

Abstract
Objectives: Renal tubular injury plays an important role in the progression of diabetic kidney disease. Previous studies demonstrated that artemether, an antimalarial agent, exerts renal tubular protection in diabetes. However, the detailed mechanisms remain unclear. Several studies have indicated that disorders of iron metabolism have a great impact on renal tubular injury. Therefore, this study was performed to explore whether the therapeutic effects of artemether on diabetic renal tubular injury are related to iron metabolism. Methods: Male C57BL/6 J mice were randomly divided into three groups. Mice in the type 1 diabetic (T1D) control and streptozotocin (STZ) groups were fed a regular diet; mice in the STZ plus artemether (STZ+Art) group were treated with artemether. Results: Artemether significantly reduced the urinary albumin:creatinine ratio and tubular injury in mice with T1D. Artemether also restored the energy imbalance and restored the changes of mitochondrial cristae in mice with T1D. Increased protein and mRNA levels of ferritin heavy chain (FTH) and ferritin light chain (FTL) were observed in renal tubules of diabetic mice. In response to iron overload, levels of iron transport-related proteins and the antioxidant system related to iron metabolism were abnormal in diabetic mice. Artemether significantly restored the protein and mRNA expression levels of both FTH and FTL. Both the iron transport and antioxidant systems were also restored by artemether to varying degrees. Conclusions: Artemether attenuates renal tubular injury in diabetic mice; this effect might be related to its regulation of iron metabolism.
Keywords: Iron overload, tubular injury, mitochondria, type 1 diabetes
Introduction
With the increasing incidence of diabetes, diabetic kidney disease (DKD) is gradually becoming the leading cause of end-stage renal disease [1]. The etiology and pathophysiology of DKD remain unclear. Effective therapeutic regimens for DKD are limited, especially in patients with type 1 diabetes (T1D). It is of great significance to explore potential pathophysiological mechanisms and develop innovative therapeutic targets for DKD prevention.
Previous evidence has highlighted the role of renal tubular injury in DKD progression [2]. Tubular injury can be caused by many factors, including hypoxia, proteinuria, toxins, metabolic disorders, and cellular senescence [3]. Multiple pathophysiological processes are involved in tubular injury, among which mitochondrial damage is a major hallmark [4]. In recent years, disorders of iron metabolism have been recognized to be closely associated with various types of kidney injury [5]. Iron is an essential element for life and participates in multiple physiological processes in the human body. Iron-containing proteins play key roles in oxygen transport, storage, and utilization. As the main sites of oxidative phosphorylation, mitochondria are inextricably linked to iron metabolism [6]. Results of a Mendelian randomization study revealed that increased systemic iron status was positively associated with type 2 diabetes [7]. However, iron status in renal tissues from patients with diabetes mellitus remains unknown. The study of renal iron metabolism in the early stages of diabetic nephropathy is beneficial to guide clinical intervention.
Artemether, an artemisinin derivative, has been routinely used for the treatment of malaria. Recent studies demonstrated that artemisinin derivatives also display anticancer, antidiabetic, renoprotective, anti-inflammatory, and immunoregulatory effects [8]. It was also reported that these biological functions are fulfilled via the regulation of mitochondrial function and iron metabolism [9-12]. However, whether artemether can improve DKD by targeting these biological processes is unknown. Therefore, we explored the hypothesis that artemether improves DKD by regulating mitochondrial function and iron metabolism and provide more evidence for the medical applications of artemether in DKD.
Materials and methods
Animals and diabetic mouse models
Male C57BL/6J mice were purchased from Guangdong Medical Laboratory Animal Center. All mice were housed in the Center Animal Facility at Shenzhen Graduate School of Peking University and exposed to a 12 h/12 h light/dark cycle. Animal studies were performed in accordance with relevant guidelines and were approved by Guangzhou University of Chinese Medicine Institutional Animal Care and Use Committee and experimental animal Ethics Committee (No. 20190301011).
T1D was established in 8-week-old C57BL/6J mice by the intraperitoneal injection of streptozotocin (STZ; Sigma-Aldrich, St. Louis, MO, USA) at a dose of 55 mg/kg body weight/day (dissolved in citrate buffer) for five consecutive days. Diabetic control (T1D-ctrl) mice received an intraperitoneal injection of an equal volume of citrate buffer. Mice were randomly divided into three groups: the T1D-ctrl group, fed a regular diet; the STZ group, fed a regular diet; mice with T1D treated with artemether (STZ+Art group), fed a regular diet supplemented with 1.6 g/kg artemether. The treatment began nine days after the last injection of STZ and lasted for 8 weeks. Artemether was purchased from Chengdu ConBon Biotech Company (Chengdu, China).
Tissue preparation
At the end of the experiment, mice were sacrificed, and blood samples were collected. Kidneys were removed, immediately snap-frozen in liquid nitrogen, and stored at -80°C for further analysis.
Light microscopy
Paraffin-embedded kidney sections were stained with periodic acid-Schiff (PAS; Baso Diagnostics, Zhuhai, China) and scanned using a digital slide scanner (3DHistech Ltd., Budapest, Hungary) to assess changes in renal morphology. For each slide, 50-80 proximal tubular areas were measured and analyzed, as previously described [13].
Urine analysis
Urine from all mice was collected using metabolic cages (Tecniplast, Buguggiate, Italy). Urinary albumin excretion was measured using an enzyme-linked immunosorbent assay (Bethyl Laboratories, Montgomery, TX, USA), according to the manufacturer’s instructions. Urinary creatinine levels were measured using an automatic biochemical analyzer (Roche, Basel, Switzerland). The urinary albumin:creatinine ratio (UACR) was then calculated.
Western blot
The renal cortex was homogenized in RIPA lysis buffer (Cell Signaling Technology, MA, USA) using an automatic homogenizer (Bertin Instruments, France). Laemmli sample buffer (Bio-Rad, Hercules, CA, USA) containing β-mercaptoethanol was added to the protein samples prior to boiling for 10 minutes, followed by protein separation using SDS-polyacrylamide gel electrophoresis and transference to polyvinylidene difluoride membranes (Merck Millipore, Danvers, MA, USA). Immunoblots were blocked in 5% skim milk at room temperature for one hour and then incubated with primary antibodies at 4°C overnight with shaking. After washing, immunoblots were incubated with horseradish peroxidase-conjugated mouse or rabbit secondary antibodies at room temperature for one hour. Protein bands were then scanned and analyzed using the ChemiDoc™ MP imaging system (Bio-Rad). The results were expressed as the integrated optical density relative to β-actin. Primary antibodies used in this study were as follows: Optic atrophy 1 (OPA1; BD Transduction Laboratories™, NY, USA), cytochrome oxidase (COX) IV, AMP-activated protein kinase (AMPK), phospho-AMPK (Thr172) (Cell Signaling Technology), transporter of outer mitochondrial membrane (TOM20), FTH, transferrin receptor (TfR), Cystine/glutamate transporter antibody (xCT; Abcam, Cambridge, UK), transcription factor A mitochondrial (Tfam; Merck Millipore), β-actin (Sigma-Aldrich), FTL (GeneTex, Irvine, CA, USA), divalent metal transporter 1 (DMT1), glutathione peroxidase 4 (GPX4) (Proteintech Group, Chicago, IL, USA).
RNA isolation, reverse transcription, and quantitative PCR analysis
Total RNA was extracted using the TRIzol reagent and an automatic homogenizer. After centrifugation, the supernatant was purified using the PureLink™ RNA Mini kit (Invitrogen, Carlsbad, CA, USA). The first strand of cDNA was synthesized using oligo(dT)12-18 primers and M-MLV reverse transcriptase (Invitrogen), according to the manufacturer’s protocol.
Real-time quantitative PCR was performed using the QuantStudio™ 5 Real-Time PCR Instrument (Applied Biosystems, Carlsbad, CA, USA). Reactions were carried out using cDNA, 1 mM primer, and SYBR green PCR master mix (Applied Biosystems). The cycling program was denatured at 95°C for 5 minutes, 45 cycles of 95°C for 15 seconds and 55°C for 15 seconds, and extension at 72°C for 20 seconds, and a melting curve was generated. Primers used in this study were synthesized by Sangon Biotechnology Company (Shanghai, China), and the sequences are listed in Table 1. Gene expression was normalized to that of β-actin, and the fold change in expression relative to the control group was calculated using the 2-ΔΔCt method.
Table 1.
List of primer sequences used for qPCR
| Gene | Primer Sequence (5’-3’) |
|---|---|
| Mouse NGAL | F: ACCACGGACTACAACCAGTTCGCC |
| R: ACTTGGCAAAGCGGGTGAAACG | |
| Mouse FTH | F: GCTGAATGCAATGGAGTGTGCA |
| R: GGCACCCATCTTGCGTAAGTTG | |
| Mouse FTL | F: CTTGCCCGGGACTTAGAGCA |
| R: ATGGCTGATCCGGAGTAGGA | |
| Mouse ISCU | F: CCTGTGAAACTGCACTGCTC |
| R: TCTCTGGCTCCTCCTTCTTG | |
| Mouse TfR | F: ATGCCGACAATAACATGAAGGC |
| R: ACACGCTTACAATAGCCCAGG | |
| Mouse DMT1 | F: CAATGTCTTTGTCGTGTCCGT |
| R: GCGACCATTTTAGGTTCAGGAAT | |
| Mouse SLC40A1 | F: ACCAAGGCAAGAGATCAAACC |
| R: AGACACTGCAAAGTGCCACAT | |
| Mouse SLC7A11 | F: GGCACCGTCATCGGATCAG |
| R: CTCCACAGGCAGACCAGAAAA | |
| Mouse GPX4 | F: CGCAGCCGTTCTTATCAATG |
| R: CACTGTGGAAATGGATGAAAGTC | |
| Mouse β-actin | F: GGACTCCTATGTGGGTGACG |
| R: AGGTGTGGTGCCAGATCTTC |
Immunohistochemical staining
Paraffin-embedded renal sections were dewaxed and rehydrated for immunohistochemical (IHC) staining. After antigen retrieval using citrate buffer or EDTA buffer, sections were incubated with 3% hydrogen peroxide for 15 minutes at room temperature to eliminate endogenous peroxidases and blocked in 10% goat serum (MXB Biotechnologies, Fuzhou, China) for 15 minutes at room temperature. The sections were stained with primary antibodies at 4°C overnight. After washing, sections were incubated with the MaxVision™ HRP-Polymer using the anti-Mouse/Rabbit IHC Kit (MXB Biotechnologies, Fuzhou, China) at room temperature for 15 minutes. The sections were then treated with diaminobenzidine (MXB Biotechnologies), followed by counterstaining with hematoxylin and mounting in neutral balsam mounting medium.
Transmission electron microscopy
Renal cortex samples (1 mm × 1 mm) were fixed in 2.5% glutaraldehyde and then postfixed in 1% osmic acid for transmission electron microscopy (TEM). The TEM images were captured using a JEM-1400 camera (JEOL, Tokyo, Japan).
Statistical analysis
Data analysis was conducted using IBM SPSS Statistics software (version 25.0). Data were expressed as the mean ± standard deviation (SD). An unpaired Student’s t test was performed for comparisons between groups. Differences among groups were analyzed using one-way analysis of variance followed by Levene’s or Dunnett’s T3 post-hoc analysis. P values less than 0.05 were considered statistically significant.
Results
Artemether reduces UACR and alleviates renal tubular injury in mice with T1D
Increased UACR is an important manifestation of diabetic nephropathy. As shown in Figure 1A, compared to the T1D-ctrl group, there was a significant increase in UACR in the STZ group, and UACR was reduced effectively by artemether treatment. As shown in Figure 1B-E, tubular hypertrophy was observed in the STZ group; Urinary neutrophil gelatinase-associated lipocalin (NGAL) mRNA levels, a tubular injury biomarker, also increased significantly in mice with T1D. In response to artemether, the hypertrophy resolved and the NGAL levels were downregulated.
Figure 1.
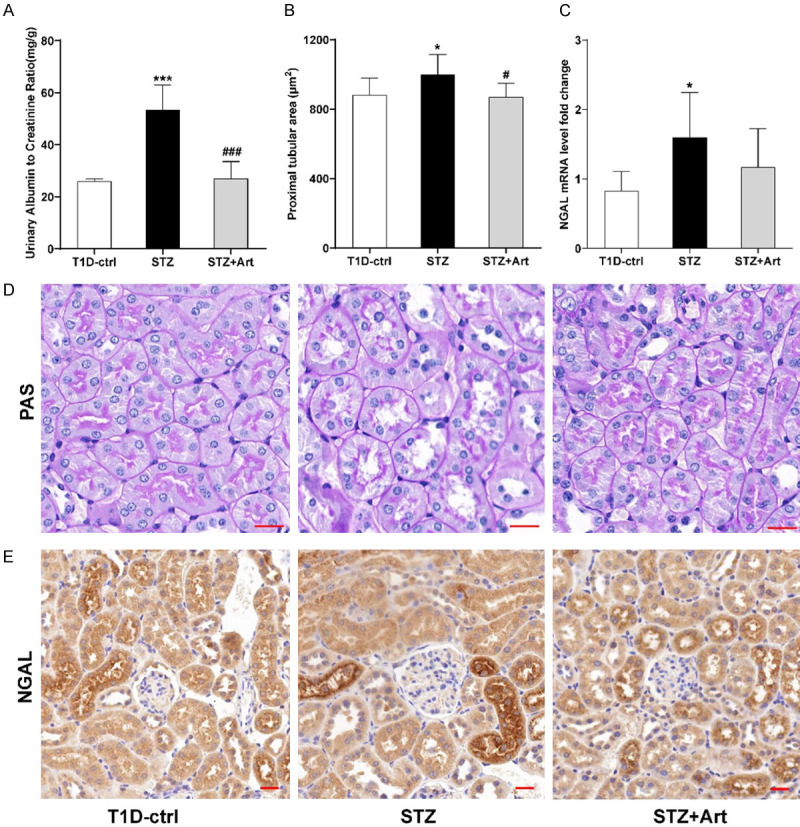
Artemether reduces UACR and alleviates renal tubular injury in mice with T1D. A: Quantification of UACR in each group. B: Proximal tubular area (μm2) of each group. C: mRNA expression of NGAL in each group. D: Representative PAS staining of the proximal tubules in each group. E: Representative immunohistochemically stained images of NGAL in renal tissue. For each group, n=4-8. Scale bar, 20 μm. *P<0.05 and ***P<0.001 vs. the T1D-ctrl group. #P<0.05 and ###P<0.001 vs. the STZ group.
Mitochondrial protein composition and biogenesis show no remarkable differences in mice with early DKD
OPA1, COX IV, and TOM20 are important components of mitochondria, and Tfam is a mitochondrial transcription factor. As shown in the Figure 2A-E, there were no significant differences in the composition of these proteins among the three groups.
Figure 2.
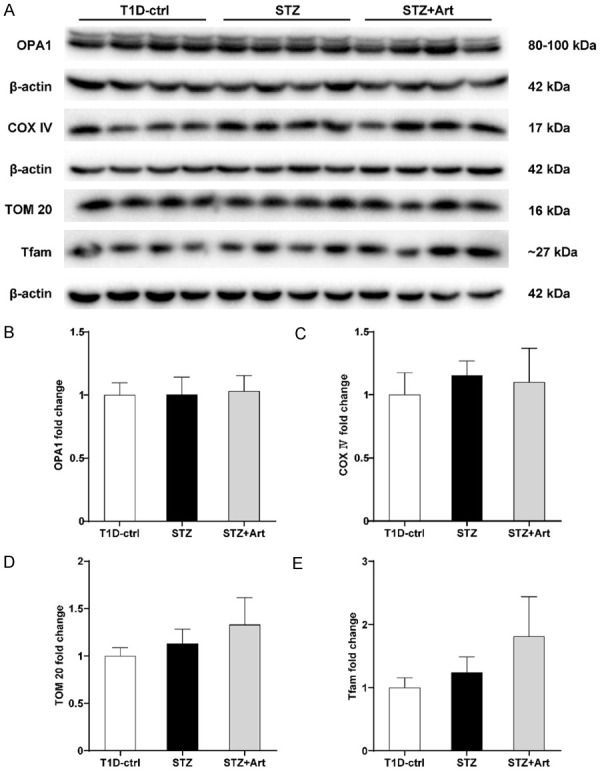
Mitochondrial protein composition and biogenesis show no remarkable differences in mice with early DKD. A-E: Representative western blot images and quantification of OPA1, COX IV, TOM20, and Tfam; n=4 per group.
Artemether helps regulate the renal energy state in mice with T1D
The expression of p-AMPK (Thr172) increased remarkably as a result of insulin deficiency in the kidneys of mice with T1D, which indicated a low energy supply. Artemether treatment led to a decrease in the expression of p-AMPK (Thr172) (Figure 3A-D).
Figure 3.

Artemether exerts effects on the regulation of the renal energy state in mice with T1D. A-C: Representative western blot images and quantification of p-AMPK (Thr172) and AMPK. D: Expression ratio of p-AMPK (Thr172): AMPK. For each group, n=4. ***P<0.001 vs. the T1D-ctrl group. ##P<0.01 and ###P<0.001 vs. the STZ group.
Artemether attenuates mitochondrial damage in renal tubules in mice with T1D
A significant reduction or disappearance of mitochondrial cristae was obvious in mice with T1D, which indicated that the low energy supply might be associated with the loss of mitochondrial cristae in the proximal tubular epithelial cells mice with T1D (Figure 4). Intriguingly, artemether treatment counteracted this change.
Figure 4.

Artemether attenuates mitochondrial damage in renal tubular cells in mice with T1D. Representative TEM images display morphological changes in mitochondrial cristae in proximal tubular epithelial cells. The corresponding scale bar of each image is labeled in the left bottom. BB, brush boarder, a symbol of proximal tubular epithelial cells. An asterisk (*) indicates the loss of mitochondrial cristae in proximal tubular epithelial cells of diabetic mice.
Artemether ameliorates effects of iron overload and restores iron homeostasis in renal tubules of mice with T1D
As shown in Figure 5A-F, increased protein and mRNA expression levels of FTH, FTL, and iron-sulfur cluster assembly enzyme (ISCU) were observed by western blot analysis and RT-PCR, respectively, in the STZ group compared with the T1D-ctrl group, and artemether downregulated these expression levels. These results were further supported by IHC staining. Of note, the staining revealed that these proteins were mainly expressed in renal tubules and only rarely expressed in glomeruli (Figure 5G, 5H).
Figure 5.
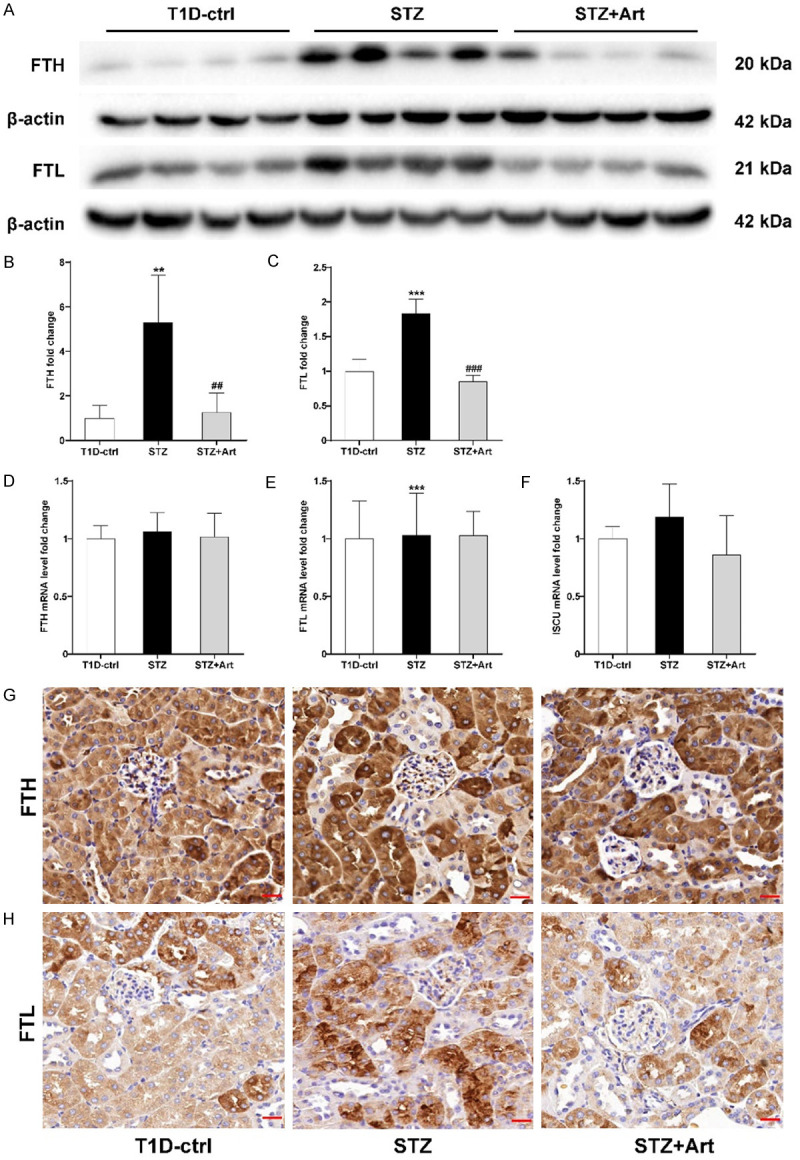
Artemether ameliorates iron overload and restores iron homeostasis in renal tubules of mice with T1D. A-C: Representative western blot images and quantification of FTH and FTL in renal tissues of mice with T1D. D-F: Relative mRNA levels of FTH, FTL, and ISCU. G: Representative images of IHC staining of FTH in each group. H: Representative images of IHC staining of FTL in each group; n=4-8 per group. Scale bar, 20 μm. **P<0.01 and ***P<0.001 vs. the T1D-ctrl group. ##P<0.01 and ###P<0.001 vs. the STZ group.
Artemether regulates iron transport in renal tubular cells in early DKD
As can be seen in Figure 6A-E, the protein and mRNA expression levels of TfR and DMT1 in the renal tissues of mice with T1D were downregulated, while the level of ferroportin (gene name: SLC40A1) mRNA increased slightly (Figure 6F), indicating that when iron overloading occurred in the renal tissues in early stages of DKD, the renal tubule uptake of iron was inhibited and iron export was enhanced. However, these levels returned to near normal after artemether treatment in the STZ+Art group, suggesting that this intervention may be conducive to stabilizing intracellular iron homeostasis.
Figure 6.
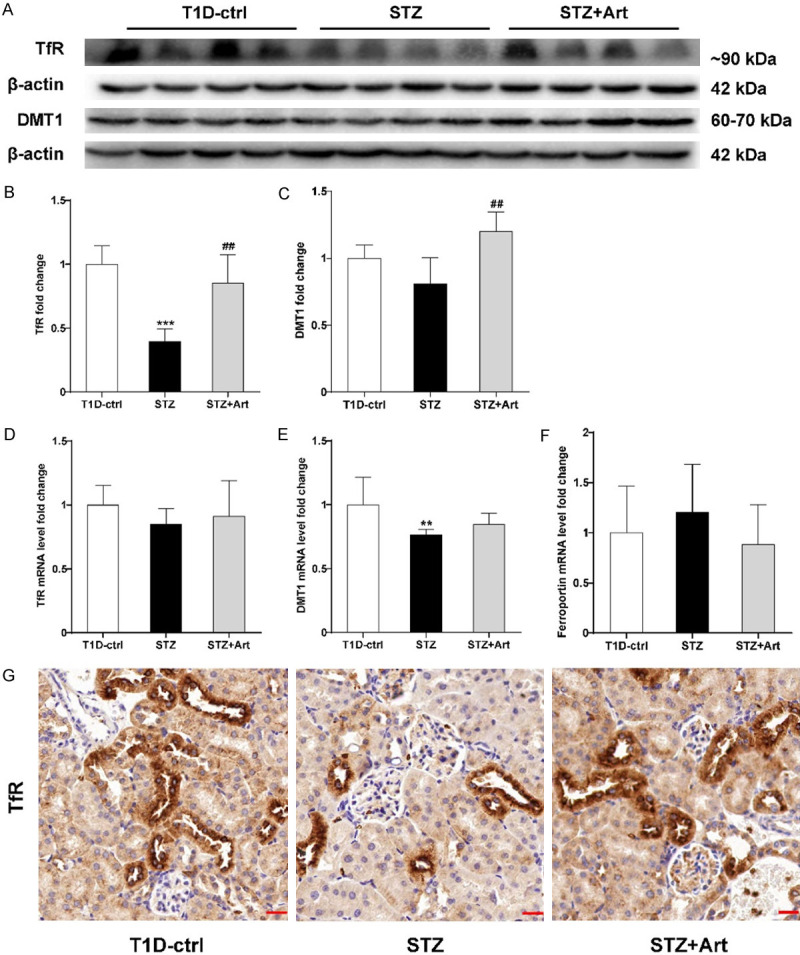
Early DKD regulates iron transport in renal tubules. A-C: Representative western blot images and quantification of TfR and DMT1. D-F: Relative mRNA levels of TfR, DMT1, and ferroportin. G: Representative images of IHC staining of TfR in each group; n=4-8 per group. Scale bar, 20 μm. **P<0.01 and ***P<0.001 vs. the T1D-ctrl group. ##P<0.01 vs. the STZ group.
Artemether enhances the antioxidant stress system to protect against iron-mediated tubular injury in mice with T1D
Compared to the T1D-ctrl group, the protein and mRNA expression levels of xCT (gene name: SLC7A11) were significantly decreased in the STZ group and increased after artemether treatment (Figure 7A, 7B, 7D, 7F). In contrast, GPX4 expression increased in the STZ group, which was further enhanced after artemether treatment in the STZ+Art group (Figure 7A, 7C, 7E, 7G).
Figure 7.
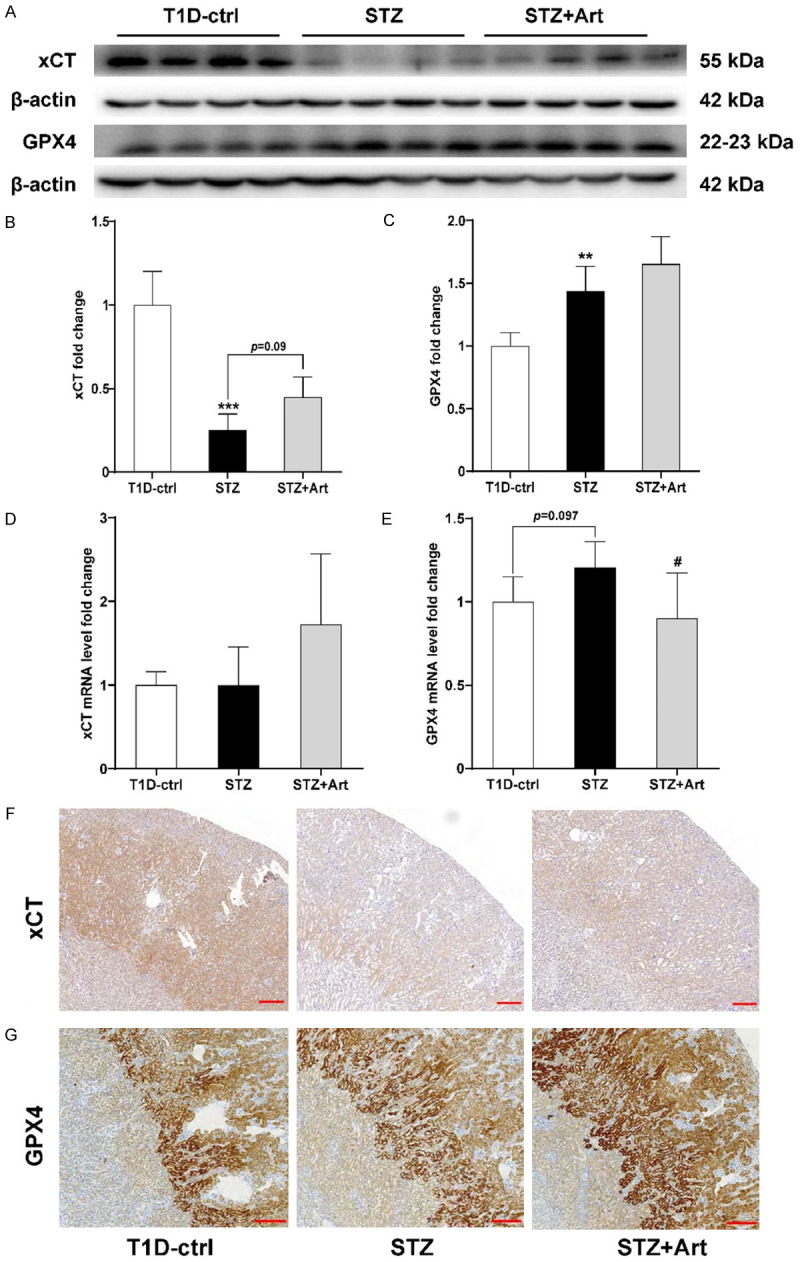
Artemether enhances the antioxidant stress system to protect against iron-mediated tubular injury in mice with T1D. A-C: Representative western blot images and quantification of xCT and GPX4. D, E: Relative mRNA levels of xCT and GPX4. F: Representative images of IHC staining of xCT in each group. G: Representative images of IHC staining of GPX4 in each group. For each group, n=4-6. Scale bar, 200 μm. **P<0.01 and ***P<0.001 vs. the T1D-ctrl group. #P<0.05 vs. the STZ group.
Discussion
The results of the present study demonstrated that artemether could alleviate renal tubular injury due to T1D, which might be associated with the regulation of iron homeostasis.
Proximal tubular injury plays an important role in the progression of DKD [14]. In recent years, with advances in DKD research, renal tubule injury is increasingly prominent in early DKD, suggesting that renal tubule lesions may occur earlier than microvascular lesions. NGAL is expressed in renal tubular epithelial cells, and increased NGAL levels are evidence of tubular injury [15,16]. In our study, lesions in diabetic kidneys manifest as proximal tubular growth, resulting from an increase in transport capacity due to diabetes mellitus [17]. Artemether ameliorated diabetic tubular injury, including decreasing NGAL levels and proximal tubular hypertrophy, which may be associated with its regulation of mitochondrial function.
Levels of mitochondrial components OPA1, COX IV, and TOM20 in diabetic and normal kidneys were not changed, implying that mitochondrial number was unaltered in the kidneys in early stages of T1D.
AMPK plays a key role in the regulation of cellular energy homeostasis. Our results show that AMPK is activated in the diabetic kidney, indicating relative low ATP production. Mitochondria are important ATP production organelles and are abundant in renal tubules. We thus speculated that mitochondrial dysfunction occurred in tubules of diabetic animals. Indeed, the TEM results provided substantial evidence that mitochondrial damage was associated with a loss of mitochondrial cristae in the proximal tubular epithelial cells of diabetic mice. The enzyme complexes of the oxidative respiratory chain are localized in the mitochondrial cristae, and the loss of the cristae leads to lower ATP production. Previous studies demonstrated that artemether could improve kidney disease in mice with STZ-induced diabetes and in db/db mice [9,10]. Our results suggest that artemether reverses AMPK activation and maintains mitochondrial structures, which may be renoprotective mechanisms. Actually, artemether was shown to ameliorate kidney injury by restoring redox imbalance and improving mitochondrial function in adriamycin-induced nephropathy in mice [11].
The kidney is also an important organ for iron filtration and reabsorption [18]. In recent years, studies have shown that lipid peroxidation induced by iron overload plays an important role in the occurrence and development of acute kidney injury [19] and chronic kidney disease [20]. DKD is more susceptible to iron homeostasis due to hyperglycemia, hyperlipidemia, and other systemic diseases. An iron-restricted diet was shown to improve mitochondrial oxidative stress and dysfunction in rats with STZ-induced diabetes by restoring mitochondrial respiration and respiratory complex activities [21]. Our previous study showed that artemether was effective in ameliorating diabetic nephropathy [9]. Therefore, we constructed a mouse model of T1D to investigate whether an imbalance of iron homeostasis accompanies diabetic nephropathy and whether the amelioration of diabetic nephropathy by artemether is related to changes in iron metabolism.
Ferritin is a marker of elevated iron stores. Our results showed that the protein and mRNA levels of FTH and FTL were significantly increased in kidney tissues of mice with T1D and were significantly decreased after artemether intervention. In addition, IHC staining showed that FTH and FTL were mainly expressed in renal tubules, suggesting iron overload and deposition were localized to the renal tubules in mice with T1D. Artemether was shown to reduce the extent of iron deposition. We then further investigated the regulation of cellular iron transport and found that protein and mRNA levels of TfR and DMT1 were significantly lowered, while ferroportin mRNA levels were slightly increased in mice with TID, suggesting that in cases of iron overload, there was a compensatory mechanism in the kidneys of mice with T1D that reduced renal tubular cell iron uptake and increased iron export. Next, we investigated defense mechanisms against iron damage.
The system xc --GSH-GPX4 pathway is one of the major compensatory mechanisms for lipid peroxidation induced by iron overload [22]. xCT is one of two subunits in system xc - that is reportedly downregulated in diabetes. For example, xCT levels are decreased in the retinas of rats with STZ-induced diabetes [23]. In addition, a decrease in xCT was observed in the aortic endothelium of db/db mice [24]. Interestingly, our results showed that the protein and mRNA levels of xCT decreased significantly in the kidneys of mice with T1D. After artemether intervention, these xCT levels were increased, suggesting that artemether might enhance xCT and GPX4 expression, thereby attenuating renal tubular injury induced by iron overload.
In conclusion, the protective effects of artemether on the kidneys of mice with T1D may be related to the regulation of iron metabolism and resolution of mitochondrial damage in proximal renal tubular cells.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (82004156), Shenzhen Science and Technology Project (JCYJ20190812183603627), Shenzhen Fund for Guangdong Provincial High Level Clinical Key Specialties.
Disclosure of conflict of interest
None.
References
- 1.DeFronzo RA, Reeves WB, Awad AS. Pathophysiology of diabetic kidney disease: impact of SGLT2 inhibitors. Nat Rev Nephrol. 2021;17:319–334. doi: 10.1038/s41581-021-00393-8. [DOI] [PubMed] [Google Scholar]
- 2.Chang J, Yan J, Li X, Liu N, Zheng R, Zhong Y. Update on the mechanisms of tubular cell injury in diabetic kidney disease. Front Med (Lausanne) 2021;8:661076. doi: 10.3389/fmed.2021.661076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu BC, Tang TT, Lv LL, Lan HY. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 2018;93:568–579. doi: 10.1016/j.kint.2017.09.033. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad AA, Draves SO, Rosca M. Mitochondria in diabetic kidney disease. Cells. 2021;10:2945. doi: 10.3390/cells10112945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Swelm RPL, Wetzels JFM, Swinkels DW. The multifaceted role of iron in renal health and disease. Nat Rev Nephrol. 2019;16:77–98. doi: 10.1038/s41581-019-0197-5. [DOI] [PubMed] [Google Scholar]
- 6.Upadhyay M, Agarwal S. Ironing the mitochondria: relevance to its dynamics. Mitochondrion. 2020;50:82–87. doi: 10.1016/j.mito.2019.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Fang X, Zheng W, Zhou J, Song Z, Xu M, Min J, Wang F. Genetic support of a causal relationship between iron status and type 2 diabetes: a Mendelian randomization study. J Clin Endocrinol Metab. 2021;106:e4641–e4651. doi: 10.1210/clinem/dgab454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma N, Zhang Z, Liao F, Jiang T, Tu Y. The birth of artemisinin. Pharmacol Ther. 2020;216:107658. doi: 10.1016/j.pharmthera.2020.107658. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Han P, Wang M, Weng W, Zhan H, Yu X, Yuan C, Shao M, Sun H. Artemether improves type 1 diabetic kidney disease by regulating mitochondrial function. Am J Transl Res. 2019;11:3879–3889. [PMC free article] [PubMed] [Google Scholar]
- 10.Han P, Wang Y, Zhan H, Weng W, Yu X, Ge N, Wang W, Song G, Yi T, Li S, Shao M, Sun H. Artemether ameliorates type 2 diabetic kidney disease by increasing mitochondrial pyruvate carrier content in db/db mice. Am J Transl Res. 2019;11:1389–1402. [PMC free article] [PubMed] [Google Scholar]
- 11.Han P, Cai Y, Wang Y, Weng W, Chen Y, Wang M, Zhan H, Yu X, Wang T, Shao M, Sun H. Artemether ameliorates kidney injury by restoring redox imbalance and improving mitochondrial function in Adriamycin nephropathy in mice. Sci Rep. 2021;11:1266. doi: 10.1038/s41598-020-80298-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiwari MK, Chaudhary S. Artemisinin-derived antimalarial endoperoxides from bench-side to bed-side: chronological advancements and future challenges. Med Res Rev. 2020;40:1220–1275. doi: 10.1002/med.21657. [DOI] [PubMed] [Google Scholar]
- 13.Sun H, Wang W, Han P, Shao M, Song G, Du H, Yi T, Li S. Astragaloside IV ameliorates renal injury in db/db mice. Sci Rep. 2016;6:32545. doi: 10.1038/srep32545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1009–1022. doi: 10.1152/ajpregu.00809.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satirapoj B. Tubulointerstitial biomarkers for diabetic nephropathy. J Diabetes Res. 2018;2018:2852398. doi: 10.1155/2018/2852398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sauriasari R, Safitri DD, Azmi NU. Current updates on protein as biomarkers for diabetic kidney disease: a systematic review. Ther Adv Endocrinol Metab. 2021;12:20420188211049612. doi: 10.1177/20420188211049612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vallon V, Thomson SC. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat Rev Nephrol. 2020;16:317–336. doi: 10.1038/s41581-020-0256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Raaij SEG, Rennings AJ, Biemond BJ, Schols SEM, Wiegerinck ETG, Roelofs HMJ, Hoorn EJ, Walsh SB, Nijenhuis T, Swinkels DW, van Swelm RPL. Iron handling by the human kidney: glomerular filtration and tubular reabsorption both contribute to urinary iron excretion. Am J Physiol Renal Physiol. 2019;316:F606–F614. doi: 10.1152/ajprenal.00425.2018. [DOI] [PubMed] [Google Scholar]
- 19.Scindia PhD Y, Leeds Md J, Swaminathan Md S. Iron homeostasis in healthy kidney and its role in acute kidney injury. Semin Nephrol. 2019;39:76–84. doi: 10.1016/j.semnephrol.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Martines AMF, Masereeuw R, Tjalsma H, Hoenderop JG, Wetzels JFM, Swinkels DW. Iron metabolism in the pathogenesis of iron-induced kidney injury. Nature Reviews Nephrology. 2013;9:385–398. doi: 10.1038/nrneph.2013.98. [DOI] [PubMed] [Google Scholar]
- 21.Pena-Montes DJ, Huerta-Cervantes M, Rios-Silva M, Trujillo X, Cortes-Rojo C, Huerta M, Saavedra-Molina A. Effects of dietary iron restriction on kidney mitochondria function and oxidative stress in streptozotocin-diabetic rats. Mitochondrion. 2020;54:41–48. doi: 10.1016/j.mito.2020.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–282. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carpi-Santos R, Calaza KC. Alterations in system xc(-) expression in the retina of type 1 diabetic rats and the role of Nrf2. Mol Neurobiol. 2018;55:7941–7948. doi: 10.1007/s12035-018-0961-8. [DOI] [PubMed] [Google Scholar]
- 24.Luo EF, Li HX, Qin YH, Qiao Y, Yan GL, Yao YY, Li LQ, Hou JT, Tang CC, Wang D. Role of ferroptosis in the process of diabetes-induced endothelial dysfunction. World J Diabetes. 2021;12:124–137. doi: 10.4239/wjd.v12.i2.124. [DOI] [PMC free article] [PubMed] [Google Scholar]