
FIGURE 1.
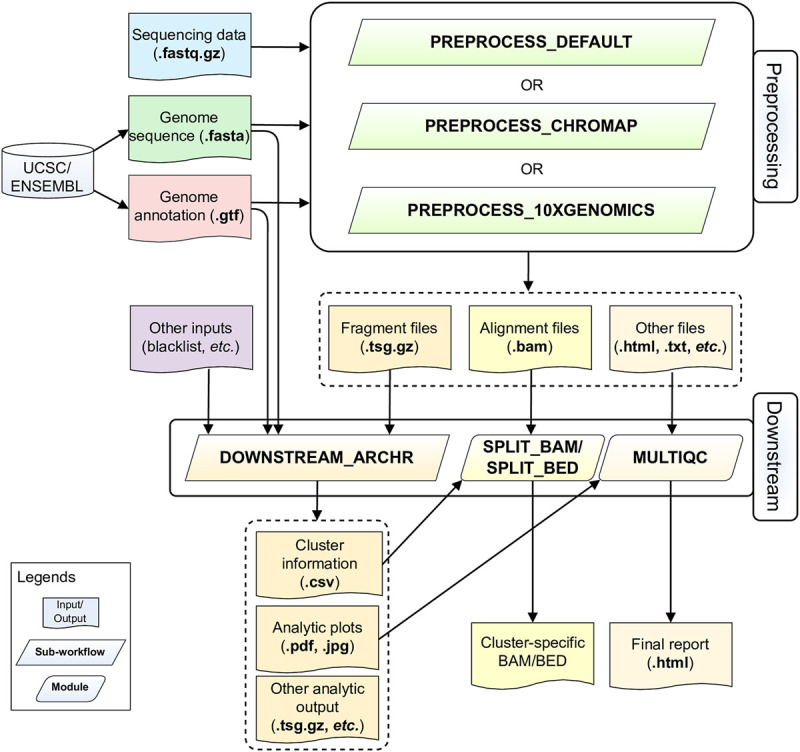
Schematic diagram of the scATACpipe workflow. The scATACpipe workflow is mainly composed of modules for the preprocessing sub-workflow and the downstream analysis sub-workflow. The preprocessing sub-workflow offers three options: the 10x Genomics Cell Ranger ATAC software-based preprocessing, the ultra-fast Chromap-based preprocessing, and the default preprocessing which implements the current best practices for scATAC-seq data analysis. The preprocessing sub-workflow takes sequencing data (.fastq.gz files), a reference genome sequence (.fasta file), and a genome annotation file (.gtf file) as inputs. The last two files can be automatically downloaded from the UCSC or Ensembl Genome Browser. Alternatively, users can specify a genome index compatible with the mapper of the desired preprocessing option. The fragment file generated from the preprocessing sub-workflow is passed along with an optional genome blacklist file and other custom inputs to the downstream analysis sub-workflow for core analyses, including batch correction, clustering, integration with scRNA-seq data, cluster annotation, peak calling, marker gene and marker peak identification, motif enrichment and deviation analysis, footprinting analysis, integrated analysis, and trajectory analysis using mainly the ArchR package. The alignment file (.bam) and fragment file (.tsv.gz) from the preprocessing sub-workflow are converted into cluster-specific BAM, BED, and/or BigWig files for visualization in genome browsers. An interactive HTML report is generated at the end with all analytic plots from the downstream analyses, and QC summaries from the default preprocessing sub-workflow if chosen.