1 and KI67 revealed highly proliferative nature of Sel1LAlb cells in the liver (Figures 1I and 1J, and quantitated in Figures S1A and S1B). Taken together, we concluded that the loss of hepatic SEL1L promotes tumorigenesis and hepatocyte proliferation in the DEN-HFD model.
Hrd1Alb mice form spontaneous liver tumors following chronic HFD feeding
As SEL1L and HRD1 may have mutually independent functions (Christianson et al., 2012; Schulz et al., 2017), we next asked whether HRD1 deficiency may also augment liver tumorigenesis. Following HFD starting at 4 weeks of age for one year, Hrd1Alb mice developed and succumbed to massive liver tumors with lower total body weight and increased liver-to-body weight ratios (Figures 2A and 2B). All Hrd1Alb livers had visible tumors, whereas none of their Hrd1f/f littermates did (Figure 2C, and quantitated in Figures 2D and 2E). Histological examination of the Hrd1Alb livers showed multiple tumors with hyperplastic parenchyma (asterisks, Figure 2F). KI67 staining confirmed hyperproliferative nature of these tumor cells (Figure 2G). Hence, similar to SEL1L deficiency, the loss of hepatic HRD1 leads to spontaneous liver cancer and hyperproliferation upon chronic HFD feeding.
Figure 2.

Loss of HRD1 leads to spontaneous liver cancer following chronic HFD feeding
Male mice were fed high-fat diet (HFD) for one year: Body weight (A), liver-to-body weight ratio (B), representative images (C arrows: visible tumors), tumor incidence (D), tumor count (E), representative images of H&E (F, asterisks: hyperplastic parenchyma with cytoplasmic inclusions), and KI67 staining with quantitation shown below (G) at the end of experiment (n = 5 per group). Values, mean ± SEM; ∗∗, p < 0.01; ∗∗∗, p < 0.001 by Student’s t test.
Importantly, both Sel1LAlb and Hrd1Alb mice remained relatively resistant to HFD feeding as described earlier (Bhattacharya et al., 2018; Wei et al., 2018), while developing tumors in their livers. This suggests that this tumorigenesis was likely independent of the HFD-induced weight gain, and was possibly accelerated by the pathologies associated with HFD feeding in the liver.
Hyperproliferation of Sel1LAlb hepatocytes following liver damage
We next explored whether ERAD plays a cell-intrinsic role in hepatocyte proliferation in two liver injury models, partial hepatectomy and treatment with a hepatotoxin carbon tetrachloride (CCL4). Post two-thirds partial hepatectomy, both Sel1Lf/f and Sel1LAlb mice recovered well and comparably (Figure 3A). However, the remaining lobes of Sel1LAlb liver showed significantly faster growth kinetics in mass restoration than those of Sel1Lf/f liver (Figure 3B). Histological examination showed larger nuclei size in Sel1LAlb livers 48 h post surgery (arrows, Figures 3C and 3D, and quantitated in Figure S2A). The signal intensity of cyclin D1 protein, the primary marker and regulator of liver re-growth (Nelsen et al., 2003; Núñez et al., 2017; Rickheim et al., 2002), was indeed markedly elevated in the nuclei of Sel1LAlb compared to Sel1Lf/f hepatocytes 48 h post surgery (Figure 3D, and quantitated in Figure S2B), although the number of cycline D1-positive nuclei was comparable (Figure 3D, and quantitated in Figure S2C).
Figure 3.

Sel1LAlb livers exhibit excessive proliferation following injury
(A–D) Two-thirds partial hepatectomy surgery of 10-week-old mice: (A and B) Body weight gain (A), and liver weight gain (B) (n = 3 per group, 2 independent repeats); (C and D) Representative images of hematoxylin-eosin (C, arrows: enlarged nuclei), and cyclin D1 staining (D) at 48-h post surgery (n = 4 per group).
(E–I) 2-month-old male mice were injected intraperitoneally with 0.2 mL/kg body weight carbon tetrachloride (CCL4) dissolved in oil thrice a week for 3 months: (E) Schematic timeline; (F) Body weight gain curves; (G) Liver-to-body weight ratio; (H) Representative liver images; (I) Representative images of KI67 staining (n = 4–5 per group). Values mean ± SEM; ∗, p < 0.05 by Student’s t test.
In the chronic CCL4 assay, 2-month-old mice on normal chow diet were injected i.p. with a very low dose (0.2 mL/kg body weight) of CCL4 three times a week for 3 months to induce mild liver injury (Figure 3E). Sel1LAlb mice gained more body weight compared to Sel1Lf/f mice (Figure 3F) with significantly enlarged livers, i.e. hepatomegaly (Figures 3G and 3H). KI67 protein level was highly elevated in Sel1LAlb livers (Figure 3I, and quantitated in Figure S2D). Serum alanine aminotransferase and aspartate aminotransferase levels were highly elevated, but comparable, between the cohorts (Figures S2E and S2F). H&E histological examination also confirmed that CCL4 caused similar level of injury in both cohorts (Figure S2G). Taken together, we concluded that SEL1L-HRD1 ERAD deficiency in the hepatocytes leads to hyperproliferation following hepatic injury, which may accelerate tumorigenesis in chemical- and HFD-induced models. These two liver injury models serve to provide a causal connection between hepatic ERAD and hepatocyte proliferation, irrespective of tumorigenic stimulus.
Proteomics identification of WNT5A in SEL1L-deficient livers
To delineate how ERAD deficiency trigger increased proliferation in hepatocytes, we performed non-biased proteomics and cDNA profiling analyses of livers from adult mice on normal chow diet without any injury stimulus. As ERAD deficiency causes an accumulation of its substrates in the ER (Bhattacharya and Qi, 2019), we performed proteomics analysis of purified ER from the livers of 2- to 3-month-old Sel1Lf/f and Sel1LAlb mice. A total of 3,273 proteins were identified, out of which 744 proteins were significantly affected with p < 0.05, with 205 significantly elevated over 2-fold in Sel1LAlb livers (top right corner, Figure 4A). Both SEL1L and HRD1 levels were reduced, while two known ERAD substrates IRE1α (Sun et al., 2014) and osteosarcoma amplified 9 (OS9) (Sha et al., 2014) were elevated (Figures 4A and 4B), thus validating the assay. Among the top 50 upregulated hits (with p < 0.05), we noted WNT5A (Figure 4B and Table S1), the only protein to have been implicated in cellular proliferation and differentiation (Asem et al., 2016; McDonald and Silver, 2009; Zhan et al., 2017; Zhou et al., 2017b). Of note, there were no other WNT proteins identified in this analysis (Table S1).
Figure 4.

WNT5A may link SEL1L-HRD1 ERAD to hepatic proliferation
(A and B) Volcano plot (A) and tabular form (B) of proteomics data obtained via TMT-LC/MS analysis of hepatic ER isolated from 2- to 3-months-old mice (n = 3 per group). Lines mark p = value 0.05 (horizontal) or 2-fold changes (vertical).
(C–F) Western blot analysis of ER and non-ER fractions from livers of 2-month-old mice (n = 3 per group) (C, black and red arrows mark WNT5A mobility shift), hepatocellular carcinoma and normal liver tissue from the DEN-HFD liver cancer model (n = 2 per group, 2 independent repeats) (D), livers of 10-week-old mice 48-h post partial hepatectomy (n = 3 per group) (E), livers from chronic CCL4 treatment (n = 2 per group) (F).
(G) Western blot analysis normal liver and tumor tissues from the year-long-HFD experiment (n = 3 per group).
(H and I) Hepatic cDNA microarray analyses of 9-week-old Sel1LAlb livers compared to Sel1Lf/f livers: (H) KEGG pathway for Gene Set Enrichment Analysis (GSEA) from differentially expressed hepatic genes with p < 0.05 and fold change >1.29, and (I) Heatmap of proliferation-associated genes as a logarithm of fold change in Sel1LAlb and Hrd1Alb livers as compared to Sel1Lf/f and Hrd1f/f livers, respectively (n = 3 per group).
(J) Western blot analysis of HepG2 and Huh7 cells treated with 500 ng/mL recombinant WNT5A protein for 30 h (2 independent repeats).
(K) Western blot analysis of Sel1Lf/f and Sel1LAlb livers 2 weeks after i.v. injection of AAV8 expressing shRNA targeting Wnt5A or luciferase control (n = 2–3 per group). Quantitation shown below each blot. Calnexin, ER fraction control; HSP90, loading and non-ER controls. Values, mean ± SEM; ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001 by Student’s t test.
Using Western blot analysis, we confirmed the 3.5-fold enrichment of WNT5A in the calnexin-positive ER fraction of the Sel1LAlb livers (Figure 4C). In contrast, in the non-ER fraction, WNT5A protein was slightly reduced and more importantly, migrated slightly faster in the Sel1LAlb livers, compared to Sel1Lf/f livers (red vs. black arrows, Figure 4C). WNT5A levels were indeed increased in the carcinomas of Sel1LAlb mice treated with DEN-HFD (Figure 4D), and in Sel1LAlb livers 48 h post partial hepatectomy (Figure 4E) or following chronic CCL4 treatment (Figure 4F). By contrast, protein levels of WNT5B and WNT1 were slightly reduced in the Sel1LAlb livers compared to Sel1Lf/f livers (Figures 4D–4F). Similarly, WNT5A protein level was elevated in the tumors from Hrd1Alb liver, as was cyclin D1 protein (Figure 4G). As Wnt5A mRNA level was unchanged in Sel1LAlb livers (Figure S3A), these data pointed to a post-transcriptional control of WNT5A by SEL1L-HRD1 ERAD in hepatocytes.
Providing further support, cDNA profiling analyses of 9-week-old Sel1LAlb livers (GSE118658) (Bhattacharya et al., 2018) revealed that processes associated with “transcription changes in cancer”, “cell cycle”, “DNA replication”, and “chemical carcinogenesis” were among the top pathways identified in KEGG analyses of Sel1LAlb livers for differentially expressed genes with fold change >1.29 (p < 0.05; Figure 4H). Moreover, in line with the elevated proliferation of Sel1LAlb and Hrd1Alb hepatocytes, several cell cycle-related genes were significantly upregulated (Figures 4I and S3B). On the other hand, several mature hepatocyte genes were downregulated in Sel1LAlb and Hrd1Alb livers (Figure S3C).
WNT5A may link SEL1L-HRD1 ERAD to hepatocyte proliferation
As previous studies have reported that WNT5A suppresses proliferation in hepatocytes (Fan et al., 2017; Wang et al., 2019; Yang et al., 2015) and many other cell types (Bitler et al., 2011; Cheng et al., 2014; Kremenevskaja et al., 2005; Li et al., 2002; Liang et al., 2003; Miyoshi et al., 2012; Roarty and Serra, 2007; Wang et al., 2018; Wu et al., 2017; Ying et al., 2008; Zhang et al., 2013; Zhou et al., 2017a), we next asked whether WNT5A may be one of the links between ERAD and hepatocyte hyperproliferation. As mentioned earlier, cyclin D1 is classically used as a readout for hepatic proliferation (Nelsen et al., 2003; Núñez et al., 2017; Rickheim et al., 2002). In line with its nature as a tumor suppressor, treatment of HepG2 and Huh7 cells with recombinant WNT5A protein reduced cyclin D1 levels (Figure 4J). Moreover, i.v. injection with adeno-associated Virus serotype 8 (AAV8) expressing shRNAs targeting Wnt5A gene (shWnt5A) reduced WNT5A levels, and markedly increased cyclin D1 levels in in both Sel1Lf/f and Sel1LAlb livers two weeks post injection (Figure 4K), suggesting that accumulated WNT5A in the latter may be non-functional. Taken together, these data demonstrated that WNT5A may link SEL1L-HRD1 ERAD to hepatocyte proliferation.
ER retention of WNT5A protein in SEL1L-deficient hepatocytes
We next further explored the mechanistic link between ERAD and WNT5A. Western blotting of total liver lysate showed a prominent faster mobility shift of WNT5A band, but not for WNT5B and WNT1 proteins, in the Sel1LAlb livers compared to Sel1Lf/f livers (red vs. black arrows, Figures 4C and 5A). To test whether this mobility shift was caused protein glycosylation, we performed the endoglycosidase H (EndoH) sensitivity assay to differentiate core glycosylated WNT5A in the ER (EndoH-sensitive) from those complex glycosylated beyond the ER (EndoH-resistant). Indeed, EndoH-sensitive WNT5A protein increased from 7% in Sel1Lf/f livers to about 50% in Sel1LAlb livers (Figure 5B). Digestion with peptide-N-glycosidase F (PNGaseF), an enzyme that cleaves all glycosylation sites, showed no difference between the two cohorts (Figure 5B), confirming that the mobility shift was indeed caused by differential glycosylation in the ER. This finding was further confirmed using an acute SEL1L deficiency model, where AAV-mediated Cre recombinase (AAV-Cre) was injected into the tail vein of Sel1Lf/f mice (Bhattacharya et al., 2018). EndoH-sensitive WNT5A protein increased from 6% in AAV-GFP-injected Sel1Lf/f livers to about 60% in AAV-Cre-injected Sel1Lf/f livers (Figure 5C). In both chronic and inducible SEL1L deficiency models, the portion of WNT5A exiting the ER was reduced from 93%–94% in WT livers to about 40%–50% in SEL1L-deficient livers (Figures 5B and 5C). Hence, SEL1L deficiency increases ER retention of WNT5A in hepatocytes.
Figure 5.

WNT5A protein is retained in the ER and forms HMW aggregates in SEL1L-deficient hepatocytes
(A and B) Western blot protein analysis of total tissue lysate (A), and EndoH/PNGaseF digestion (B) of 2-month-old mouse livers (n = 5 per group, 2 independent repeats).
(C) EndoH digestion analysis of total liver lysates from 6-week-old Sel1Lf/f mice 2 weeks after tail vein injection with either AAV8-Cre or control AAV8-GFP (n = 3 per group).
(D) Western blot analysis of detergent (NP40) fractionation (S, soluble; P, pellet) of total liver lysates from 9-week-old mice (n = 3 per group). Black and red arrows mark WNT5A mobility shift. H2A and HSP90, loading controls for soluble (NP40S) and pellet (NP40P) fractions.
(E–H) Reducing and non-reducing Western blot analyses of sucrose-gradient fractionated liver lysates (E) and quantitation (F); EndoH digestion analysis of fractions #7 (G) and #13 (H) (n = 3 per group, 2 independent repeats). r and s, EndoH-resistant and -sensitive.
Elevated WNT5A aggregation in SEL1L-deficient hepatocytes
We next explored the consequence of increased ER retention of WNT5A. We first performed detergent NP40-based fractionation of total liver lysates. WNT5A, but no other WNT proteins, accumulated in the NP40-insoluble or pellet (NP40P) fraction of Sel1LAlb liver lysate (lane 4, Figure 5D), pointing to the possibility of forming high-molecular weight (HMW) aggregates in the absence of SEL1L. To visualize these aggregates, we performed sucrose gradient fractionation of liver lysates followed by non-reducing SDS-PAGE, which separated WNT5A into monomers, dimers, and HMW complexes or aggregates. In Sel1Lf/f livers, WNT5A was present as a mixture of monomers and oligomers, with a small fraction as dimers (Figure 5E). By contrast, in Sel1LAlb livers, WNT5A-containing HMW aggregates were significantly elevated, especially in the fraction #13—the pellet at the bottom of the sucrose gradient column (Figure 5E, and quantitated in Figure 5F). All HMW complexes and aggregates were reduced to the monomers under reducing conditions with β-mercaptoethanol incorporated into sample buffer (Figure 5E), suggesting that HMW is formed via intermolecular disulfide bonds. Unlike WNT5A, SEL1L deficiency did not result in protein aggregation of WNT1 and WNT5B (Figures S4A and S4B).
To determine the status of WNT5A maturation for each fraction, we performed EndoH analysis of fractions #7 and #13. Interestingly, in the fraction #7, 20% WNT5A was present in the ER in the absence of SEL1L vs. 6% in WT livers (Figure 5G). In the fraction #13, WNT5A was entirely retained in the ER of SEL1L-deficient livers, while undetectable in WT livers (Figure 5H). Hence, these data suggested that SEL1L deficiency enhances the disulfide bond-mediated aggregation and ER-retention of WNT5A.
WNT5A is an endogenous SEL1L-HRD1 ERAD substrate
WNT5A is a 380 amino acids-long secreted protein containing 11 confirmed (black) and one putative (gray) intramolecular disulfide bonds in humans (Figure 6A). We next explored how ERAD regulates WNT5A maturation. We first tested whether WNT5A is an endogenous ERAD substrate. In transfected HEK293T cells, WNT5A interacted with SEL1L and HRD1 (Figures S5A and S5B). WNT5A was ubiquitinated by HRD1, requiring the activity of HRD1 E3-ligase activity as shown with the help of the HRD1 RING finger domain C2A mutant (Kikkert et al., 2004) (Figure 6B). Next, using primary mouse hepatocytes treated with a proteasomal inhibitor bortezomib (BTZ), our data showed that WNT5A was highly ubiquitinated and degraded by proteasomes in a SEL1L-dependent manner (Figure 6C). Consequently, in primary hepatocytes treated with a translation inhibitor cycloheximide, WNT5A protein was significantly stabilized in the absence of SEL1L (Figure 6D). Together, these data demonstrated that WNT5A is a bona fide endogenous ERAD substrate in hepatocytes.
Figure 6.

Hepatic WNT5A is a bona fide ERAD substrate
(A) Schematic map of WNT5A protein domains; sp, signal peptide; pro, pro-peptide; bracket, disulfide bonds; red flag, glycosylation.
(B) Western blot analysis of WNT5A ubiquitination in HEK293T cells transfected with indicated plasmids: cells were treated with proteasomal inhibitor bortezomib (BTZ) for 4 h prior to harvest (3 independent repeats).
(C) Western blot analysis of endogenous WNT5A ubiquitination in primary mouse hepatocytes treated with BTZ for 4 h as indicated (2 independent repeats).
(D) Western blot analysis of endogenous WNT5A protein half-life, with quantitation shown below, in primary mouse hepatocytes, pre-treated with Brefeldin A for 2 h to block secretion, and after treatment with CHX as indicated (2 independent repeats). HSP90, loading control.
SEL1L-HRD1 ERAD in human liver cancer
Lastly, we examined the expression of SEL1L-HRD1 ERAD in human hepatocellular carcinoma samples. In a correlation analysis done using Spearman method on RNA sequencing data, HRD1 expression showed co-occurrence with SEL1L in both hepatocellular carcinoma and normal liver samples (Figures 7A and 7B). In the cBio Portal for Cancer Genome, by analyzing the TCGA Pan Cancer Atlas dataset, we found that SEL1L and HRD1 genomic loci or expression levels were altered in 4% and 8% of liver hepatocellular carcinoma samples, respectively (Figure S6A). This analysis showed several instances of deep deletions and a few amplifications (Figure S6A), and 3 SEL1L and 4 HRD1 single nucleotide polymorphisms (SNPs), majority of which were missense mutations resulting in amino acid change (Figures S6B and S6C). This suggests that while SEL1L-HRD1 ERAD is important in the pathogenesis of human liver cancers, the role of disease mutants and the detailed downstream mechanism require further investigation.
Figure 7.

SEL1L-HRD1 ERAD expression in human liver cancer
(A and B) Spearman correlation of HRD1 and SEL1L in 369 tumors samples (A), and 160 normal samples (B).
(C–E) Kaplan-Meier survival curves in a cohort of 364 TCGA hepatocellular carcinoma patients with hazard ratios (HR) with 95% confidence intervals and p values (log rank test, Mantel-Haenszel), based on following gene expressions: (C) SEL1L, (D) HRD1, and (E) SEL1L + HRD1 combined.
(F and G) Western blot analysis (F) and quantitation (G) of SEL1L and HRD1 protein levels in tumor and adjacent non-tumor tissue obtained from human liver cancer patients, with αTUBULIN as a loading control (n = 14 per group).
(H and I) Representative images of HRD1 stain (H) and quantitation (I) on human hepatocellular carcinoma sections (n = 85 per group). T, tumor; N, adjacent non-tumor tissues. Values mean ± SEM; ∗∗, p < 0.01 by Student’s t test.
We next analyzed ERAD expression at both mRNA and protein levels in liver tumors. Kaplan-Meier survival analysis of 364 patients from TCGA liver hepatocellular carcinoma samples showed that higher expression of SEL1L or HRD1 genes was associated with better overall survival (Figures 7C and 7D). Higher expression levels of both genes together showed an even stronger positive prognosis for overall survival (Figure 7E). Furthermore, analysis of 14 pairs of tumor and adjacent normal tissues excised from human liver cancer patients (University of Michigan Pathology) revealed that both SEL1L and HRD1 protein levels were elevated, albeit not to a significant extent for HRD1, in tumor tissues compared to adjacent normal tissues (Figure 7F, and quantitated in Figure 7G). WNT5A distribution in these samples could not be properly assessed owing to poor antibody performance in human tissues. Lastly, immunostaining analysis of HRD1 in normal and tumor tissue sections obtained from 85 human liver cancer patients (US Biomax Inc.) revealed a significant increase in HRD1 protein level in tumors compared to adjacent normal tissues (Figure 7H, and quantitated in Figure 7I).
Discussion
Hepatocellular carcinoma (HCC) is an aggressive malignancy of the liver and the leading cause of cancer mortality all over the world (Ferenci et al., 2010). Here, we have uncovered a causal relationship between SEL1L-HRD1 ERAD, hepatocyte proliferation, and the pathogenesis of HCC, where SEL1L-HRD1 ERAD suppresses liver cell proliferation and tumorigenesis. Mechanistically, using non-biased high-throughput approaches, we identified WNT5A, a tumor suppressor in the liver, as a bona fide endogenous substrate of hepatic SEL1L-HRD1 ERAD. In the absence of SEL1L-HRD1 ERAD, WNT5A accumulated, formed disulfide-bonded HMW aggregates, and was largely retained in the ER. This potentially led to reduced tumor suppressive activity of WNT5A and as a result, contributed, in part, to increased cyclin D1 levels and hepatocyte hyperproliferation. In human liver cancer patients, there was indeed a strong correlation between SEL1L-HRD1 expression and disease prognosis. We propose that ERAD levels are elevated in tumor tissues as an adaptive mechanism to the excess proliferation. Overall, these data suggest that SEL1L-HRD1 may play a critical role in liver tumorigenesis in humans.
The wingless or WNT pathway is one of the most frequently perturbed pathways in HCC and hence a popular therapeutic target in liver cancer (Gedaly et al., 2014; Lee et al., 2014; Polakis, 2000; Vilchez et al., 2016). In humans and mice, 19 WNT family ligands exist, which bind to frizzled (Fzd) and low-density lipoprotein receptor-related protein (Lrp) receptors, thereby activating either β-catenin-mediated canonical signaling or largely uncharacterized non-canonical signaling pathway (van Amerongen and Nusse, 2009). WNT5A is a non-canonical WNT family ligand and has widespread functions in cancer or normal stem cell self-renewal, proliferation, and metastasis (Asem et al., 2016; McDonald and Silver, 2009; Zhan et al., 2017). With regards to liver cancer, studies have reported WNT5A to act as a tumor suppressor in hepatocytes (Fan et al., 2017; Wang et al., 2019; Yang et al., 2015). While the downstream signaling pathway of WNT5A in hepatocytes remains unclear, our data showed that treatment of hepatocytes with recombinant WNT5A reduced the cyclin D1 levels and conversely, knockdown of WNT5A in the livers using the AAV-shRNA system had the opposite effect. Therefore, WNT5A activity in hepatocytes seems to be inversely correlated with proliferation. This is consistent with other studies that show WNT5A to inhibit cellular proliferation, often acting as a tumor suppressor, in various developmental and oncogenic contexts (Bitler et al., 2011; Cheng et al., 2014; Fan et al., 2017; Kremenevskaja et al., 2005; Li et al., 2002; Liang et al., 2003; Miyoshi et al., 2012; Roarty and Serra, 2007; Wang et al., 2018, 2019; Wu et al., 2017; Yang et al., 2015; Ying et al., 2008; Zhang et al., 2013; Zhou et al., 2017a).
To date, cellular processes underlying the biogenesis and maturation of nascent WNT proteins have not been explored. The maturation process of WNT5A protein starts within the ER, and involves multiple glycosylation events and the formation of 11–12 intramolecular disulfide bridges for a stable globular structure (Willert and Nusse, 2012). These complexities render the folding and assembly of WNT5A a complex and error-prone process, and making it an ideal substrate for ERAD. Indeed, our data show that WNT5A is ubiquitinated by and targeted for proteasomal degradation by SEL1L-HRD1 ERAD in the liver under basal conditions. In the absence of ERAD, nascent WNT5A protein accumulates in the ER and, for some, form disulfide-bonded HMW aggregates caused by aberrant disulfide bonds. The maturation defects of WNT5A in the ER of ERAD-deficient cells may contribute to its suppressive effect on proliferation. Therefore, our data identify and implicate the “ERAD-WNT5A″ axis, at least partially, in hepatocyte proliferation and tumorigenesis in the liver.
The effect of SEL1L-HRD1 ERAD on WNT5A is akin to some other substrates previously reported such as prohormones POMC (with 2 intramolecular disulfide bonds) (Kim et al., 2018) and proAVP (with 8 intramolecular disulfide bonds) (Shi et al., 2017). ERAD deficiency triggers disulfide-bond-mediated protein aggregation in the ER. Interestingly, our data show that SEL1L-HRD1 ERAD has negligible effect on the maturation of other WNT proteins such as WNT5B and WNT1 in the adult mouse liver. However, how such substrate specificity for ERAD is defined, potentially with the help of various ER chaperones, remains to be an intriguing question.
In human liver tumor samples, we found that there is indeed a correlation between SEL1L-HRD1 mRNA levels and better prognosis. Interestingly, we also found that both SEL1L and HRD1 protein levels were increased in tumor tissues compared to adjacent normal tissues. We speculate that this increased SEL1L-HRD1 levels could be an adaptive response to elevated protein synthesis in the ER during tumorigenesis (Kim et al., 2015). This change may enhance ERAD-mediated clearance of misfolded WNT5A in the ER, thereby increasing WNT5A suppressive activity and leading to better prognosis. The answers to these questions will be explored in future studies. Nonetheless, this study provides an important framework for future studies to directly assess the relevance, significance, and molecular mechanism of ERAD in various cancers in vivo.
Limitations of the study
Future investigations are required on establishing the full causal link between SEL1L-HRD1 ERAD in the liver and hepatic hyperproliferation, and the extent of the contribution of WNT5A misfolding in this axis needs to be carefully assessed. Furthermore, human mutants of WNT5A, SEL1L, and HRD1 need to be explored in the context of liver cancer pathogenesis. Last but not the least, exploring the crosstalk of UPR with ERAD deficiency in this system is an exciting future question.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| KI67 | Abcam | ab16667 |
| CYCLIND1 | Abcam | ab16663 |
| c-MYC | MilliporeSigma | C3956 |
| V5 | Invitrogen | R960-25 |
| HA | MilliporeSigma | H9658 |
| WNT5A | Proteintech | 55184-1-AP |
| WNT5B | Abclonal | A8313 |
| WNT1 | Proteintech | 27935-1-AP |
| E-CADHERIN | BD | 610181 |
| α-TUBULIN | Santa Cruz | sc-5286 |
| HSP90 | Abcam | ab13492 |
| CALNEXIN | Enzo | ADI-SPA-860-F |
| SEL1L | Abcam | ab78298 |
| HRD1 (for Western blot) | Dr. Richard Wojcikiewicz, SUNY Upstate Medical University | Pearce et al., 2007 |
| HRD1 (for IHC) | MilliporeSigma | HPA024300 |
| Bacterial and virus strains | ||
| AAV8 | University of Michigan Vector Core | pAAV8-D(+)-U6-siRNA-CMV-GFP |
| Biological samples | ||
| Frozen biopsies of hepatocellular carcinoma tumor samples and adjacent normal liver tissue | University of Michigan Pathology | https://www.pathology.med.umich.edu/ |
| Human hepatocellular carcinoma tissue section microarray | US Biomax, Inc. | HLivH180Su14 |
| Chemicals, peptides, and recombinant proteins | ||
| DEN | MilliporeSigma | N0258 |
| CCL4 | MilliporeSigma | 289116 |
| Recombinant Wnt5A | R&D systems | 645-WN-010 |
| Critical commercial assays | ||
| TMTsixplex™ Isobaric Label Reagent Set | ThermoFisher | PI90061 |
| Deposited data | ||
| Proteomics analysis of purified microsomes | PRIDE ProteomeXchange | PXD035243 |
| cDNA profiling data of total liver | GEO | GSE118658 |
| Liver Hepatocellular Carcinoma TCGA Pan Cancer Atlas dataset | cBio Portal for Cancer Genomics | https://www.cbioportal.org/ |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-3216 |
| HepG2 | ATCC | HB-8065 |
| Huh7 | Dr. Kezhong Zhang, Wayne State University | Dr. Christopher M. Schonhoff, Tufts University Cummings School of Veterinary Medicine |
| Experimental models: Organisms/strains | ||
| Mice: Sel1Lf/f;Alb-Cre/+ and Sel1Lf/f | Dr. Ling Qi, University of Michigan | Bhattacharya et al. (2018) |
| Mice: Hrd1f/f;Alb-Cre/+ and Hrd1f/f | Dr. Deyu Fang, Northwestern University | Wei et al. (2018) |
| Oligonucleotides | ||
| Primers and siRNA | See Table S2 for sequences | N/A |
| Recombinant DNA | ||
| Active-WNT5A-V5 | Addgene | 43813 |
| pAAV8-D(+)-U6-siRNA-CMV-GFP | Addgene | 85451 |
| HRD1 plasmid (MYC-tagged WT and mutant C2A) | Dr. Yihong Ye, NIDDK | Kikkert et al. (2004) |
| pcDNA3-HA-Ub | Dr. Hideki Nishitoh, University of Miyazaki | Sun et al. (2015) |
| Software and algorithms | ||
| Kaplan-Meier Plotter | http://kmplot.com/analysis, | Menyhart et al. (2018) |
| Gene Expression Profiling Interactive Analysis (GEPIA) | http://gepia.cancer-pku.cn/index.html | Tang et al. (2017) |
Resource availability
Lead contact
Further information for resources and reagents should be directed to the lead contact, Ling Qi (lingq@med.umich.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
Generation of Sel1LAlb mice and their Sel1Lf/f littermates has been described previously (Bhattacharya et al., 2018). Hrd1Alb and their Hrd1f/f littermate mice have also been previously described (Wei et al., 2018). Mice were fed normal chow diet (LabDiet 5LOD, 13% fat, 30% protein and 57% carbohydrate). In high-fat diet (HFD) studies, Sel1L cohorts were fed with 60% HFD (60% fat, 20% protein, 20% carbohydrate, Research Diets D12492) and the Hrd1 cohorts were placed on 45% HFD (45% fat, 20% protein, 35% carbohydrate Research Diets D12451). All mice procedures were in compliance with IACUC at the University of Michigan (PRO00008989) or Cornell University (2007-0051) or Northwestern University (IS00001833). Littermates of same sex were randomly assigned to the experimental groups. Isoflurane anesthesia before major organ removal was the method used for euthanasia. All tissues were either fixed or frozen in liquid nitrogen right upon isolation.
Cell lines and transfection
HEK293T and HepG2 from ATCC, and Huh7 from Dr. Kezhong Zhang, Wayne State University, were cultured in DMEM (Corning, NY) with heat inactivated 10% FBS (GIBCO), 1% sodium pyruvate and 1% penicillin/streptomycin. Transfection of cells was carried out 16–24 h after plating with Lipofectamine 2000 reagent and harvested in about another 24 h for analysis. Recombinant WNT5A was purchased from R&D systems (645-WN-010).
Method details
DEN model
2-week-old male Sel1LAlb mice were injected i.p. with a single dose of 25 mg/kg diethylnitrosamine (DEN; Sigma N0258), then put on high fat diet at 4 weeks of age, and finally euthanized for analysis at 28 weeks of age.
CCL4 model
8-week-old male Sel1LAlb mice were injected i.p. with 0.2 mL/kg carbon tetrachloride (CCL4; Sigma 289,116) dissolved in oil thrice a week for up to 12 weeks, and finally euthanized for analysis at 20 weeks of age.
Partial hepatectomy
Two-third of the adult mouse liver was surgically resected to observe the regeneration of hepatocytes thereafter. During this procedure, 2–3 months old mice were anesthetized via isoflurane inhalation and placed on a heating pad. Abdominal hair was shaved, followed by the surgical area being wiped with betadine and 70% alcohol and draped. A transverse bilateral skin incision was made, followed by entry into the peritoneal cavity. A stitch was placed just above the xiphoid process and taped to the nosepiece to allow proper visualization of the liver. The body cavity was intermittently moistened with sterile saline during the procedure. The ligament connected to the median lobe was gently cut away and the left lobe mobilized with a cotton tip. A suture knot was tied just around the base of the lobe and tightened, darkening it. This lobe was then excised, leaving a small stump preventing knot slippage. This procedure was repeated to transect the median lobe next, taking care not to ligate too close to the vena cava. The peritoneal cavity was checked with cotton tip to ensure no bleeding and closed using interrupted sutures. The skin was then closed with 7 mm would clips. Mice were kept solitary in a cage supplied with soft food, water and heating pad until fully recovered and ambulatory. All surgical animals were given i.p. injection of carprofen 10 mg/kg preemptively and every 24 h postoperatively for 48 h, and monitored daily. All animals recovered steadily and fully after the surgery without any overt signs of pain or inflammation.
Proteomics of purified liver microsomes
Fresh liver tissue was homogenized manually using Dounce pestle in homogenization buffer (0.3 M sucrose, 0.02 M HEPES pH7.4) followed by centrifugation at 20,000 g for 20 min. After this, the supernatant was centrifuged at 100,000 g for 1.5 h. The pellet was purified further via a sucrose gradient centrifugation (the sucrose layers loaded being 2, 1.35, 1.28 M containing microsomes, 1.15 and 0.3 M from bottom to top) overnight. The final ER fraction was collected from between 2 and 1.35 M layers, and protein concentration was measured using Bradford assay. 75 μg protein from each sample was submitted for mass spectrometry using Tandem Mass Tag technique (TMTsixplex Isobaric Label Reagent Set, Fisher PI90061) at University of Michigan Proteomics core on a fee-for-service basis. The general procedure for the proteomics analysis can be found in the following publications (McAlister et al., 2014; Tank et al., 2018). The entire dataset along with full experimental details are available via PRIDE (Perez-Riverol et al., 2022) ProteomeXchange with identifier PXD035243.
Western blot and antibodies
Protein extraction from cells and tissues and analysis via SDS-PAGE and Western blotting were carried out as described previously (Sha et al., 2009; Yang et al., 2010). Quantification of signals was done using BioRad ImageLab software. Glycosylation status was assessed by incubation with EndoH and PNGaseF enzymes for 1 h at 37°C (NEB). Total protein levels were normalized with the help of loading controls. The following primary antibodies were used: KI67 (1:200, Abcam ab16667); CYCLIN D1 (1:100 for staining, 1:2000 for Western blot, Abcam ab16663); c-MYC (1:1000, MilliporeSigma C3956); V5 (1:5000 for Western blot, 1:250 for immunoprecipitation, Invitrogen R960-25); HA (1:3,000, Sigma H9658); WNT5A (1:2000 for Western blot, 1:100 for immunoprecipitation, Proteintech 55184-1-AP); WNT5B (1:1000, Abclonal A8313); WNT1 (1:1000, Proteintech 27935-1-AP); E-CADHERIN (1:1000, BD 610181); α-TUBULIN (1:2,000, Santa Cruz sc-5286); HSP90 (1:1,000, Abcam ab13492); CALNEXIN (1:5000, Enzo ADI-SPA-860-F); SEL1L (1:2,000, Abcam ab78298); HRD1 (1:300, kindly gifted by Dr. Richard Wojcikiewicz, SUNY Upstate Medical University for Western blot; 1:1000, Sigma HPA024300 for immunostaining of human liver tumors). The following secondary antibodies were used: goat anti-mouse and goat anti-rabbit IgG-HRP (1:5,000; BioRad).
Immunoprecipitation
Lysis of cells was carried out, as described previously (Bhattacharya et al., 2018), in a buffer that contained 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 10 mM N-ethylmaleimide, protease- and phosphatase-inhibitors. This was followed by overnight incubation with antibody and Protein A agarose beads at 4°C with gentle rocking. The next day, immuno-complexes were washed in a buffer that contained 20 mM Tris-HCl pH 7.5, 137 mM NaCl, 10% glycerol and 2 mM EDTA, followed by elution by boiling for 5 min at 95°C in 1X SDS-sample buffer.
Detergent solubility analysis
Frozen liver tissue was homogenized as described previously (Sun et al., 2015) in NP40 lysis buffer. The volume of lysate was normalized by protein concentration measured via Bradford assay. Then, the lysate was centrifuged at 12,000g for 10 min and the supernatant collected (NP40-Soluble fraction), which was finally mixed with 5X SDS-sample buffer and heated for 15 min at 65°C. The pellet (NP40-Insoluble/Pellet fraction) was sonicated and boiled in 1X SDS-sample buffer for 30 min at 95°C.
RNA extraction, RT and qPCR
Total RNA was isolated from tissues and cells with the help of Trizol and BCP reagent, followed by RNA quality measurement via Nanodrop. qPCR analysis was performed using SYBR-Green based master mix, oilgo-dT primer, Taq polymerase, and the Applied Biosystems machine. All qPCR data were normalized to expression levels of the ribosomal L32 gene. The primer sequences are as follows:
Wnt5A F: CAAATAGGCAGCCGAGAGAC, R: CTCTAGCGTCCACGAACTCC;
Wnt5B F: CTGCTTGCGTAATGAGACCA, R: AAAGCAACACCAGTGGAACC;
CycD1 F: CACAACGCACTTTCTTTCCA, R: ACCAGCCTCTTCCTCCACTT;
CycD3 F: TAGGCGCCTGCTCTATGTCT, R: ATCTGTGGGAGTGCTGGTCT;
p21 F: ACAAGAGGCCCAGTACTTCC, R: GGGCACTTCAGGGTTTTCTC;
L32 F: GAGCAACAAGAAAACCAAGCA, R: TGCACACAAGCCATCTACTCA.
Immunostaining and histology
For staining and histology, tissues were directly immersed in 10% neutral buffered formalin after dissection and stored at 4°C. These were then paraffin embedded and sectioned (and also H&E stained) by the Michigan Histology Core for fee-for-service. For immunostaining, antigen retrieval was done by boiling in sodium citrate buffer for 20 min. Blocking was done at room temperature in 1% donkey serum containing 0.03% Triton X-100 blocking solution for an hr, followed by primary antibody incubation at 4°C, overnight, in humidified chambers. For immunohistochemistry, peroxidase quenching was carried out before antigen retrieval, and avidin-biotin kit was used to amplify the signal obtained using the DAB substrate kit. The next day, sections were washed thrice in PBS-0.03% Triton X-100 and treated with secondary antibody at room temperature for 2 h. Counterstaining and/or mounting were done using Permount. Trichrome staining was carried out using the TRM-1 kit from ScyTek. H&E and DAB stained sections were scanned with the help of Aperio Scanscope (Leica Biosystems). All images were analyzed using ImageJ plugin (FIJI).
Primary hepatocyte culture
Mice were anesthetized and their livers perfused with pre-warmed Ca/Mg-free HBSS first containing 25 mM HEPES, 0.171mM EDTA and 1% penicillin-streptomycin, followed by perfusion with pre-warmed Ca/Mg-free HBSS containing 0.75 mg/mL collagenase (Sigma-Aldrich C8051), 25 mM HEPES, 2 mM CaCl2 and 1% penicillin-streptomycin. After digestion, hepatocytes were manually shaken out, washed in cold DMEM containing 10% heat-inactivated FBS, 1 μM dexamethasone, 0.1 μM insulin, 2% Sodium pyruvate and 2% penicillin-streptomycin, and filtered through a 70 μm cell strainer. Cells were pelleted by centrifuging at 60 g for 3 min at 11°C. Hepatocytes were isolated by centrifuging through buffered Percoll at 800 g for 4 min at 11°C. Finally, the cells were washed 2–3 times in the same medium, counted (with trypan blue exclusion at >80%) and plated. After 2–3 h, the medium was replaced with DMEM supplemented with 2% Sodium pyruvate, 0.2% BSA and 2% penicillin-streptomycin.
Sucrose gradient
Protein extracted from sonicating liver tissue was layered onto a sucrose gradient column (12–36%) constructed 6 h earlier by layering lower on top of higher density sucrose fractions (prepared in 150 mM NaCl, 50 mM Tris-Cl, 1 mM EDTA, protease inhibitors) in 6% increments. After ultra-centrifuging for 16 h overnight at 4°C and 48,000 rpm using Beckman Coulter SW50Ti rotors, the column was divided into 12 fractions. The pellet at the bottom of the column was dissolved by sonication in the same volume of lysis buffer as the 13th fraction. Equal volumes from each fraction were then mixed with SDS sample buffer with or without β-mercaptoethanol, and heated at 65°C (for non-reducing) or 100°C (for reducing) for 5 min.
Plasmids
Plasmid constructs for Active-WNT5A-V5 and pAAV8-D(+)-U6-siRNA-CMV-GFP were purchased from Addgene. HRD1 WT, C2A mutant and pcDNA3-HA-Ub plasmids was previously described (Sun et al., 2015). HRD1 plasmid constructs (MYC-tagged WT and mutant C2A) were kind gifts from Dr. Yihong Ye (NIDDK). pcDNA3-HA-Ub was kindly shared by Dr. Hideki Nishitoh (University of Miyazaki, Japan).
AAV-mediated gene delivery
siRNA sequences targeted against mouse Wnt5A (CCACTTGTATCAGGACCACAT and GAGTTCGTGGACGCTAGAGAA) and control luciferase (GTTGCGCGGAGGAGTTGTG) were generated, and cloned into the pAAV8-D(+)-U6-siRNA-CMV-GFP plasmid between the BamHI and EcoRI restriction sites. These constructs were used to generate AAV at the University of Michigan Vector Core on a fee-for-service basis. 14-weeks-old mice were injected once with AAV8 via the tail vein at a dose of 2–5 x 1011 genome copies/mouse, and analyzed after 2 weeks.
Human liver cancer samples
University of Michigan Pathology provided 14 pairs of frozen biopsies of hepatocellular carcinoma tumor samples and adjacent normal liver tissue obtained from liver cancer patients for Western blot analysis. Human hepatocellular carcinoma tissue section microarray was obtained from US Biomax, Inc. (HLivH180Su14). Immunostaining intensity and extent scores were assigned as per the standard German semi-quantitative scoring system. Each specimen was assigned a score according to the intensity of the nucleic, cytoplasmic and membrane staining (no staining = 0; weak staining = 1; moderate staining = 2; strong staining = 3), and also according to the extent of stained cells (0–5% = 0, 5–25% = 1, 26–50% = 2, 51–75% = 3 and 76–100% = 4). The final immunoreactivity score was determined by multiplying the intensity score by the score for extent of stained cells, generating a final score ranging from 0 (minimum) to 12 (maximum).
Quantification and statistical analysis
Liver hepatocellular carcinoma (LIHC) survival, correlation plots and mutation analysis
Kaplan-Meier Plotter (http://kmplot.com/analysis) (Menyhart et al., 2018) was used to perform Kaplan Meier survival analysis to evaluate the influence of SEL1L or HRD1 on clinical prognosis. 364 TCGA hepatocellular carcinoma patients were split by the expression level of selected genes into two groups – high or low. For multigene survival analysis, the mean expression of SEL1L and HRD1 was calculated and used. The interactive database Gene Expression Profiling Interactive Analysis (GEPIA) (http://gepia.cancer-pku.cn/index.html) (Tang et al., 2017) was used to perform gene expression correlation analysis between HRD1 and SEL1L. The Spearman method was used to determine the correlation coefficient in 369 TCGA liver hepatocellular carcinoma samples, using RNA sequencing expression. For normal tissue gene correlation analysis, Spearman method was used in 50 normal samples from TCGA combined with 110 normal liver samples from the Geno-type-Tissue Expression (GTEx) Project (GTEx Consortium, 2013). Non-log scale was used for correlation coefficient calculations and log scale for visualization. Analysis of single nucleotide polymorphisms (SNPs) for SEL1L and HRD1 was done from the Liver Hepatocellular Carcinoma TCGA Pan Cancer Atlas dataset (353 samples) using cBio Portal for Cancer Genomics (www.cbioportal.org).
Statistical analysis
All results in this study have been expressed in the form of mean ± SEM unless otherwise depicted. Groups of data were compared via the paired two-tailed Student’s t test or the two-way ANOVA as necessary. All experiments were repeated at least twice, and executed using multiple independent biological samples out of which the most representative data have been shown here.
Acknowledgments
We sincerely thank Dr. Dafydd Thomas for sharing tissue biopsies from human liver cancer patients; Dr. Richard Wojcikiewicz for reagents; Erik Peterson and Megan Schaller for technical assistance; and Drs. Robert Weiss, Peter Arvan, Natasza Kurpios and Kenneth Simpson for discussion and insightful comments; the Histology, Pathology, Vision, Animal testing and Vector Cores at the University of Michigan Medical School for assistance; Dr. Venkatesha Basrur and the Proteomics Core at the University of Michigan for proteomics analysis; and other members of the Qi laboratory for critique, discussion, and support. This work was supported by Ministry of Science and Technology of the People's Republic of China National Key R&D Programs 2018YFA0506903 (L.C.), American Diabetes Association 1-19-IBS-235 (L.Q.) and NIH 1R01DK120330 (L.Q. and D.F.). A.B. was in part supported by AHA Predoctoral Fellowship #16PRE29750001.
Author contributions
A.B. designed and performed most experiments; J.W., B.G., and D.F. designed and performed experiments involving liver-specific HRD1 knockout mice; C.Y. and J.W. performed human tissue array staining and scoring; W.S. and L.C. performed statistical analyses of human liver cancer data; S.A.W. assisted in experiments; L.Q. directed and supervised the study; A.B. and L.Q. wrote the manuscript; everybody commented on and approved the manuscript. None of the authors have any competing interests.
Declaration of interests
The authors declare no competing interests.
Published: October 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105183.
Contributor Information
Deyu Fang, Email: fangd@northwestern.edu.
Ling Qi, Email: lingq@med.umich.edu.
Supplemental information
Data and code availability
The proteomics screen data is deposited at the public repository (ProteomeXchange PRIDE: PXD035243). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Asem M.S., Buechler S., Wates R.B., Miller D.L., Stack M.S. Wnt5a signaling in cancer. Cancers. 2016;8:E79. doi: 10.3390/cancers8090079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashktorab H., Green W., Finzi G., Sessa F., Nouraie M., Lee E.L., Morgano A., Moschetta A., Cattaneo M., Mariani-Costantini R., et al. SEL1L, an UPR response protein, a potential marker of colonic cell transformation. Dig. Dis. Sci. 2012;57:905–912. doi: 10.1007/s10620-011-2026-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A., Qi L. ER-associated degradation in health and disease - from substrate to organism. J. Cell Sci. 2019;132:jcs232850. doi: 10.1242/jcs.232850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A., Sun S., Wang H., Liu M., Long Q., Yin L., Kersten S., Zhang K., Qi L. Hepatic Sel1L-Hrd1 ER-associated degradation (ERAD) manages FGF21 levels and systemic metabolism via CREBH. EMBO J. 2018;37:e99277. doi: 10.15252/embj.201899277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitler B.G., Nicodemus J.P., Li H., Cai Q., Wu H., Hua X., Li T., Birrer M.J., Godwin A.K., Cairns P., Zhang R. Wnt5a suppresses epithelial ovarian cancer by promoting cellular senescence. Cancer Res. 2011;71:6184–6194. doi: 10.1158/0008-5472.CAN-11-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazanin N., Son J., Craig-Lucas A.B., John C.L., Breech K.J., Podolsky M.A., Glick A.B. ER stress and distinct outputs of the IRE1alpha RNase control proliferation and senescence in response to oncogenic Ras. Proc. Natl. Acad. Sci. USA. 2017;114:9900–9905. doi: 10.1073/pnas.1701757114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrovnikova-Marjon E., Grigoriadou C., Pytel D., Zhang F., Ye J., Koumenis C., Cavener D., Diehl J.A. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29:3881–3895. doi: 10.1038/onc.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardano M., Diaferia G.R., Cattaneo M., Dessì S.S., Long Q., Conti L., Deblasio P., Cattaneo E., Biunno I. mSEL-1L (Suppressor/enhancer Lin12-like) protein levels influence murine neural stem cell self-renewal and lineage commitment. J. Biol. Chem. 2011;286:18708–18719. doi: 10.1074/jbc.M110.210740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo M., Fontanella E., Canton C., Delia D., Biunno I. SEL1L affects human pancreatic cancer cell cycle and invasiveness through modulation of PTEN and genes related to cell-matrix interactions. Neoplasia. 2005;7:1030–1038. doi: 10.1593/neo.05451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Iliopoulos D., Zhang Q., Tang Q., Greenblatt M.B., Hatziapostolou M., Lim E., Tam W.L., Ni M., Chen Y., et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508:103–107. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng R., Sun B., Liu Z., Zhao X., Qi L., Li Y., Gu Q. Wnt5a suppresses colon cancer by inhibiting cell proliferation and epithelial-mesenchymal transition. J. Cell. Physiol. 2014;229:1908–1917. doi: 10.1002/jcp.24566. [DOI] [PubMed] [Google Scholar]
- Christianson J.C., Olzmann J.A., Shaler T.A., Sowa M.E., Bennett E.J., Richter C.M., Tyler R.E., Greenblatt E.J., Harper J.W., Kopito R.R. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 2012;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium The genotype-tissue expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Wei Q., Liao J., Zou Y., Song D., Xiong D., Ma C., Hu X., Qu X., Chen L., et al. Noncanonical Wnt signaling plays an important role in modulating canonical Wnt-regulated stemness, proliferation and terminal differentiation of hepatic progenitors. Oncotarget. 2017;8:27105–27119. doi: 10.18632/oncotarget.15637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.X., Sokol E.S., Del Vecchio C.A., Sanduja S., Claessen J.H.L., Proia T.A., Jin D.X., Reinhardt F., Ploegh H.L., Wang Q., Gupta P.B. Epithelial-to-mesenchymal transition activates PERK-eIF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 2014;4:702–715. doi: 10.1158/2159-8290.CD-13-0945. [DOI] [PubMed] [Google Scholar]
- Ferenci P., Fried M., Labrecque D., Bruix J., Sherman M., Omata M., Heathcote J., Piratsivuth T., Kew M., Otegbayo J.A., et al. Hepatocellular carcinoma (HCC): a global perspective. J. Clin. Gastroenterol. 2010;44:239–245. doi: 10.1097/MCG.0b013e3181d46ef2. [DOI] [PubMed] [Google Scholar]
- Francisco A.B., Singh R., Li S., Vani A.K., Yang L., Munroe R.J., Diaferia G., Cardano M., Biunno I., Qi L., et al. Deficiency of suppressor enhancer Lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. J. Biol. Chem. 2010;285:13694–13703. doi: 10.1074/jbc.M109.085340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedaly R., Galuppo R., Daily M.F., Shah M., Maynard E., Chen C., Zhang X., Esser K.A., Cohen D.A., Evers B.M., et al. Targeting the Wnt/beta-catenin signaling pathway in liver cancer stem cells and hepatocellular carcinoma cell lines with FH535. PLoS One. 2014;9:e99272. doi: 10.1371/journal.pone.0099272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granelli P., Cattaneo M., Ferrero S., Bottiglieri L., Bosari S., Fichera G., Biunno I. SEL1L and squamous cell carcinoma of the esophagus. Clin. Cancer Res. 2004;10:5857–5861. doi: 10.1158/1078-0432.CCR-04-0075. [DOI] [PubMed] [Google Scholar]
- Guerriero C.J., Brodsky J.L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnoss J.M., Le Thomas A., Shemorry A., Marsters S.A., Lawrence D.A., Lu M., Chen Y.C.A., Qing J., Totpal K., Kan D., et al. Disruption of IRE1alpha through its kinase domain attenuates multiple myeloma. Proc. Natl. Acad. Sci. USA. 2019;116:16420–16429. doi: 10.1073/pnas.1906999116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkert M., Doolman R., Dai M., Avner R., Hassink G., van Voorden S., Thanedar S., Roitelman J., Chau V., Wiertz E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- Kim G.H., Shi G., Somlo D.R., Haataja L., Song S., Long Q., Nillni E.A., Low M.J., Arvan P., Myers M.G., Jr., Qi L. Hypothalamic ER-associated degradation regulates POMC maturation, feeding, and age-associated obesity. J. Clin. Invest. 2018;128:1125–1140. doi: 10.1172/JCI96420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Bhattacharya A., Qi L. Endoplasmic reticulum quality control in cancer: friend or foe. Semin. Cancer Biol. 2015;33:25–33. doi: 10.1016/j.semcancer.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremenevskaja N., von Wasielewski R., Rao A.S., Schöfl C., Andersson T., Brabant G. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene. 2005;24:2144–2154. doi: 10.1038/sj.onc.1208370. [DOI] [PubMed] [Google Scholar]
- Lee J.M., Yang J., Newell P., Singh S., Parwani A., Friedman S.L., Nejak-Bowen K.N., Monga S.P. beta-Catenin signaling in hepatocellular cancer: implications in inflammation, fibrosis, and proliferation. Cancer Lett. 2014;343:90–97. doi: 10.1016/j.canlet.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Xiao J., Hormi K., Borok Z., Minoo P. Wnt5a participates in distal lung morphogenesis. Dev. Biol. 2002;248:68–81. doi: 10.1006/dbio.2002.0729. [DOI] [PubMed] [Google Scholar]
- Liang H., Chen Q., Coles A.H., Anderson S.J., Pihan G., Bradley A., Gerstein R., Jurecic R., Jones S.N. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell. 2003;4:349–360. doi: 10.1016/s1535-6108(03)00268-x. [DOI] [PubMed] [Google Scholar]
- Liu L., Yu L., Zeng C., Long H., Duan G., Yin G., Dai X., Lin Z. E3 ubiquitin ligase HRD1 promotes lung tumorigenesis by promoting sirtuin 2 ubiquitination and degradation. Mol. Cell Biol. 2020;40:e00257-19. doi: 10.1128/MCB.00257-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Chen J., Mai B., Amos C., Killary A.M., Sen S., Wei C., Frazier M.L. A single-nucleotide polymorphism in tumor suppressor gene SEL1L as a predictive and prognostic marker for pancreatic ductal adenocarcinoma in Caucasians. Mol. Carcinog. 2012;51:433–438. doi: 10.1002/mc.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B., Lee A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–818. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G.C., Nusinow D.P., Jedrychowski M.P., Wühr M., Huttlin E.L., Erickson B.K., Rad R., Haas W., Gygi S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald S.L., Silver A. The opposing roles of Wnt-5a in cancer. Br. J. Cancer. 2009;101:209–214. doi: 10.1038/sj.bjc.6605174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert M., Sommermeyer F., Berger M., Kumar Lakshmipathy S., Gauss R., Aebi M., Jarosch E., Sommer T. The interplay of Hrd3 and the molecular chaperone system ensures efficient degradation of malfolded secretory proteins. Mol. Biol. Cell. 2015;26:185–194. doi: 10.1091/mbc.E14-07-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menyhárt O., Nagy Á., Győrffy B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R. Soc. Open Sci. 2018;5:181006. doi: 10.1098/rsos.181006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H., Ajima R., Luo C.T., Yamaguchi T.P., Stappenbeck T.S. Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338:108–113. doi: 10.1126/science.1223821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller B., Klemm E.J., Spooner E., Claessen J.H., Ploegh H.L. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA. 2008;105:12325–12330. doi: 10.1073/pnas.0805371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelsen C.J., Rickheim D.G., Tucker M.M., Hansen L.K., Albrecht J.H. Evidence that cyclin D1 mediates both growth and proliferation downstream of TOR in hepatocytes. J. Biol. Chem. 2003;278:3656–3663. doi: 10.1074/jbc.M209374200. [DOI] [PubMed] [Google Scholar]
- Niederreiter L., Fritz T.M.J., Adolph T.E., Krismer A.M., Offner F.A., Tschurtschenthaler M., Flak M.B., Hosomi S., Tomczak M.F., Kaneider N.C., et al. ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. J. Exp. Med. 2013;210:2041–2056. doi: 10.1084/jem.20122341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez K.G., Gonzalez-Rosario J., Thevenot P.T., Cohen A.J. Cyclin D1 in the liver: role of noncanonical signaling in liver steatosis and hormone regulation. Ochsner J. 2017;17:56–65. [PMC free article] [PubMed] [Google Scholar]
- Orlandi R., Cattaneo M., Troglio F., Casalini P., Ronchini C., Ménard S., Biunno I. SEL1L expression decreases breast tumor cell aggressiveness in vivo and in vitro. Cancer Res. 2002;62:567–574. [PubMed] [Google Scholar]
- Pearce M.M., Wang Y., Kelley G.G., Wojcikiewicz R.J. SPFH2 mediates the endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphosphate receptors and other substrates in mammalian cells. J. Biol. Chem. 2007;282:20104–20115. doi: 10.1074/jbc.M701862200. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- Pytel D., Gao Y., Mackiewicz K., Katlinskaya Y.V., Staschke K.A., Paredes M.C.G., Yoshida A., Qie S., Zhang G., Chajewski O.S., et al. PERK is a haploinsufficient tumor suppressor: gene dose determines tumor-suppressive versus tumor promoting properties of PERK in melanoma. PLoS Genet. 2016;12:e1006518. doi: 10.1371/journal.pgen.1006518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L., Tsai B., Arvan P. New insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell Biol. 2017;27:430–440. doi: 10.1016/j.tcb.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickheim D.G., Nelsen C.J., Fassett J.T., Timchenko N.A., Hansen L.K., Albrecht J.H. Differential regulation of cyclins D1 and D3 in hepatocyte proliferation. Hepatology. 2002;36:30–38. doi: 10.1053/jhep.2002.33996. [DOI] [PubMed] [Google Scholar]
- Roarty K., Serra R. Wnt5a is required for proper mammary gland development and TGF-beta-mediated inhibition of ductal growth. Development. 2007;134:3929–3939. doi: 10.1242/dev.008250. [DOI] [PubMed] [Google Scholar]
- Schulz J., Avci D., Queisser M.A., Gutschmidt A., Dreher L.S., Fenech E.J., Volkmar N., Hayashi Y., Hoppe T., Christianson J.C. Conserved cytoplasmic domains promote Hrd1 ubiquitin ligase complex formation for ER-associated degradation (ERAD) J. Cell Sci. 2017;130:3322–3335. doi: 10.1242/jcs.206847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira S.J., Ranganathan A.C., Adam A.P., Iglesias B.V., Farias E.F., Aguirre-Ghiso J.A. Inhibition of proliferation by PERK regulates mammary acinar morphogenesis and tumor formation. PLoS One. 2007;2:e615. doi: 10.1371/journal.pone.0000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha H., He Y., Chen H., Wang C., Zenno A., Shi H., Yang X., Zhang X., Qi L. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009;9:556–564. doi: 10.1016/j.cmet.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha H., Sun S., Francisco A.B., Ehrhardt N., Xue Z., Liu L., Lawrence P., Mattijssen F., Guber R.D., Panhwar M.S., et al. The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell Metab. 2014;20:458–470. doi: 10.1016/j.cmet.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X., Nenseth H.Z., Qu S., Kuzu O.F., Frahnow T., Simon L., Greene S., Zeng Q., Fazli L., Rennie P.S., et al. IRE1alpha-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019;10:323. doi: 10.1038/s41467-018-08152-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G., Somlo D.R., Kim G.H., Prescianotto-Baschong C., Sun S., Beuret N., Long Q., Rutishauser J., Arvan P., Spiess M., Qi L. ER-associated degradation is required for vasopressin prohormone processing and systemic water homeostasis. J. Clin. Invest. 2017;127:3897–3912. doi: 10.1172/JCI94771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha N., Liu T., Ji Y., Reinert R., Torres M., Li X., Zhang M., Tang C.A., Hu C.A., Liu C., et al. Sel1L-Hrd1 ER-associated degradation maintains beta-cell identity via TGFbeta signaling. J. Clin. Invest. 2020;130:3499–3510. doi: 10.1172/JCI134874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Shi G., Han X., Francisco A.B., Ji Y., Mendonça N., Liu X., Locasale J.W., Simpson K.W., Duhamel G.E., et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA. 2014;111:E582–E591. doi: 10.1073/pnas.1318114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Shi G., Sha H., Ji Y., Han X., Shu X., Ma H., Inoue T., Gao B., Kim H., et al. IRE1alpha is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015;17:1546–1555. doi: 10.1038/ncb3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z., Li C., Kang B., Gao G., Li C., Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank E.M., Figueroa-Romero C., Hinder L.M., Bedi K., Archbold H.C., Li X., Weskamp K., Safren N., Paez-Colasante X., Pacut C., et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 2018;9:2845. doi: 10.1038/s41467-018-05049-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen R., Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–3214. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- Vashistha N., Neal S.E., Singh A., Carroll S.M., Hampton R.Y. Direct and essential function for Hrd3 in ER-associated degradation. Proc. Natl. Acad. Sci. USA. 2016;113:5934–5939. doi: 10.1073/pnas.1603079113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez V., Turcios L., Marti F., Gedaly R. Targeting Wnt/beta-catenin pathway in hepatocellular carcinoma treatment. World J. Gastroenterol. 2016;22:823–832. doi: 10.3748/wjg.v22.i2.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Liu X., Wang J. Up-regulation of Wnt5a inhibits proliferation and migration of hepatocellular carcinoma cells. J. Cancer Res. Ther. 2019;15:904–908. doi: 10.4103/jcrt.JCRT_886_18. [DOI] [PubMed] [Google Scholar]
- Wang W., Yu X., Wu C., Jin H. Differential effects of Wnt5a on the proliferation, differentiation and inflammatory response of keratinocytes. Mol. Med. Rep. 2018;17:4043–4048. doi: 10.3892/mmr.2017.8358. [DOI] [PubMed] [Google Scholar]
- Wei J., Chen L., Li F., Yuan Y., Wang Y., Xia W., Zhang Y., Xu Y., Yang Z., Gao B., et al. HRD1-ERAD controls production of the hepatokine FGF21 through CREBH polyubiquitination. EMBO J. 2018;37:e98942. doi: 10.15252/embj.201898942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K., Nusse R. Wnt proteins. Cold Spring Harb. Perspect. Biol. 2012;4:a007864. doi: 10.1101/cshperspect.a007864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Liang W., Guan H., Liu J., Liu L., Li H., He X., Zheng J., Chen J., Cao X., Li Y. Exendin-4 promotes pancreatic beta-cell proliferation via inhibiting the expression of Wnt5a. Endocrine. 2017;55:398–409. doi: 10.1007/s12020-016-1160-x. [DOI] [PubMed] [Google Scholar]
- Xie H., Tang C.H.A., Song J.H., Mancuso A., Del Valle J.R., Cao J., Xiang Y., Dang C.V., Lan R., Sanchez D.J., et al. IRE1alpha RNase-dependent lipid homeostasis promotes survival in Myc-transformed cancers. J. Clin. Invest. 2018;128:1300–1316. doi: 10.1172/JCI95864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagishita N., Ohneda K., Amano T., Yamasaki S., Sugiura A., Tsuchimochi K., Shin H., Kawahara K.I., Ohneda O., Ohta T., et al. Essential role of synoviolin in embryogenesis. J. Biol. Chem. 2005;280:7909–7916. doi: 10.1074/jbc.M410863200. [DOI] [PubMed] [Google Scholar]
- Yamasaki S., Yagishita N., Sasaki T., Nakazawa M., Kato Y., Yamadera T., Bae E., Toriyama S., Ikeda R., Zhang L., et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase 'Synoviolin'. EMBO J. 2007;26:113–122. doi: 10.1038/sj.emboj.7601490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Cusimano A., Monga J.K., Preziosi M.E., Pullara F., Calero G., Lang R., Yamaguchi T.P., Nejak-Bowen K.N., Monga S.P. WNT5A inhibits hepatocyte proliferation and concludes beta-catenin signaling in liver regeneration. Am. J. Pathol. 2015;185:2194–2205. doi: 10.1016/j.ajpath.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Xue Z., He Y., Sun S., Chen H., Qi L. A Phos-tag-based approach reveals the extent of physiological endoplasmic reticulum stress. PLoS One. 2010;5:e11621. doi: 10.1371/journal.pone.0011621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Kong S., Zhang Y., Melo-Cardenas J., Gao B., Zhang Y., Zhang D.D., Zhang B., Song J., Thorp E., et al. The endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 controls a critical checkpoint in B cell development in mice. J. Biol. Chem. 2018;293:12934–12944. doi: 10.1074/jbc.RA117.001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying J., Li H., Yu J., Ng K.M., Poon F.F., Wong S.C.C., Chan A.T.C., Sung J.J.Y., Tao Q. WNT5A exhibits tumor-suppressive activity through antagonizing the Wnt/beta-catenin signaling, and is frequently methylated in colorectal cancer. Clin. Cancer Res. 2008;14:55–61. doi: 10.1158/1078-0432.CCR-07-1644. [DOI] [PubMed] [Google Scholar]
- Zhan T., Rindtorff N., Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–1473. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Li Y., Wu Y., Yang T., Yang K., Wang R., Yang J., Guo H. Wnt5a inhibits the proliferation and melanogenesis of melanocytes. Int. J. Med. Sci. 2013;10:699–706. doi: 10.7150/ijms.5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N., Cao J., Xu L., Tang Q., Dobrolecki L.E., Lv X., Talukdar M., Lu Y., Wang X., Hu D.Z., et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Invest. 2018;128:1283–1299. doi: 10.1172/JCI95873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Chen D., Huang X.M., Long F., Cai H., Yao W.X., Chen Z.C., Liao Z.J., Deng Z.Z., Tan S., et al. Wnt5a promotes cortical neuron survival by inhibiting cell-cycle activation. Front. Cell. Neurosci. 2017;11:281. doi: 10.3389/fncel.2017.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Kipps T.J., Zhang S. Wnt5a signaling in normal and cancer stem cells. Stem Cell. Int. 2017;2017:5295286. doi: 10.1155/2017/5295286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Torres M., Sha H., Halbrook C.J., Van den Bergh F., Reinert R.B., Yamada T., Wang S., Luo Y., Hunter A.H., et al. Endoplasmic reticulum-associated degradation regulates mitochondrial dynamics in brown adipocytes. Science. 2020;368:54–60. doi: 10.1126/science.aay2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomics screen data is deposited at the public repository (ProteomeXchange PRIDE: PXD035243). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.