

Abstract

Second coordination sphere interactions of cyanido complexes with hydrogen-bonding solvents and Lewis acids are known to influence their electronic structures, whereby the non-labile attachment of B(C6F5)3 resulted in several particularly interesting new compounds lately. Here, we investigate the effects of borylation on the properties of two FeII cyanido complexes in a systematic manner by comparing five different compounds and using a range of experimental techniques. Electrochemical measurements indicate that borylation entails a stabilization of the FeII-based t2g-like orbitals by up to 1.65 eV, and this finding was confirmed by Mössbauer spectroscopy. This change in the electronic structure has a profound impact on the UV–vis absorption properties of the borylated complexes compared to the non-borylated ones, shifting their metal-to-ligand charge transfer (MLCT) absorption bands over a wide range. Ultrafast UV–vis transient absorption spectroscopy provides insight into how borylation affects the excited-state dynamics. The lowest metal-centered (MC) excited states become shorter-lived in the borylated complexes compared to their cyanido analogues by a factor of ∼10, possibly due to changes in outer-sphere reorganization energies associated with their decay to the electronic ground state as a result of B(C6F5)3 attachment at the cyanido N lone pair.
Short abstract
Borylation in the second coordination sphere of two well-known FeII cyanido complexes leads to isocyanoborato complexes. The effects of borylation on their electronic structure and photophysical properties are thoroughly investigated with a range of experimental techniques.
Introduction
Lewis acid–base interactions between different boron-containing compounds and the terminal N-atoms of cyanido complexes have been known for over 50 years.1−3 Recently, the idea of exploiting such interactions in the second coordination sphere of metal complexes received more interest again, and the borylation of mixed-ligand complexes of IrIII,4 OsII,5 ReI,6−9 RuII,10−12 FeII,10,13 CuI,14,15 NiII,16 PdII,17 PtII,18 and AgI14 has resulted in new compounds with enhanced photophysical, electrochemical, and photochemical properties. The boosted properties of the isocyanoborato complexes compared to their cyanido precursors typically originate from the energetic stabilization of the metal-based t2g-like (dπ) orbitals upon cyanido borylation, entailing a rise in the energy of metal-to-ligand charge transfer (MLCT) excited states as well as a lower tendency to undergo oxidation in the electronic ground state. Due to these beneficial effects, isocyanoborato complexes of 5d metals often have longer MLCT excited-state lifetimes and increased photoluminescence quantum yields when compared to the cyanido parent compounds. Isocyanoborato complexes have become promising candidates as emitters for organic light emitting diodes (OLEDs)4,5,7 or sensors,8 some of them feature uncommon electrochemical properties,10,11,13 and others have been used as photocatalysts for challenging photoredox and triplet–triplet energy transfer (TTET) reactions.12,19 In addition, iscocyanoborato complexes of IrIII and RuII were shown to be exceptionally photorobust under catalytic conditions.12,19
Research on isocyanoborato complexes so far has mainly focused on 4d and 5d metals, with only a few studies investigating the influence of borylation on the photophysical and electrochemical properties of mixed-ligand FeII α-diimine cyanido complexes.10,13 FeII plays a very special role in modern photophysics and photochemistry because it can adopt the same low-spin d6 valence electron configuration as RuII and IrIII, from which many of the most widely used photoactive transition metal compounds are made. Considering in addition that iron is the most abundant d-metal element in Earth’s crust, it seems unsurprising that complexes of FeII are currently intensely researched.20−34 Against this background, it seemed meaningful to investigate the effects of borylation on the properties and the electronic structure of well-known heteroleptic FeII complexes with cyanido and 2,2′-bipyridine ligands. Specifically, we focused on the five complexes shown in Figure 1 because they represent a useful platform to rationalize the different effects that cyanido- and isocyanoborato ligands exert on their electronic structure. Among them, [Fe(bpy)3]2+,29,30,35−38 [Fe(bpy)2(CN)2],39−41 and [Fe(bpy)(CN)4]2–40,42−44 are well known, whereas [Fe(bpy)(BCF)4]2– has been reported recently10 and [Fe(bpy)2(BCF)2] is new. We find that the attachment of B(C6F5)3 in the second coordination sphere of the FeII cyanido complexes leads to a drastic increase of their oxidation potential, in line with prior reports.10,13 UV–vis studies illustrate how the MLCT absorption bands can be shifted over a large portion of the visible spectrum by varying the number of isocyanoborato/cyanido ligands. X-ray crystal structure analysis along with infrared (IR) and Mössbauer spectroscopic studies provide insight into the molecular and electronic (ground-state) structures of the complexes, whereas ultrafast time-resolved UV–vis absorption spectroscopy was used to investigate excited-state dynamics.
Figure 1.

Chemical structures of [Fe(bpy)3]2+ (a), [Fe(bpy)2(CN)2] and [Fe(bpy)2(BCF)2] (b), and [Fe(bpy)(CN)4]2– and [Fe(bpy)(BCF)4]2– (c) (BCF = CNB(C6F5)3; bpy = 2,2′-bipyridine).
Results and Discussion
Synthesis, Characterization, IR Spectroscopy, and Crystal Structure
The two complexes (PPN)2[Fe(bpy)(BCF)4] (PPN+ = bis(triphenylphosphine)iminium; bpy = 2,2′-bipyridine; BCF = CNB(C6F5)3) and [Fe(bpy)2(BCF)2] were synthesized by reacting the precursor complexes [Fe(bpy)(CN)4]2– and [Fe(bpy)2(CN)2] with 4.4 or 2.2 equivalents of B(C6F5)3, respectively. The PPN+ counter-cation gave good solubility in CH2Cl2 (in which the borylation reaction worked well), and furthermore, PPN+ afforded an easier to purify and to characterize compound than the more widely known TBA+ (tetra-n-butylammonium) cation. The borylated complexes were characterized by 1H, 13C, 11B, and 19F NMR spectroscopy as well as by infrared (IR) spectroscopy, elemental analysis (EA), and high-resolution mass spectrometry (HRMS). Suitable crystals for X-ray diffraction analysis of [Fe(bpy)2(BCF)2] (CCDC deposition number 2159475) were obtained by slow evaporation from a mixture of CHCl3 and CH2Cl2, and the obtained crystal structure is displayed in Figure 2.
Figure 2.

X-ray crystal structure of [Fe(bpy)2(BCF)2]. Individual atoms shown as 50% thermal ellipsoids. Hydrogen atoms are omitted for clarity.
Relative to [Fe(bpy)2(CN)2], borylation entails a shortening of both the Fe–C and the C≡N bonds in [Fe(bpy)2(BCF)2] (Table 1), which we attribute to two effects. On the one hand, the Lewis-acidic B(C6F5)3 unit lowers the energy of the relevant π-bonding orbitals in the C≡N bond,13 thereby strengthening the bonding interaction and shortening the C–N distance by 0.015(5) Å. However, the C≡N π*-orbitals are stabilized in parallel with the respective π orbitals,13 resulting in increased π-backbonding from FeII to the C≡N π*-orbitals and causing a 0.030(4) Å shorter Fe–C bond in [Fe(bpy)2(BCF)2] compared to [Fe(bpy)2(CN)2].45 A combination of steric effects and (partial) population of the C≡N π*-orbitals through π-backbonding is furthermore responsible for the slightly bent C≡N—B angle (Table 1).13
Table 1. Selected Average Bond Lengths and Angles and Infrared Spectral Data for [Fe(bpy)2(CN)2] and [Fe(bpy)2(BCF)2].
| [Fe(bpy)2(CN)2]a | [Fe(bpy)2(BCF)2] | |
|---|---|---|
| C≡N | 1.168(5) Å | 1.153(3) Å |
| Fe–C | 1.907(4) Å | 1.877(2) Å |
| Fe–Ntrans | 1.996(3) Å | 1.995(2) Å |
| Fe–Ncis | 1.958(3) Å | 1.964(2) Å |
| C≡N–B | 171.9(2)° | |
| νC≡N | 2070 and 2077 cm–1 | 2166 and 2181 cm–1 |
Crystal structure data for [Fe(bpy)2(CN)2] was obtained from ref (45).
Furthermore, the Lewis-acidity of B(C6F5)3 results in a weakened σ-bonding interaction between the isocyanoborato ligand and the FeII center, which in principle could be expected to lead to a longer Fe–C bond. However, according to the data in Table 1, this effect is overcompensated by the increased π-backbonding into the C≡N-based π* orbitals described above, resulting in an overall shorter Fe–C bond in [Fe(bpy)2(BCF)2] relative to [Fe(bpy)2(CN)2].
In the IR spectrum of [Fe(bpy)2(CN)2] (Figure S20), C≡N stretching modes are observed at 2070 and 2077 cm–1, and these bands are shifted to higher wavenumbers in [Fe(bpy)2(BCF)2] (2166 and 2181 cm–1). This effect is commonly observed upon the borylation of cyanido complexes and is a consequence of the stronger C≡N bond in the borylated complexes compared to their cyanido precursors.10,13,16 A similar frequency shift (Figure S21) can also be observed when going from [Fe(bpy)(CN)4]2– (νC≡N = 2054 cm–1) to [Fe(bpy)(BCF)4]2– (νC≡N = 2162 cm–1).
Electrochemistry
Cyclic voltammograms (CVs) and differential pulse voltammograms (DPVs) were recorded for all five complexes. The DPVs of the key complexes [Fe(bpy)2(BCF)2] and (PPN)2[Fe(bpy)(BCF)4] as well as the reference compound [Fe(bpy)3](PF6)2 are displayed in Figure 3 to illustrate the influence of exchanging bpy ligands with isocyanoborato ligands. In the following, we first focus on the changes in the electronic structure associated with the exchange of bpy ligands with cyanido ligands. In a second step, we then discuss the effects of borylation of the cyanido ligands.
Figure 3.
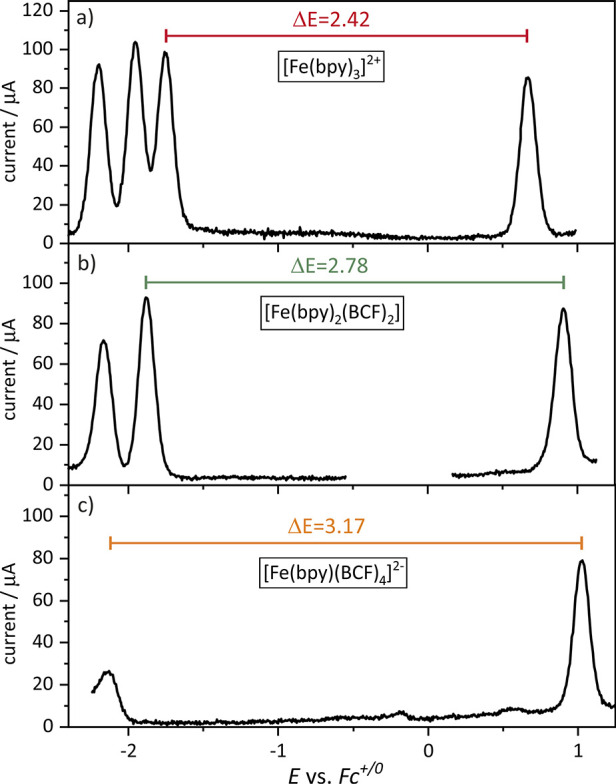
Differential pulse voltammograms of 1.1 mM [Fe(bpy)3](PF6)2 (a), 1.3 mM [Fe(bpy)2(BCF)2] (b), and 1.0 mM (PPN)2[Fe(bpy)(BCF)4] (c) in dry, argon-saturated CH3CN at 293 K with 0.1 M (NBu4)(PF6) as supporting electrolyte. In all three cases, the step height was 5 mV, the pulse height was 100 mV, the pulse width was 100 ms, and the step width was 50 ms.
In the DPV of [Fe(bpy)3]2+, three reduction features at −1.74, −1.93, and −2.19 V vs Fc+/0, attributable to three consecutive one-electron reduction events of the bpy-ligands, as well as the oxidation of FeII to FeIII at 0.68 V vs Fc+/0 are observed (Figure 3a). When one bpy ligand is replaced by two cyanido ligands ([Fe(bpy)2(CN)2]), the metal-based oxidation wave is shifted cathodically from 0.68 V vs Fc+/0 to 0.06 V vs Fc+/0 (Figure S25 and Figure 4). This is due to an increase in electron density at the FeII center, caused by the stronger σ-donation from the two anionic cyanido ligands compared to the charge-neutral bpy ligand.46 Furthermore, only two (instead of three) reduction features are observable in this case, attributable to one-electron reduction of the two remaining bpy ligands. The first reduction potential is shifted cathodically compared to the [Fe(bpy)3]2+ complex (Figure S25) by 0.32 V, signaling that the increased electron density at the metal center affects bpy reduction. For simplicity, we assume that the change in the ligand environment affects the energy of the t2g-like orbitals of the FeII and the FeIII complexes to the same extent. Even though an increased charge of the central metal ion is in principle expected to lead to increased d-orbital interactions between the metal and the ligands, our crude approach is sufficient to account for the observable shifts in the MLCT absorption bands upon ligand exchange (vide infra). In the following discussion, we will further use experimentally determined redox potentials to draw conclusions concerning the relative energies of frontier orbitals, which is evidently a simplistic approach that will however allow for an integrated and comparative discussion of data originating from different experimental techniques.
Figure 4.

Energy level diagram of the key complexes as determined by cyclic voltammetry and differential pulse voltammetry. Dotted lines are guides to the eye. The metal-centered oxidation process illustrated in the lower part is a measure of the HOMO energy. The bpy-ligand-based reduction process illustrated in the upper part is a measure of the energy of the lowest-lying π* orbital on the bpy ligands.
When going from [Fe(bpy)3]2+ to [Fe(bpy)(CN)4]2–, an even larger cathodic shift of 1.33 V for the first oxidation potential, from 0.68 V vs Fc+/0 to −0.65 V vs Fc+/0, is observed. Furthermore, the first reduction potential is shifted cathodically from −1.74 ([Fe(bpy)3]2+) to −2.51 ([Fe(bpy)(CN)4]2–) V vs Fc+/0 (Figure S32 and Figure 4). These observations are in line with previous electrochemical studies of [Fe(bpy)3]2+,29 [Fe(bpy)2(CN)2],47 and [Fe(bpy)(CN)4]2–.10
Upon borylation of the two cyanido ligands in [Fe(bpy)2(CN)2] to give [Fe(bpy)2(BCF)2], the respective metal oxidation potential is shifted anodically from 0.06 V ([Fe(bpy)2(CN)2]) to 0.89 V vs Fc+/0 ([Fe(bpy)2(BCF)2]) (Figure 3b). The same effects are also observed in [Fe(bpy)(BCF)4]2–; however, due to the presence of four cyanido/isocyanoborato ligands instead of only two, the observed effect doubles in magnitude. Specifically, upon going from [Fe(bpy)(CN)4]2– to [Fe(bpy)(BCF)4]2–, a shift of the oxidation potential by 1.65 V from −0.65 to 1.00 V vs Fc+/0 is observed. The bpy0/·- reduction potential is shifted to a lesser extent (0.34 V) from −2.51 to −2.17 V vs Fc+/0 (Figure 4). Thus, in both comparative cases investigated here, the borylation has a much stronger influence on the electrochemical potential of the metal-based oxidation than on the bpy-based reduction, which is understandable on the basis that the borylation occurs much closer to the FeII center than to the bpy ligands.
The difference between the oxidation potential and the first reduction feature (ΔE) is 2.42 V for [Fe(bpy)3]2+ (Figure 3a), and this value increases with greater number of BCF ligands. The ΔE increases to 2.78 V in [Fe(bpy)2(BCF)2] (Figure 3b) and further rises to 3.17 V in [Fe(bpy)(BCF)4]2– (Figure 3c). The effects of exchanging bpy ligands with cyanido ligands and their subsequent borylation are summarized in Figure 4, which captures the changes starting from [Fe(bpy)3]2+ and going to the cyanido complexes (black lines) and onwards to the BCF complexes (red lines). The dotted red and black lines are guides to the eye. The orange labels and dotted lines indicate the extent of the stabilization of the metal-based t2g-like (dπ) orbitals (bottom) and of the energetically lowest-lying π* orbital on the bpy ligands (top).
Based on the available electrochemical data and a Randles–Ševčík analysis, diffusion coefficients (D0; Table 2) on the order of 10–6 cm2 s–1 were estimated for all five compounds (see the SI for details). For the previously reported compounds, the diffusion coefficients obtained here are in good agreement with prior studies.10 Based on the structural changes upon borylation, a decrease in the D0 values of the borylated complexes compared to their cyanido precursors might in principle be expected. However, on the contrary, we find larger values for D0 in the borylated complexes, possibly due to stronger interactions of the cyanido complexes with the solvent as well as stronger ion pairing effects in the cyanido compounds.10
Table 2. Summary of the UV–Vis Absorption,a Electrochemical,b and Mössbauerc Properties of the Key Complexes.
| λmax, abs, MLCT (ε) (nm M–1 cm–1) | E1/2ox (V vs Fc+/0) | E1/2red (V vs Fc+/0) | ΔEoxd (mV) | ΔEredd (mV) | D0e (cm2 s–1) | δ (mm s–1) | ΔEQ (mm s–1) | |
|---|---|---|---|---|---|---|---|---|
| [Fe(bpy)2(CN)2] | 613 (7000) | 0.06 | –2.06, −2.33 | 70 | 70, 73 | 6.7 × 10–6 | 0.25 | 0.62 |
| [Fe(bpy)2(BCF)2] | 482 (5500) | 0.89 | –1.89, −2.15 | 75 | 69, 73 | 7.5 × 10–6 | 0.21 | 0.57 |
| [Fe(bpy)(CN)4]2– | 666 (1100) | –0.65 | –2.51 | 73 | 1.6 × 10–6 | 0.14 | 0.66 | |
| [Fe(bpy)(BCF)4]2– | 426 (3000) | 1.00 | –2.17 | 73 | 96 | 4.1 × 10–6 | 0.07 | 0.70 |
| [Fe(bpy)3]2+ | 520 (8000) | 0.68 | –1.74, −1.93, −2.19 | 75 | 60, 65, 72 | 4.1 × 10–6 | 0.3949 | 0.3449 |
UV–vis data were obtained in dry, N2-saturated CH3CN at 293 K.
Electrochemical data were obtained in dry, argon-saturated CH3CN at room temperature with 0.1 M (NBu4)(PF6) as supporting electrolyte.
Zero-field 57Mössbauer data were obtained in the solid state at 80 K.
ΔEox/red denotes peak-to-peak separation and was determined for a scan rate of 0.1 V s–1.
D0 values were calculated on the basis of a Randles–Ševčík analysis (see the SI for details).
Mössbauer Spectroscopy
In order to gain further insight into the electronic ground state structure of the new complexes and to further understand the effects of borylation, we employed 57Fe Mössbauer spectroscopy on solid samples at 80 K.
Upon borylation of [Fe(bpy)2(CN)2] to [Fe(bpy)2(BCF)2], the isomer shift (δ) decreased from 0.25 to 0.21 mm s–1 due to the increased s-electron density at the FeII nucleus resulting from less d-shielding in the more electron-withdrawing borylated complex (Figure 5a). Similarly, δ decreased from 0.14 mm s–1 ([Fe(bpy)(CN)4]2–) to 0.07 mm s–1 ([Fe(bpy)(BCF)4]2–), and as already observed in the electrochemical measurements, the magnitude of the change upon fourfold borylation is roughly by a factor of 2 larger compared to the twofold borylation of [Fe(bpy)2(CN)2] (Figure 5b). All parameters are in accordance with a ferrous low-spin configuration.48 Quadrupole splittings (ΔEQ) are rather small and compatible with only small lattice contributions due to the non-symmetric ligand environment.
Figure 5.

(a) Solid state zero-field 57Fe Mössbauer spectra of [Fe(bpy)2(CN)2] (black line) and [Fe(bpy)2(BCF)2] (red line) at 80 K. (b) Solid state zero-field 57Fe Mössbauer spectra of (PPN)2[Fe(bpy)(CN)4] (black line) and (PPN)2[Fe(bpy)(BCF)4] (red line) at 80 K.
UV–Vis Absorption Spectroscopy
In the visible to near-UV range of the optical absorption spectrum of [Fe(bpy)3]2+ (Figure 6a), two prominent bands with maxima at 350 and 520 nm are observed, both of which are attributed to metal-to-ligand charge transfer (MLCT) transitions.35,50
Figure 6.

UV–vis absorption spectra of [Fe(bpy)3]2+ (a), [Fe(bpy)2(CN)2] and [Fe(bpy)2(BCF)2] (b), and [Fe(bpy)(CN)4]2– and [Fe(bpy)(BCF)4]2– in dry CH3CN at 293 K.
When replacing one bpy ligand by two cyanido ligands to obtain [Fe(bpy)2(CN)2], the maximum of the lower-energy MLCT band is shifted from 520 to 613 nm (dashed red trace in Figure 6b). This is in line with the cathodic shift of the oxidation potential observed in the electrochemical measurements presented above (Figure 4); as the relevant bpy-based π*-orbitals are destabilized much less than the t2g-like orbitals, the respective energy gap decreases, leading to a redshifted MLCT absorption band. However, when B(C6F5)3 groups are attached to the two cyanido ligands of [Fe(bpy)2(CN)2], the low-energy MLCT absorption band is blueshifted by ca. 0.54 eV (green trace in Figure 6b), in line with the observed stabilization of the t2g-like orbitals in the electrochemical measurements presented above (Figure 4). Compared to [Fe(bpy)2(CN)2], the metal-based t2g-like orbitals in [Fe(bpy)2(BCF)2] are stabilized by 0.83 eV, whereas the bpy-based π*-orbitals are stabilized by 0.17 eV (Figure 4 and Table 2). Taking both effects into account, an increase of the t2g– π* energy gap of 0.66 eV is expected based on the electrochemical measurements, and this is in good agreement with the observed shift (0.54 eV) of the MLCT band between [Fe(bpy)2(CN)2] and [Fe(bpy)2(BCF)2].
A similar trend is observed when comparing the UV–vis absorption spectra of [Fe(bpy)3]2+, [Fe(bpy)(CN)4]2– and [Fe(bpy)(BCF)4]2–. From [Fe(bpy)3]2+ to [Fe(bpy)(CN)4]2–, a redshift of the low-energy MLCT absorption band by 0.52 eV is observed, and when the B(C6F5)3 groups are attached to the four cyanido ligands, the respective MLCT absorption band is blueshifted by 1.01 eV (Figure 6c). This is in line with the electrochemical measurements presented above, where an increase of 1.31 V in the t2g–π* energy gap was determined (Figure 4). These results are in line with previous reports, in which a correlation between the relevant redox potentials and the energy of the respective MLCT transitions in RuII complexes was observed.51−53 Furthermore, the effects of borylation on the UV–vis absorption spectra of [Fe(bpy)2(CN)2] and [Fe(bpy)(CN)4]2– are comparable to the effects observed upon methylation of [Ru(bpy)(CN)4]2– to yield [Ru(bpy)(CNMe)4]2+, which leads to a blueshift of the MLCT absorption band maximum of ∼1.35 eV in CH3CN.54
A qualitatively similar but less pronounced effect is also observed for the precursor cyanido complexes [Fe(bpy)2(CN)2] and [Fe(bpy)(CN)4]2– as well as their RuII analogues when changing from non-hydrogen bonding solvents (DMSO and CH3CN) to hydrogen-bonding H2O or MeOH.40,42,55−57 The Gutmann–Becket acceptor number (AN) is a frequently employed measure for the hydrogen-bond acceptor capability of solvents and as such quantifies the solvents’ Lewis acidity. Strongly Lewis-acidic solvents have higher values (AN = 54.8 for H2O) than non-Lewis-acidic solvents (AN = 1 for hexane).58 Solvents with large AN lead to reduced electron density on the FeII and RuII metal centers of mixed-ligand α-diimine cyanido complexes, similar to the effect of borylation.
Transient Absorption Spectroscopy
As noted in the introduction, the photophysics of iron complexes are currently intensely studied, in part because FeII would be a very attractive substitute for precious-metal-based photoactive d6 metal complexes,20−34,59−63 though there are now attractive alternative options based on isoelectronic MnI and Cr0.64−66 The excited-state dynamics of [Fe(bpy)2(CN)2] and [Fe(bpy)(CN)4]2– were previously investigated and were strongly solvent-dependent because the energy of the lowest MLCT excited state depends on whether solvents with high AN (H2O and MeOH) or low AN (DMSO and CH3CN) are used.39,43,44,67
In the transient absorption spectrum of the [Fe(bpy)2CN2] complex recorded in CH3CN (Figure S46), an excited state absorption feature (ESA) at 315 nm as well as two negative signals centered at ca. 400 nm (partly masked by scattered excitation light) and 606 nm are observed. The negative signals coincide with the bands observed in the absorption spectrum of [Fe(bpy)2(CN)2] (red trace in Figure 6b). After a very short time (<1 ps), no ESA feature around 370 nm (typically attributed to bpy·– and usually diagnostic for an MLCT excited state) is observed,68 indicating that the initially populated MLCT state undergoes fast relaxation (<1 ps) to a metal-centered (MC) state, which is then probed in our measurements. Recent findings suggest that in the Lewis-acidic solvent MeOH, the 5MC state of [Fe(bpy)2CN2] is populated on the fs timescale,39 and this also seems plausible for our measurements in CH3CN, although this aspect is not of core interest for the present study. The observable metal-centered state decays with a lifetime of 650 ps (Figure S47). The excited-state dynamics of [Fe(bpy)2CN2] therefore seem somewhat comparable to [Fe(bpy)3]2+, whose MLCT-excited state has a lifetime on the order of 50–100 fs and subsequently undergoes relaxation through different processes to the lowest-excited MC state, which in turn decays to the ground state with a comparably long lifetime of 1.05 ns29,30,69 and can become exploitable in photoredox catalysis.70−74
The TA spectrum of [Fe(bpy)2(BCF)2] recorded in CH3CN 1 ps after excitation (Figure 7a) shows very similar features as the precursor complex [Fe(bpy)2CN2], namely, an ESA band centered at 315 nm as well as a ground-state bleach with local minima around 350 nm and ca. 450 nm. Analogously to [Fe(bpy)2(CN)2], no spectral signature of an MLCT excited state was observable for [Fe(bpy)2(BCF)2] after times over 1 ps, again signaling rapid relaxation to a metal-centered excited state. The three most prominent TA features in Figure 7a decay with a lifetime of 67 ps (Figure 7b), which is ca. 10 times shorter compared to the non-borylated [Fe(bpy)2(CN)2] complex under identical conditions (see Figures S48–S50 and Table S2 for the fit details). According to a recent study on [Fe(bpy)3]2+, the outer-sphere reorganization energy plays an important role in the relaxation of the 5MC excited state to the electronic ground state, and this could also be true for related FeII polypyridine compounds.69 Given that solvent molecules are known to interact strongly with the cyanido ligands of [Fe(bpy)2(CN)2],39,40,42 the borylation likely has a substantial impact on the interaction between the solvent and the metal complex, in line with the differences in diffusion coefficients discussed above. Consequently, it seems plausible that the outer-sphere reorganization energies associated with MC excited state relaxation in [Fe(bpy)2(CN)2] and [Fe(bpy)2(BCF)2] are significantly different, which in turn could contribute to their very different MC lifetimes (650 vs 67 ps in N2-saturated CH3CN at 23 °C).
Figure 7.

(a, c) Transient absorption spectra recorded after different time delays and inverted absorption spectra (designated as (−)abs in the legends) of [Fe(bpy)2(BCF)2] (a) and [Fe(bpy)(BCF)4]2– (c). (b, d) Measured kinetics at selected wavelengths (symbols) and the results of a global fit (solid lines; see Figures S48–S50 and Table S2 for the fit details) for [Fe(bpy)2(BCF)2] (b) and [Fe(bpy)(BCF)4]2– (d). All measurements were performed in dry, N2-saturated CH3CN at 296 K. The samples were excited at 455 nm ((a) and (b)) or 400 nm ((c) and (d)).
The excited state dynamics of the intensely studied [Fe(bpy)(CN)4]2– complex in CH3CN have been reported to be fundamentally different to [Fe(bpy)3]2+ and [Fe(bpy)2(CN)2],22,44,67 because the probed excited state has relatively clear MLCT-character, manifesting by the presence of an ESA band around 370 nm that is commonly associated with a one-electron reduced bpy ligand.67 This behavior is likely the consequence of the strongly σ-donating nature of the four cyanido ligands, which destabilize the metal-centered states to such an extent that the MLCT decay is no longer ultrafast.44,67 However, the situation changes when the solvent is changed from non-hydrogen-bonding CH3CN to hydrogen-bonding H2O. In water, [Fe(bpy)(CN)4]2– was reported to undergo rapid (<100 fs) deactivation to an MC state before returning to the ground state with a time constant of 13 ps.44
When the four cyanido ligands of [Fe(bpy)(CN)4]2– are borylated to give the [Fe(bpy)(BCF)4]2– complex, no spectral signature of an MLCT state is observed anymore in a TA spectrum recorded in CH3CN (Figure 7c). This finding is in line with a recent literature report,10 and we attribute this to the 1.01 eV blueshift of the MLCT absorption in Figure 6c, whereas the effect of borylation on the MC states is likely substantially smaller. Thus, with the MLCT energy rising whereas the relevant MC-state energies remain comparatively constant, MLCT relaxation to MC states becomes easier in [Fe(bpy)(BCF)4]2– than in [Fe(bpy)(CN)4]2–.10 Borylation of the cyanido ligands therefore seems to have a similar effect as solvent environments with high Gutmann–Becket acceptor numbers (AN), and this is in line with the reported AN values for B(C6F5)3 (78.9) and H2O (54.8) compared to CH3CN (19.3).10,39,58
The transient absorption spectrum of [Fe(bpy)(BCF)4]2– features an MLCT ground-state bleach at ∼410 nm (Figure 7c), along with an ESA band at around 560 nm decaying with identical kinetics (τ0 = 28 ps; Figure 7d). A similar ESA band at 560 nm has been previously observed for [Fe(bpy)(CN)4]2– and was attributed to a 3MC state.44 Given the spectral resemblance of the ESA band at 560 nm in Figure 7c and the close chemical relationship between [Fe(bpy)(CN)4]2– and [Fe(bpy)(BCF)4]2–, it seems plausible that their lowest MLCT states relax into a 3MC state in both cases. This stands in contrast to [Fe(bpy)2(CN)2] and [Fe(bpy)2(BCF)2], in which the 5MC state population seems more plausible. Prior studies specifically addressed the issue of the 3MC/5MC crossover point in FeII complexes.34,75,76
Photostability
Isocyanoborato complexes of RuII and IrIII have been shown to be exceptionally photorobust,12,19 and therefore, it seemed interesting to conduct similar photostability measurements with the FeII isocyanoborato complexes. To that end, CH3CN solutions of [Fe(bpy)2(BCF)2] and [Fe(bpy)(BCF)4]2– as well as their precursors [Fe(bpy)2(CN)2] and [Fe(bpy)(CN)4]2– were irradiated with a blue continuous-wave (cw) laser (447 nm, 1.1 W). The initial absorbance of each of the solutions was adjusted to 0.1 at the excitation wavelength (447 nm), ensuring that all the different complexes absorb roughly the same amount of photons in a given time interval at least at the beginning of the irradiation period. The photodegradation of the four complexes was investigated by recording UV–vis absorption spectra of the solutions after different time intervals and by tracking the absorbance at their respective MLCT-absorption band maxima (Table S3). Using the Lambert–Beer law and the known molar extinction coefficients at the MLCT band maxima of the four complexes (Table 2), the change in absorbance during the irradiation was converted to change in concentration (Δc). Because all samples absorb (roughly) the same number of photons in a given time interval, plotting Δc as a function of irradiation time results in a reasonable graphical representation of the photostability of the respective complexes (Figure 8).
Figure 8.

Photodegradation of the four key complexes, determined in dry, N2-saturated CH3CN at 293 K. Δc is the change in concentration of intact complex.
Similar to a recently published procedure, we determined the photodegradation quantum yield (Φdegr) (Table 3), defined as the number of decomposed complexes divided by the number of absorbed photons (see the SI for details).12 Taking the individual excited-state lifetimes into account (see the SI), it follows that [Fe(bpy)(BCF)4]2– exhibits a roughly 150 times slower photodegradation than [Fe(bpy)(CN)4]2–, whereas [Fe(bpy)2(BCF)2] and its precursor [Fe(bpy)2(CN)2] photodegrade with similar rate constants (Table S4).
Table 3. Photodegradation Quantum Yields (Φdegr) and Excited-State Lifetimes (τ0) of the Four Key Complexesa.
| τ0 | Φdegr (%) | |
|---|---|---|
| [Fe(bpy)2(CN)2] | (650 ± 25) ps(MC) | 8.8 × 10–5 |
| [Fe(bpy)2(BCF)2] | (67 ± 2) ps(MC) | 1.5 × 10–5 |
| [Fe(bpy)(CN)4]2– | 18 ps (MLCT)22 | 4.9 × 10–3 |
| [Fe(bpy)(BCF)4]2– | (28 ± 3) ps(MC) | 4.9 × 10–5 |
The dominant character of their lowest excited state is indicated in parentheses.
Conclusions
The second coordination sphere interaction between boron-based Lewis-acids and cyanido complexes of different metals was actively explored in recent years.4−10,13−18 Until now, the focus was mostly on isocyanoborato complexes of 4d and 5d metals, with a few exceptions of first-row transition metals including NiII,16,17 Cu,I14,15 and FeII10,13 complexes. This study shows that the attachment of B(C6F5)3 to two easily accessible, well-known FeII cyanido complexes has a strong impact on their structural, electrochemical, and spectroscopic properties. By comparison with the archetypal [Fe(bpy)3]2+ complex, the influence of the cyanido ligands and their borylation can be readily rationalized. In a simplified picture, the exchange of bpy ligands by cyanido ligands entails a destabilization of the metal-based HOMO compared to [Fe(bpy)3]2+, as determined by cyclic voltammetry and differential pulse voltammetry. However, this effect is overcompensated by the borylation of the respective cyanido complexes to obtain their BCF congeners, which show unusually large separations of their first reduction and oxidation potentials. This has a pronounced impact on the UV–vis spectra of the borylated complexes, in which the MLCT absorption bands are shifted to substantially higher energies compared to the cyanido precursor compounds, similar to what is known for cyanido complexes of RuII and FeII upon increasing the Gutmann–Becket acceptor number (AN) of the solvent.40,42−44,55−57,77−80
Mössbauer spectroscopy confirms the decrease of d-electron density at the metal center associated with the borylation of the cyanido ligands. Infrared spectra of the cyanido complexes and their borylated congeners as well as X-ray crystal structure analyses of [Fe(bpy)2(CN)2] and [Fe(bpy)2(BCF)2] provide further insight into metal–ligand binding. Most importantly, the borylation entails shorter (i.e., stronger) C≡N bonds as well as a shortened Fe–C bond distances.
As complexes of FeII and FeIII were in the focus of recent research aiming at replacing photoactive complexes of the precious metals RuII and IrIII,81,82 the excited-state dynamics of the isocyanoborato complexes were investigated and compared to their cyanido precursor complexes. Whereas [Fe(bpy)2(CN)2] and [Fe(bpy)2(BCF)2] behave similarly to each other in that the lowest MLCT excited state relaxes to an MC state within less than 1 ps, the MLCT lifetime of [Fe(bpy)(CN)4]2– in CH3CN is 18 ps according to previous reports22 but shortens to less than 1 ps upon borylation. This suggests that the attachment of B(C6F5)3 exerts a similar effect on the electronic excited-state structure and the excited-state dynamics as a solvent environment with a high Gutmann–Becket acceptor number caused by the Lewis acid–base interaction between the cyanido ligands and their respective Lewis-acidic solvents.22,39,44 In principle, it seems conceivable to elongate the MLCT excited-state lifetimes of FeII complexes through second coordination sphere interactions; however, the borylation of the cyanido complexes investigated herein is not well-suited for this purpose. Perhaps the idea of exploiting second coordination sphere interactions would deserve more attention in the design of new photoactive first-row transition metal complexes.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c01667.
Synthetic protocols and characterization data, electrochemical characterization, Mössbauer spectra, time-resolved data, equipment and methods, NMR data and details to photostability studies (PDF)
This work was funded by the Swiss National Science Foundation through grant number 200021_207329 (to O.S.W.) and by the Deutsche Forschungsgemeinschaft (DFG) in the framework of the SPP 2102 through project 404391096 (Me1313/15-2 to F.M.). I.R. thanks the Fonds der Chemischen Industrie (PhD fellowship). P.P. acknowledges support from the Swedish Research Council through grant number 2021-05313.
The authors declare no competing financial interest.
Supplementary Material
References
- Shriver D. F. Preparation and Structures of Metal Cyanide-Lewis Acid Bridge Compounds. J. Am. Chem. Soc. 1963, 85, 1405–1408. 10.1021/ja00893a011. [DOI] [Google Scholar]
- Shriver D. F. Bridge Adducts-the Interaction of BF3 with Transition Metal Cyanide Complexes. J. Am. Chem. Soc. 1962, 84, 4610–4611. 10.1021/ja00882a061. [DOI] [Google Scholar]
- Woodcock C.; Shriver D. F. Electrochemistry and spectroscopy of dicyanobis(phenanthroline)iron, tetracarbonyltetrakis(cyclopentadienyl)tetrairon and dicarbonylcyano(cyclopentadienyl)iron in an acidic molten salt. Inorg. Chem. 1986, 25, 2137–2142. 10.1021/ic00233a008. [DOI] [Google Scholar]
- Chan K.-C.; Chu W.-K.; Yiu S.-M.; Ko C.-C. Synthesis, characterization, photophysics and electrochemical study of luminescent iridium(III) complexes with isocyanoborate ligands. Dalton Trans. 2015, 44, 15135–15144. 10.1039/C5DT00904A. [DOI] [PubMed] [Google Scholar]
- Chu W.-K.; Yiu S.-M.; Ko C.-C. Neutral Luminescent Bis(bipyridyl) Osmium(II) Complexes with Improved Phosphorescent Properties. Organometallics 2014, 33, 6771–6777. 10.1021/om500630c. [DOI] [Google Scholar]
- Chu W. K.; Wei X. G.; Yiu S. M.; Ko C. C.; Lau K. C. Strongly Phosphorescent Neutral Rhenium(I) Isocyanoborato Complexes: Synthesis, Characterization, and Photophysical, Electrochemical, and Computational Studies. Chem. – Eur. J. 2015, 21, 2603–2612. 10.1002/chem.201405291. [DOI] [PubMed] [Google Scholar]
- Chu W.-K.; Ko C.-C.; Chan K.-C.; Yiu S.-M.; Wong F.-L.; Lee C.-S.; Roy V. A. L. A Simple Design for Strongly Emissive Sky-Blue Phosphorescent Neutral Rhenium Complexes: Synthesis, Photophysics, and Electroluminescent Devices. Chem. Mater. 2014, 26, 2544–2550. 10.1021/cm4038654. [DOI] [Google Scholar]
- Xiao Y.; Chu W.-K.; Ng C.-O.; Cheng S.-C.; Tse M.-K.; Yiu S.-M.; Ko C.-C. Design and Synthesis of Luminescent Bis(isocyanoborato) Rhenate(I) Complexes as a Selective Sensor for Cyanide Anion. Organometallics 2020, 39, 2135–2141. 10.1021/acs.organomet.0c00204. [DOI] [Google Scholar]
- Chan K. C.; Tong K. M.; Cheng S. C.; Ng C. O.; Yiu S. M.; Ko C. C. Design of Luminescent Isocyano Rhenium(I) Complexes: Photophysics and Effects of the Ancillary Ligands. Inorg. Chem. 2018, 57, 13963–13972. 10.1021/acs.inorgchem.8b02536. [DOI] [PubMed] [Google Scholar]
- Ngo D. X.; Del Ciello S. A.; Barth A. T.; Hadt R. G.; Grubbs R. H.; Gray H. B.; McNicholas B. J. Electronic Structures, Spectroscopy, and Electrochemistry of [M(diimine)(CN-BR3)4]2– (M = Fe, Ru; R = Ph, C6F5) Complexes. Inorg. Chem. 2020, 59, 9594–9604. 10.1021/acs.inorgchem.0c00632. [DOI] [PubMed] [Google Scholar]
- Ngo D. X.; Del Ciello S. A.; McNicholas B. J.; Sanders B. C.; Fajardo J. Jr.; Gray H. B.; Winkler J. R. Cyano-ambivalence: Spectroscopy and photophysics of [Ru(diimine)(CN-BR3)4]2– complexes. Polyhedron 2020, 188, 114692. 10.1016/j.poly.2020.114692. [DOI] [Google Scholar]
- Schmid L.; Kerzig C.; Prescimone A.; Wenger O. S. Photostable Ruthenium(II) Isocyanoborato Luminophores and Their Use in Energy Transfer and Photoredox Catalysis. JACS Au 2021, 1, 819–832. 10.1021/jacsau.1c00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas B. J.; Grubbs R. H.; Winkler J. R.; Gray H. B.; Despagnet-Ayoub E. Tuning the formal potential of ferrocyanide over a 2.1 V range. Chem. Sci. 2019, 10, 3623–3626. 10.1039/C8SC04972F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakkaradhari G.; Eskelinen T.; Degbe C.; Belyaev A.; Melnikov A. S.; Grachova E. V.; Tunik S. P.; Hirva P.; Koshevoy I. O. Oligophosphine-thiocyanate Copper(I) and Silver(I) Complexes and Their Borane Derivatives Showing Delayed Fluorescence. Inorg. Chem. 2019, 58, 3646–3660. 10.1021/acs.inorgchem.8b03166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.-C.; Cheng S.-C.; Lo L. T.-L.; Yiu S.-M.; Ko C.-C. Luminescent Charge-Neutral Copper(I) Phenanthroline Complexes with Isocyanoborate Ligand. Eur. J. Inorg. Chem. 2018, 2018, 897–903. 10.1002/ejic.201701205. [DOI] [Google Scholar]
- Lancaster S. J.; Walker D. A.; Thornton-Pett M.; Bochmann M. New weakly coordinating counter anions for high activity polymerisation catalysts: [(C6F5)3B–CN–B(C6F5)3]− and [Ni{CNB(C6F5)3}4]2–. Chem. Commun. 1999, 1533–1534. 10.1039/a903788h. [DOI] [Google Scholar]
- Zhou J.; Lancaster S. J.; Walker D. A.; Beck S.; Thornton-Pett M.; Bochmann M. Synthesis, Structures, and Reactivity of Weakly Coordinating Anions with Delocalized Borate Structure: The Assessment of Anion Effects in Metallocene Polymerization Catalysts. J. Am. Chem. Soc. 2001, 123, 223–237. 10.1021/ja002820h. [DOI] [PubMed] [Google Scholar]
- Na H.; Maity A.; Teets T. S. Postsynthetic Systematic Electronic Tuning of Organoplatinum Photosensitizers via Secondary Coordination Sphere Interactions. Organometallics 2016, 35, 2267–2274. 10.1021/acs.organomet.6b00332. [DOI] [Google Scholar]
- Schmid L.; Glaser F.; Schaer R.; Wenger O. S. High Triplet Energy Iridium(III) Isocyanoborato Complex for Photochemical Upconversion, Photoredox and Energy Transfer Catalysis. J. Am. Chem. Soc. 2022, 144, 963–976. 10.1021/jacs.1c11667. [DOI] [PubMed] [Google Scholar]
- Chábera P.; Kjaer K. S.; Prakash O.; Honarfar A.; Liu Y.; Fredin L. A.; Harlang T. C. B.; Lidin S.; Uhlig J.; Sundström V.; Lomoth R.; Persson P.; Wärnmark K. FeII Hexa N-Heterocyclic Carbene Complex with a 528 ps Metal-to-Ligand Charge-Transfer Excited-State Lifetime. J. Phys. Chem. Lett. 2018, 9, 459–463. 10.1021/acs.jpclett.7b02962. [DOI] [PubMed] [Google Scholar]
- Harlang T. C. B.; Liu Y.; Gordivska O.; Fredin L. A.; Ponseca C. S.; Huang P.; Chábera P.; Kjaer K. S.; Mateos H.; Uhlig J.; Lomoth R.; Wallenberg R.; Styring S.; Persson P.; Sundström V.; Wärnmark K. Iron Sensitizer Converts Light to Electrons with 92% Yield. Nat. Chem. 2015, 7, 883–889. 10.1038/nchem.2365. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Kjaer K. S.; Fredin L. A.; Chábera P.; Harlang T.; Canton S. E.; Lidin S.; Zhang J.; Lomoth R.; Bergquist K.-E.; Persson P.; Wärnmark K.; Sundström V. A heteroleptic ferrous complex with mesoionic bis(1,2,3-triazol-5-ylidene) ligands: taming the MLCT excited state of iron(II). Chem. – Eur. J. 2015, 21, 3628–3639. 10.1002/chem.201405184. [DOI] [PubMed] [Google Scholar]
- Chábera P.; Fredin L. A.; Kjær K. S.; Rosemann N. W.; Lindh L.; Prakash O.; Liu Y.; Wärnmark K.; Uhlig J.; Sundström V.; Yartsev A.; Persson P. Band-selective dynamics in charge-transfer excited iron carbene complexes. Faraday Discuss. 2019, 216, 191–210. 10.1039/C8FD00232K. [DOI] [PubMed] [Google Scholar]
- Kjær K. S.; Van Driel T. B.; Harlang T. C. B.; Kunnus K.; Biasin E.; Ledbetter K.; Hartsock R. W.; Reinhard M. E.; Koroidov S.; Li L.; Laursen M. G.; Hansen F. B.; Vester P.; Christensen M.; Haldrup K.; Nielsen M. M.; Dohn A. O.; Pápai M. I.; Møller K. B.; Chabera P.; Liu Y.; Tatsuno H.; Timm C.; Jarenmark M.; Uhlig J.; Sundstöm V.; Wärnmark K.; Persson P.; Németh Z.; Szemes D. S.; Bajnóczi É.; Vankó G.; Alonso-Mori R.; Glownia J. M.; Nelson S.; Sikorski M.; Sokaras D.; Canton S. E.; Lemke H. T.; Gaffney K. J. Finding intersections between electronic excited state potential energy surfaces with simultaneous ultrafast X-ray scattering and spectroscopy. Chem. Sci. 2019, 10, 5749–5760. 10.1039/C8SC04023K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger O. S. Is Iron the New Ruthenium?. Chem. – Eur. J. 2019, 25, 6043–6052. 10.1002/chem.201806148. [DOI] [PubMed] [Google Scholar]
- Braun J. D.; Lozada I. B.; Kolodziej C.; Burda C.; Newman K. M. E.; van Lierop J.; Davis R. L.; Herbert D. E. Iron(II) Coordination Complexes with Panchromatic Absorption and Nanosecond Charge-Transfer Excited State Lifetimes. Nat. Chem. 2019, 11, 1144–1150. 10.1038/s41557-019-0357-z. [DOI] [PubMed] [Google Scholar]
- Leis W.; Argüello Cordero M. A.; Lochbrunner S.; Schubert H.; Berkefeld A. A Photoreactive Iron(II) Complex Luminophore. J. Am. Chem. Soc. 2022, 144, 1169–1173. 10.1021/jacs.1c13083. [DOI] [PubMed] [Google Scholar]
- Paulus B. C.; Adelman S. L.; Jamula L. L.; McCusker J. K. Leveraging excited-state coherence for synthetic control of ultrafast dynamics. Nature 2020, 582, 214–218. 10.1038/s41586-020-2353-2. [DOI] [PubMed] [Google Scholar]
- Carey M. C.; Adelman S. L.; McCusker J. K. Insights into the excited state dynamics of Fe(II) polypyridyl complexes from variable-temperature ultrafast spectroscopy. Chem. Sci. 2019, 10, 134–144. 10.1039/C8SC04025G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker J. K. Electronic structure in the transition metal block and its implications for light harvesting. Science 2019, 363, 484–488. 10.1126/science.aav9104. [DOI] [PubMed] [Google Scholar]
- Fatur S. M.; Shepard S. G.; Higgins R. F.; Shores M. P.; Damrauer N. H. A Synthetically Tunable System To Control MLCT Excited-State Lifetimes and Spin States in Iron(II) Polypyridines. J. Am. Chem. Soc. 2017, 139, 4493–4505. 10.1021/jacs.7b00700. [DOI] [PubMed] [Google Scholar]
- Dierks P.; Vukadinovic Y.; Bauer M. Photoactive iron complexes: more sustainable, but still a challenge. Inorg. Chem. Front. 2022, 9, 206–220. 10.1039/D1QI01112J. [DOI] [Google Scholar]
- Cebrián C.; Pastore M.; Monari A.; Assfeld X.; Gros P. C.; Haacke S. Ultrafast Spectroscopy of Fe(II) Complexes Designed for Solar-Energy Conversion: Current Status and Open Questions. ChemPhysChem 2022, 23, e202100659. 10.1002/cphc.202100659. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Ash R.; Girolami G. S.; Vura-Weis J. Tracking the Metal-Centered Triplet in Photoinduced Spin Crossover of Fe(phen)32+ with Tabletop Femtosecond M-Edge X-ray Absorption Near-Edge Structure Spectroscopy. J. Am. Chem. Soc. 2019, 141, 17180–17188. 10.1021/jacs.9b07332. [DOI] [PubMed] [Google Scholar]
- Auböck G.; Chergui M. Sub-50-fs Photoinduced Spin Crossover in [Fe(bpy)3]2+. Nat. Chem. 2015, 7, 629–633. 10.1038/nchem.2305. [DOI] [PubMed] [Google Scholar]
- Sutin N.; Creutz C. Light induced electron transfer reactions of metal complexes. Pure Appl. Chem. 1980, 52, 2717–2738. 10.1351/pac198052122717. [DOI] [Google Scholar]
- Lemke H. T.; Kjær K. S.; Hartsock R.; van Driel T. B.; Chollet M.; Glownia J. M.; Song S.; Zhu D.; Pace E.; Matar S. F.; Nielsen M. M.; Benfatto M.; Gaffney K. J.; Collet E.; Cammarata M. Coherent structural trapping through wave packet dispersion during photoinduced spin state switching. Nat. Commun. 2017, 8, 15342. 10.1038/ncomms15342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker J. K.; Walda K. N.; Dunn R. C.; Simon J. D.; Magde D.; Hendrickson D. N. Subpicosecond 1MLCT → 5T2 Intersystem Crossing of Low-spin Polypyridyl Ferrous Complexes. J. Am. Chem. Soc. 1993, 115, 298–307. 10.1021/ja00054a043. [DOI] [Google Scholar]
- Kjær K. S.; Zhang W.; Alonso-Mori R.; Bergmann U.; Chollet M.; Hadt R. G.; Hartsock R. W.; Harlang T.; Kroll T.; Kubiček K.; Lemke H. T.; Liang H. W.; Liu Y.; Nielsen M. M.; Robinson J. S.; Solomon E. I.; Sokaras D.; van Driel T. B.; Weng T.-C.; Zhu D.; Persson P.; Wärnmark K.; Sundström V.; Gaffney K. J. Ligand manipulation of charge transfer excited state relaxation and spin crossover in [Fe(2,2′-bipyridine)2(CN)2]. Struct. Dyn. 2017, 4, 044030 10.1063/1.4985017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilt A. A. Mixed Ligand Complexes of Iron(II) and (III) with Cyanide and Aromatic Di-imines. J. Am. Chem. Soc. 1960, 82, 3000–3005. 10.1021/ja01497a007. [DOI] [Google Scholar]
- Kotowski M.; Van Eldik R.; Bin Ali R.; Burgess J.; Radulović S. Piezochromic and thermochromic behaviour of ternary diimine–cyanide–Iron(II) and −Iron(III) complexes. Inorg. Chim. Acta 1987, 131, 225–228. 10.1016/S0020-1693(00)96029-8. [DOI] [Google Scholar]
- Schilt A. A. Proton Affinities of Some Cyanide and Aromatic Diimine Complexes of Iron, Ruthenium and Osmium. J. Am. Chem. Soc. 1963, 85, 904–908. 10.1021/ja00890a016. [DOI] [Google Scholar]
- Zhang W.; Kjær K. S.; Alonso-Mori R.; Bergmann U.; Chollet M.; Fredin L. A.; Hadt R. G.; Hartsock R. W.; Harlang T.; Kroll T.; Kubiček K.; Lemke H. T.; Liang H. W.; Liu Y.; Nielsen M. M.; Persson P.; Robinson J. S.; Solomon E. I.; Sun Z.; Sokaras D.; van Driel T. B.; Weng T.-C.; Zhu D.; Wärnmark K.; Sundström V.; Gaffney K. J. Manipulating Charge Transfer Excited State Relaxation and Spin Crossover in Iron Coordination Complexes with Ligand Substitution. Chem. Sci. 2017, 8, 515–523. 10.1039/C6SC03070J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjær K. S.; Kunnus K.; Harlang T. C. B.; Van Driel T. B.; Ledbetter K.; Hartsock R. W.; Reinhard M. E.; Koroidov S.; Li L.; Laursen M. G.; Biasin E.; Hansen F. B.; Vester P.; Christensen M.; Haldrup K.; Nielsen M. M.; Chabera P.; Liu Y.; Tatsuno H.; Timm C.; Uhlig J.; Sundstöm V.; Németh Z.; Szemes D. S.; Bajnóczi É.; Vankó G.; Alonso-Mori R.; Glownia J. M.; Nelson S.; Sikorski M.; Sokaras D.; Lemke H. T.; Canton S. E.; Wärnmark K.; Persson P.; Cordones A. A.; Gaffney K. J. Solvent control of charge transfer excited state relaxation pathways in [Fe(2,2′-bipyridine)(CN)4]2–. Phys. Chem. Chem. Phys. 2018, 20, 4238–4249. 10.1039/C7CP07838B. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Ma X.; Hu S.; Wen Y.; Xue Z.; Zhu X.; Zhang X.; Sheng T.; Wu X. Syntheses, crystal structures, MMCT and magnetic properties of four one-dimensional cyanide-bridged complexes comprised of M(II)-CN-Fe(III) (M = Fe, Ru, Os). Dalton Trans. 2014, 43, 17453–17462. 10.1039/C4DT02272F. [DOI] [PubMed] [Google Scholar]
- Sutton G. D.; Olumba M. E.; Nguyen Y. H.; Teets T. S. The diverse functions of isocyanides in phosphorescent metal complexes. Dalton Trans. 2021, 50, 17851–17863. 10.1039/D1DT03312C. [DOI] [PubMed] [Google Scholar]
- Wang X.; Stanbury D. M. Oxidation of iodide by a series of Fe(III) complexes in acetonitrile. Inorg. Chem. 2006, 45, 3415–3423. 10.1021/ic052022y. [DOI] [PubMed] [Google Scholar]
- Gütlich P.; Bill E.; Trautwein A. X.. Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications; Springer Berlin Heidelberg: Berlin, Heidelberg, 2011. [Google Scholar]
- Sato H.; Tominaga T. Mössbauer Studies of the Thermal Decomposition of Tris(2,2′-bipyridine)iron(II) Chloride and the Structures of the Isomers of 2,2′-Bipyridineiron(II) Chloride. Bull. Chem. Soc. Jpn. 1976, 49, 697–700. 10.1246/bcsj.49.697. [DOI] [Google Scholar]
- Ferguson J.; Herren F.; Krausz E. R.; Maeder M.; Vrbancich J. Electronic spectroscopy of M(bpy)2+3 (M = Fe, Ru, Os), Cr(bpy)33+ and related compounds. Coord. Chem. Rev. 1985, 64, 21–39. 10.1016/0010-8545(85)80039-4. [DOI] [Google Scholar]
- Dodsworth E. S.; Lever A. B. P. Correlations between electrochemical potentials and optical charge transfer energies in ruthenium bipyridine derivatives. Chem. Phys. Lett. 1986, 124, 152–158. 10.1016/0009-2614(86)85135-1. [DOI] [Google Scholar]
- Gorelsky S. I.; Kotov V. Y.; Lever A. B. P. Vertical Ionization Energies and Electron Affinities of Ions in Solution from Outer-Sphere Charge Transfer Transition Energies. Inorg. Chem. 1998, 37, 4584–4588. 10.1021/ic980217i. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Lever A. B. P.. Inorganic Electronic Structure and Spectroscopy, Applications and Case Studies; Volume II.; Wiley-Interscience: Hoboken, N.J, 2006; p. 672. [Google Scholar]
- Indelli M. T.; Bignozzi C. A.; Marconi A.; Scandola F. Ruthenium(II) 2,2′-bipyridine complexes containing methyl isocyanide ligands. Extreme effects of nonchromophoric ligands on excited-state properties. J. Am. Chem. Soc. 1988, 110, 7381–7386. 10.1021/ja00230a018. [DOI] [Google Scholar]
- Scandola F.; Indelli M. T. Second sphere donor acceptor interactions in excited states of coordination compounds. Ruthenium(II) bipyridine cyano complexes. Pure Appl. Chem. 1988, 60, 973–980. 10.1351/pac198860070973. [DOI] [Google Scholar]
- Indelli M. T.; Ghirotti M.; Prodi A.; Chiorboli C.; Scandola F.; McClenaghan N. D.; Puntoriero F.; Campagna S. Solvent Switching of Intramolecular Energy Transfer in Bichromophoric Systems: Photophysics of (2,2’-Bipyridine)tetracyanoruthenate(II)/Pyrenyl Complexes. Inorg. Chem. 2003, 42, 5489–5497. 10.1021/ic034185x. [DOI] [PubMed] [Google Scholar]
- Balzani V.; Sabbatini N.; Scandola F. ″Second-sphere″ photochemistry and photophysics of coordination compounds. Chem. Rev. 1986, 86, 319–337. 10.1021/cr00072a002. [DOI] [Google Scholar]
- Mayer U.; Gutmann V.; Gerger W. The acceptor number — A quantitative empirical parameter for the electrophilic properties of solvents. Monatsh. Chem. 1975, 106, 1235–1257. 10.1007/BF00913599. [DOI] [Google Scholar]
- Chábera P.; Liu Y.; Prakash O.; Thyrhaug E.; Nahhas A. E.; Honarfar A.; Essén S.; Fredin L. A.; Harlang T. C. B.; Kjær K. S.; Handrup K.; Ericson F.; Tatsuno H.; Morgan K.; Schnadt J.; Häggström L.; Ericsson T.; Sobkowiak A.; Lidin S.; Huang P.; Styring S.; Uhlig J.; Bendix J.; Lomoth R.; Sundström V.; Persson P.; Wärnmark K. A Low-Spin Fe(III) Complex with 100-ps Ligand-to-Metal Charge Transfer Photoluminescence. Nature 2017, 543, 695–699. 10.1038/nature21430. [DOI] [PubMed] [Google Scholar]
- Kjær K. S.; Kaul N.; Prakash O.; Chábera P.; Rosemann N. W.; Honarfar A.; Gordivska O.; Fredin L. A.; Bergquist K.-E.; Häggström L.; Ericsson T.; Lindh L.; Yartsev A.; Styring S.; Huang P.; Uhlig J.; Bendix J.; Strand D.; Sundström V.; Persson P.; Lomoth R.; Wärnmark K. Luminescence and reactivity of a charge-transfer excited iron complex with nanosecond lifetime. Science 2019, 363, 249–253. 10.1126/science.aau7160. [DOI] [PubMed] [Google Scholar]
- Rosemann N. W.; Chábera P.; Prakash O.; Kaufhold S.; Wärnmark K.; Yartsev A.; Persson P. Tracing the Full Bimolecular Photocycle of Iron(III)-Carbene Light Harvesters in Electron-Donating Solvents. J. Am. Chem. Soc. 2020, 142, 8565–8569. 10.1021/jacs.0c00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydogan A.; Bangle R. E.; Cadranel A.; Turlington M. D.; Conroy D. T.; Cauët E.; Singleton M. L.; Meyer G. J.; Sampaio R. N.; Elias B.; Troian-Gautier L. Accessing Photoredox Transformations with an Iron(III) Photosensitizer and Green Light. J. Am. Chem. Soc. 2021, 143, 15661–15673. 10.1021/jacs.1c06081. [DOI] [PubMed] [Google Scholar]
- Moll J.; Förster C.; König A.; Carrella L. M.; Wagner M.; Panthöfer M.; Möller A.; Rentschler E.; Heinze K. Panchromatic Absorption and Oxidation of an Iron(II) Spin Crossover Complex. Inorg. Chem. 2022, 61, 1659–1671. 10.1021/acs.inorgchem.1c03511. [DOI] [PubMed] [Google Scholar]
- Herr P.; Kerzig C.; Larsen C. B.; Häussinger D.; Wenger O. S. Manganese(I) complexes with metal-to-ligand charge transfer luminescence and photoreactivity. Nat. Chem. 2021, 13, 956–962. 10.1038/s41557-021-00744-9. [DOI] [PubMed] [Google Scholar]
- Wegeberg C.; Häussinger D.; Wenger O. S. Pyrene-Decoration of a Chromium(0) Tris(diisocyanide) Enhances Excited State Delocalization: A Strategy to Improve the Photoluminescence of 3d6 Metal Complexes. J. Am. Chem. Soc. 2021, 143, 15800–15811. 10.1021/jacs.1c07345. [DOI] [PubMed] [Google Scholar]
- Wegeberg C.; Wenger O. S. Luminescent chromium(0) and manganese(I) complexes. Dalton Trans. 2022, 51, 1297–1302. 10.1039/D1DT03763C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. R.; Creutz C.; Sutin N. Solvent tuning of the excited-state properties of (2,2′-bipyridine)tetracyanoferrate(II): direct observation of a metal-to-ligand charge-transfer excited state of iron(II). J. Am. Chem. Soc. 1987, 109, 3470–3471. 10.1021/ja00245a053. [DOI] [Google Scholar]
- Heath G. A.; Yellowlees L. J.; Braterman P. S. Spectro-electrochemical studies on tris-bipyridyl ruthenium complexes; ultra-violet, visible, and near-infrared spectra of the series [Ru(bipyridyl)3]2+/1+/0/1−. J. Chem. Soc., Chem. Commun. 1981, 287. 10.1039/c39810000287. [DOI] [Google Scholar]
- Miller J. N.; McCusker J. K. Outer-sphere effects on ligand-field excited-state dynamics: solvent dependence of high-spin to low-spin conversion in [Fe(bpy)3]2+. Chem. Sci. 2020, 11, 5191–5204. 10.1039/D0SC01506G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse M. D.; McCusker J. K. Mechanistic Origin of Photoredox Catalysis Involving Iron(II) Polypyridyl Chromophores. J. Am. Chem. Soc. 2020, 142, 16229–16233. 10.1021/jacs.0c08389. [DOI] [PubMed] [Google Scholar]
- Gualandi A.; Marchini M.; Mengozzi L.; Natali M.; Lucarini M.; Ceroni P.; Cozzi P. G. Organocatalytic Enantioselective Alkylation of Aldehydes with [Fe(bpy)3]Br2 Catalyst and Visible Light. ACS Catal. 2015, 5, 5927–5931. 10.1021/acscatal.5b01573. [DOI] [Google Scholar]
- Zhou W.-J.; Wu X.-D.; Miao M.; Wang Z.-H.; Chen L.; Shan S.-Y.; Cao G.-M.; Yu D.-G. Light runs across iron catalysts in organic transformations. Chem. – Eur. J. 2020, 26, 15052–15064. 10.1002/chem.202000508. [DOI] [PubMed] [Google Scholar]
- Parisien-Collette S.; Hernandez-Perez A. C.; Collins S. K. Photochemical Synthesis of Carbazoles Using an [Fe(phen)3](NTf2)2/O2 Catalyst System: Catalysis toward Sustainability. Org. Lett. 2016, 18, 4994–4997. 10.1021/acs.orglett.6b02456. [DOI] [PubMed] [Google Scholar]
- Larsen C. B.; Wenger O. S. Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements. Chem. – Eur. J. 2018, 24, 2039–2058. 10.1002/chem.201703602. [DOI] [PubMed] [Google Scholar]
- Britz A.; Gawelda W.; Assefa T. A.; Jamula L. L.; Yarranton J. T.; Galler A.; Khakhulin D.; Diez M.; Harder M.; Doumy G.; March A. M.; Bajnóczi É.; Németh Z.; Pápai M.; Rozsályi E.; Sárosiné Szemes D.; Cho H.; Mukherjee S.; Liu C.; Kim T. K.; Schoenlein R. W.; Southworth S. H.; Young L.; Jakubikova E.; Huse N.; Vankó G.; Bressler C.; McCusker J. K. Using Ultrafast X-ray Spectroscopy To Address Questions in Ligand-Field Theory: The Excited State Spin and Structure of [Fe(dcpp)2]2+. Inorg. Chem. 2019, 58, 9341–9350. 10.1021/acs.inorgchem.9b01063. [DOI] [PubMed] [Google Scholar]
- Jamula L. L.; Brown A. M.; Guo D.; McCusker J. K. Synthesis and Characterization of a High-Symmetry Ferrous Polypyridyl Complex: Approaching the 5T2/3T1 Crossing Point for FeII. Inorg. Chem. 2014, 53, 15–17. 10.1021/ic402407k. [DOI] [PubMed] [Google Scholar]
- Ward M. D. Structural and photophysical properties of luminescent cyanometallates [M(diimine)(CN)4]2– and their supramolecular assemblies. Dalton Trans. 2010, 39, 8851–8867. 10.1039/c0dt00312c. [DOI] [PubMed] [Google Scholar]
- Ward M. D. [Ru(bipy)(CN)4]2– and its derivatives: Photophysical properties and its use in photoactive supramolecular assemblies. Coord. Chem. Rev. 2006, 250, 3128–3141. 10.1016/j.ccr.2006.02.012. [DOI] [Google Scholar]
- Lazarides T.; Easun T. L.; Veyne-Marti C.; Alsindi W. Z.; George M. W.; Deppermann N.; Hunter C. A.; Adams H.; Ward M. D. Structural and photophysical properties of adducts of [Ru(bipy)(CN)4]2− with different metal cations: metallochromism and its use in switching photoinduced energy transfer. J. Am. Chem. Soc. 2007, 129, 4014–4027. 10.1021/ja068436n. [DOI] [PubMed] [Google Scholar]
- Adams H.; Alsindi W. Z.; Davies G. M.; Duriska M. B.; Easun T. L.; Fenton H. E.; Herrera J.-M.; George M. W.; Ronayne K. L.; Sun X.-Z.; Towrie M.; Ward M. D. New members of the [Ru(diimine)(CN)4]2– family: structural, electrochemical and photophysical properties. Dalton Trans. 2006, 39–50. 10.1039/B509042C. [DOI] [PubMed] [Google Scholar]
- Förster C.; Heinze K. Photophysics and photochemistry with Earth-abundant metals - fundamentals and concepts. Chem. Soc. Rev. 2020, 49, 1057–1070. 10.1039/C9CS00573K. [DOI] [PubMed] [Google Scholar]
- Wegeberg C.; Wenger O. S. Luminescent First-Row Transition Metal Complexes. JACS Au 2021, 1, 1860–1876. 10.1021/jacsau.1c00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.