

SUMMARY
Fungal microorganisms (mycobiota) comprise a small but immunoreactive component of the human microbiome, yet little is known about their role in human cancers. Pan-cancer analysis of multiple body sites revealed tumor-specific mycobiomes at up to 1 fungal per 104 tumor cells. In lung cancer, Blastomyces was associated with tumor tissues. In stomach cancers, high rates of Candida were linked to the expression of proinflammatory immune pathways, while in colon cancers Candida was predictive of metastatic disease and attenuated cellular adhesions. Across multiple GI sites, live Candida species were enriched in tumor samples and tumor-associated Candida DNA was predictive of decreased survival. The presence of Candida in human GI tumors was confirmed by external ITS sequencing of tumor samples and by culture-dependent analysis in an independent cohort. These data implicate the mycobiota in the pathogenesis of GI cancers and suggest that tumor-associated fungal DNA may serve as diagnostic or prognostic biomarkers.
Keywords: Cancer, tumor-associated fungi, mycobiome, Candida, Blastomyces, Malassezia, lung cancer, stomach cancer, colon cancer
Graphical Abstract
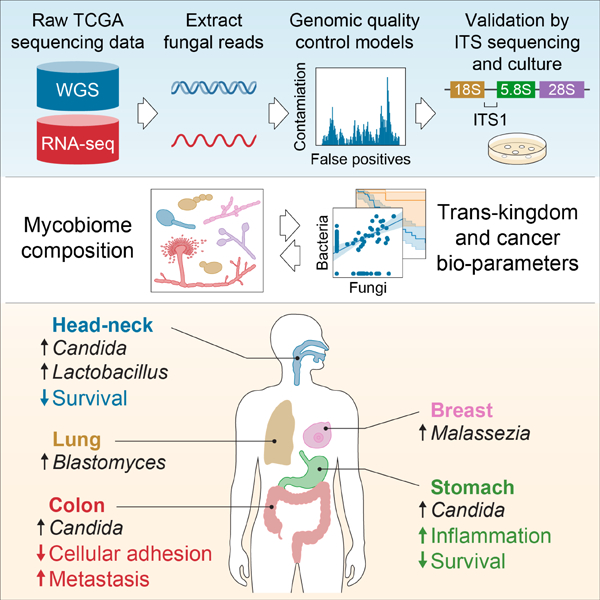
INTRODUCTION
Cancer is among the leading causes of death worldwide. Host-bacterial immune interactions profoundly influence tumorigenesis, cancer progression, and response to therapy (Davar et al., 2021; Dzutsev et al., 2017; Finlay et al., 2020; Garrett, 2019; Grivennikov et al., 2010; Iida et al., 2013; Routy et al., 2018; Sharma et al., 2017; Shiao et al., 2021; Spencer et al., 2021; Tanoue et al., 2019). Nevertheless, the role of fungi (mycobiota) in these processes remains largely unexplored, missing a potential avenue for developing novel diagnostic and preventative strategies. Fungi and bacteria co-colonize the mammalian GI tract, skin epithelium, respiratory tract, and reproductive organs, forming a complex ecosystem of microbe-microbe and host-microbe interactions with significant implications for human health ( Findley et al., 2013; Hoarau et al., 2016; Leonardi et al., 2020; Doron et al., 2021; Lewis et al., 2015; Liguori et al., 2016; Sokol et al., 2017; Tipton et al., 2018; Zhai et al., 2020; Zuo et al., 2018). Despite comprising around just 0.1% of the microbial DNA present in the gut (Qin et al., 2010), fungal infections are responsible for more than 1.5 million global deaths per year (Brown et al., 2012) and species from this kingdom have a disproportionate influence on the overall microbiome and host immunity (Huffnagle and Noverr, 2013).
A growing body of evidence links the human microbiome to cancer and cancer outcomes, including viruses, bacteria, and fungi (Helmink et al., 2019; Vogtmann and Goedert, 2016). Recent years have seen several bacterial species linked to cancer development and progression, including overall survival (Dohlman et al., 2020; Sepich-Poore et al., 2021). Helicobacter pylori is responsible for approximately 75% of attributable risk for gastric cancer (Polk and Peek, 2010), while in the lower GI tract, genotoxic Escherichia coli, Bacteroides fragilis, Streptococcus bovis/gallolyticus and Fusobacterium nucleatum have been implicated in the pathogenesis of colorectal cancer (Sepich-Poore et al., 2021). Common among these cancer-associated bacteria is their ability to modulate host immunity and provoke chronic inflammation, features which are proposed to contribute to their tumorigenic capacity. Recent reports have also suggested that bacterial DNA circulates in the blood of cancer patients and may serve as a predictive biomarker (Poore et al., 2020; Dohlman et al., 2020), while intracellular bacteria have been identified in numerous tumor types (Nejman et al., 2020). Nevertheless, conclusive links between the fungal microbiome and cancer remain elusive.
The mycobiome plays a key role in activation of innate, Type 17 and B-cell mediated immunity in the gut during health and disease. Fungal toxins and bioactivated amines have been linked to carcinogenesis (Chang et al., 1992; Yang, 1980), while trans-kingdom features have been recently linked with colorectal cancers across cohorts (Coker et al., 2018; Liu et al., 2022). Recent experimental studies support fungal involvement in cancers under specific contexts (Alam et al., 2022; Malik et al., 2018; Shiao et al., 2021; Wang et al., 2018). Previously, we demonstrated that next-generation sequencing (NGS) data of tumors from The Cancer Genome Atlas (TCGA) contained high rates of microbial sequencing reads (Dohlman et al., 2020) which can be leveraged to characterize the intratumoral metagenome and understand host-microbe interactions. However, the fungal composition of TCGA sequencing data has remained unexamined.
Analyzing multiple cancer types from TCGA, we extracted profiles of tumor-associated mycobiomes with species-level resolution. We then analyzed the distribution of reads aligning to these fungal genomes to thoroughly screen for contamination and false-positive signals. After removing such taxa, we found that fungal compositions varied by cancer type, with GI sites and non-GI sites each harboring disease-specific fungi. Overall, we found up to 1 fungal cell per 104 human tumor cells, a rate consistent with (1) fungi representing 0.1–1% of the microbiome (Sender et al., 2016), and (2) estimates that bacteria comprise just below 1% of the cells found in tumors (Nejman et al., 2020; Sepich-Poore et al., 2021).
Across GI samples, we find that several Candida species, Saccharomyces cerevisiae, and Cyberlindnera jadinii are highly abundant in GI tumor mycobiome communities, while Blastomyces and Malassezia species are abundant in lung and breast tumors respectively. We demonstrate that Candida is living and transcriptionally active at the tumor site and predictive of host tumor gene expression, disease state, and survival. Taken together, these results not only implicate Candida spp. in the pathogenesis of GI cancers, but also indicate its potential as a therapeutic target and prognostic tool.
Finally, we provide the normalized, decontaminated mycobiota profiles we uncovered from TCGA sequencing data to the research community. This curated dataset consists of fungal community profiles from 883 sequencing runs on 767 primary tumor samples from a total of 671 individuals and is accompanied by detailed histological and clinical annotations, including tumor stage and patient survival.
RESULTS
Fungal DNA is abundant in GI tumor samples from TCGA
To explore tumor-associated mycobiomes across different cancers we employed a metagenomic analysis of whole-genome sequencing (WGS) data from multiple tumor samples across different cancers available in TCGA. We selected cancer types based on previously reported presence of mycobiota, including GI tissues (head-neck/HNSC, n = 338; esophagus/ESCA, n = 142; stomach/STAD, n = 321; colon/COAD, n = 300; rectum/READ, n = 127), non-GI external sites (breast/BRCA, n = 229), as well as non-GI internal sites (lung/LUSC, n = 100; brain/LGG, n = 183), and used PathSeq (Walker et al., 2018) to determine their fungal composition. The mycobiomes detected in these tissues were then screened for contamination and false-positive signals (See “Identification and removal of contaminant fungi and false-positive signals”).
This approach led to the detection of fungal sequences across multiple cancer patient’s tissue types, with higher rates of fungal DNA in tissues of the lung and specific sites of the GI tract (Figure 1A). As the brain is canonically described as a sterile organ (fungal brain infections are usually lethal) and few fungal sequences were detected in brain tissue (LGG, Figure 1A), we reasoned that such microbial reads likely represented biological contamination and/or false-positive signals, suggesting it can be used as a presumptive “negative control” for identifying spurious signals in other sample types. Across the GI tract, fungal DNA was particularly abundant in tissues from head-neck (HNSC), colorectal (COAD and READ) and stomach (STAD) tissues, and less abundant in the esophagus (ESCA) (Figure 1A, Figure S1A). Samples from lower GI tissues harbored a greater density of fungi than upper GI tissues did, in a pattern consistent with bacteria (Figure 1B). As expected, fungal sequences represented a much smaller proportion of microbial sequences in tissues when compared to bacterial DNA (Figure 1B), consistent with previous reports of intestinal human samples (Coker et al., 2018; Hoarau et al., 2016; Leonardi et al., 2022; Liguori et al., 2016; Liu et al., 2022; Nash et al., 2017; Proctor et al., 2021; Sokol et al., 2017).
Figure 1. Fungal DNA is present in multiple cancer types not explained by contamination, See also Figure S1.
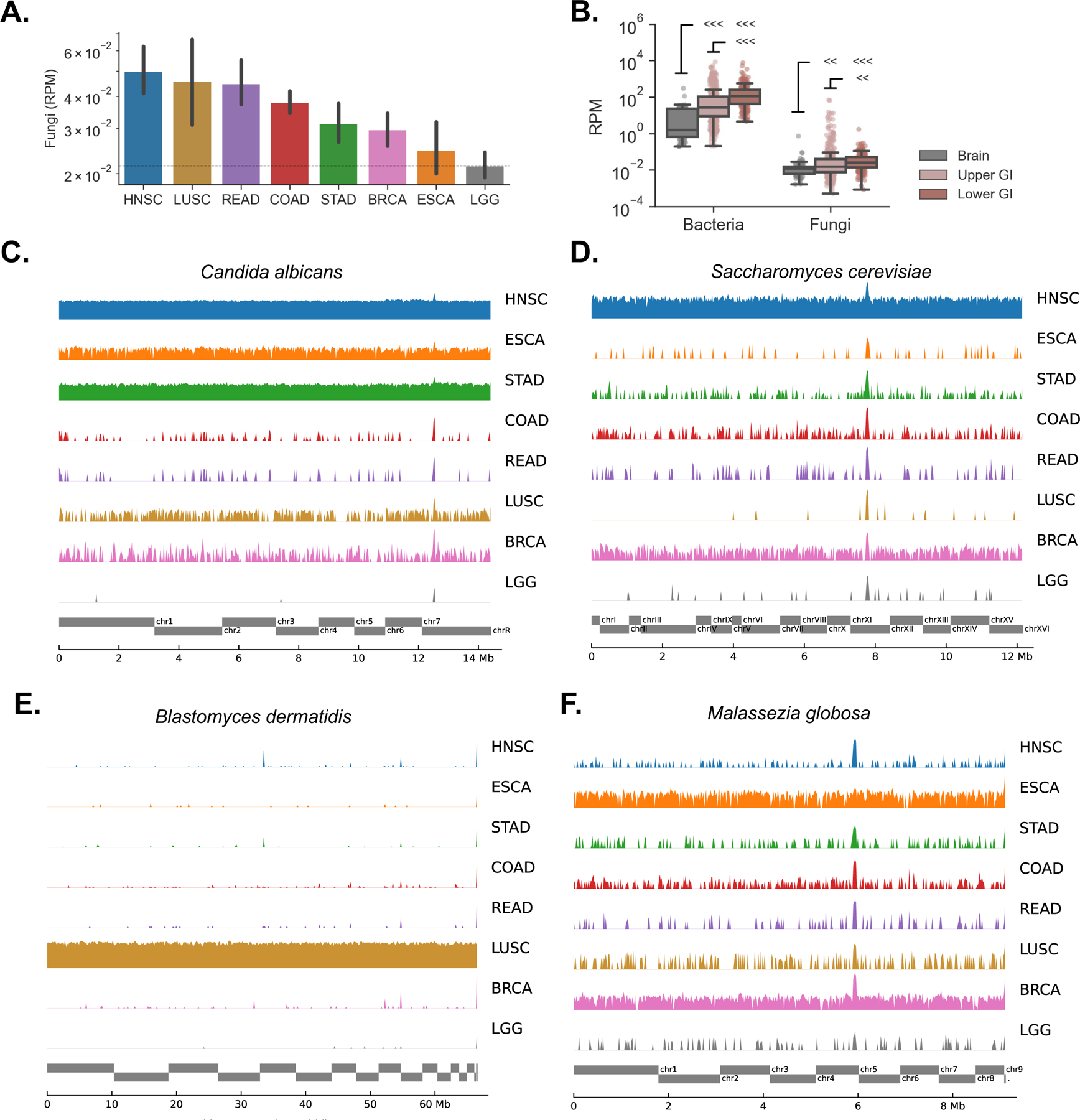
(A) Geometric mean of reads per million (RPM) of fungal DNA detected in tumor and tumor-associated tissue samples from head-neck (HNSC), lung (LUSC), rectum (READ), colon (COAD), stomach (STAD), breast (BRCA), esophageal (ESCA) and brain (LGG) cancers.
(B) Both bacterial and fungal reads were more abundant in the lower GI tract (COAD, READ) than the upper GI tract (HNSC, ESCA, STAD), and were more abundant in both GI groups compared to the brain (LGG) that was used here as a negative control.
(C – D)Genome alignments to C. albicans (C) and S. cerevisiae (D) are largely absent in brain but present at high rates across other tumor types, especially upper GI.
(E) Genome alignments to B. dermatidis are found at high rates in lung tumors, but not elsewhere.
(F) The distribution of sequencing reads aligning to M. globosa displays similar depth across sequencing projects including brain. Reads are distributed randomly, a signature of biological contamination.
Identification and removal of contaminant fungi and false-positive signals
Contamination is a plausible source of fungal DNA in metagenomic profiling experiments (Davis et al., 2018), particularly in studies of low biomass tissue sites (Glassing et al., 2016). Additionally, incorrect assignment of microbial or non-microbial sequencing reads can lead to reporting of spurious signals (Ye et al., 2019). To ensure accurate capture of the mycobiome of these samples, we first applied a prevalence-based decontamination model to identify and remove (1) fungal species and genera whose presence was associated with specific sequencing batches and could not be explained by biological variation, and (2) samples from multi-well sequencing plates with strong evidence of contamination (See Methods). This analysis identified 23 species and 12 genera meeting these criteria (Table S1). Additionally, we removed 18 samples from a single sequencing plate which displayed evidence of significant fungal contamination (Figure S1B).
While tracking the presence of taxa across sequencing batches can effectively identify contaminants, such a strategy is unable to identify contamination events that span sequencing batches, nor is it capable of identifying signals which may be the result of false-positive alignments. To address these possibilities, we performed a genome-wide analysis of sequence alignments for the fungal species detected in each tumor type (See Methods, Table S1). For each cancer type, we compared the genome coverage depth (“Vertical QC model”) as well as the distribution of sequencing reads across the length of each genome (“Horizontal QC model”). The use of orthogonal models in this case allows for the identification of different categories of false-positive signals. Species truly present at the time of sequencing but not in the original biopsies are referred to as biological contaminants and are likely to have similar levels of coverage depth across tissue types and a random distribution of read alignments across the span of their genome. Conversely, false-positive alignments are likely to occur at conserved or highly mobile genes from other fungal or non-fungal genomes, generating similar patterns of sequence alignments across tissue types.
For example, these analyses found that reads aligning to specific Malassezia genomes displayed similar coverage depth across sequencing projects but a horizontal read distribution that was generally random (Figure 1F, Figure S1C). Malassezia spp. are frequently found on the skin surface (Findley et al., 2013; Saheb Kashaf et al., 2022) and were likely transferred to samples during handling. Meanwhile, reads aligning to the genome of Agaricus bisporus (common mushroom or portabello) displayed a consistent horizontal distribution pattern across sequencing projects (Figure S1D). Thus, Malassezia restricta and Agaricus bisporus were respectively removed by our vertical and horizontal QC models (Table S1).
Overall, our decontamination and QC analyses resulted in the removal of 97.27% of species detected in GI tumors, 99.26% of species detected in lung tumors, and 95.53% of species detected in breast tumors. Remaining were a set of commensal and pathogenic fungi, including Candida albicans (Figure 1C), C. tropicalis, C. dubliniensis, C. glabrata, C. lusitaniae, C. guilliermondii and food-associated Saccharomyces cerevisiae (Figure 1D), Cyberlindnera jadinii, and Pichia membranifaciens which were abundant in GI tumors and Blastomyces dermitidis/gilchristii (Figure 1E) which are abundant in lung tumors and causative agents of blastomycosis, a disease that primarily affects the lungs (Brown et al., 2013). Many of the species classified as contaminants and/or false-positive signals were not known to colonize humans, including plant pathogens Alternaria alternata and Bipolaris oryzae (Table S1), while Malassezia spp. were classified as probable contaminants in all tumor types except for breast tissue (Figure 1F, Table S1), suggesting that reads from Malassezia spp. may have originated from both endogenous and contaminant sources as we have previously shown for E. coli in CRC samples (Dohlman et al., 2020). Finally, we validated the abundance of several of these species with a secondary metagenomic analysis using TaxaTarget (Commichaux et al., 2021), a tool specifically designed for the detection of eukaryotic marker genes (Figure S1E).
TCGA tissue samples are composed of disease-specific fungi
Our approach generated species-level resolution data allowing the identification of specific fungi across various tumor types. Principal coordinate analysis (PCoA) and hierarchical clustering of species abundances across TCGA cancer types revealed that head-neck, colon, and rectal tumors had highly similar fungal compositions, as did stomach and esophageal tumors, while the fungal composition of non-GI tumors were largely distinct (Figure 2A-B). Differences in the fungal communities we observed across GI sites could be affected by variations in pH, oxygen availability, or bacterial biogeography across the GI tract, among a few key factors driving microbial variation. In addition to environmental factors, the detection of fungal species in these samples is affected by the availability of reference genomes, meaning there may be additional unknown fungal species not detected by our analysis.
Figure 2. Primary tumor samples harbor disease-specific mycobiomes, See also Figure S2.
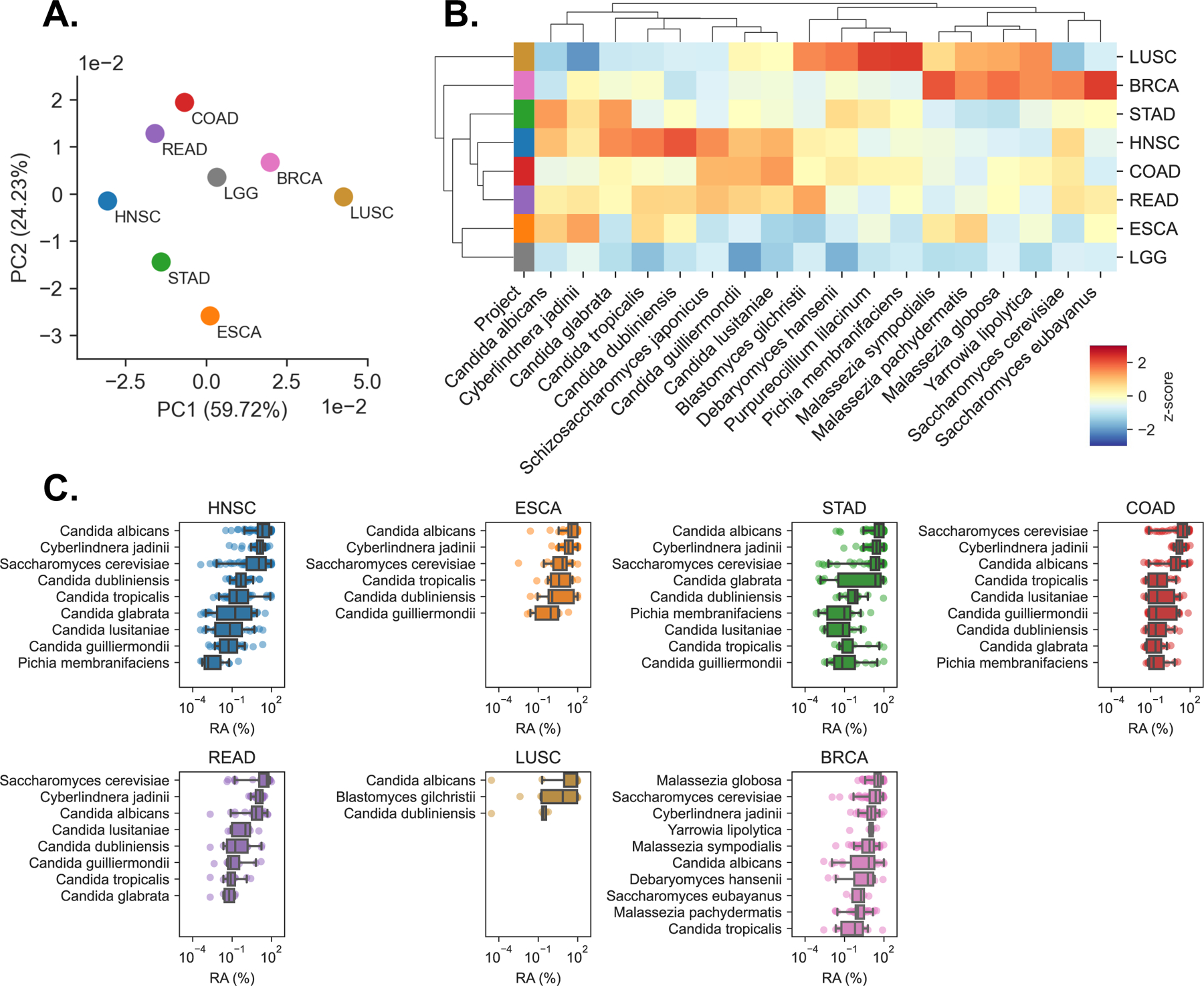
(A) Principal coordinate analysis (PCoA) of normalized species abundances from head-neck (HNSC), esophageal (ESCA), stomach (STAD), colon (COAD), rectal (READ), lung (LUSC), breast (BRCA), and brain (LGG) reveal clustering by tumor type, after filtering contaminants and false-positive signals.
(B) Clustered heatmap showing difference in relative fungal species abundances (RPM) between tissues from each TCGA cancer type, after filtering. Species are included if classified as tissue-associated in any of GI, lung, or breast samples, even if they were classified as contaminants in others. Heatmap values are z-scored by species abundance to highlight tissue-specific differences.
(C) Boxplots showing distribution of relative abundances (RA) from the 10 or fewer most abundant species detected in each cancer type, after removing low-prevalence and contaminant species.
We found that tumor-associated fungal communities were characterized by high abundance and prevalence of Saccharomycetales, consistent with previous gut mycobiome studies relying on metagenomics, culture-dependent analyses, and ITS-amplicon sequencing (Hoarau et al., 2016; Leonardi et al., 2020; Li et al., 2022; Liguori et al., 2016; Nash et al., 2017; Proctor et al., 2021; Sokol et al., 2017). In addition to these more common fungi, deeper analysis revealed the presence of sequences from multiple fungal species and genera as well as their distribution across different cancer types (Figure 2C, Figure S2A, Table S2).
The growing consensus on the importance of intestinal mycobiota has prompted the investigation of (1) which fungi are capable of surviving, residing, and replicating in the GI tract (fungal symbionts or commensals) to influence the host over a prolonged period, and (2) which are transient passengers, contaminants, or represent environmental fungi (non-commensal fungi) that can impact immunosuppressed individuals (Fiers et al., 2019). Candida spp. were more abundant across the GI tract as compared to other body sites, consistent with their known commensal status in this part of the body and ability to expand during disease (Figure 2C, Figure S2A) (Aggor et al., 2020; Break et al., 2021; Fan et al., 2015; Hoarau et al., 2016; Kumamoto et al., 2020; Leonardi et al., 2020; Li et al., 2022; Liguori et al., 2016; Sokol et al., 2017; Zhai et al., 2020). Species-level analysis determined that C. albicans was the most abundant representative of the Candida genus; C. albicans was highly abundant in multiple cancers and particularly abundant in cancers of the GI tract (Figure 2B-C), consistent with previous studies. Species C. tropicalis, C. dubliniensis, C. glabrata, C. lusitaniae, C. guilliermondii, C. parapsilosis, and P. membranifaciens were also present, but at lower abundance and prevalence across samples (Figure 2B-C, Figure S2A). Saccharomyces spp. were primarily represented by S. cerevisiae. Among fungi broadly assigned as non-commensal, we also detected C. jadinii in multiple GI tissues, a species that rarely infects people and is found in processed food products, presumably arriving via diet. Lung tissues carried B. dermitidis/gilchristii. Interestingly, we detected evidence of Blastomyces DNA in 6 out of 50 patients with squamous cell lung carcinomas. In the general population, the incidence of blastomycosis is 1–2 cases per 100,000 (Benedict et al., 2012). Together, these findings indicated the presence of biologically meaningful associations linking the presence of fungal DNA to tissues from specific body sites.
Emergence of Candida and Saccharomyces co-abundance groups is associated with GI cancers
Microbiota participate in a complex web of interspecies ecological interactions and the dynamics of these interaction networks can profoundly influence human health (Dohlman and Shen, 2019; Faust and Raes, 2012). To explore the potential presence of fungal interaction networks and co-abundant taxa, we applied a bootstrapping procedure SparCC (Friedman and Alm, 2012) and found that C. albicans and S. cerevisiae were each at the center of two anticorrelated co-abundance clusters across GI cancer types (Figure 3A). The co-abundance group associated with C. albicans included C. dubliniensis, C. tropicalis, and C. guilliermondii, while the group associated with S. cerevisiae was comprised of taxa including S. eubayanus, C. jadinii, P. membranifaciens, as well as C. parapsilosis and C. glabrata. Additionally, we found that these two co-abundance clusters were predictive of host gene expression across head-neck, stomach, and colon cancers (Figure 3B-D). These findings suggested that cancers of the GI tract may segregate into Candida- and Saccharomyces-associated tumors. Notably, many of the species in each of these clusters are taxonomically related, thus the degree to which they are driven by biological or phylogenetic factors (or both) warrants further exploration.
Figure 3. Trans-kingdom analysis reveals Candida- and Saccharomyces-associated GI cancer coabundance groups, See also Figure S3.
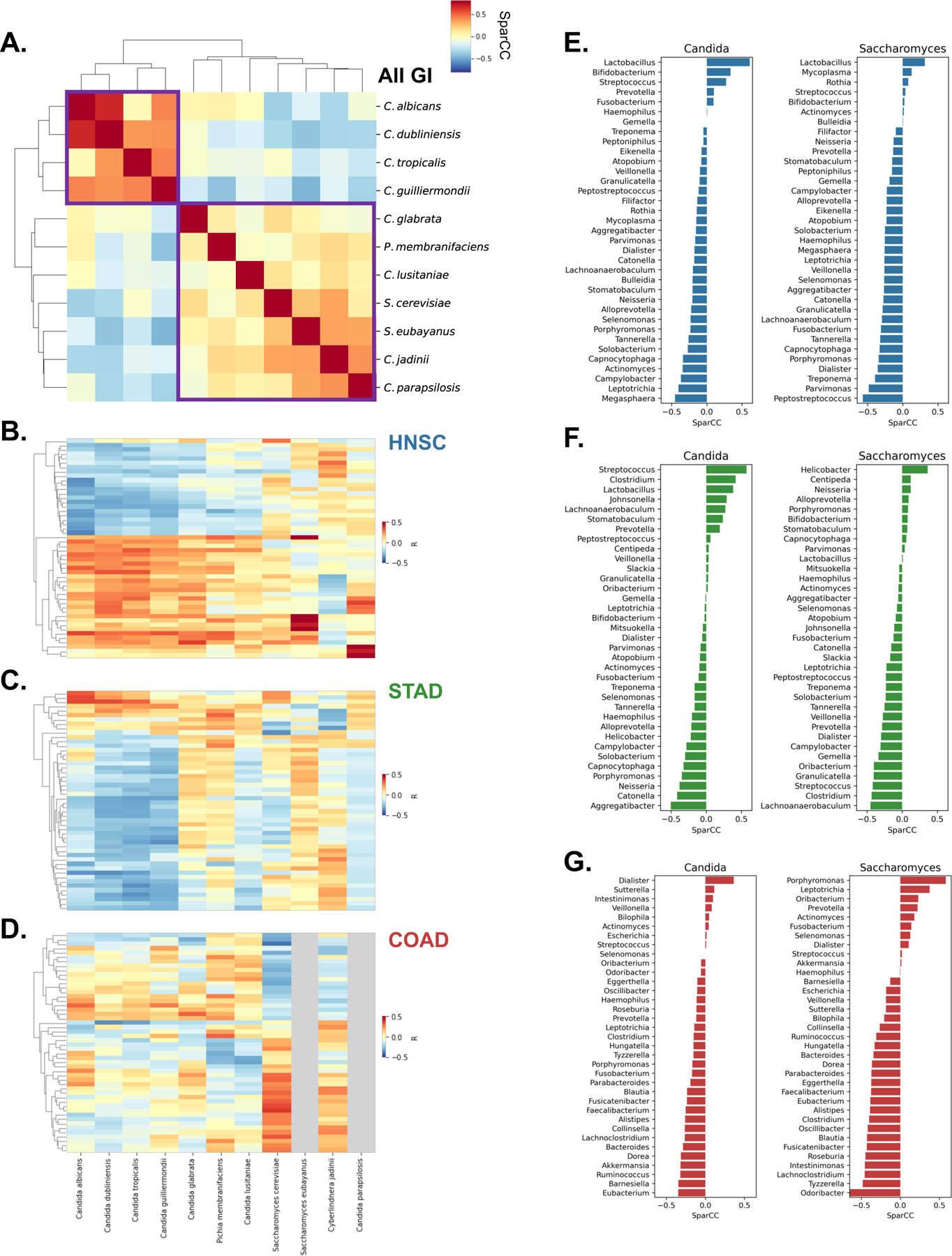
(A) Clustered heatmap showing SparCC co-abundance among fungal species reveals species associated with C. albicans and S. cerevisiae (purple boxes).
(B – D) Clustered heatmaps showing gene expression patterns in head-neck (HNSC; B), stomach (STAD; C), and colon (COAD; D) cancers. Heatmaps are clustered by row, while column clustering is determined by (A). Gray columns indicate species not detected in certain cancer types
(E - G) SparCC co-abundance between Candida and Saccharomyces and bacterial genera found in matched tumor samples from TCMA, across head-neck (HNSC; E), stomach (STAD; F), and colon (COAD; G) cancers.
Trans-kingdom analysis reveals co-abundance groups associated with Candida and Saccharomyces in GI cancers
To further explore the microbial communities associated with the Candida and Saccharomyces tumor co-abundance clusters and their relevance to disease, we examined bacterial populations associated with Candida and Saccharomyces and applied the same correlation approach to identify associations among GI tumor-associated fungi and matched, decontaminated, tumor-associated bacterial communities from The Cancer Microbiome Atlas (TCMA) (Dohlman et al., 2020). This analysis identified several interesting bacterial subpopulations that were correlated with Candida and Saccharomyces in each cancer type.
In head-neck tumors, Candida and Saccharomyces were associated with similar bacteria (Figure 3E, Figure S3A). Lactobacillus spp. and especially Lactobacillus gasseri were frequently found in the presence of Candida and, to a lesser extent, Saccharomyces (Figure S3D-F). This observation is consistent with reports that Lactobacillus spp. interact extensively with Candida to influence its pathogenicity (Ballou et al., 2016; MacAlpine et al., 2021; Zeise et al., 2021). Bifidobacterium, which is known to support intestinal barrier function (Ewaschuk et al., 2008) was also positively associated with Candida in head-neck cancers. In stomach tumors, we also observed that Candida was strongly associated with Lactobacillus (Figure 3F, Figure S3B,E). However unlike in head-neck cancer, Candida and Saccharomyces in stomach tumors were largely associated with dissimilar clusters of bacteria. Most notably, we observed that Candida-associated tumors were less likely to harbor Helicobacter pylori, while Saccharomyces was more likely to be found alongside H. pylori. A similar pattern was identified for the genera Streptococcus and Clostridium, which were positively associated with Candida and negatively associated with Saccharomyces. In lower GI tumors, Candida and Saccharomyces were also co-abundant with distinct bacterial populations (Figure 3G, Figure S3C). Unlike upper GI cancers, we did not observe any association between L. gasseri and Candida in colon tumors (Figure S3F). However, we found that among colon cancers, Candida was positively associated with Dialister, and was negatively associated with Ruminococcus, Akkermansia municiphila, and Barnesiella intestinihominis (Figure 3G, Figure S3C) , some of which known to promote beneficial host-microbe interactions. Interestingly, the presence of Candida and Saccharomyces were also associated with differing species of Fusobacterium spp. in colon cancer (Figure S3C). In addition to providing insight into tumor-associated microbiomes, such trans-kingdom ecological interactions may be relevant for disease detection and potentially inform strategies for modulating tumor microbiomes for therapeutic benefit.
Candida and Saccharomyces are predictive of gene expression patterns in GI cancers
To better understand the effect of Candida and Saccharomyces co-abundance groups on GI cancers, we next sought to compare the rates of Candida and Saccharomyces across GI tumors. Across cancer types, we discovered that Candida-to-Saccharomyces ratios displayed striking bimodality, corroborating our previous observations of Candida and Saccharomyces co-abundance clusters and suggesting that GI tumors could be reliably organized into subgroups of Candida- and Saccharomyces-associated cancers (Figure 4A, Figure S4A). To understand the relevance of these two subgroups, we divided GI tumors into Candida-dominant (Ca-type) and Saccharomyces-dominant (Sa-type) clusters and compared them.
Figure 4. Candida is associated with late-stage and metastatic GI cancers, See also Figure S4.

(A) Kernel density estimation (KDE) of Candida-to-Saccharomyces ratios in head-neck (HNSC), stomach (STAD), and colon (COAD) cancers.
(B) Volcano plot showing genes differentially expressed in Candida-negative (blue) and Candida-high (red) tumor samples head-neck, stomach, and colon cancers.
(C) Boxplots depicting Candida-to-Saccharomyces ratios in early-stage (I-III) and late-stage (IV) for head-neck (HNSC), stomach (STAD), and colon (COAD) cancers.
(D) KDE analysis of Candida-to-Saccharomyces ratios in metastatic (orange) and non-metastatic (blue) tumor samples finds that Ca-type colon tumors are significantly more likely to be metastatic.
(E) Violin-plots showing Bray-Curtis distances between fungal species compositions of patient-matched tumor and blood samples (blue) and unmatched tumor and blood samples (orange).
To see if Ca-type and Sa-type tumors harbored functional differences, we used RNA-seq data from TCGA to analyze gene expression between tumor samples that were highly abundant in Candida or Saccharomyces with tumors in which these taxa were not detected (Figure 4B, Figure S4B). This analysis identified several interesting changes in gene expression that were associated with Candida status. In head-neck cancer, we found that tumor-suppressors TP53 and CDKN2A were expressed at lower rates in Ca-type tumors, along with fibronectin (FN1), a marker of epithelial-to-mesenchymal transition (EMT) in head-neck cancers. Interestingly, we also saw that IL22, IL24, CARD10, and CD44 were up-regulated in Ca-type tumors, but not Sa-type tumors. Gene-set enrichment analysis (GSEA) of this expression signature demonstrated that the presence of Candida was associated with decreased expression of genes relating to cell adhesion molecules (q < 0.001) in head-neck cancers. In stomach cancers, we found that genes related to cytokine interactions, host immunity, and inflammation were positively enriched in Ca-type tumors, including IL1A, IL1B, IL6, IL8, CXCL1, CXCL2, and IL17C. This pro-inflammatory immune signature is consistent with previous reports that C. albicans invokes IL-1β, neutrophils and Th17 cell infiltration in the gut (Li et al., 2022). By contrast, these genes were differentially expressed to a lesser degree or were not differentially expressed at all in Sa-type tumors. Genes down-regulated in Ca-type tumors included ALAD, FTL, IL17D, CST5, ELN, and TREM2. This gene expression pattern was associated with significant up-regulation of genes involved in cytosolic DNA sensing (q = 0.008), Toll-like receptor (q = 0.033) signaling, Nod-like receptor (q = 0.033) signaling, and cytokine-cytokine receptor interactions (q = 0.035). In colon cancers, we found that tumor suppressor genes and genes regulating cellular adhesion pathways were downregulated in Ca-type tumors, including PTK2B, CDKN2C, and NET1, while genes such as BMP15, PFN3, CCL27, PIP, and SAGE1 were up-regulated in Ca-type tumors. Moreover, GSEA identified significant down-regulation of genes involved in ECM-receptor interactions (q = 0.036) and focal adhesion (q = 0.101) pathways in Ca-type colon tumors. Thus, the presence of Candida in head-neck and colon tumors appears to be associated with pro-tumorigenic and cellular adhesion-related gene pathways, while Candida appears to be associated with a robust immune response in stomach tumors. However, additional analyses are needed to determine whether Candida plays a causative role in these gene expression changes or is merely responding to them.
A Candida-to-Saccharomyces ratio is associated with late-stage, metastatic colon cancer
The observation that Candida is associated with down-regulation of genes involved in cellular adhesion pathways and epithelial barrier function in head-neck and colon tumors led us to explore if ratios between these two genera were predictive of cancer outcomes. We found that Candida-to-Saccharomyces (C/S) ratios were generally low among early-stage colon cancers but were dramatically increased in stage IV disease (Figure 4C, Table S3). These ratios did not vary significantly by stage in head-neck, stomach, or other cancers (Figure 4C, Figure S4C). The association with late-stage colon cancer led us to examine rates of metastases among Ca-type and Sa-type tumors. Comparing Candida-to-Saccharomyces ratios in metastatic and non-metastatic groups, we found that Ca-type colon tumors were significantly more likely to be metastatic than tumors with higher rates of Saccharomyces (Figure 4D; p = 8.49E-3). Similar analyses did not find significant differences in other cancer types (Figure S4D). Thus, Candida-to-Saccharomyces ratios may capture a clinically relevant shift in tumor mycobiomes with potential prognostic value for colon cancer.
Our observation that tumor mycobiomes were predictive of metastatic colon cancer and deregulation of genes involved in epithelial barrier function led us to question if fungi or fungal DNA might transfer into the bloodstream from the barrier surfaces in which these fungi normally reside. To explore this possibility, we examined the composition of patient-matched tumor and blood samples from cancer types of the lower and upper GI tracts. We found statistically significant similarities in the composition of patient-matched tumor and blood samples from patients with upper GI cancers (p = 3.27E-2) and lower GI cancers (p = 3.72E-5) compared to unmatched samples (Figure 4E). The same was not true for other tumors, suggesting that the GI tract might be a possible entrance point for fungi or fungal DNA into the bloodstream. Together these data indicate that Candida may be linked to loss of gut epithelial barrier function, metastasis, and the translocation of fungal cell components from the GI tract into the bloodstream. However, whether Candida cells or other fungal DNA can consistently be detected in the blood of GI cancer patients requires additional examination.
Live, transcriptionally active Candida species are associated with GI tumors
To further examine the role of Candida, we next analyzed the distribution of fungi across the lower GI tract. Consistent with previous studies focused on fecal mycobiota (Chehoud et al., 2015; Hoarau et al., 2016; Leonardi et al., 2020; Sokol et al., 2017), the Ascomycota phylum was more prevalent in the ascending colon (Figure 5A, Figure S5). A targeted, species-level analysis determined that C. albicans is likely driving the abundance of Ascomycota in the ascending colon (Figure 5B).
Figure 5. Live, transcriptionally active Candida species are associated with GI tumors, See also Figure S5.
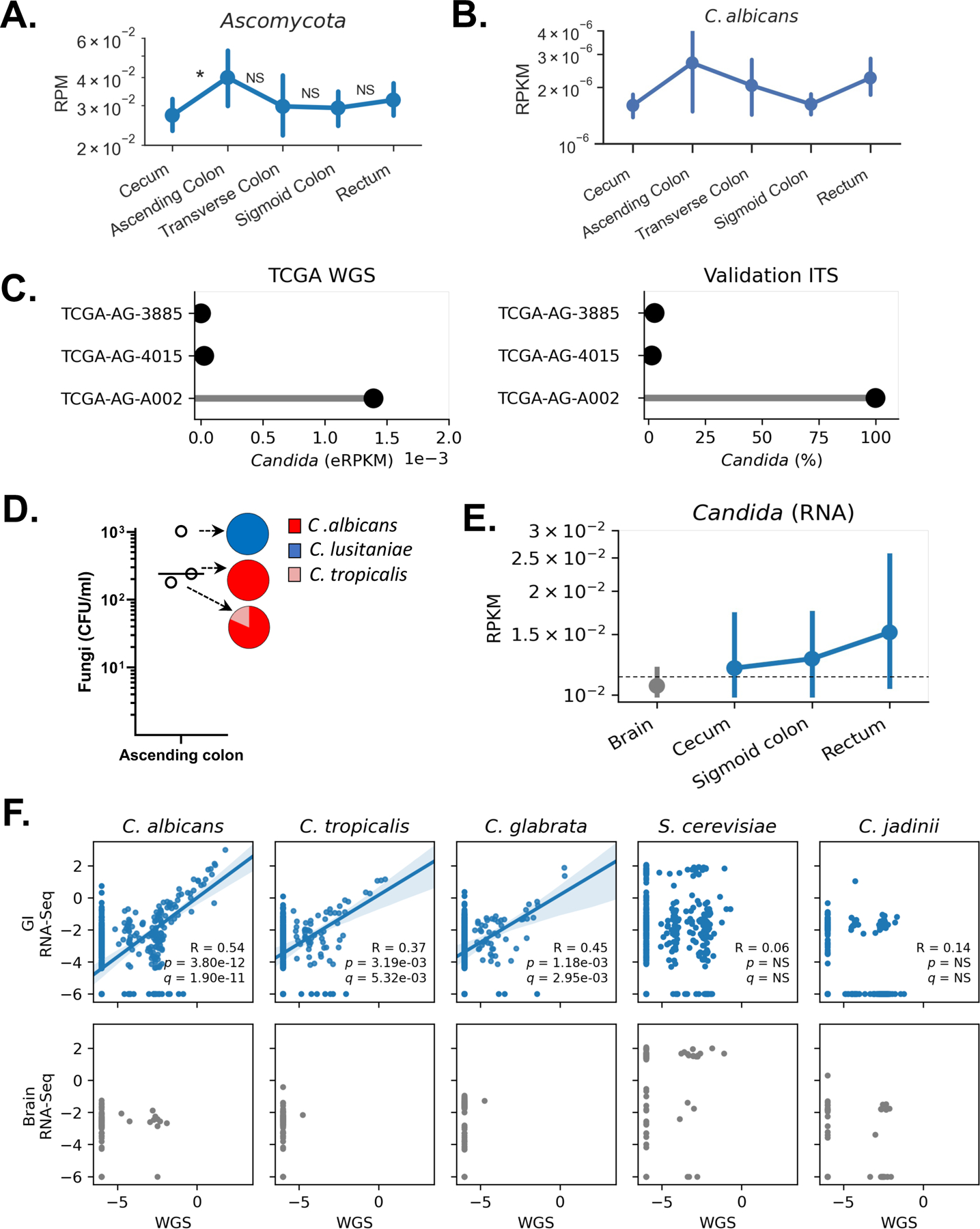
(A) Spatial distribution of Ascomycota abundance along the colorectal tract. Significance was calculated between adjacent tumor sites.
(B) Targeted analysis showing spatial distribution of C. albicans abundance (RPKM).
(C) Comparison of Candida abundance detected in TCGA WGS data (eRPKM; left) and matched original tissues by independent ITS sequencing (relative abundance; right).
(D) Live C. albicans, C. lusitaniae, and C. tropicalis were isolated from the mucosa of adenocarcinomas from ascending colon of three individuals, Viable colony forming units (CFU) per mL of sample were determined by MALDI-TOF.
(E) Abundance of RNA transcripts aligning to Candida in brain (gray) and sites across the lower GI tract (blue) from solid tissues in the HCMI cohort; no solid tumor samples were available from the ascending or transverse colon.
(F) Correlation between fungal species abundances (log10-eRPKM) determined analysis of TCGA WGS and RNA-seq data in GI samples (blue) and brain samples (gray).
We next sought to experimentally validate the presence of Candida in lower GI cancer tissues. To do so, we obtained three primary colorectal tumor samples from an original TCGA tissue provider. Two of these samples were classified as Candida-positive (TCGA-AG-A002) and two as Candida-negative (TCGA-AG-4015, TCGA-AG-3885). We performed independent, ITS sequencing of these three samples and confirmed the presence of high rates of Candida in TCGA-AG-A002 (98.89% of reads), while Candida appeared to be much less abundant in TCGA-AG-4015 and TCGA-AG-3885 (<2% of reads) (Figure 5C).
Notably, a culture-dependent analysis (Li et al., 2022) of colorectal adenocarcinomas from a separate cohort found that live C. albicans, C. lusitaniae and C. tropicalis are present in the mucosa of adenocarcinomas from ascending colon (Figure 5D, Table S3). No live S. cerevisiae, M. sympodialis or M. globosa were isolated from these samples. In a third cohort from the Human Cancer Model Initiative (HCMI), we screened for the presence of Candida RNA in solid tumor samples, finding that the distribution of Candida RNA along the length of the lower GI tract (Figure 5E) matched the anatomical distribution of Candida DNA in TCGA cohort (Figure 5B).
The detection of live Candida and Candida RNA in GI tumors prompted us to examine if RNA from Candida or other species could be detected in GI tumors profiled by TCGA. Comparing the abundance of fungal sequences from matched tumors analyzed using both WGS and RNA-seq, we found that rates of genomic Candida DNA were highly correlated with the presence of Candida RNA transcripts (Figure 5F), indicating that these Candida species were transcriptionally active across GI tumors. In comparison, no such correlations were observed for other species such as S. cerevisiae and C. jadinii, suggesting that DNA and RNA obtained from these species do not represent living fungi in these tumor tissues. Together, these data demonstrate that live, transcriptionally active Candida species are present in tissues associated with GI tumors and that fungal DNA detected in the blood of patients with lower GI tumors may originate from the gut.
Targeted analysis of Candida and Saccharomyces spp.
To further evaluate the prevalence of specific fungi across different cancer types, we performed targeted analyses of C. albicans, C. tropicalis, and S. cerevisiae. This analysis revealed that C. albicans, C. tropicalis, and S. cerevisiae were more prevalent in GI tract tumors than breast tumors or brain tumor controls (Figure 6A-B).
Figure 6. Candida species are present in GI cancers and high abundance is associated with early-stage stomach cancer.

(A – B) Targeted analysis measuring abundance (RPKM) of C. albicans and C. tropicalis (A) and S. cerevisiae (B) across TCGA cancer types.*
(C – D)Abundance of C. albicans, C. tropicalis (C), and S. cerevisiae (D) are elevated in stage 1 stomach cancer tumors and stage 4 colon cancer tumors. Significance was calculated between stage 1 tumors and each subsequent stage.*
* The direction of the inequality symbol indicates which sample group is greater, while the number of symbols indicates the degree of statistical significance, determined by a two-sided Wilcoxon rank-sum statistic (1: p < 0.05, 2: p < 0.01, 3: p < 0.001).
Given our finding that Candida-to-Saccharomyces ratios may be prognostic of GI cancer outcomes (Figure 4C-D), we next used our targeted approach to examine associations between specific fungi and tumor stage. Consistent with our observation of Candida-to-Saccharomyces ratios, we found that C. albicans, C. tropicalis, and S. cerevisiae were significantly associated with stage IV colon cancer (Figure 6C-D, Table S3). Notably, both C. albicans and C. tropicalis were more abundant in stage I stomach cancer specifically (Figure 6C-D). None of the fungal species we examined were associated with a specific tumor stage in head-neck samples. Collectively, these data indicate that increased abundance of Candida in late-stage, metastatic colon tumors may be directly or indirectly involved in the deregulation of genes mediating cellular adhesion (Figure 4B), thereby leading to a deteriorated epithelial barrier, metastasis, and potential translocation of fungal cell components associated with the primary tumor site into the bloodstream (Figure 4E). Alternatively, increased abundance in late-stage colon tumors might instead be the result of deregulations in the tumor’s immune system, which would allow the unhindered growth of Candida and other pathogens.
Cancer-associated mycobiota and clinical outcomes highlight predictive value of Candida
Having observed that higher rates of Candida were associated with increased expression of immune/inflammatory genes in GI cancers (Figure 4B-D), we sought to further explore associations between specific fungi and GI cancer types by comparing abundance of Candida between tumor samples and normal tissue. We found that Candida was significantly and uniquely enriched in stomach tumor samples compared to patient-matched normal tissue (p = 4.23E-3, Figure 7A-B), while Cyberlindnera was significantly enriched in normal tissue (p = 2.15E-5). Notably, a similar analysis determined that Blastomyces (p = 8.80E-3) was similarly enriched in lung tumors compared to matched adjacent normal tissue (Figure S7A).
Figure 7. Cancer-associated fungal mycobiota and clinical outcomes highlight predictive value of Candida, See also Figure S6.
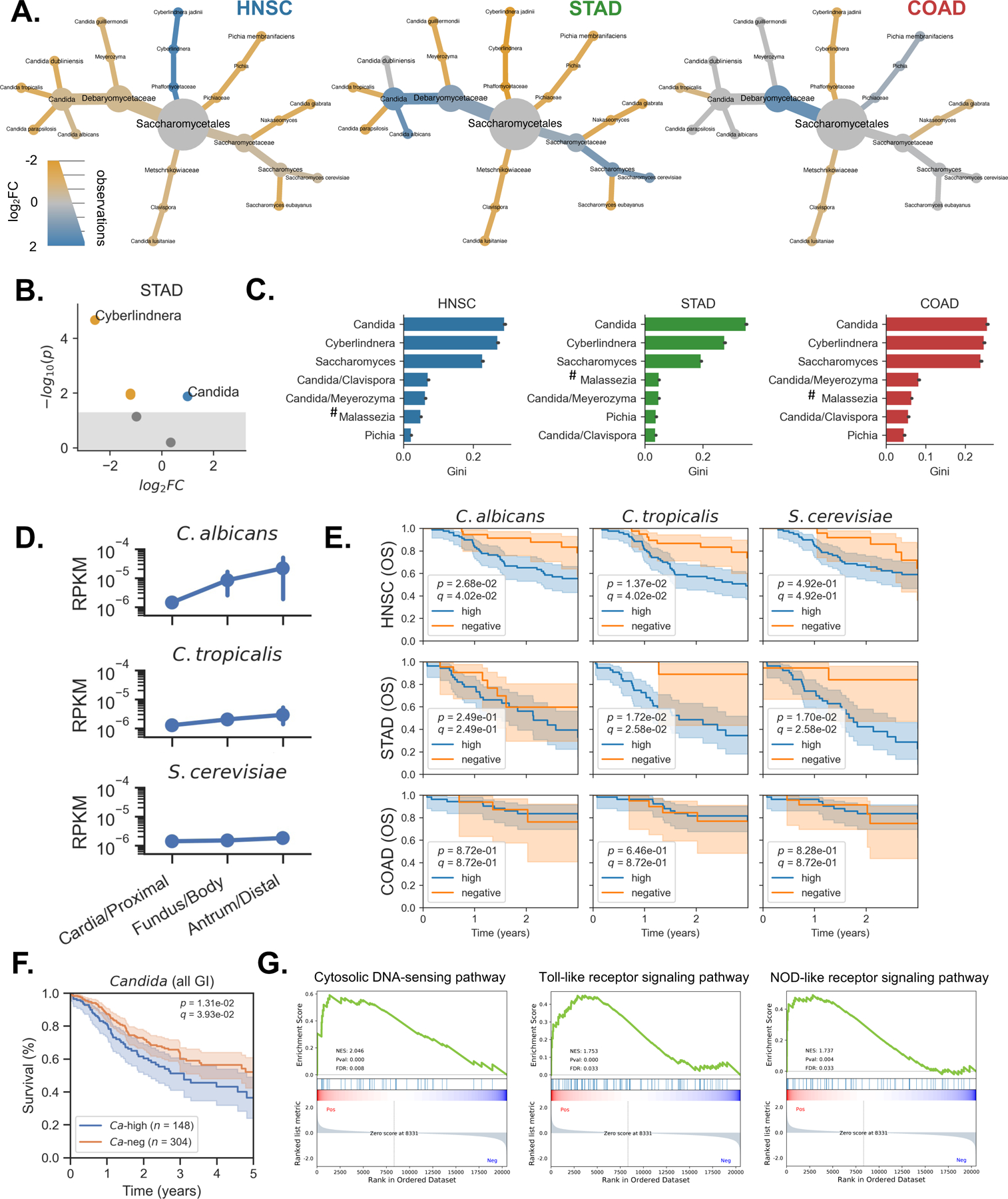
(A) Heat-tree depicting differential abundance of genera between tumor (blue) and matched adjacent normal tissue (yellow) in head-neck (HNSC), stomach (STAD), and colon (COAD) cancers.
(B) Volcano plot showing differential abundance of genera between tumor (blue) and matched adjacent normal tissue (yellow) in stomach cancer.
(C) Genera identified as important for distinguishing head-neck, stomach, and colon tumors from other tumor types, based on the Gini coefficient from RF classifiers. Site specific contaminants (#) were set to 0 prior to running the analysis and therefore may be predictive due to their absence.
(D) Targeted analysis of Candida spp. shows that C. albicans and C. tropicalis increases in abundance from the proximal to distal stomach, while S. cerevisiae abundance remains relatively stable.
(E) Survival analysis comparing outcomes for cancer patients with high rates of tumor-associated C. albicans, C. tropicalis, and S. cerevisiae, compared to patients whose head-neck, stomach, or colon tumors were negative for these species.
(F) Across GI cancer types, patients with high levels of tumor-associated Candida experience decreased survival compared to Candida-negative patients.
(G) GSEA reveals that genes related to cytosolic DNA sensing, Toll-like receptor, and Nod-like receptor signaling are up-regulated in stomach cancers with higher rates of Candida spp.
Our analyses of GI tumor samples suggested that Candida DNA may have potential as a prognostic biomarker. To examine this possibility, we employed a non-parametric machine learning ensemble method known as a random forests (RF) classifier. This approach found that Candida was by far the most important feature for distinguishing GI tumors from other cancer types, followed by Cyberlindnera and Saccharomyces (Figure 7C). Additional targeted analyses of C. albicans and C. tropicalis revealed that the abundance of both Candida species increased steadily from the proximal to distal stomach, with the lowest abundance in the cardia and the greatest abundance in the antrum (Figure 7D). Interestingly, these results mirror the colonization pattern of H. pylori, which preferentially infects the antrum (Suerbaum and Michetti, 2002).
Enrichment of Candida in tumor samples and its predictive power for GI cancer led us to question if Candida might be predictive of disease outcomes. Using survival data from TCGA, we found that high rates of tumor-associated C. tropicalis DNA were significantly associated with decreased survival among stomach cancer patients (p = 1.72E-2) and head-neck cancers (p =1.37E-2), indicating that the presence of Candida DNA at the tumor site might represent a prognostic biomarker for GI cancers (Figure 7E).
We next sought to determine if these associations extended beyond specific cancer types. To explore this possibility, we performed a pan-cancer analysis, incorporating fungal abundance and survival information from all GI cancer types. This analysis found that GI cancer patients with high rates of Candida at the tumor site had significantly decreased survival rates compared to patients who were Candida-negative (p = 1.31E-2, Figure 7F). Saccharomyces was not associated with survival (Figure S6B). The associations between Candida, GI cancer, and reduced survival were particularly pronounced in stomach cancer and consistent with the results of our pathway analysis, which found that the presence of Candida was associated with the expression of genes involved in cytosolic DNA sensing, Toll-like receptor signaling, and Nod-like receptor signaling in stomach cancers (Figure 7G). Together, these data not only contribute to a growing body of evidence suggesting that Candida contributes to GI cancer severity, but also suggest that Candida may serve as a promising biomarker for predicting disease outcomes.
DISCUSSION
In this pan-cancer analysis of tumor mycobiomes, we screened NGS data from TCGA to extract and characterize the fungal DNA presence and composition of hundreds of tissue and blood samples from GI and non-GI cancer types. To precisely determine the fungal composition of these samples, we applied orthogonal QC models to identify and remove potential contaminant fungi and false-positive signals, showing that thorough examinations of genome coverage patterns can identify both biological contamination and false-positive assignments. This approach, in conjunction with previous metagenomic studies of publicly available NGS data (Dohlman et al., 2020; Poore et al., 2020), indicates that careful analysis of existing sequencing data yields cost-effective and biologically meaningful metagenomic profiles which can be leveraged to study multi-kingdom microbe-microbe and host-microbe interactions at the cellular interface between microorganisms and the body sites they inhabit. The capacity to simultaneously profile microbial and tumoral DNA should be taken into consideration when designing such experiments.
Our analysis of tumor mycobiomes revealed both pan-cancer and cancer-specific associations between tumor-associated fungi and human cancers. Moreover, community analysis showed that Candida and Saccharomyces spp. could act as “keystone taxa” in the tumor microbiome, driving ecological interactions and overall variation in multi-kingdom microbial composition. Such changes in tumor-associated microbial communities are likely to have effects on the tumor immune environment and therefore influence the course of tumorigenesis and tumor progression. Accordingly, we found that Candida was associated with increased expression of pro-inflammatory immune pathways, particularly in stomach cancer. In the lower GI tract, we found that Candida was associated with metastasis and deregulation of genes involved in maintaining cellular focal adhesions. In lower GI cancers, we found that tumor and blood samples from the same patient harbored highly similar fungal compositions, raising the possibility that fungal DNA translocates from the GI tumor site to the bloodstream. The same was not true for non-GI tumors.
Increased tight junction permeability and loss of epithelial barrier function are common features of lower GI cancers in particular (Soler et al., 1999), and are significant risk factor for metastasis (Martin and Jiang, 2009). Transformation of intestinal epithelial cells to a mesenchymal-like state is encouraged by chronic inflammation (Ricciardi et al., 2015), a process enhanced by dysbiotic, pro-inflammatory microbiota (Hofman and Vouret-Craviari, 2012; Vergara et al., 2019). As C. albicans potentiates intestinal inflammation via IL-1-dependent mechanisms (Li et al., 2022), it is reasonable to hypothesize that Candida spp. contribute to inflammatory tumorigenesis in cooperation with other microorganisms in the GI tract (Ramirez-Garcia et al., 2016). Inflammation has been shown to strongly promote Candida colonization; Candida maintains this pro-inflammatory environment by itself augmenting inflammation (Jawhara et al., 2008). Thus, effective management of Candida infections and associated inflammation might be a reasonable co-therapeutic option during cancer treatment.
Given our findings that Candida is correlated with worse survival outcomes, pro-inflammatory gene expression, and metastasis, it is apparent that future work is needed to better understand the intricacies of Candida species interaction with the host during tumor development and progression. Additional studies may help to clarify whether tumor-associated Candida is driving these signatures (Tjalsma et al., 2012). Regardless, Candida’s associations with patient survival and enrichment in tumor samples compared to uninvolved tissues indicate that the identification of fungal DNA at the tumor site may provide a predictive biomarker for GI cancers.
Limitations of the Study
Here, we propose that fungi are involved in multiple human tumor types targeting the barrier surfaces, and that specific fungi are predictive of survival. While this data is based on tumor samples from an ethnically and geographically diverse TCGA cohort and samples from a validation cohort, the associations with survival, metastasis, and gene expression presented here should nevertheless be examined in additional settings. Further, while we found many interesting associations between GI tumors and Candida spp., the scope of this study is not capable of addressing whether Candida is contributing to these phenotypes, or instead is enriched because of them. Although the scope of this study is unable to determine if the tumor-associated fungi we found are intratumoral or come from the mucosa associated with these tissues, our data so far suggest the latter possibility. Future work should be done to better understand the role that the mycobiome, or specific fungal species and strains play in cancer development and progression.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Iliyan Iliev (iliev@med.cornell.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This paper analyzes existing, publicly available data from TCGA and HCMI. Information for accessing these datasets can be found in the key resources table.
Original code for generating fungal compositions from TCGA and HCMI datasets has been deposited at https://github.com/abdohlman/tcma_code and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Original TCGA tissue | Indivumed | N/A |
| Colon cancer mucosal samples (adenocarcinoma) | Weill Cornell Medicine | N/A |
| Critical Commercial Assays | ||
| Sabouraud dextrose broth | VWR | Cat# 89406–400 |
| Sabouraud 4% dextrose agar | VWR | Cat# EM1.05438.0500 |
| Glycerol monostearate (Alfa Aesar) | Thermo Fisher Scientific | Cat# AA4388330 |
| modified Dixon (mDixon) | ATCC protocol | N/A |
| Deposited Data | ||
| TCGA WGS bam files | GDC API (Legacy) | https://portal.gdc.cancer.gov/legacy-archive |
| TCGA RNA-seq bam files | GDC API | https://api.gdc.cancer.gov/ |
| HCMI RNA-seq bam files | GDC API | https://api.gdc.cancer.gov/ |
| TCGA sequencing metadata | GDC API | https://api.gdc.cancer.gov/ |
| TCGA sample metadata (biotab) | GDC web portal | https://portal.gdc.cancer.gov/ |
| TCGA patient metadata | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| TCGA clinical data resource outcomes | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Human and microbe reference genomes | PathSeq bundle | ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/pathseq/ |
| Human and microbe reference genomes | PathSeq bundle | ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/pathseq/ |
| TCGA mRNA-seq data | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Gene sets for GSEA (KEGG) | MSigDB v7.1 | https://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp |
| TCMA bacterial profiles (COAD, READ, HNSC, ESCA, STAD) | TCMA database | https://doi.org/10.7924/r4rn36833 |
| Software and Algorithms | ||
| GATK 4.2.0 (PathSeq) | (Walker et al., 2018) | https://github.com/broadinstitute/gatk/ |
| TaxaTarget | (Commichaux et al., 2021) | https://github.com/SethCommchaux/taxaTarget |
| SAMtools v1.9, v1.14 | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| Burrows-Wheeler Aligner | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/ |
| BEDTools | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/ |
| deepTools2 | (Ramirez et al., 2016) | https://deeptools.readthedocs.io/ |
| pyGenomeTracks | (Lopez-Delisle et al., 2021) | https://github.com/deeptools/pyGenomeTracks |
| phyloseq 1.30.0 | (McMurdie and Holmes, 2013) | https://github.com/joey711/phyloseq |
| metacoder 0.3.3 | (Foster et al., 2017) | https://grunwaldlab.github.io/metacoder_documentation/ |
| STAR 2.7.3a | (Dobin et al., 2013) | https://github.com/alexdobin/STAR/ |
| SparCC | (Friedman and Alm, 2012) | https://bitbucket.org/yonatanf/sparcc/src/default/ |
| lifelines 0.23.8 | (Davidson-Pilon et al., 2020) | https://github.com/CamDavidsonPilon/lifelines/tree/0.24.6 |
| GSEA 4.0.3 | (Subramanian et al., 2007) | https://www.gsea-msigdb.org/gsea/ |
| Other | ||
| Decontamination analysis | This paper | Table S1, Related to Figure 1 |
| Fungal composition of TCGA samples | This paper | Table S2, Related to Figures 1–7 |
| False-discovery rate adjustments | This paper | Table S3, Related to Figures 1–7 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Information on the age/developmental stage, sex, and gender identity of all subjects from the TCGA and HCMI cohorts are publicly available from the GDC website. Links for accessing this information is available in the key resources table. Colon tumor samples (adenocarcinomas of ascending colon) used for Candida cultures originated from deidentified individuals according to the institutional review board approved protocol from the Weill Cornell Medicine in accordance with the Helsinki Declaration. TCGA and HCMI sequencing data were used in accordance with the TCGA and HCMI Data Use Certification Agreement, dbGaP authorization to access controlled data and under authorization of Duke University and Weill Cornell Medicine institutional review boards.
METHOD DETAILS
Detection and quantification of mycobiomes in TCGA and HCMI sequencing data
The TCGA project collected biospecimens including primary tumors, normal tissue, and blood samples from cancer patients both prospectively and retrospectively until 2013. Requirements for sample collection included (1) a minimum size of 200mg, (2) a minimum of 80% tumor nuclei, (3) a maximum of 50% necrosis, and (4) availability of matched germline DNA (Cancer Genome Atlas Research, 2008). Analyte-, sample-, and patient-level metadata (including information on tumor stage, location, metastasis, etc.) associated with each sequencing run were obtained from the NCI Genomic Data Commons (GDC). Raw TCGA WGS data were obtained from the GDC’s legacy archive (https://portal.gdc.cancer.gov/legacy-archive/), while raw HCMI RNA-seq data was obtained directly from GDC (https://portal.gdc.cancer.gov/). Overall, we analyzed data from 1,759 sequencing runs for HNSC (n = 338), ESCA (n = 143), STAD (n = 321), COAD (n = 300), READ (n = 127), BRCA (n = 230), LUSC (n = 100), and LGG (n = 200) projects from TCGA with WGS data available. From HCMI, we analyzed data from 34 sequencing runs on solid tissue samples from brain (n = 13) and lower Gi sites (n = 21).
All WGS and RNA-seq data from TCGA and HCMI were screened for fungal content using PathSeq (Walker et al., 2018), which is made available as part of the Broad Institute’s Genome Analysis Toolkit (GATK version 4.0.3) and relies on the Burrows-Wheeler Aligner (BWA-MEM) (Li and Durbin, 2009). Prior to screening for microbial alignments, PathSeq performs multiple, iterative subtractive alignments of reads previously unaligned to a host genome reference. The core host reference genome used was GRCh38 (hg38); this host reference is supplemented by (1) highly variable sequences from the immunohistocompatibility complex (MHC) from the Immuno-Polymorphisms Database (IDP), (2) Cloning vector sequences from NCBI UniVec, (3) mammalian consensus repetitive sequences from RepBase, (3) a curated database of human transcripts (human v25) from Gencode, and (4) human breakpoint sequences from GenBank (KY503218, KY5808060). Reference genomes for this analysis were obtained from the PathSeq resource bundle. These files were accessed via ftp from the Broad Institute (ftp.broadinstitute.org/bundle/beta/PathSeq/). PathSeq was used with default settings, except for the “minClippedReadLength” parameter, which was set to 50 for WGS and 45 for RNA-seq (a read length of 50bp is used for most TCGA RNA-seq data). All sequencing data were analyzed on a local high-performance computing (HPC) cluster with 60 compute nodes, 1,512 CPU cores, and approximately 15TB of RAM.
To isolate the endogenous fungal composition of these samples, sequencing reads from taxa at the genus and species level were normalized (1) by genome size (i.e. per kilobase of mapped fungal genome), (2) by the expected accuracy of the taxonomic assignment (i.e. weights are divided by the number of ambiguous alignments), and then (3) by the total library size (i.e. per million primary sequencing reads, regardless of alignment). These normalizations produced an “expected reads per kilobase of genome, per million primary reads” statistic (eRPKM). Kingdom- and phylum-level read counts were normalized to the library size (reads per million, RPM), as these alignments are much less prone to ambiguous assignment or significant fluctuations in genome size. Relative abundance (%) values were calculated by scaling eRPKM values, such that the sum of taxa abundances from a given taxonomic rank and sample sum to 100.
QC by removal of fungi associated with TCGA sequencing batches
To mitigate the possibility of fungal contamination in the mycobiomes we analyzed, we performed a screen to identify species and genera that showed signs of technical variation, but not biological variation. We therefore devised a two-step prevalence-based decontamination model (See Methods) to identify and remove (1) fungal taxa whose presence was associated with specific sequencing batches and could not be explained by biological variation, and (2) samples from multi-well sequencing plates with strong evidence of contamination.
We calculated prevalence of species and genera across each sequencing batch (plate id), then for each tumor type (TCGA sequencing project) and compared these to their expected frequencies assuming a random distribution. Specifically, expected frequency distributions for each species were calculated by multiplying the total number of samples in each project or sequencing plate by the species prevalence across the entire dataset; these values were compared to the observed prevalence across projects or plates. We used these observed and expected frequencies to compute p-values for a Chi-square statistic, which was then adjusted for multiple comparisons using the Benjamini-Hochberg false-discovery rate correction (FDR, q-values). Species and genera that were associated with sequencing batch (q < 0.1) but not tumor type (q > 0.1) were classified as potential contaminants and removed from downstream analysis. Lastly, we screened samples to determine if there were sequencing plates with significant evidence of contamination that needed to be excluded from the analysis entirely. This analysis identified a single sequencing plate (A19H) with significant contamination. Samples from this plate harbored fungal reads at rates around five magnitudes greater than samples from different plates, independent of sample type.
QC by vertical and horizontal analyses of fungal genome coverage
To further address the possibility of contamination or false-positive alignments, we sought to characterize the genomic coverage of the most frequently detected species in our PathSeq analysis of WGS data from TCGA. We selected any species detected in more than 5 sequencing runs (eRPKM > 0) in any of TCGA sequencing projects we analyzed (HNSC, ESCA, STAD, COAD, READ, LUSC, BRCA) that remained our precursory decontamination analysis of sequencing batches, as well as several closely related species with NCBI reference genomes available. For sequenced tumor samples from each cancer type, the human subtracted PathSeq BAM file outputs were converted back to their raw, unmapped, reads using SAMtools v1.14 (Li et al., 2009) . Raw reads were aligned using the Burrows-Wheeler Aligner (BWA) (Li and Durbin, 2009) to each species’ reference genome to create a new BAM file containing only reads mapped to that reference. Genome coverage statistics were stored in bedgraph files using BEDTools (Quinlan and Hall, 2010) genomeCoverageBed with the -bg flag. Each tumor type’s bedgraphs were then pooled together and their genome coverage was assessed using deepTools2 (Ramirez et al., 2016) bamCoverage command.
We used the resulting bedgraphs to analyze the coverage depth and horizontal read distribution for each genome. Coverage depth (Vertical QC model) was assessed by calculating the average log10-coverage per-base per-sample. We then calculated the ratio of average log10-coverage per-base per-sample between each sequencing project and brain tumor samples to estimate the fraction of reads that could be the result of contamination. To assess horizontal distribution (Horizontal QC model) for each species and cancer type, we generated a genome-length Boolean vector indicating whether reads had aligned to each base. The hamming distance between the vector generated for brain tissues and the vector for each cancer type was then calculated to determine the base-wise horizontal similarity of alignments across each genome. For the vertical QC model, species were classified as possible contaminants if the average log10-coverage per-base per-sample coverage for each tumor type was greater than 30% that of brain tumors. For the horizontal QC model, species were classified as possible false-positive signals if the hamming distance to brain was less than 0.02. Species which were classified as possible contaminants or false-positive signals by either model were removed from downstream analysis. Genome alignments were visualized using pyGenomeTracks (Ramirez et al., 2018).
Validation with TaxaTarget
We used TaxaTarget (Commichaux et al., 2021) to analyze eukaryotic marker genes and validate the presence of key species from our PathSeq analysis. Human-filtered PathSeq output BAM files from TCGA were converted to raw, unaligned forward and reverse fastq formats using samtools. The taxaTarget results were then screened for marker genes aligning to Homo sapiens to determine the degree of contamination by human DNA, as well Candida, Saccharomyces, and Malassezia species to validate fungal presence in TCGA tumor samples.
Targeted analysis and quantification of Candida and Saccharomyces species of interest
We identified several species of interest that were abundant across TCGA tissue samples. To better quantify these species, we performed a targeted analysis by mapping fungal genomes to libraries of putative microbial reads generated for each TCGA sequencing run after stringent filtering of human sequences with PathSeq. Representative genomes for C. albicans (GCA_003454735.1), C. tropicalis (GCA_000633855.1), and S. cerevisiae (GCA_000146045.2) were downloaded from GenBank and mapped to these libraries using STAR (Dobin et al., 2013) without allowing for spliced alignments (--alignIntronMax=1). Raw read counts for each species were then normalized by genome size and total library size as previously described to calculate an empirical reads per kilobase of genome, per million primary reads (RPKM).
Estimation of intra- and inter-kingdom co-abundance groups and associated gene expression signatures
Compositional effects complicate robust calculation of correlations between microbiota (Gloor et al., 2017). To address these effects, we used SparCC (Friedman and Alm, 2012) to estimate taxa that are frequently found together across each cancer type. This method relies on a bootstrapping procedure to reduce for spurious results. Prior to calculating correlations, we filtered out low-abundance samples and selected the 20 most abundant fungal species from each cancer type. We then ran SparCC for 1000 iterations with default parameters to identify fungal co-abundance groups within head-neck (HNSC), stomach (STAD), and colon (COAD) tumor samples.
Our trans-kingdom analysis was used to identify associations between fungi and bacteria and was performed by comparing the decontaminated fungal compositions generated in the current work with decontaminated bacterial compositions from matched samples in TCMA (Dohlman et al., 2020). To accurately quantify associations across kingdoms and control for the significant difference in their respective abundances, we applied a scaling factor to the fungal compositions to obtain similar distributions for each kingdom and allow robust estimation of bacterial-fungal co-abundance associations. The most abundant fungal and bacterial taxa were selected from each cancer type prior to running SparCC.
Acquisition and analysis of original TCGA tumor samples
For validation of Candida presence in lower GI tumors, we obtained original, matched tissue and plasma samples from three CRC patients from Indivumed, an original TCGA tissue provider. Tumor tissues were minced, homogenized and treated with 200 U/mL lyticase (Sigma) followed by bead beating, and processing using the Quick-DNA Fungal/Bacterial Kit (Zymo Research) as in (Li et al., 2022). Fungal DNA presence was validated by RT-PCR for fungal 18S and fungal ITS1–2 regions were amplified by PCR using primers with sample barcodes and sequencing adaptors.
Fungal primers: ITS1F-CTTGGTCATTTAGAGGAAGTAA, ITS2R-GCTGCGTTCTTCATCGATGC
Forward overhang: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐[locus-specific sequence]
Reverse overhang: 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG‐[locus-specific sequence]
ITS amplicons were generated with 35 cycles using Invitrogen AccuPrime PCR reagents (Carlsbad). Amplicons were then used in the second PCR reaction, using Illumina Nextera XT v2 (Illumina) barcoded primers to uniquely index each sample. DNA was amplified using the following PCR protocol: Initial denaturation at 94°C for 10 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 2 min, followed by an elongation step at 72°C for 30 min. All libraries were subjected to QC using DNA 1000 Bioanalyzer (Agilent), and Qubit (Life Technologies) to validate and quantify library construction prior to preparing a Paired-End flow cell. Samples were randomly divided among flow cells to minimize sequencing bias. Clonal bridge amplification (Illumina) was performed using a cBot (Illumina). 2 × 250 bp sequencing-by-synthesis was performed on Illumina MiSeq platform (Illumina).
Quantification, isolation and characterization of live fungi in primary colorectal tumor samples
Adenocarcinoma-associated tissues were collected from ascending colon surgical resections that were then weighed, minced, homogenized, diluted in sterile PBS and plated onto Sabouraud dextrose agar (SDA) and modified Dixon media (mDixon with glycerol monostearate), and inhibitory mold agar (Hardy Diagnostics), all supplemented with both penicillin/streptomycin (Sigma), inhibitory mold agar (Hardy Diagnostics) and modified Dixon broth with glycerol monostearate. SDA plates were incubated at 37°C for 48 hours. Inhibitory mold agar plates and modified Dixon media were incubated at 30°C for up to a week. Isolated fungal colonies from each individual subject were identified by matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometer.
Identification of Candida- and Saccharomyces-type TCGA tumor samples and associated signatures
To identify Candida- and Saccharomyces-associated tumors, we calculated a log-normalized Candida-to- Saccharomyces abundance ratio (log2(C/S)) across all tumor samples for which either genus was detected. Tumors were classified as Ca-type or Sa-type if they had a log2(C/S) value above 1 or below −1, respectively, i.e. samples for which neither genus was detected at more than twice the rate of the other were excluded. To test associations between gene expression and the presence of Candida and Saccharomyces, we performed differential gene expression analysis using batch-normalized gene expression data from the PanCanAtlas publication page (https://gdc.cancer.gov/about-data/publications/pancanatlas). For each cancer type, we calculated log2-fold changes (log2FC) in gene expression between tumors that were negative for Candida or Saccharomyces (eRPKM < 1E-6) and tumors which were high in Candida or Saccharomyces (eRPKM > 1E-6). All taxonomic abundance profiles were collapsed to the sample level by calculating the geometric mean of taxon abundances across the available tumor sequencing data for each tumor sample. We then estimated the significance of gene expression changes using Student’s independent two-sample t-test. Differential gene expression values generated by this analysis were then used perform GSEA (Subramanian et al., 2005) and analyze gene expression pathways enriched in Candida- and Saccharomyces-associated cancers based on gene lists obtained from MSigDB v7.1. Using pre-ranked differential gene expression values, we ran GSEA for 1000 iterations to identify enriched KEGG biological pathways (Kanehisa and Goto, 2000).
To compare rates of metastasis in Ca- and Sa-type tumors, we used TNM-stage classifications of each TCGA tumor sample to determine metastatic (M1) and non-metastatic (M0) status. Samples for which no metastatic information was available (MX) were excluded. A contingency table for each cancer type was generated, comparing metastatic status (M0/M1) and tumor mycobiome classification (Ca-type v.s. Sa-type). Fisher’s exact test was then used to determine if Ca-type or Sa-type tumors were more likely to be metastatic.
Differential abundance analysis between tumor and adjacent normal tissue
Associations between fungal genera and sample type (tumor v.s. matched adjacent normal tissue) were calculated in R, using a custom paired analysis function written for metacoder (Foster et al., 2017). For each cancer type we analyzed the 20 most abundant taxa were selected, provided they were present in at least 30 samples overall. Such filters were applied to remove low-abundance and low-prevalence fungi. Pseudocounts were added and data were transformed to relative abundance for each sequencing run. Across all patients with matched tumor and normal tissue, we then calculated the log2 median ratio of relative abundance values in tumor samples compared to matched adjacent normal tissue for each taxon. Significance values were calculated for log2 median ratios using Wilcoxon’s rank-sums test. Taxa with significant p-values (p < 0.05) were selected for downstream analysis.
Survival analysis
The survival analyses was performed using the log-rank test, as implemented by the lifelines survival analysis python package (Davidson-Pilon et al., 2020). Information on TCGA patient survival outcomes were collected from the PanCanAtlas clinical follow-up data (Liu et al., 2018). Survival analysis was performed at both the species and genus level. For the species-level analysis, we used normalized fungal abundances from our targeted analysis (RPKM for C. albicans, C. tropicalis, and S. cerevisiae). For each species of interest and cancer type, we compared survival between patients whose tumors did not harbor the species (“negative”; 0th percentile) with patients whose tumors were abundant in the species (“high”; top 50th percentile). The genus-level analysis was performed using fungal abundances determined by our PathSeq analysis (eRPKM for Candida and Saccharomyces) and used the same set of criteria for assigning patients as “negative” or “high” as the differential gene expression analysis. Taxonomic abundances were collapsed to the patient level using the geometric mean of taxon abundances across the tumor sequencing data available for each patient.
Random forest classification of cancer types using fungal compositions of tumor and blood samples
To identify fungal genera predictive of cancer location, we used a decision-tree based ensemble machine learning method known as random forest classifiers (Breiman, 2001), as implemented by the python package sklearn (Abraham et al., 2014). A separate classifier model was trained on the mycobacterial compositions of tumor samples from seven TCGA cancer types (HNSC, ESCA, STAD, COAD, READ, LUSC, and BRCA). For each cancer type, we implemented a one-versus-all classification strategy which sought to identify genera capable of distinguishing a specific cancer type (e.g. stomach tumors) from all others (e.g. non-stomach tumors). Prior to classification, taxa that were detected in fewer than 1% of samples were removed. Species abundances were log-normalized after the addition of a pseudocount to achieve a gaussian distribution. For each classifier a forest of 400 estimators was used with a maximum depth of 30 features per tree and a minimum of 5 samples per split. Default values were used for all other hyperparameters. To bootstrap the estimation of feature importances, we used a repeated, stratified cross-fold cross validation strategy with 10 folds and 10 repeats.
QUANTIFICATION AND STATISTICAL ANALYSIS
For removal of fungi associated with TCGA sequencing batches we used observed and expected frequencies to compute p-values for a Chi-square statistic, which was then adjusted for multiple comparisons using the Benjamini-Hochberg false-discovery rate correction (FDR, q-values). All statistical comparisons between sample groups were done using Wilcoxon’s rank-sums test, unless otherwise specified. To accurately quantify associations across kingdoms and control for significant differences in their respective abundances, we applied a scaling factor to the fungal compositions to obtain similar distributions for each kingdom and allow robust estimation of bacterial-fungal co-abundance associations. To identify Candida- and Saccharomyces-type TCGA tumor samples and associated gene expression changes, we estimated significance using Student’s independent two-sample t-test. Differential gene expression values generated by this analysis were then used perform GSEA and analyze gene expression pathways enriched in Candida- and Saccharomyces-associated cancers. Significance values for GSEA were computed by permuting gene labels. Across all patients with matched tumor and normal tissue, we calculated the log2 median ratio of relative abundance values in tumor samples compared to matched adjacent normal tissue for each taxon. Taxa with significant p-values (p < 0.05) were selected for downstream analysis. Feature importances were estimated by averaging Gini impurity measures for each of the 100 resulting sub-models. The survival analyses was performed using the log-rank test. Additional details on the statistical analysis are provided in the “Methods Details”.
Supplementary Material
Figure S1. Fungal DNA is present in multiple cancer types not explained by contamination, Related to Figure 1
(A) Boxplot showing the distribution of reads per million (RPM) of fungal DNA detected in tumor and tumor-associated tissue samples from head-neck (HNSC), lung (LUSC), rectum (READ), colon (COAD), stomach (STAD), breast (BRCA), esophageal (ESCA) and brain (LGG) cancers, ordered by upper quartiles.
(B) Multi-well aliquot plate A19H shows significant signs of contamination; samples shown in orange were discarded.
(C) The distribution of sequencing reads aligning to the M. restricta genome displays similar depth across sequencing projects including brain, but reads are distributed randomly, a signature of biological contamination.
(D) The distribution of sequencing reads aligning to the A. bisporus genome displays uneven depth, but reads are horizontally distributed in a predictable manner, a signature of false-positive assignments.
(E) Re-analysis of human-subtracted sequencing data using TaxaTarget (Commichaux et al., 2021) validates the presence of Candida spp, S. cerevisiae, and Malassezia spp. across TCGA samples. R- and p-values indicate the result of a spearman rank correlation test.
Figure S2. Primary tumor samples harbor disease-specific mycobiomes, Related to Figure 2
(A) Prevalence of the top 10 fungal species detected in tissue from each cancer type, after removing potential contaminants.
Figure S3. Trans-kingdom analysis reveals Candida- and Saccharomyces-associated GI cancer co-abundance groups, Related to Figure 3
(A – C)SparCC correlations between Candida and Malassezia and the most abundant bacterial species found in matched tumor samples from TCMA in head-neck (HNSC; A), stomach (STAD; B), and colon (COAD; C) cancers.
(D – F) Scatterplots and fitted regression lines showing correlations between Candida, Saccharomyces, and Lactobacillus in head-neck (HNSC; D), stomach (STAD; E), and colon (COAD; F) cancers. R- and p-values indicate the result of a spearman rank correlation test.
Figure S4. Candida is associated with late-stage and metastatic GI cancers, Related to Figure 4
(A) Kernel density estimation (KDE) of Candida-to-Saccharomyces ratios in esophageal (ESCA), rectum (READ), breast (BRCA), and brain tumors.
(B) Volcano plot showing genes differentially expressed Saccharomyces-negative (blue) and Saccharomyces-high (red) tumor samples head-neck, stomach, and colon cancers.
(C) Boxplots depicting the ratio of Candida-to-Saccharomyces in early-stage (I-III) and late-stage (IV)
(D) KDE analysis showing the distribution of Candida-to- Saccharomyces ratios in metastatic (orange) and non-metastatic (blue) tumor samples across cancer types.
Figure S5. Alive and transcriptionally active Candida species are associated with GI tumors, Related to Figure 5
(A) Spatial distribution of Ascomycota and Basidiomycota abundance along the colorectal tract.
Figure S6. Cancer-associated fungal mycobiota and clinical outcomes highlight predictive value of Candida, Related to Figure 7
(A) Volcano plot showing differential abundance of genera between tumor (blue) and matched adjacent normal tissue (yellow) in lung cancer. Blastomyces was enriched in lung tumors compared to matched adjacent normal tissue.
(B) Across GI cancer types, comparison of survival between patients with high rates of Saccharomyces in their tumors compared to patients whose tumors are negative for this taxon.
Table S1. Identification and removal of fungal contamination in TCGA sequencing data, Related to Figure 1
Screening of TCGA mycobiomes identifies contaminant and false-positive species and genera, and multi-well sequencing plate IDs with significant evidence of contamination. This table includes a list of the most prevalent contaminants/false-positives identified, as well as a list of genomes used in the genome-based QC models.
Table S2. Pan-cancer fungal microbiomes, Related to Figures 1–7
Normalized (eRPKM) and filtered fungal compositions from each TCGA cancer type, as well as associated metadata, including Ca/Sa tumor status. This table also includes a list of fungal genomes included in the PathSeq reference.
Table S3. False-discovery rate adjustments, Related to Figures 1–7
False-discovery rate (FDR) adjusted p-values for statistical tests performed, where not already indicated in the text or figures.
HIGHLIGHTS.
A pan-cancer analysis reveals human samples harbor tumor-associated mycobiota
Fungal genome coverage analysis removes contamination and false-positive alignments
Alive, transcriptionally active Candida is associated with gastrointestinal cancers
Candida is enriched in tumors and predictive of reduced survival in GI cancers
Pan-cancer analyses of multiple body sites identify tumor-specific fungi including an enrichment of Candida with gastrointestinal cancers. Tumor-associated fungal DNA may also serve as potential prognostic markers in this context.
Acknowledgments
Research in the Iliev laboratory is supported by US National Institutes of Health (R01DK113136, R01DK121977, and R01AI163007), the Leona M. and Harry B. Helmsley Charitable Trust, the Irma T. Hirschl Career Scientist Award, the Research Corporation for Science Advancement Award, the Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease (PATH) Award and the Cancer Research Institute Lloyd J. Old STAR Award. I.D.I is a fellow of the CIFAR program Fungal Kingdom: Threats and Opportunities. The authors thank Katia Manova-Todorova, Maria Pulina, Eric Rosiek and members of the Molecular Cytology Core at MSKCC (support through P30 CA008748) for technical assistance. Research in the Shen Lab is supported by the National Institutes of Health (R35GM122465, DK119795, and NIH-U01CA214300)..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
X.S. is the co-founder and CEO of Xilis, Inc. This study and its findings do not have any overlap or implications over Xilis’ commercial interests. A.B.D, X.L. and I.D.I. are inventors on a US provisional patent application, covering inventions described in this manuscript. The authors declare that they have no conflicts of interest with the contents of this article.
References:
- Abraham A, Pedregosa F, Eickenberg M, Gervais P, Mueller A, Kossaifi J, Gramfort A, Thirion B, and Varoquaux G (2014). Machine learning for neuroimaging with scikit-learn. Front Neuroinform 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggor FEY, Break TJ, Trevejo-Nunez G, Whibley N, Coleman BM, Bailey RD, Kaplan DH, Naglik JR, Shan W, Shetty AC, et al. (2020). Oral epithelial IL-22/STAT3 signaling licenses IL-17-mediated immunity to oral mucosal candidiasis. Sci Immunol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A, Levanduski E, Denz P, Villavicencio HS, Bhatta M, Alhorebi L, Zhang Y, Gomez EC, Morreale B, Senchanthisai S, et al. (2022). Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell 40, 153–167 e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballou ER, Avelar GM, Childers DS, Mackie J, Bain JM, Wagener J, Kastora SL, Panea MD, Hardison SE, Walker LA, et al. (2016). Lactate signalling regulates fungal beta-glucan masking and immune evasion. Nat Microbiol 2, 16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict K, Roy M, Chiller T, and Davis JP (2012). Epidemiologic and Ecologic Features of Blastomycosis: A Review. Current Fungal Infection Reports 6, 327–335. [Google Scholar]
- Break TJ, Oikonomou V, Dutzan N, Desai JV, Swidergall M, Freiwald T, Chauss D, Harrison OJ, Alejo J, Williams DW, et al. (2021). Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L (2001). Random Forests. Machine Learning 45, 5–32. [Google Scholar]
- Brown EM, McTaggart LR, Zhang SX, Low DE, Stevens DA, and Richardson SE (2013). Phylogenetic analysis reveals a cryptic species Blastomyces gilchristii, sp. nov. within the human pathogenic fungus Blastomyces dermatitidis. PLoS One 8, e59237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, and White TC (2012). Hidden killers: human fungal infections. Sci Transl Med 4, 165rv113. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang F, Syrjanen S, Wang L, and Syrjanen K (1992). Infectious agents in the etiology of esophageal cancer. Gastroenterology 103, 1336–1348. [DOI] [PubMed] [Google Scholar]
- Chehoud C, Albenberg LG, Judge C, Hoffmann C, Grunberg S, Bittinger K, Baldassano RN, Lewis JD, Bushman FD, and Wu GD (2015). Fungal Signature in the Gut Microbiota of Pediatric Patients With Inflammatory Bowel Disease. Inflamm Bowel Dis 21, 1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coker OO, Nakatsu G, Dai RZ, Wu WKK, Wong SH, Ng SC, Chan FKL, Sung JJY, and Yu J (2018). Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commichaux S, Javkar K, Muralidharan HS, Ramachandran P, Ottesen A, Rand H, and Pop M (2021). TaxaTarget: Fast, Sensitive, and Precise Classification of Microeukaryotes in Metagenomic Data (Research Square) [Google Scholar]
- Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, Deblasio RN, Menna C, Ding Q, Pagliano O, et al. (2021). Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 371, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis NM, Proctor DM, Holmes SP, Relman DA, and Callahan BJ (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman AB, Arguijo Mendoza D, Ding S, Gao M, Dressman H, Iliev ID, Lipkin SM, and Shen X (2020). The cancer microbiome atlas: a pan-cancer comparative analysis to distinguish tissue-resident microbiota from contaminants. Cell Host Microbe [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman AB, and Shen X (2019). Mapping the microbial interactome: Statistical and experimental approaches for microbiome network inference. Exp Biol Med (Maywood) 244, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doron I, Mesko M, Li XV, Kusakabe T, Leonardi I, Shaw DG, Fiers WD, Lin WY, Bialt-DeCelie M, Roman E, et al. (2021). Mycobiota-induced IgA antibodies regulate fungal commensalism in the gut and are dysregulated in Crohn’s disease. Nat Microbiol 6, 1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzutsev A, Badger JH, Perez-Chanona E, Roy S, Salcedo R, Smith CK, and Trinchieri G (2017). Microbes and Cancer. Annu Rev Immunol 35, 199–228. [DOI] [PubMed] [Google Scholar]
- Ewaschuk JB, Diaz H, Meddings L, Diederichs B, Dmytrash A, Backer J, Looijer-van Langen M, and Madsen KL (2008). Secreted bioactive factors from Bifidobacterium infantis enhance epithelial cell barrier function. Am J Physiol Gastrointest Liver Physiol 295, G1025–1034. [DOI] [PubMed] [Google Scholar]
- Fan D, Coughlin LA, Neubauer MM, Kim J, Kim MS, Zhan X, Simms-Waldrip TR, Xie Y, Hooper LV, and Koh AY (2015). Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat Med 21, 808–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust K, and Raes J (2012). Microbial interactions: from networks to models. Nat Rev Microbiol 10, 538–550. [DOI] [PubMed] [Google Scholar]
- Fiers WD, Gao IH, and Iliev ID (2019). Gut mycobiota under scrutiny: fungal symbionts or environmental transients? Curr Opin Microbiol 50, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, Schoenfeld D, Nomicos E, Park M, Program, N.I.H.I.S.C.C.S., et al. (2013). Topographic diversity of fungal and bacterial communities in human skin. Nature 498, 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay BB, Goldszmid R, Honda K, Trinchieri G, Wargo J, and Zitvogel L (2020). Can we harness the microbiota to enhance the efficacy of cancer immunotherapy? Nat Rev Immunol 20, 522–528. [DOI] [PubMed] [Google Scholar]
- Foster ZS, Sharpton TJ, and Grunwald NJ (2017). Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput Biol 13, e1005404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, and Alm EJ (2012). Inferring correlation networks from genomic survey data. PLoS Comput Biol 8, e1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS (2019). The gut microbiota and colon cancer. Science 364, 1133–1135. [DOI] [PubMed] [Google Scholar]
- Glassing A, Dowd SE, Galandiuk S, Davis B, and Chiodini RJ (2016). Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog 8, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloor GB, Macklaim JM, Pawlowsky-Glahn V, and Egozcue JJ (2017). Microbiome Datasets Are Compositional: And This Is Not Optional. Front Microbiol 8, 2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, and Karin M (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, and Wargo JA (2019). The microbiome, cancer, and cancer therapy. Nat Med 25, 377–388. [DOI] [PubMed] [Google Scholar]
- Hoarau G, Mukherjee PK, Gower-Rousseau C, Hager C, Chandra J, Retuerto MA, Neut C, Vermeire S, Clemente J, Colombel JF, et al. (2016). Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. Mbio 7, e01250–01216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman P, and Vouret-Craviari V (2012). Microbes-induced EMT at the crossroad of inflammation and cancer. Gut Microbes 3, 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffnagle GB, and Noverr MC (2013). The emerging world of the fungal microbiome. Trends Microbiol 21, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, et al. (2013). Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342, 967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawhara S, Thuru X, Standaert-Vitse A, Jouault T, Mordon S, Sendid B, Desreumaux P, and Poulain D (2008). Colonization of mice by Candida albicans is promoted by chemically induced colitis and augments inflammatory responses through galectin-3. J Infect Dis 197, 972–980. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto CA, Gresnigt MS, and Hube B (2020). The gut, the bad and the harmless: Candida albicans as a commensal and opportunistic pathogen in the intestine. Curr Opin Microbiol 56, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi I, Gao IH, Lin WY, Allen M, Li XV, Fiers WD, De Celie MB, Putzel GG, Yantiss RK, Johncilla M, et al. (2022). Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell 185, 831–846 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi I, Paramsothy S, Doron I, Semon A, Kaakoush NO, Clemente JC, Faith JJ, Borody TJ, Mitchell HM, Colombel JF, et al. (2020). Fungal Trans-kingdom Dynamics Linked to Responsiveness to Fecal Microbiota Transplantation (FMT) Therapy in Ulcerative Colitis. Cell Host Microbe 27, 823–829 e823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman ES, Hoffmann C, et al. (2015). Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 18, 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XV, Leonardi I, Putzel GG, Semon A, Fiers WD, Kusakabe T, Lin WY, Gao IH, Doron I, Gutierrez-Guerrero A, et al. (2022). Immune regulation by fungal strain diversity in inflammatory bowel disease. Nature 603, 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguori G, Lamas B, Richard ML, Brandi G, da Costa G, Hoffmann TW, Di Simone MP, Calabrese C, Poggioli G, Langella P, et al. (2016). Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. Journal of Crohn’s & colitis 10, 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu NN, Jiao N, Tan JC, Wang Z, Wu D, Wang AJ, Chen J, Tao L, Zhou C, Fang W, et al. (2022). Multi-kingdom microbiota analyses identify bacterial-fungal interactions and biomarkers of colorectal cancer across cohorts. Nat Microbiol 7, 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine J, Daniel-Ivad M, Liu Z, Yano J, Revie NM, Todd RT, Stogios PJ, Sanchez H, O’Meara TR, Tompkins TA, et al. (2021). A small molecule produced by Lactobacillus species blocks Candida albicans filamentation by inhibiting a DYRK1-family kinase. Nat Commun 12, 6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik A, Sharma D, Malireddi RKS, Guy CS, Chang TC, Olsen SR, Neale G, Vogel P, and Kanneganti TD (2018). SYK-CARD9 Signaling Axis Promotes Gut Fungi-Mediated Inflammasome Activation to Restrict Colitis and Colon Cancer. Immunity 49, 515–530 e515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TA, and Jiang WG (2009). Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta 1788, 872–891. [DOI] [PubMed] [Google Scholar]
- Nash AK, Auchtung TA, Wong MC, Smith DP, Gesell JR, Ross MC, Stewart CJ, Metcalf GA, Muzny DM, Gibbs RA, et al. (2017). The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 5, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, Rotter-Maskowitz A, Weiser R, Mallel G, Gigi E, et al. (2020). The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polk DB, and Peek RM Jr. (2010). Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer 10, 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, Kosciolek T, Janssen S, Metcalf J, Song SJ, et al. (2020). Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor DM, Dangana T, Sexton DJ, Fukuda C, Yelin RD, Stanley M, Bell PB, Baskaran S, Deming C, Chen Q, et al. (2021). Integrated genomic, epidemiologic investigation of Candida auris skin colonization in a skilled nursing facility. Nat Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Bhardwaj V, Arrigoni L, Lam KC, Gruning BA, Villaveces J, Habermann B, Akhtar A, and Manke T (2018). High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Garcia A, Rementeria A, Aguirre-Urizar JM, Moragues MD, Antoran A, Pellon A, Abad-Diaz-de-Cerio A, and Hernando FL (2016). Candida albicans and cancer: Can this yeast induce cancer development or progression? Crit Rev Microbiol 42, 181–193. [DOI] [PubMed] [Google Scholar]
- Ricciardi M, Zanotto M, Malpeli G, Bassi G, Perbellini O, Chilosi M, Bifari F, and Krampera M (2015). Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br J Cancer 112, 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. (2018). Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97. [DOI] [PubMed] [Google Scholar]
- Saheb Kashaf S, Proctor DM, Deming C, Saary P, Holzer M, Program NCS, Taylor ME, Kong HH, Segre JA, Almeida A, et al. (2022). Integrating cultivation and metagenomics for a multi-kingdom view of skin microbiome diversity and functions. Nat Microbiol 7, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R, Fuchs S, and Milo R (2016). Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol 14, e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, and Knight R (2021). The microbiome and human cancer. Science 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiao SL, Kershaw KM, Limon JJ, You S, Yoon J, Ko EY, Guarnerio J, Potdar AA, McGovern DPB, Bose S, et al. (2021). Commensal bacteria and fungi differentially regulate tumor responses to radiation therapy. Cancer Cell 39, 1202–1213 e1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, Landman C, Cohen D, Liguori G, Bourrier A, Nion-Larmurier I, et al. (2017). Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler AP, Miller RD, Laughlin KV, Carp NZ, Klurfeld DM, and Mullin JM (1999). Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis 20, 1425–1431. [DOI] [PubMed] [Google Scholar]
- Spencer CN, McQuade JL, Gopalakrishnan V, McCulloch JA, Vetizou M, Cogdill AP, Khan MAW, Zhang X, White MG, Peterson CB, et al. (2021). Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 374, 1632–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerbaum S, and Michetti P (2002). Helicobacter pylori infection. N Engl J Med 347, 1175–1186. [DOI] [PubMed] [Google Scholar]
- Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, Narushima S, Vlamakis H, Motoo I, Sugita K, et al. (2019). A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605. [DOI] [PubMed] [Google Scholar]
- Tipton L, Muller CL, Kurtz ZD, Huang L, Kleerup E, Morris A, Bonneau R, and Ghedin E (2018). Fungi stabilize connectivity in the lung and skin microbial ecosystems. Microbiome 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalsma H, Boleij A, Marchesi JR, and Dutilh BE (2012). A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol 10, 575–582. [DOI] [PubMed] [Google Scholar]
- Vergara D, Simeone P, Damato M, Maffia M, Lanuti P, and Trerotola M (2019). The Cancer Microbiota: EMT and Inflammation as Shared Molecular Mechanisms Associated with Plasticity and Progression. J Oncol 2019, 1253727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogtmann E, and Goedert JJ (2016). Epidemiologic studies of the human microbiome and cancer. Br J Cancer 114, 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MA, Pedamallu CS, Ojesina AI, Bullman S, Sharpe T, Whelan CW, and Meyerson M (2018). GATK PathSeq: a customizable computational tool for the discovery and identification of microbial sequences in libraries from eukaryotic hosts. Bioinformatics 34, 4287–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Fan C, Yao A, Xu X, Zheng G, You Y, Jiang C, Zhao X, Hou Y, Hung MC, et al. (2018). The Adaptor Protein CARD9 Protects against Colon Cancer by Restricting Mycobiota-Mediated Expansion of Myeloid-Derived Suppressor Cells. Immunity 49, 504–514 e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CS (1980). Research on esophageal cancer in China: a review. Cancer Res 40, 2633–2644. [PubMed] [Google Scholar]
- Ye SH, Siddle KJ, Park DJ, and Sabeti PC (2019). Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 178, 779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeise KD, Woods RJ, and Huffnagle GB (2021). Interplay between Candida albicans and Lactic Acid Bacteria in the Gastrointestinal Tract: Impact on Colonization Resistance, Microbial Carriage, Opportunistic Infection, and Host Immunity. Clin Microbiol Rev 34, e0032320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai B, Ola M, Rolling T, Tosini NL, Joshowitz S, Littmann ER, Amoretti LA, Fontana E, Wright RJ, Miranda E, et al. (2020). High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo T, Wong SH, Cheung CP, Lam K, Lui R, Cheung K, Zhang F, Tang W, Ching JYL, Wu JCY, et al. (2018). Gut fungal dysbiosis correlates with reduced efficacy of fecal microbiota transplantation in Clostridium difficile infection. Nat Commun 9, 3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Fungal DNA is present in multiple cancer types not explained by contamination, Related to Figure 1
(A) Boxplot showing the distribution of reads per million (RPM) of fungal DNA detected in tumor and tumor-associated tissue samples from head-neck (HNSC), lung (LUSC), rectum (READ), colon (COAD), stomach (STAD), breast (BRCA), esophageal (ESCA) and brain (LGG) cancers, ordered by upper quartiles.
(B) Multi-well aliquot plate A19H shows significant signs of contamination; samples shown in orange were discarded.
(C) The distribution of sequencing reads aligning to the M. restricta genome displays similar depth across sequencing projects including brain, but reads are distributed randomly, a signature of biological contamination.
(D) The distribution of sequencing reads aligning to the A. bisporus genome displays uneven depth, but reads are horizontally distributed in a predictable manner, a signature of false-positive assignments.
(E) Re-analysis of human-subtracted sequencing data using TaxaTarget (Commichaux et al., 2021) validates the presence of Candida spp, S. cerevisiae, and Malassezia spp. across TCGA samples. R- and p-values indicate the result of a spearman rank correlation test.
Figure S2. Primary tumor samples harbor disease-specific mycobiomes, Related to Figure 2
(A) Prevalence of the top 10 fungal species detected in tissue from each cancer type, after removing potential contaminants.
Figure S3. Trans-kingdom analysis reveals Candida- and Saccharomyces-associated GI cancer co-abundance groups, Related to Figure 3
(A – C)SparCC correlations between Candida and Malassezia and the most abundant bacterial species found in matched tumor samples from TCMA in head-neck (HNSC; A), stomach (STAD; B), and colon (COAD; C) cancers.
(D – F) Scatterplots and fitted regression lines showing correlations between Candida, Saccharomyces, and Lactobacillus in head-neck (HNSC; D), stomach (STAD; E), and colon (COAD; F) cancers. R- and p-values indicate the result of a spearman rank correlation test.
Figure S4. Candida is associated with late-stage and metastatic GI cancers, Related to Figure 4
(A) Kernel density estimation (KDE) of Candida-to-Saccharomyces ratios in esophageal (ESCA), rectum (READ), breast (BRCA), and brain tumors.
(B) Volcano plot showing genes differentially expressed Saccharomyces-negative (blue) and Saccharomyces-high (red) tumor samples head-neck, stomach, and colon cancers.
(C) Boxplots depicting the ratio of Candida-to-Saccharomyces in early-stage (I-III) and late-stage (IV)
(D) KDE analysis showing the distribution of Candida-to- Saccharomyces ratios in metastatic (orange) and non-metastatic (blue) tumor samples across cancer types.
Figure S5. Alive and transcriptionally active Candida species are associated with GI tumors, Related to Figure 5
(A) Spatial distribution of Ascomycota and Basidiomycota abundance along the colorectal tract.
Figure S6. Cancer-associated fungal mycobiota and clinical outcomes highlight predictive value of Candida, Related to Figure 7
(A) Volcano plot showing differential abundance of genera between tumor (blue) and matched adjacent normal tissue (yellow) in lung cancer. Blastomyces was enriched in lung tumors compared to matched adjacent normal tissue.
(B) Across GI cancer types, comparison of survival between patients with high rates of Saccharomyces in their tumors compared to patients whose tumors are negative for this taxon.
Table S1. Identification and removal of fungal contamination in TCGA sequencing data, Related to Figure 1
Screening of TCGA mycobiomes identifies contaminant and false-positive species and genera, and multi-well sequencing plate IDs with significant evidence of contamination. This table includes a list of the most prevalent contaminants/false-positives identified, as well as a list of genomes used in the genome-based QC models.
Table S2. Pan-cancer fungal microbiomes, Related to Figures 1–7
Normalized (eRPKM) and filtered fungal compositions from each TCGA cancer type, as well as associated metadata, including Ca/Sa tumor status. This table also includes a list of fungal genomes included in the PathSeq reference.
Table S3. False-discovery rate adjustments, Related to Figures 1–7
False-discovery rate (FDR) adjusted p-values for statistical tests performed, where not already indicated in the text or figures.
Data Availability Statement
This paper analyzes existing, publicly available data from TCGA and HCMI. Information for accessing these datasets can be found in the key resources table.
Original code for generating fungal compositions from TCGA and HCMI datasets has been deposited at https://github.com/abdohlman/tcma_code and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Original TCGA tissue | Indivumed | N/A |
| Colon cancer mucosal samples (adenocarcinoma) | Weill Cornell Medicine | N/A |
| Critical Commercial Assays | ||
| Sabouraud dextrose broth | VWR | Cat# 89406–400 |
| Sabouraud 4% dextrose agar | VWR | Cat# EM1.05438.0500 |
| Glycerol monostearate (Alfa Aesar) | Thermo Fisher Scientific | Cat# AA4388330 |
| modified Dixon (mDixon) | ATCC protocol | N/A |
| Deposited Data | ||
| TCGA WGS bam files | GDC API (Legacy) | https://portal.gdc.cancer.gov/legacy-archive |
| TCGA RNA-seq bam files | GDC API | https://api.gdc.cancer.gov/ |
| HCMI RNA-seq bam files | GDC API | https://api.gdc.cancer.gov/ |
| TCGA sequencing metadata | GDC API | https://api.gdc.cancer.gov/ |
| TCGA sample metadata (biotab) | GDC web portal | https://portal.gdc.cancer.gov/ |
| TCGA patient metadata | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| TCGA clinical data resource outcomes | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Human and microbe reference genomes | PathSeq bundle | ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/pathseq/ |
| Human and microbe reference genomes | PathSeq bundle | ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/pathseq/ |
| TCGA mRNA-seq data | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Gene sets for GSEA (KEGG) | MSigDB v7.1 | https://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp |
| TCMA bacterial profiles (COAD, READ, HNSC, ESCA, STAD) | TCMA database | https://doi.org/10.7924/r4rn36833 |
| Software and Algorithms | ||
| GATK 4.2.0 (PathSeq) | (Walker et al., 2018) | https://github.com/broadinstitute/gatk/ |
| TaxaTarget | (Commichaux et al., 2021) | https://github.com/SethCommchaux/taxaTarget |
| SAMtools v1.9, v1.14 | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| Burrows-Wheeler Aligner | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/ |
| BEDTools | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/ |
| deepTools2 | (Ramirez et al., 2016) | https://deeptools.readthedocs.io/ |
| pyGenomeTracks | (Lopez-Delisle et al., 2021) | https://github.com/deeptools/pyGenomeTracks |
| phyloseq 1.30.0 | (McMurdie and Holmes, 2013) | https://github.com/joey711/phyloseq |
| metacoder 0.3.3 | (Foster et al., 2017) | https://grunwaldlab.github.io/metacoder_documentation/ |
| STAR 2.7.3a | (Dobin et al., 2013) | https://github.com/alexdobin/STAR/ |
| SparCC | (Friedman and Alm, 2012) | https://bitbucket.org/yonatanf/sparcc/src/default/ |
| lifelines 0.23.8 | (Davidson-Pilon et al., 2020) | https://github.com/CamDavidsonPilon/lifelines/tree/0.24.6 |
| GSEA 4.0.3 | (Subramanian et al., 2007) | https://www.gsea-msigdb.org/gsea/ |
| Other | ||
| Decontamination analysis | This paper | Table S1, Related to Figure 1 |
| Fungal composition of TCGA samples | This paper | Table S2, Related to Figures 1–7 |
| False-discovery rate adjustments | This paper | Table S3, Related to Figures 1–7 |