
Figure 6.

Proposed model in which UA activates mammalian target of rapamycin complex 1 (mTORC1). In several cell models, high UA concentration causes an increase in AKT activity and/or a decrease in AMPK activity causing a potentiation of the regulatory pathway leading to mTORC1 activation. Activated AKT phosphorylates tuberous sclerosis complex 2 (TSC2) leading to the release of the dimer tuberous sclerosis complex 1/2 (TSC1/TSC2) from the mTORC1 complex. Since the dimer is an activator of the GTPase activity of RAS homologous enriched in brain (RHEB), this G-protein binds GTP for long time, thereby activating mTORC1. AKT also directly activates mTORC1 by phosphorylating the mTOR inhibitor proline-rich AKT substrate of 40 kDa (PRAS40), which is then released from the complex. On the contrary, phosphorylation of TSC2 by AMPK increases the concentration of the dimer TSC1/TSC2, thus favoring the GTPase activity of RHEB and therefore inactivating mTORC1. The inhibitory activity of AMPK is also exerted by phosphorylation and consequent release of the mTOR activator regulatory associated protein of mTOR (RAPTOR). Therefore, activation of AKT and inhibition of AMPK cause an increase in mTORC1 activity leading to an increase in the rate of synthesis of proteins, lipids, and nucleotides, while autophagy is inhibited. mTORC1 activation also promotes mitochondria proliferation, producing an increase in intracellular ROS, responsible for an increase in cytokines and chemokines production, leading to inflammation.  : activation;
: activation;  : inhibition;
: inhibition;  : increase;
: increase; 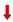 : decrease.
: decrease.