

Supplemental Digital Content is Available in the Text.
Reduced spinal cord blood flow leads to a hypoxia-mediated activation of hypoxia inducible factor 1α in dorsal horn neurons. This results in a carbonic anhydrase–dependent neuropathic pain.
Keywords: Pain, Neuron, Endothelial, Spinal cord, Hypoxia, Diabetes, VEGF
Abstract
Neuropathic pain, such as that seen in diabetes mellitus, results in part from central sensitisation in the dorsal horn. However, the mechanisms responsible for such sensitisation remain unclear. There is evidence that disturbances in the integrity of the spinal vascular network can be causative factors in the development of neuropathic pain. Here we show that reduced blood flow and vascularity of the dorsal horn leads to the onset of neuropathic pain. Using rodent models (type 1 diabetes and an inducible endothelial-specific vascular endothelial growth factor receptor 2 knockout mouse) that result in degeneration of the endothelium in the dorsal horn, we show that spinal cord vasculopathy results in nociceptive behavioural hypersensitivity. This also results in increased hypoxia in dorsal horn neurons, depicted by increased expression of hypoxia markers such as hypoxia inducible factor 1α, glucose transporter 3, and carbonic anhydrase 7. Furthermore, inducing hypoxia through intrathecal delivery of dimethyloxalylglycine leads to the activation of dorsal horn neurons as well as mechanical and thermal hypersensitivity. This shows that hypoxic signalling induced by reduced vascularity results in increased hypersensitivity and pain. Inhibition of carbonic anhydrase activity, through intraperitoneal injection of acetazolamide, inhibited hypoxia-induced pain behaviours. This investigation demonstrates that induction of a hypoxic microenvironment in the dorsal horn, as occurs in diabetes, is an integral process by which neurons are activated to initiate neuropathic pain states. This leads to the conjecture that reversing hypoxia by improving spinal cord microvascular blood flow could reverse or prevent neuropathic pain.
1. Introduction
Chronic pain is a significant burden faced by patients, with pain not restricted to a single condition but developing because of an array of differing health and disease-related afflictions. Forty-three percent of the United Kingdom17 and 25% of the global population20 suffer from chronic pain, which can arise either idiopathically, as a consequence of another disease (eg, diabetes68), or as a consequence of therapyo (eg, chemotherapy46). Neuropathic pain arises due to damage to the somatosensory sensory system with the modulation of intrinsic sensory neural circuits fundamental to the maintenance of nociception4 and presented as a hypersensitivity to evoked sensory stimuli (eg, allodynia or hyperalgesia) as well as ongoing pain18,30. There is a severe lack of effective analgesic management in the clinic.6
Dorsal horn neurons possess the ability to adapt to neuronal stress through key mechanisms that underlie maladaptive plasticity32 A balance between excitatory and inhibitory processing is maintained, but during chronic pain, excitatory pathways predominate40 with GABAergic excitation of a contributing factor.33,51 How these sensory neuronal networks alter in times of stress is unknown. Neural tissues are susceptible to depleted oxygen availability, although they can reprogram metabolic pathways to support continued neuronal function.29 Perturbed vascular function accompanies the development of neurodegenerative conditions such as Alzheimer disease,45 and a causal relationship between peripheral limb ischaemia and onset of pain has been shown.22,39 Previous work has explored systems' level modulation of blood flow and the resulting impact on nociceptive processing and modulation of pain behaviours.7,70 Reduced dorsal horn spinal cord blood flow in a rodent model of diabetes, potentially due to vascular rarefaction, is associated with the onset of pain hypersensitivity.70 Vascular integrity is mediated by the vascular endothelial growth factor (VEGF) family of proteins, signalling through VEGF receptor 2 (VEGFR2). Vascular endothelial growth factor is a master regulator of angiogenic function, with fluctuations in VEGF expression associated with distinct endothelial cell activity relating to physiological function and pathogenesis. High VEGF expression and VEGFR2-mediated signalling drives aberrant vascular expansion,24,65 whereas diminished VEGFR2 signalling leads to vascular pruning.31 Interestingly, a systemic endothelial specific knockout of VEGFR2, predicted to reduce vascular density in the spinal cord, reduced nociceptive behavioural hypersensitivity in an animal model of arthritis, by reducing spinal cord microglial activation.7 We, therefore, set out to determine whether vascular rarefaction in the spinal cord per se initiates neuropathic pain phenotypes.
Here we show that the spinal cord microvasculature is critical in maintaining sensory neuronal modulation and demonstrate that alterations in vascular support to the dorsal horn contribute to diabetic neuropathic pain development. Type I diabetic and transgenic rodent models (endothelial-specific VEGFR2 knockout) of reduced spinal cord vascularity demonstrate that localised vascular degeneration induces a hypoxic microenvironment in the dorsal horn, leading to neuron activation and initiation of neuropathic pain. The initiation of neuropathic pain is dependent on the hypoxia response, in particular the upregulation of hypoxia inducible genes and the resultant change in bicarbonate metabolism, which can be prevented by carbonic anhydrase inhibition. These results show a potential new therapeutic strategy for analgesia development.
2. Methods
2.1. Ethical approval and animals used
All experiments using animals were performed in accordance with the UK Home Office Animals (Scientific Procedures) Act 1986 and EU Directive 2010/63/EU, in line with the ARRIVE guidelines, with experimental design and procedures reviewed by the local Animal Welfare and Ethics Review Boards (University of Nottingham and Nottingham Trent University). Animals had ad libitum access to standard chow and were housed under 12:12 hours light:dark conditions.
2.2. Rodent models of diabetes
Sprague–Dawley female rats (∼250 g) and Db/db mice were purchased from Charles River. Db/db mice are rodent models of type II diabetes, which have impaired leptin receptor signalling to induce obesity and hyperglycaemia (Charles River; vs lean controls). Streptozotocin-induced rodent model of type 1 diabetes; Sprague–Dawley female rats were used and treated with either vehicle (saline; naive Group, n = 15) or streptozotocin (50 mg/kg; STZ in saline; n = 15) administered by intraperitoneal injection. Hyperglycaemia was determined as >15 mmol in samples of blood extracted by tail prick (end of study week 8). Under isoflurane anaesthesia (∼2% oxygen), one third of an insulin pellet (LinShin, Canada) was implanted in the scruff of the neck using a trocar, in STZ-treated animals. Experimental group animal body weights and blood glucose measurements were recorded during the study. Measures (mean ± SEM) recorded at the end of the study (week 8) are presented below: blood glucose (mmol/dL): naive = 7.44 ± 0.29, diabetic =28.5 ± 1.82, animal body weight (grams): naive = 330.6 ± 4.49, and diabetic = 284.2 ± 4.86.
2.3. Transgenic mice
Tie2CreERT2 mice (European Mutant Mouse Archive Tg [Tek-cre/ERT2] 1Arnd) were interbred with homozygous vegfr2fl/fl mice (as previously described).2,7,63 This generates tamoxifen inducible endothelial-specific vegfr2 knockout adult mice. Mice (both genders) used were vegfr2fl/fl and were either Tie2CreERT2 negative (n = 83, termed WT) or positive (n = 83, termed VEGFR2CreERT2). Mice were treated with either intraperitoneal injection of 1 mg tamoxifen per mouse (10% ethanol in sunflower oil) once daily for 5 days consecutively (n = 116)7,70 referred to as VEGFR2ECKO or a single intrathecal injection of 1 μM 4-hydroxytamoxifen (OHT; 10% ethanol in sunflower oil) (n = 34) referred to as VEGFR2scECKO. The remaining 16 mice were used for endothelial cell culture. All transgenic animals were taken 8 days after first tamoxifen or OHT injection. All intrathecal injections were performed under brief isoflurane anaesthesia (∼2% oxygen).
2.4. Chemically induced hypoxia
For the hypoxia mimetic study, 1 mM dimethyloxalylglycine (DMOG, in PBS, Tocris, 22 mice) or vehicle (16 mice) was intrathecally injected into C57Bl6 adult male mice (total 38) under recovery anaesthesia (isoflurane ∼2% oxygen). Acetazolamide (ACZ) was delivered through intraperitoneal injection (40 mg/kg, in 1% DMSO in PBS, Tocris).
2.5. Intravital imaging
C57Bl6 male mice (∼30 g; n = 12) were deeply terminally anaesthetised and anaesthesia maintained (ip ketamine and medetomidine). Body temperature was maintained (∼ 37°C) using a feedback-controlled heating blanket with a rectal probe. Midline incision was made along the thoracic–lumbar region of the mouse, and muscle was dissected from the vertebrae. A laminectomy was performed between thoracic (T10) and lumbar vertebra (L5) to expose the lumbar spinal cord. In house stainless steel spinal vertebrae clamps were used to stabilise the vertebrae while an in house window chamber was attached to the spinal cord vertebra.16 The spinal cord hydration was maintained with physiological saline before the application of Kwik-Sil silicone elastomer (World Precision Instruments) and a 5 mm coverslip number 1 (Harvard Apparatus). Following this, the mouse was placed on to the confocal microscope (Leica SPE) and images were captured by 10x objective. Mice were administered Alexa Fluor 555 Conjugated Wheat Germ Agglutinin (1 mg/mL, Thermo Scientific) by tail vein injection. On identification of the endothelium, sodium fluorescein (i.p. 100 mg/mL) was administered intraperitoneally.3 Image acquisition began immediately after sodium fluorescein injection. Spinal cords were treated with either vehicle (PBS) or 2.5 nM VEGF-A165a with or without VEGFR2 inhibitor ZM323881 (100 nM) for 5 minutes before fluorescein injection (as described by Wardlaw et al.72). Sodium fluorescein permeability was measured across multiple microvessels, as previously described in the mesentery,9 which involved determining a region of interest extending across the vessel width and encompassing the region outside the vessel wall with solute permeability determined. Animals were killed by anaesthetic overdose on completion of the study.
2.6. Nociceptive behaviour
Animals were habituated to the testing environment before nociceptive behavioural experimentation, which were performed as previously described.70 Mechanical withdrawal thresholds were determined by application of von Frey (vF) monofilaments to the hind paw plantar surface. von Frey filaments of increasing diameter were applied (a maximum of 5 seconds or until paw withdrawal), with each vF filament applied a total of 5 times to generate force response curves. Withdrawal frequencies of 50% at each weight were used to determine the mechanical withdrawal threshold (grams).8 The Hargreaves test was performed to measure thermal nociceptive behaviour.21 A radiant heat source was focused on the plantar surface of the hind paw. The time taken for the mouse to withdraw their paws was recorded. The latency to hind paw response was measured 3 times. For motor behavioural assessment, mice were placed in Perspex enclosures and were left undisturbed for 5 minutes while being video recorded to explore the environment.48 After experimentation, animals were returned to housing cages and data were analysed offline. All assays were undertaken by observers blinded to treatment or genotype. Animals were killed by perfusion fixation.
Animals that had been terminally anaesthetised with sodium pentobarbital (60 mg/kg, Sigma-Aldrich) and transcardially perfused with PBS followed by 4% PFA (pH7.4) had tissue dissected, cryoprotected (30% sucrose), and stored at −80°C until spinal cords were cryosectioned (40 μm thickness).70 Slides were incubated in primary antibodies (Table 1) in blocking solution (5% bovine serum albumin, 10% fetal calf serum, and 0.2% Triton-X) at 4°C for 72 hours and then washed in PBS. Slides were subsequently incubated in secondary antibodies in PBS + 0.2% Triton X-100 at room temperature for 2 hours. Confocal microscopy of the dorsal horn of the lumbar region of the spinal cord was performed using a Leica SPE confocal microscope.
Table 1.
List of primary and secondary antibodies for immunofluorescence.
| Biotin-conjugated isolectin B4 (IB4; 1:100, Sigma Aldrich) |
| PECAM (1:100, Santacruz) |
| Rabbit anti-CD31 (1:00, Abcam) |
| Chicken anti-NeuN (1:500, Synaptic systems) |
| Chicken anti Map2 (1:500, Millipore) |
| Rabbit FI10:13C-conjugated antihypoxyprobe (1:200, Hypoxyprobe, Omni kit) |
| Mouse anti-CD31 (1:100, Abcam) |
| Rabbit anti-fos (1:100, Santa Cruz) |
| Mouse anti-NeuN (1:200, Millipore) |
| Guinea pig anti-NeuN (1:100, Synaptic Systems) |
| Mouse anti-Hif1a (1:100, BD Bioscience) |
| Rabbit anti-collagen IV (1:200; Abcam) |
| Glut3 (1 in 200: Alomone) |
| Biotinylated anti-rabbit IgG (1:500, Jackson Laboratories) (for-cfos) |
| Secondary antibodies (1:500, all Invitrogen, United Kingdom) used were Alexa Fluor 488-conjugated chicken anti-mouse, Alexa Fluor 555-conjugated donkey anti-rabbit, and streptavidin-conjugated Alexa Fluor-555 |
GLUT3, glucose transporter 3; HIF1α, hypoxia-inducible factor 1α.
2.7. Hypoxyprobe assay
Hypoxyprobe staining was performed as outlined in the manufacturer's instructions (Hypoxyprobe Omni Kit).58 Fiveik Tie2CreERT2 vegfr2fl/fl (VEGFR2scECKO) mice and 5 vegfr2fl/fl (WT) were used 8 days after intrathecal injection of hydroxytamoxifen was used, as well as 5 STZ-induced diabetic and 5 vehicle-treated naive rats were used. Experimental animals were administered 60 mg/kg hypoxyprobe by intraperitoneal injection. Animals were terminally anaesthetised with sodium pentobarbital (60 mg/kg, Sigma-Aldrich) 30 minutes later and transcardially perfused with PBS followed by 4% paraformaldehyde (PFA; pH7.4). Lumbar spinal cords were extracted and cryosectioned as outlined above. Anti-pimonidazole rabbit antisera (PAb2627AP) was added to slides at 1 in 200 dilution overnight at 4°C and subsequently incubated in Alexafluor-555-conjugated donkey anti-rabbit in PBS + 0.2% Triton X-100 at room temperature for 2 hours. Confocal imaging of the dorsal horn of the lumbar spinal cord was performed on a Leica TCS SPE confocal microscope.
2.8. Tetramethylrhodamine isothiocyanate dextran perfusion assay
Five Tie2CreERT2 vegfr2fl/fl (VEGFR2scECKO) mice and 5 vegfr2fl/fl (WT) mice were terminally anaesthetised 8 days after intrathecal administration of 1 μM hydroxytamoxifen; and anaesthesia was maintained with sodium pentobarbital (60 mg/kg, Sigma-Aldrich). The external jugular vein was cannulated, and 1 mg per 20 g tetramethylrhodamine isothiocyanate (TRITC) dextran (76 Mw; Sigma-Aldrich) was injected.47,69 Mice were monitored for 30 minutes after TRITC dextran infusion. Subsequently animals were transcardially perfused with 4% PFA (pH 7.4), and lumbar spinal cords were extracted, and samples were prepared for immunofluorescent confocal imaging as outlined previously.
2.9. Immunocytochemistry
Spinal cord neurons were isolated as outlined above and plated on glass coverslips coated in poly-l-lysine (24 hours, Sigma-Aldrich) followed by laminin (5 mg/mL 2 hours, Sigma-Aldrich). They were cultured in media for 24 hours and subsequently washed in PBS, before incubation in 4% PFA for 15 minutes at room temperature. Neurons were permeabilised with 0.4% Triton X-100 in PBS for 15 minutes at room temperature. Cells were incubated in blocking solution (5% bovine serum albumin, 10% fetal calf serum, and 0.2% Triton-X), mixed gently, and incubated for 30 minutes at room temperature. Cells were incubated in primary antibodies for identification of neurons (chicken anti-MAP2, SySy) overnight at 4OC. Cells were subsequently washed in PBS and secondary antibodies (Alexa Fluor 555-conjugated donkey anti-mouse and Alexa Fluor 488-conjugated donkey anti-chicken) added for 2 hours at room temperature. Glass coverslips were mounted on slides using Vector Shield (H-1000, Vector Labs).
2.10. qPCR method
Whole lumbar spinal cord lysates (5 Tie2CreERT2 vegfr2fl/fl [VEGFR2scECKO] mice and 5 vegfr2fl/fl [WT] mice) and isolated lumbar spinal cord neurons (5 C57.bl6 mice) were used (killed via cervical dislocation) to extract RNA in experimental groups using TRIzol reagent (Invitrogen) from 10 C57.bl6 mice. One microgram RNA was reverse transcribed to cDNA using PrimeScriptRT reagent kit (TaKaRa, RR037A). Quantitative PCR was performed using a LightCycler 480 SYBR Green I Mastermix (Roche 04707516001) following the manufacturer's instructions. All primers were obtained from Eurofins and outlined in Table 2. All other primers were obtained from Sigma-Aldrich. The reference gene used was β-actin. The primer sequences were as follows (5′-3′).
Table 2.
List of primers used.
| B-actin F – 5′ ATTGCCAATGAGCGGTTC-3′ |
| B-actin R–5′ GGATGCCACAGGACTCCA-3′ |
| CD31 F–5′ GAAATGCTCTCGAAGCCCAG-3′ |
| CD31 R–5′ ACCTCGAGAGTCTGGAAGTC-3′ |
| VE-cadherin F–5′ TCCCTGGACTATGAAGTCAT-3′ |
| VE-cadherin R–5′ GAAGACAGGGGGCTCATCCA-3′ |
| VEGFR2 F–5′ GGATCTGAAAAGACGCTTGG-3′ |
| VEGFR2 R–5′ TGCTCCAAGGTCAGGAAGTC-3′ |
| Carbonic anhydrase II-F–5′ CAAGCACAACGGACCAGA-3′ |
| Carbonic anhydrase II-R–5′ ATGAGCAGAGGCTGTAGG-3′ |
| Carbonic anhydrase VII F–5′ CAATGACAGTGATGACAGAA-3′ |
| Carbonic anhydrase VII R–5′ TCCAGTGAACCAGATGTAG-3′ |
| Carbonic anhydrase F–5′ CTGAAGACAGGATGGAGAAG-3′ |
| Carbonic anhydrase R–5′ GCAGAGTGCGGCAGAATG-3′ |
| GLUT1 F–5′ CATCCTTATTGCCCAGGTGTTT-3′ |
| GLUT1 R–5′ GAAGACGACACTGAGCAGCAGA-3′ |
| GLUT3 F: 5′ GGAGGAAGACCAAGCTACAGAG-3′ |
| GLUT3 R–5′ GAGCTCCAGCACAGTCACCT-3′ |
| HIF1-alpha F–5′ TGCTCATCAGTTGCCACTTC-3′ |
| HIF1-alpha R–5′ TCCTCACACGCAAATAGCTG-3′ |
GLUT3, glucose transporter 3; HIF1α, hypoxia-inducible factor 1α; VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2.
2.11. Multielectrode arrays
The mouse lumbar spinal cord was extracted from 5 C57.Bl6 male mice (Charles River; killed by cervical dislocation) and maintained in neurobasal media (supplemented with B27 and L-Glutamine, Invitrogen). Lumbar spinal cord tissue was cut to 400 μm thickness and incubated on high-density microelectrode arrays (3Brain, 4096 electrodes). The neuronal electrical activity was recorded digitally by the Biocam X after 24 hours treatment with either vehicle or 1 mM DMOG application. Data were recorded offline using BrainWave analysis software.
2.12. Western blotting
Protein was extracted from lumbar spinal cord tissue collected as above and L3-L5 dorsal root ganglion as previously described8,70 from experimental animals (killed by cervical dislocation)–experimental groups (VEGFR2KO; 5 Tie2CreERT2 vegfr2fl/fl [VEGFR2scECKO] mice and 5 vegfr2fl/fl [WT] mice as well as intrathecal DMOG; 5 saline-treated mice and 5 DMOG-treated mice) studies compared with vehicle experimental controls as above. In additional experiments, protein lysate samples were also extracted from in vitro lumbar spinal cord slices prepared from 6 C57bl6 male adult mice and slices maintained in Neurobasal media (supplemented with L-Glutamine, Invitrogen) and exposed to vehicle or 1 mM DMOG for 24 hours. Tissue was lysed using RIPA buffer (ThermoFisher) containing protease inhibitor cocktail (20 μL/mL buffer; 1 mM phenylmethylsulfonyl fluoride, 10 mM sodium orthovanadate, and 50 mM sodium fluoride, Sigma Aldrich). Equal protein lysate concentrations were loaded on a 4% to 20% precast Mini-Protean gradient TGX gel (Biorad), separated by SDS-PAGE electrophoresis, and transferred using Trans-blot turbo transfer system (Bio-Rad) to a PVDF membrane. The membrane was incubated in 5% BSA in tris-buffered saline-Tween 0.1% (TBST) for 1 hour at room temperature, followed by incubation with primary antibodies (Table 3) overnight at 4°C. The membrane was then incubated in secondary antibodies in TBST-0.1% to 1% BSA. Membranes were washed 3 times with TBST and visualised on the Licor Odyssey Fc.
Table 3.
List of primary and secondary antibodies for Western blotting.
| HIF1-alpha anti-mouse (1 in 100, BD Bioscience) |
| Actin–Rabbit polyclonal anti-Beta-Actin (1 in 100 Santa Cruz) |
| Actin–Goat polyclonal anti-Beta-Actin (1 in 200 Santa Cruz) |
| Tubulin–anti-rabbit anti-α tubulin (1 in 1000, Cell Signalling, #2144) |
| ZO-1–rabbit anti-ZO1 (1 in 500, Invitrogen, Cat #61-7300) |
| Occludin–rabbit anti‐occludin (5 μg/mL, Invitrogen) |
| CD-31 mouse monoclonal anti-CD31 (2 μg/mL, Abcam; AB24590), |
| CA-7 rabbit (1 in 500, cell signalling, #PA5-103703) |
| VEGFR2–rabbit anti VEGFR2 (1 in 500, Cell Signalling, #2479) |
| Licor donkey anti-goat, anti-rabbit, and anti-mouse antibodies 1:10,000 |
HIF1α, hypoxia-inducible factor 1α; VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2.
2.13. Spinal cord endothelial cell culture
Whole lumbar spinal cords were dissected from 16 Tie2CreERT2 vegfr2fl/fl mice (culled by cervical dislocation), and spinal cord endothelial cells were isolated as previously described.70 Spinal cords were incubated in 1.25% collagenase in endothelial cell media (M199 media, 60 mg/mL endothelial cell growth supplement and 50 μg/mL Heparin; Sigma-Aldrich) for 30 minutes at 37°C, 5% oxygen. The cell suspension was centrifuged at 240 g for 5 minutes. The supernatant was discarded, and the pellet was resuspended in media with an equal volume of 1% BSA and centrifuged at 1200 g for 20 minutes. The BSA or myelin fraction was aspirated and discarded. The remaining cell pellet resuspended in endothelial cell media and plated onto a 96-well sterile plates coated with 1% gelatin. On 80% cell confluency, spinal cord endothelial cells were treated for 24 hours with either media supplemented with either vehicle (10% ethanol in sunflower oil) or OHT. Cell viability was determined with Neutral Red (Sigma-Aldrich).
2.14. Spinal-isolated neuronal cell culture
Spinal cord neurons were dissected as described by Brewer and Torricelli.10 Whole spinal cords were isolated from 10 C57Bl6 mice (Charles River; killed by cervical dislocation) and digested in papain (2 mg/mL, Worthington, cat. no. 3119) for 30 minutes at 30°C. The cell suspension was triturated using Pasteur pipette. To purify neurons, a density gradient separation preparation was performed using OptiPrep (Sigma-Aldrich) density gradient. The neuronal containing fraction was collected and centrifuged (1100 rpm 2 minutes) to obtain a pellet. The pellet was resuspended in Neurobasal media (supplemented with B27 and L-Glutamine, Invitrogen) and plated on sterile 6-well or 24-well plates coated with poly-D-Lysine (sterile lyophilized 135 kD; Sigma) and 1 mg/mL laminin. Cells were exposed to hypoxia (1% O2) or normoxia for 24 hours.
2.15. Statistical analysis
All data are represented as mean ± SEM stated, and the experimenter was blinded to group treatment during experimental procedures. Data were acquired or quantified using Microsoft Excel and GraphPad Prism 8. Quantification of immunofluorescence was performed by obtaining 5 random images from 5 random nonsequential sections (Z stacks) per animal and a mean value calculated per animal. Images were analysed using ImageJ (https://imagej.nih.gov/ij/) or Imaris 8.11 (Bitplane) to determine vessel diameter and volume. The dorsal horn neuronal number was determined according to lamina I-V as previously characterised.23 Densitometry analysis of Western blot was performed using gel quantification plugin in ImageJ. The mean fluorescence intensity was measured over time enabling visualisation of vessel perfusion and permeability. Nociceptive behaviour and the neuron number in the dorsal horn of the spinal cord were analysed using 2-way ANOVA with post hoc analysis. Western blot densitometry, QPCR, MEA recordings, and TRITC dextran perfusion were analysed using the Mann–Whitney U test. Video analysis of rodent motor behaviour was performed using MATLAB48 and analysed using the Mann–Whitney U test.
3. Results
3.1. Dorsal horn hypoxia induction
To determine whether increased diabetic neuropathic pain was associated with vascular loss and hypoxia, we used a rat model of type 1 (streptozotocin induced and insulin supplemented) that led to the development of mechanical (Fig. 1A) and heat (Fig. 1B) nociceptive behavioural hypersensitivity. This was associated with an increase in collagen sleeves (collagen positive microvessels, negative for IB4-labelled endothelium) in diabetic animals compared with age-matched vehicle control animals (Figs. 1C and D) indicative of loss of endothelium and vascular rarefaction. In addition, there was a significant increase in the number of hypoxic dorsal horn neurons (labelled with hypoxyprobe) in diabetic animals when compared with age-matched vehicle control animals (Figs. 1E and F; supplemental Figure 1A and 1B, available at http://links.lww.com/PAIN/B597) and a small decrease in the total number of identified neurons in the dorsal horn (NeuN labelled; Figs. 1G–I). These results indicate that hypoxia caused by loss of blood vessels could play a part in induction of diabetic neuropathic pain.
Figure 1.
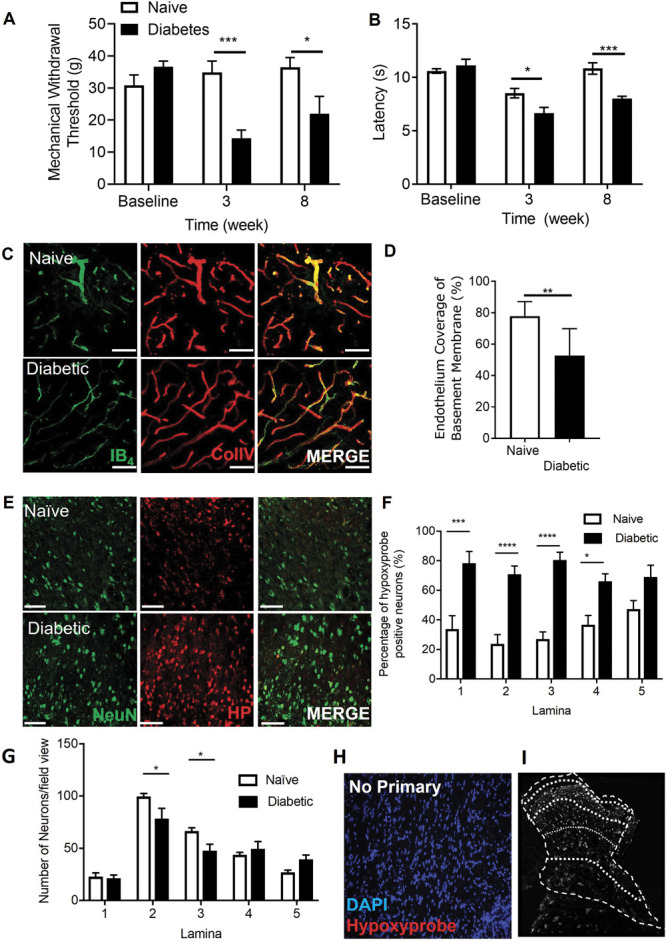
Dorsal horn hypoxia develops in a rodent model of type 1 diabetic neuropathic pain. Type 1 diabetes was induced in rats by intraperitoneal injection of streptozotocin supplemented with insulin. Diabetic rodents developed (A) decreases in mechanical withdrawal threshold and (B) reduced latencies to heat withdrawal compared with age-matched control animals (Naive). (C) Cryosections of the dorsal horn from naive and diabetic experimental groups were stained for basement membrane (Collagen IV) and endothelium (IB4). (D) In the diabetic dorsal horn, there was reduction in endothelial coverage of the basement membrane (ie, increased sleeves) compared with age-matched naive animals (n = 5 per group, ** P < 0.01 unpaired t Test). (E) Representative confocal microscopy images of hypoxyprobe (HP)-positive neurons (NeuN labelled) in the dorsal horn of the spinal cord in naive and diabetic cryosections (colabelled with NeuN, scale bar = 50 μm). (F) Mapping of the dorsal horn laminae demonstrates increased hypoxyprobe positive neurons or lamina in diabetic animals when compared with naive control animals (*P < 0.05, ***P < 0.001 2-way ANOVA with Tukey multiple comparison, n = 5 per group). (G) In the diabetic rat dorsal horn, there were reductions in neuronal number in lamina 2 and 3 of the dorsal horn when compared with naive Sprague–Dawley female rats controls (*P < 0.05 2-ay ANOVA with Tukey multiple comparison, n = 10 per group). (H) Representative image of images acquired after omission of primary antibody; negative control and mapping of (I) dorsal horn lamina.
We have previously shown that VEGFR2ECKO animals had no change in nociceptive processing compared with wild type animals and were actually resistant to hyperalgesia and allodynia induced by induction of arthritis. To determine whether these mice had a change in vessel density, hypoxia, and cell number, we stained the spinal cord of these animals after knockout induction with tamoxifen. VEGFR2ECKO animals showed an increase in the number of collagen sleeves (decrease in endothelial covered collagen positive vessels) compared with age-matched litter mate control wild type animals (Figs. 2A and B), indicating vascular rarefaction. Although VEGFR2ECKO did not affect the number of neurons (NeuN labelled) in the dorsal horn across lamina 1-5 (supplemental Figure 2A–C, available at http://links.lww.com/PAIN/B597). There was a significantly increase in the number of hypoxyprobe-labelled neurons in the dorsal horn compared with control animals (Figs. 2C and D) demonstrating hypoxia as a consequence of VEGFR2ECKO. VEGFR2ECKO mice had a lower withdrawal latency to heat when compared with WT mice (Fig. 2E).
Figure 2.
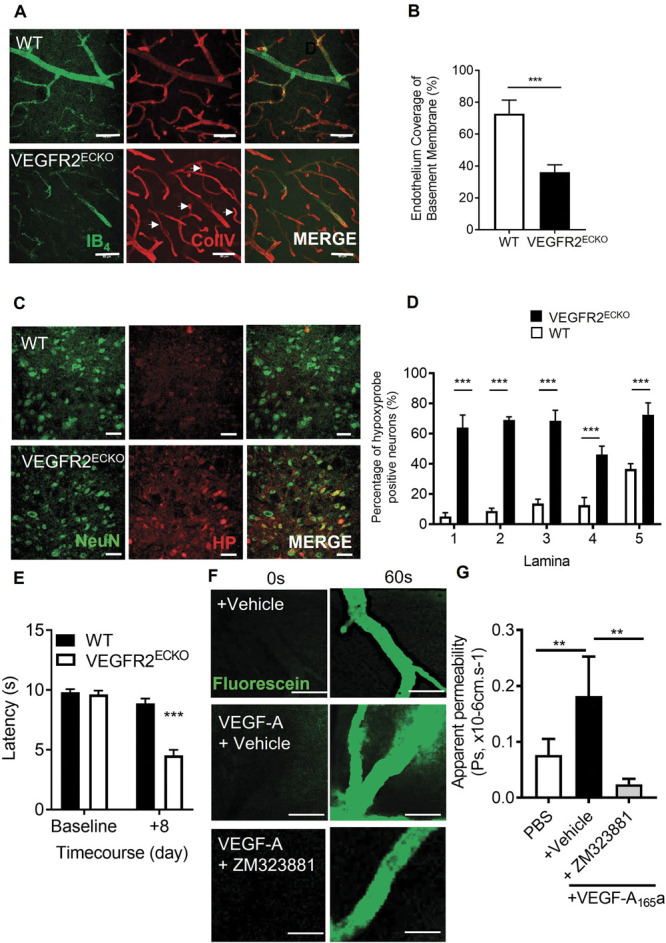
Vascular endothelial growth factor modulation of the spinal cord blood vasculature. (A) Dorsal horn cryosections were imaged by confocal microscopy to identify microvessels (Collagen IV–basement membrane–and IB4–endothelium, scale bar = 50 μm) in WT and VEGFR2ECKO mice, a systemic transgenic model that was induced with intraperitoneal administration of tamoxifen. (B) In VEGFR2ECKO mice, there was a decreased percentage of microvessels positive for both endothelium (IB4) and collagen IV (ie, increased sleeves) compared with WT mice (n = 5 per group, ** P < 0.01 unpaired t test). Both WT and VEGFR2ECKO mice were treated by intraperitoneal injection of hypoxyprobe. (C) Confocal microscopy of the dorsal horn was used to identify hypoxyprobe (HP)-labelled neurons (colabelled with NeuN, scale bar = 50 μm) in WT and VEGFR2ECKO mice. (D) VEGFR2ECKO mice had increased percentage of hypoxic neurons across dorsal horn laminae 1-5 compared with WT mice (*P < 0.05, **P < 0.01, ***P < 0.001 2-way ANOVA with Tukey multiple comparison, n = 5 per group). (E) Heat hypersensitivity developed in VEGF2ECKO mice when compared with WT controls (***P < 0.001 2-way ANOVA with Tukey multiple comparison, n = 10 per group). (F) To confirm VEGFR2 signalling in the spinal cord, microvessels were identified in the dorsal horn of the lumbar region of the spinal cord in C57 Bl6 mice. 100 mg/mL sodium fluorescein was administered by intraperitoneal injection, with data acquired by time lapse confocal imaging allowing for recording of solute permeability (Ps, scale bar = 20 μm). Representative images of fluorescein-filled microvessels in the spinal cord from experimental groups—PBS control and VEGF-A165a (VEGF-A65a, 2.5 nM) with either vehicle (0.01% DMSO in saline) or VEGFR2-specific antagonist ZM323221 (100 nM). (G) VEGF-A65a treatment led to increased fluorescein leakage from the dorsal horn microvessels (**P < 0.01 1-way ANOVA with Tukey multiple comparison, PBS n = 5 and VEGF-A65a +vehicle n = 6). Treatment with ZM323881 attenuated VEGF-A165–induced vascular leakage (**P < 0.01 1-way ANOVA with Tukey multiple comparison, VEGF-A165 + ZM323881 n = 7). VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2.
Blood vessel survival has previously been shown to require endogenous VEGFR2 activation in tissues including the oesophagus, skin, and salivary gland as well as in other tissues.27,34 However, it is not known whether VEGFR2 signalling is required for vessels in the spinal cord, which form a part of the blood–brain barrier. We therefore sought to determine whether spinal cord VEGFR2 signalling is intact in spinal cord microvessels. VEGF-A165a was administered directly to the dorsal horn of the spinal cord and vascular permeability measured using intravital imaging. VEGF-A165 increased solute permeability of the blood–spinal cord barrier when compared with vehicle control (Figs. 2F and G) which was blocked by treatment with VEGFR2 inhibitor ZM323881 (Figs. 2F and G).
3.2. Onset of chronic pain due to the induction of spinal cord restricted dorsal horn vasculopathy
The previous explored experimental models were systemic rodent models of vasculopathy, which means that pain processing could be altered in both the brain, and peripherally which could affect sensitivity. To determine whether spinal cord vascularity per se could regulate nociceptive processing, we developed a refined model that induced vasculopathy restricted to the spinal cord. Endothelial cells (CD31 positive) were extracted from WT and VEGFR2CreERT2 mice spinal cords (Fig. 3A). Cell viability was determined after OHT treatment. In a concentration-dependent manner, OHT treatment led to a significant reduction in endothelial cell viability in VEGFR2CreERT2 mice spinal cord endothelial cells when compared with WT mice spinal cord endothelial cells (Fig. 3B). To determine whether this was also apparent in vivo, spinal cord endothelial-specific VEGFR2 knockout (VEGFR2scECKO) mice were generated by intrathecal injection of OHT into the lumbar enlargement. This was confirmed by a reduction in VEGFR2 expression (Fig. 3C QPCR quantification and Fig. 3G. immunofluorescence) in the spinal cord of VEGFR2scECKO mice when compared with WT mice. In addition, endothelial markers VE-Cadherin (Fig. 3D), CD31 (Fig. 3E), and glucose transporter 1 (GLUT1, Fig. 3F) were also reduced in the spinal cord of VEGFR2scECKO mice when compared with WT mice. Furthermore, VEGFR2scECKO mice had reduced protein expression of endothelial markers (representative Western blot, Fig. 3H, and quantification of ZO1 [Fig. 3I], CD31 [Fig. 3J], and occluding [Fig. 3K]) in the spinal cord. Notably, there was no change in protein expression of these endothelial markers (ZO-1 and CD31) in the lumbar 3- 5 dorsal root ganglia of either WT or VEGFR2scECKO mice, showing that this induction was limited to the spinal cord (supplemental Figure 3, available at http://links.lww.com/PAIN/B597).
Figure 3.
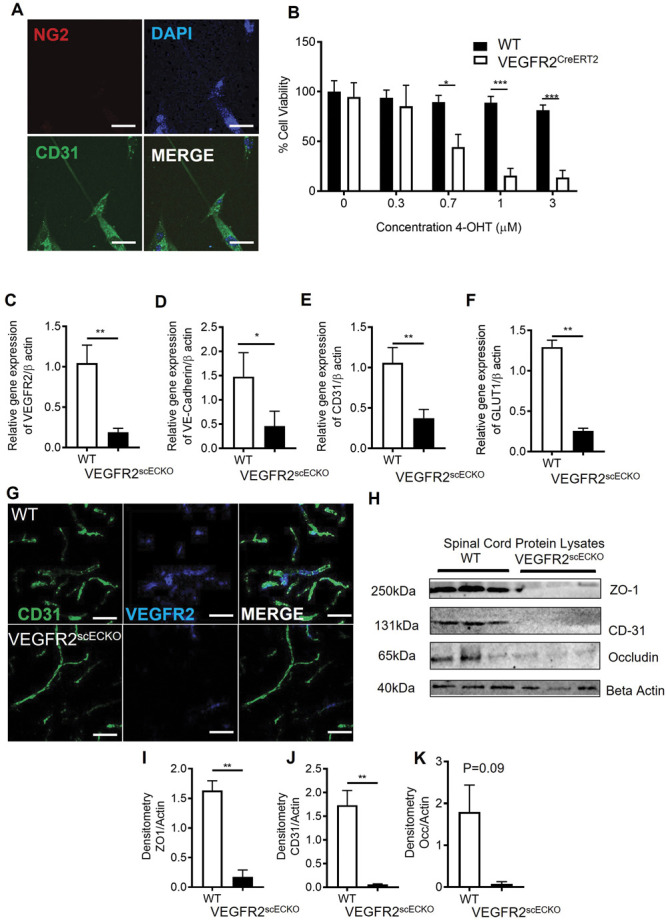
VEGFR2 knockout in the spinal cord induces localized vasculopathy. (A) Spinal cord endothelial cells (NG2−ve and CD31+ve) were isolated from WT and VEGFR2CreERT2 transgenic mice (scale bar = 10 μm), and (B) cell viability was determined after treatment with hydroxytamoxifen (OHT). Endothelial cells extracted from WT mice were unaffected by OHT treatment, whereas those from VEGFR2CreERT2 mice demonstrated a concentration dependent reduction in cell viability(*P < 0.05, ***P < 0.001 2-way ANOVA with Tukey multiple comparison, n = 3 per group). (C) Intrathecal injection of 1 μM OHT into the lumbar region of the spinal cord of VEGFR2CreERT2 mice (VEGFR2scECKO) led to reduced VEGFR2 mRNA expression in the lumbar spinal cord compared with similarly treated WT mice. There were reductions in mRNA expression of endothelial-specific markers (D) VE-Cadherin, (E) CD31, and (F) GLUT1 (*P < 0.05, **P < 0.01, unpaired t test, n = 5 per group). (G) VEGFR2 immunoreactivity was reduced in CD31-labelled microvessels (scale bar = 50 μm) in the dorsal horn of VEGFR2scECKO mice. (H) Representative immunoblots show reductions in protein expression of endothelial markers in the lumbar region of the spinal cord from VEGFR2scECKO mice. Densitometric quantification demonstrates reduced protein expression of (I) ZO1, (J) CD31, and (K) occludin in VEGFR2scECKO mice (**P < 0.01, unpaired t test, n = 5 per group). VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2.
Intrathecal OHT treatment led to significant vascular degeneration (Fig. 4A) of the microvasculature in the dorsal horn of VEGFR2scECKO mice, shown as reduced vessel diameter (Fig. 4B) and decreased volume of total vessel coverage (Fig. 4C). In addition, there was a reduction in dorsal horn vessel perfusion in VEGFR2scECKO mice indicated by decreased TRITC-Dextran–labelled vessels (Fig. 4D). This reduction in vessel perfusion was accompanied by an increase in hypoxyprobe-labelled neurons (Fig. 4F, hypoxyprobe labelled with NeuN, supplemental Figure 4A, available at http://links.lww.com/PAIN/B597) in laminae 1- 5 in the dorsal horn (Fig. 4G). There were no alterations in total neuron number in the dorsal horn in either WT or VEGFR2scECKO mice (supplemental Figure 4B and C, available at http://links.lww.com/PAIN/B597). VEGFR2scECKO mice had heat hypersensitivity (Fig. 4H) but no alterations in mechanical withdrawal thresholds (Fig. 4I) or motor behaviour (supplemental Figure 4D, available at http://links.lww.com/PAIN/B597). No change in heat withdrawal latency or mechanical withdrawal threshold was identified in WT mice after OHT treatment (Figs. 4H and I).
Figure 4.
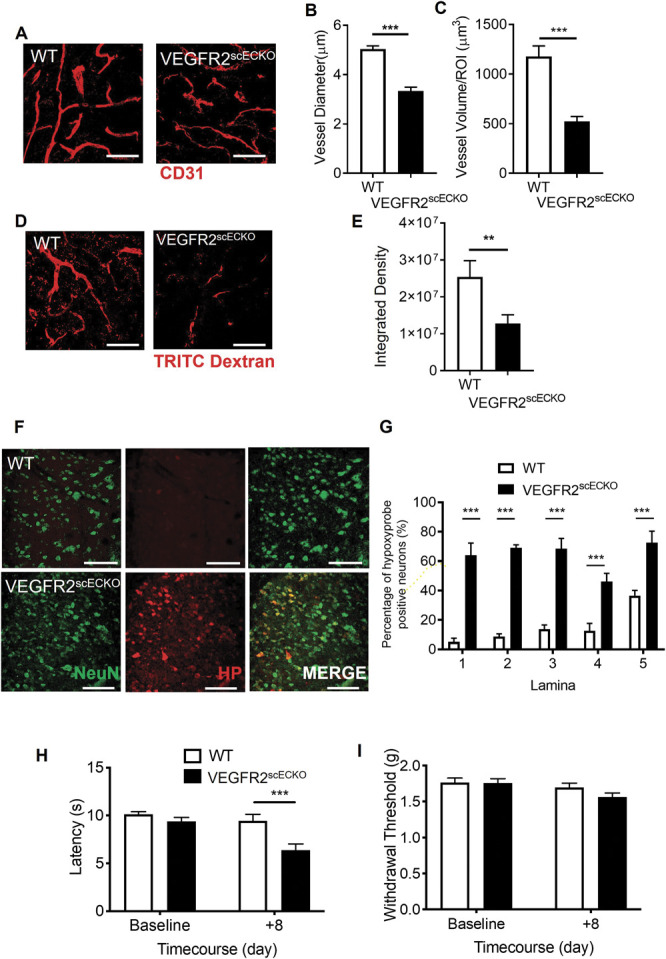
Spinal cord VEGFR2 knockout induced vasculopathy causes hypoxia and chronic pain. (A) VEGFR2scECKO mice have degeneration of the dorsal horn endothelium 8 days after intrathecal injection of hydroxytamoxifen (OHT), highlighted by the representative confocal images of the CD31-labelled endothelium (scale bar = 50 μm). OHT treatment led to (B) reduced vessel diameter and (C) decreased vessel volume in VEGFR2scECKO mice (*P < 0.05, ***P < 0.001, unpaired t test, n= 4 per group). (D) TRITC dextran was intravenously injected into terminally anaesthetized WT and VEGFR2scECKO mice and TRITC Dextran-positive perfused vessels identified as outlined in the representative confocal images (scale bar = 50 μm). (E) In VEGFR2scECKO mice, there was less TRITC Dextran in the dorsal horn compared with WT mice (**P < 0.01, unpaired t test, n= 5 per group). (F) Representative confocal images of hypoxyprobe-labelled neurons (colabelled with NeuN) in the dorsal horn of WT and VEGFR2scECKO mice (high-power images = 50 μm). (G) There was an increased percentage of neurons positive for hypoxyprobe, in each laminae 1- 5 across the dorsal horn in VEGFR2scECKO mice (***P < 0.001 2-way ANOVA with Tukey multiple comparison, n = 5 per group). (H) VEGFR2scECKO mice developed reduced withdrawal latencies to heat (***P < 0.001 2-way ANOVA with Tukey multiple comparison, n = 16 per experimental group). (I) No change to mechanical withdrawal threshold was observed (n = 16 per experimental group). TRITC, tetramethylrhodamine isothiocyanate; VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2.
3.3. Induction of hypoxia signalling in the dorsal horn neurons
VEGFR2scECKO mice demonstrated an increase in expression of hypoxia-inducible factor 1α (HIF1α) and glucose transporter 3 (GLUT3, supplemental Figure 5A and B, available at http://links.lww.com/PAIN/B597) in dorsal horn neurons (colabelled with neuronal marker NeuN, Fig. 5 A and 5B; supplemental Figure 5C and D, available at http://links.lww.com/PAIN/B597) and a significant increase in HIF1α mRNA (Fig. 5C) and that of hypoxia responsive genes carbonic anhydrase VII (CA7, Fig. 5D) and GLUT3 (Fig. 5E). Interestingly, increased HIF1α was also increased in a rodent type 1 (STZ induced, Fig. 5F-I) and type 2 (db/db obese mice, Fig. 5J) models of diabetes.
Figure 5.
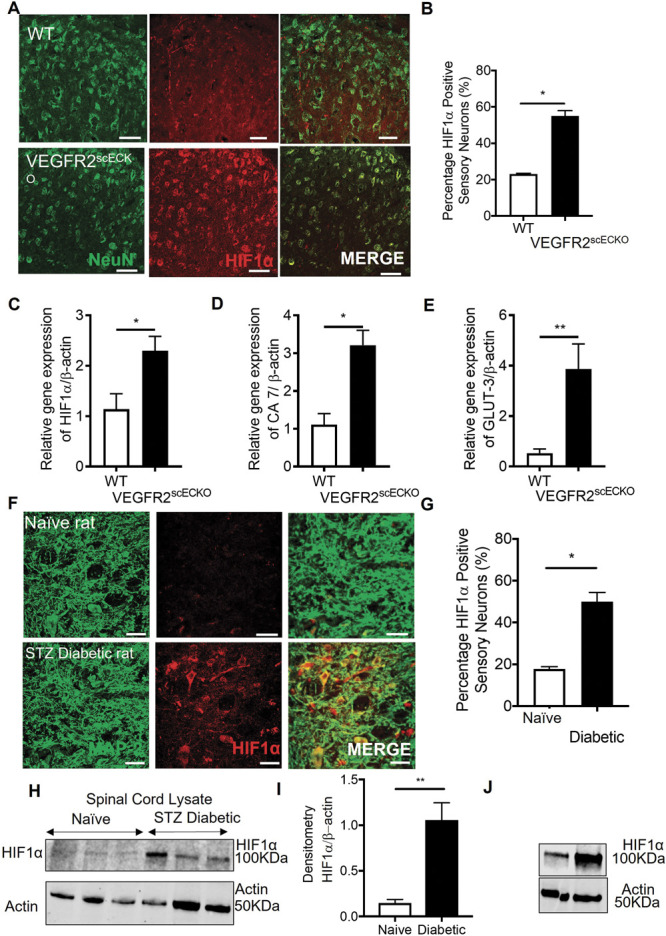
Spinal cord vasculopathy induces HIF1α expression in neurons in the dorsal horn. (A) 8 days after OHT treatment, HIF1α protein immunoreactivity was increased in neurons of the dorsal horn in VEGFR2scECKO mice (representative confocal images of WT and VEGFR2scECKO colabelled with NeuN, high-power images = 50 μm). (B) There was an increase in the percentage of dorsal horn neurons expressing HIF1α in VEGFR2scECKO (*P < 0.05, unpaired t test, n= 5 per group). mRNA quantification from lumbar spinal cord samples demonstrated increased expression of (C) HIF1α (*P < 0.05, unpaired t test, n= 5 per group), (D) carbonic anhydrase 7 (*P < 0.05, unpaired t test, n= 5 per group), and (E) glucose transporter 3 (**P < 0.01, unpaired t test, n= 5 per group) in VEGFR2scECKO mice. (F) In a rat model of type 1 diabetes and age-matched controls, HIF1α protein immunoreactivity was determined in the dorsal horn neurons (representative confocal images of naive and diabetic colabelled with MAP2, high-power images = 50 μm). (G) There was an increased percentage of dorsal horn neurons expressing HIF1α in diabetic rats when compared with age-matched naive control animals (*P < 0.05, unpaired t test, n= 5 per group). (H) Representative Western blots showing an increase HIF1α protein expression in lumbar spinal cord protein lysates extracted from types 1 (STZ*induced) diabetic rats. (I) Densitometry quantification demonstrated an increase HIF1α protein expression (**P < 0.05, unpaired t test, n= 5 per group). (I) Immunoblot of protein from the spinal cord of type 2 diabetic mouse (db/db) compared with controls. HIF1α, hypoxia-inducible factor 1α; VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2.
3.4. Hypoxia induction and onset of chronic pain
These results all suggest that hypoxia due to vascular loss can induce behavioural hypersensitivity. To test this, the chemical hypoxia mimetic DMOG was intrathecally injected into WT mice. Twenty-four hours after intrathecal injection of DMOG, there was a significant reduction in mechanical withdrawal threshold (Fig. 6A) and a decrease in heat withdrawal latency (Fig. 6B) compared with vehicle treatment. CA7 expression was increased in the spinal cord neurons as was GLUT3 and HIF1α. In addition, there was an increased number of dorsal horn neurons expressing fos immunoreactivity after intrathecal DMOG injection (Figs. 6C and D). Furthermore, isolated spinal cord neurons presented increased potassium chloride–induced calcium influx after 24 hours treatment to hypoxia mimetic (DMOG) when compared with vehicle-treated samples (supplemental Figure 6A, available at http://links.lww.com/PAIN/B597). Using multielectrode arrays on spinal cord slices (Fig. 6E), we showed increased baseline neuronal activity after DMOG treatment in the dorsal horn aspect (Fig. 6F) compared with vehicle-treated lumbar spinal cord slices (Fig. 6G). In isolated neurons from the lumbar region of the spinal cord (supplemental Figure 6B, available at http://links.lww.com/PAIN/B597) exposed to differing environmental oxygen conditions, normoxia and hypoxia (1%) resulted in an increase of HIF1α (Fig. 6H), CA7 (Fig. 6I), and GLUT3 (Fig. 6J). In addition, KCC2 was downregulated (supplemental Figure 6C, available at http://links.lww.com/PAIN/B597).
Figure 6.
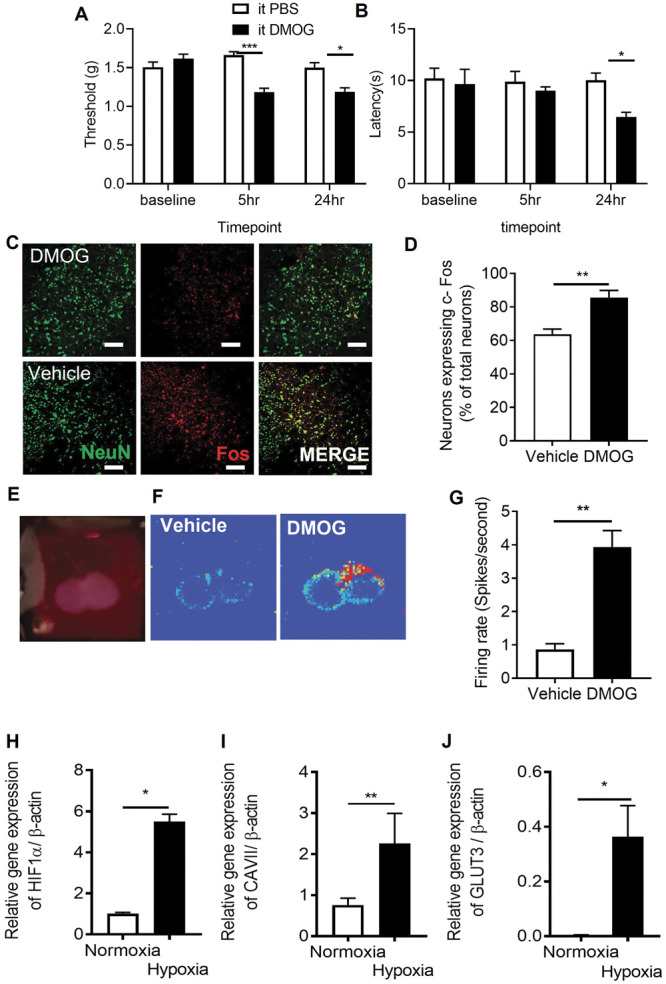
Chemically induced hypoxia causes sensory neuronal activation in dorsal horn and initiation of pain hypersensitivity. Intrathecal injection of 1 mM DMOG in C57Bl6 male mice led to (A) lower mechanical withdrawal thresholds at 5 and 24 hours as well as (B) reductions in heat withdrawal latency when compared with vehicle-treated mice (*P < 0.05, ***P < 0.001 2-way ANOVA with Tukey multiple comparison, n = 10 per group). (C–D) Fos, a marker of neuron activation, was increased in dorsal horn neurons (colabelled with neuronal marker NeuN, scale bar = 50 μm) after intrathecal DMOG injection (**P < 0.01, unpaired t test, n = 5 per group). (E) Neuronal activity was recorded in vitro from lumbar spinal cord slices using multielectrode arrays and (F) basal neuronal activity heat maps 24 hours after vehicle and DMOG-treated samples generated. (G) DMOG-treated lumbar spinal cord slices demonstrate increased basal neuronal activity compared with vehicle-treated samples (**P < 0.01, unpaired t test, n = 5). Isolated neurons were exposed to normoxic and hypoxic conditions. Hypoxia led to the increased expression of induced (H) HIF1α, (I) carbonic anhydrase 7, and (J) glucose transporter 3. DMOG, dimethyloxalylglycine; HIF1α, hypoxia-inducible factor 1α.
3.5. Carbonic anhydrase mediated hypoxia-induced chronic pain
We identified that CA7 was upregulated after induction of hypoxia by VEGFR2 knockout in spinal cord endothelial cells. To determine whether this mediated neuronal hypersensitivity, we reproduced this by treatment with DMOG for 24 hours, which showed increased protein expression (Fig. 7A representative Western blot) of HIF1α (Fig. 7B) and CA7 (Fig. 7C) compared with vehicle treatment. Similarly, intrathecally administered DMOG led to increased protein expression (Fig. 7D representative Western blot) of HIF1α (Fig. 7E) and CA7 (Fig. 7F). The nonselective carbonic anhydrase inhibitor ACZ delivered by intraperitoneal injection attenuated DMOG-induced nociceptive behavioural hypersensitivity. Acetazolamide increased mechanical withdrawal thresholds returning to comparable thresholds with the control group (Fig. 7G). Similarly, heat withdrawal latencies were increased to normal values in the DMOG-treated experimental cohort (Fig. 7H). Acetazolamide also increased withdrawal latencies in VEGFR2scECKO mice, partially restoring them towards vehicle treatment (Fig. 7I).
Figure 7.
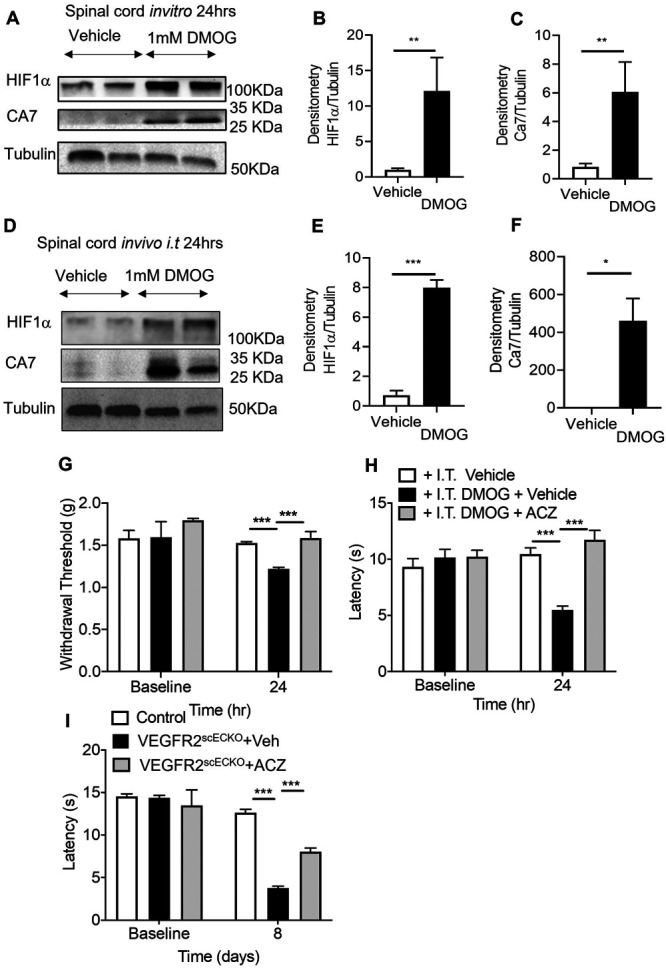
Inhibition of carbonic anhydrase 7 attenuates hypoxia-induced pain hypersensitivity. (A) In vitro lumbar spinal cord slices were treated with either vehicle or 1 mM DMOG for 24 hours to determine protein expression of HIF1α and CA7 (representative Western blots), with (B) DMOG treatment increasing expression of (B) HIF1α and (C) CA7 compared with vehicle treatment (**P < 0.01, unpaired t test, n = 5 per group). In C57Bl6 mice, lumbar spinal cord protein lysate samples ([D] representative Western blots) 24 hours after intrathecal delivery of 1 mM DMOG demonstrated an increase in protein expression of (E) HIF1α and (F) CA7 compared with vehicle-treated animals (*P < 0.05, ***P < 0.001, unpaired t test, n = 3 per group). (G) Intraperitoneal treatment of acetazolamide (ACZ) led to attenuation of DMOG-induced reduction in mechanical withdrawal thresholds and (H) heat withdrawal latencies (**P < 0.01, ***P < 0.001 2-way ANOVA with Tukey multiple comparison, n = 6 per group). ACZ treatment led to nociceptive withdrawal thresholds returning to baseline values and comparable with vehicle-treated animals. (I) Intraperitoneal ACZ inhibited heat hypersensitivity in VEGFR2scECKO mice with heat withdrawal latencies increased compared with VEGFR2scECKO mice treated with vehicle (***P < 0.001 2-way ANOVA with Tukey multiple comparison, n = 5 per group). CA7, carbonic anhydrase 7; DMOG, dimethyloxalylglycine; HIF1α, hypoxia-inducible factor 1α; VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2.
4. Discussion
Chronic pain is induced through alterations in neuronal activity, with the vascular system playing a key role in the manifestation of long-term pain hypersensitivity through induction of proinflammatory signalling.12 Here we have explored how vascular dysfunction through curtailed blood flow and subsequent hypoxia at the level of the spinal cord could modulate nociceptive processing. Currently, those mechanisms associated with induction of central sensitisation are fundamental to the development of neuropathic pain, with chloride and bicarbonate homeostasis integral to GABA-mediated excitation. In this study, we identify that reduced blood perfusion of the spinal cord induces hypoxia-mediated activation of dorsal horn spinal neurons and chronic pain manifestation. Importantly, chronic pain behaviours were suppressed by inhibition of carbonic anhydrase activity.
4.1. Vasculopathy, VEGFR2 signalling, and nociception
Impairment in nervous system blood flow is highly associated with the development of neurological disease, eg, Alzheimer and dementia,45,57 and is also a fundamental cause of ischaemic disease of the nervous system such as stroke.73 The underlying mechanisms by which the somatosensory system residing in the central nervous system adapts to such fluctuations in the microenvironment remain unclear. Investigations presented here suggest that a reduction of the vascular supply to the spinal cord is sufficient to initiate pain.
Chronic pain is a highly prevalent pathological condition, affecting large proportions of the population, with the onset and severity correlated with progression in age or disease.17 Incidentally capillary health is also greatly influenced by age and disease, with a decline in vessel number and increased frequency of vasoconstrictive tone associated with neurological pathology.37,45 Endothelial degeneration in the spinal cord capillary network is implicated in the development of neuropathic pain,70 with loss of endothelial-specific VEGFR2 signalling highly attributable to the manifestation of pain hypersensitivity. Vascular endothelial growth factor 2 signalling is integral for the development and maintenance of vascular function in health and disease through maturation of the capillary network19 and regulation of vessel permeability.60 In neurological disease, alterations in VEGFR2 signalling results in vascular defects and consequently a decline neuronal function.52,53,67 Furthermore, supplementation with VEGF-A has been shown to prevent the development of nervous system vasculopathy leading to the suppression of neurological disease.7,67,70 In relation to the spinal cord, VEGFR2 expression and activity were attributable to spinal cord endothelium function by manipulation of vascular permeability. Consequently, we showed that the loss of VEGFR2 expression in the spinal cord endothelium led to a reduction in the capillary network throughout the dorsal horn, supporting the integral role of VEGFR2 in maintaining capillary health. Previous work70 has explored systemic VEGFR2 knockdown, plausibly influencing a wider context in relation to the somatosensory network.25 Current understanding demonstrates that microvessel functionality within the spinal cord induces pain hypersensitivity through the development of a pronounced proinflammatory environment within the dorsal horn.13 Recent evidence highlights that loss of endothelial-derived tight junctional proteins and enhanced inflammatory cell adhesion drives increased spinal cord vascular permeability.43 Consequently, impairment in spinal cord endothelial cell VEGFR2-dependent function prevents vascular permeability and inhibits the onset of proinflammatory-derived pain hypersensitivity.7 Here in this study, VEGFR2 knockdown was restricted to the spinal cord endothelium, enabling interrogation of the interplay between the vascular and neuronal systems in relation to pain modulation. Induction of VEGFR2 knockdown in specifically the spinal cord endothelium leads to thermal nociceptive hypersensitivity.
4.2. Hypoxia-induced chronic pain
The loss of spinal cord microvasculature in the dorsal horn and subsequent loss of capillary perfusion was associated with the induction of hypoxia signalling in rodent models of chronic pain. Neuronal tissue is highly metabolically active; therefore, an established blood supply to neural tissues is pivotal, with neuronal activity coupled to provision of nutritional support.14 As a consequence of this, neurons are highly sensitive to fluctuations in oxygen and nutrient delivery,15,49 with neuronal integrity compromised by diminished nutritional support. Changes in the neural tissue environment such as diminished tissue oxygen level are detrimental to neural tissue integrity, altering neuronal activity leading to pathological neurological disabilities, seizures, and cognitive impairments.44 After induction of vascular degeneration and induction of hypoxia mechanisms, neurons in the dorsal horn increased levels of neuronal activity. This activity has been observed in rodent models of chronic pain66 and in spinal cord damage observed in patients with diabetic neuropathic pain.1,62 In addition, hypoxia as highlighted in this study is a significant causative factor in the onset of pain hypersensitivity. However, other factors are also culpable in neuropathic pain development, including metabolic disturbances and inflammation, which need to be considered during the time course of diabetic neuropathic pain.
Impaired blood flow to the peripheral sensory nervous system induces exacerbated pain behaviours in rodents that is accompanied by enhanced peripheral sensory activity56,64 typifying onset of chronic pain states after vascular occlusion. Currently, there are few published investigations into the relationship between the dorsal horn neuron at the level of the spinal cord and reduced blood flow. However, reductions in blood flow and consequently tissue oxygen level impair motor neuron activity, underlying the significance of blood flow to neuron function.37 In this study using rodent models of vasculopathy that induce chronic pain, we demonstrated pronounced hypoxia signalling in the dorsal horn of the spinal cord, in association with neuronal activation. Increased expression of C-fos (marker of neuronal activation) was accompanied by elevated expression levels of hypoxia markers such as HIF1α, GLUT3, and carbonic anhydrase 7 (CA7). Previous exploration of the impact of hypoxia on the sensory neuron demonstrated the dorsal root ganglia sensory neuron is activated under such low oxygen conditions.28,55 Furthermore, development of diabetic neuropathic pain is inherently associated with a neurovascular disruption with reductions in oxygen attributable to sensory neurodegeneration.54 Dorsal horn neuronal activity has previously been demonstrated to be influenced by hypoxia and HIF1α activity.28 Reductions of HIF1α expression in dorsal root ganglia sensory neurons was associated with a decline in the number of c-fos expressing neurons after peripheral sensory neuron activation.28 However, hypoxia-induced expression of neuron-specific GLUT3 and CAR7 highlights a putative adaptation of the dorsal horn neuron to alterations in the tissue microenvironment, underlying plausible avenues by which synaptic plasticity may evolve to underlie chronic pain manifestation. Of interest, we saw no change in DRG vessel markers in the VEGFR2scECKO, and yet, changes in pain behaviour were seen, suggesting that while DRG may contribute, there is also significant spinal cord involvement.
Identifying those neuronal adaptation mechanisms responsible for modulation of neuronal function and synaptic plasticity are integral in the understanding how chronic pain develops.40,50 Dysregulation of GABAergic signalling is widely recognised in the development of chronic pain states through differential regulation of chloride, potassium, and bicarbonate homeostasis.41 Crucial loss of potassium chloride cotransporter 2 (KCC2) in the spinal cord leads to the onset of chronic pain and dorsal horn neuronal excitation.38,41 Potassium chloride cotransporter 2 inhibition or downregulation (such as in times of pathological pain and hypoxia reduced KCC2 activity) leads to accumulation of intracellular chloride. Subsequent activation of GABA receptor A (GABAA) leads to efflux of chloride leading to excitation of the dorsal horn neuron. Disturbances in GABAA function, involving KCC2 expression and activity,26 have been implicated at the spinal cord level to contribute to diabetic neuropathic pain development.35 However, recent evidence identifies that regulation in the transport of chloride and bicarbonate influences spinal cord neuronal activation and chronic pain states.5 GABA receptor A not only induces chloride efflux but bicarbonate is also transported out of the neuron. Consequently, suppression of bicarbonate synthesis by carbonic anhydrase activity through administration of ACZ inhibits dorsal horn neuron activity and nociception at the level of the spinal cord.33,42 Acetazolamide inhibits dorsal horn neuron function and is implicated in the inhibition of diabetic neuropathic pain states.36 Similarly, our work shows that hypoxia-induced pain can be ameliorated by ACZ treatment, which in part is driven by hypoxia induced upregulation of CA7 expression. Carbonic anhydrase 7 is a cytoplasmic isoform that uses carbon dioxide and water to synthesise bicarbonate. In relation to neuronal function, it has been established that in the hippocampus, GABAA-mediated neural excitation is dependent on intracellular bicarbonate levels maintained by cytoplasmic carbonic anhydrase activity,71 with CA7 associated with aberrant hippocampal neural activity responsible for seizures.59 Furthermore, CA7 is expressed in the spinal cord59 in predominantly excitatory neurons, along with membrane-bound CA12.11,61 However, a CA7 knockout mouse demonstrated no alterations in nociceptive behaviour in comparison with a control mouse. Of note, that study explored naive animals and our results highlight that CA7 is expressed at low levels in such an instance. It is plausible that induction of GABA disinhibition such as in a pathological pain state driven by hypoxia, such as in diabetes, would be needed to induce CA7-dependent sensory neural excitation.
This study presents a novel mechanism that activates excitatory neuronal networks in the spinal cord to initiate chronic pain. By understanding the intrinsic plasticity of the sensory neural network in relation to the impact of specific disease states on the somatosensory nervous system enables greatly informed development of novel analgesics therapies.
Conflict of interest statement
The authors have no conflicts of interest to declare in relation to this work.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B597.
Supplementary Material
Acknowledgments
R. P. Hulse, M. E. Da Vitoria Lobo, N. Weir, R. Madden, L. Hardowar, and S. M. Bestall performed the experimental work. R. P. Hulse, M. E. Da Vitoria Lobo, N. Weir, R. Madden, L. Hardowar, S. M. Bestall, C. Schaffer, L. F. Donaldson, and D. O. Bates contributed to the conception or design of the work in addition to acquisition, analysis, or interpretation of data for the work. R. P. Hulse, M. E. Da Vitoria Lobo, N. Weir, R. Madden, L. Hardowar, S. M. Bestall, L. F. Donaldson, and D. O. Bates drafted the work or revised it critically for important intellectual content. R. P. Hulse drafted the manuscript with contributions from all authors. All authors approved the final version of the manuscript, agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. The authors thank Graham Hickman of the Imaging Suite at Nottingham Trent University for their support and assistance in this work. This work was supported by the European Foundation for the Study of Diabetes Microvascular Programme supported by Novartis to R. P. Hulse (Nov 2015_2 to R. P. Hulse), the EFSD/Boehringer Ingelheim European Research Programme in Microvascular Complications of Diabetes (BI18_5 to R. P. Hulse), the Rosetrees Trust (A1360 to R. P. Hulse), and Diabetes United Kingdom (11/0004192 to L. F. Donaldson) and Arthritis Research United Kingdom (grant numbers 20400 to L. F. Donaldson).
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
Contributor Information
Marlene E. Da Vitoria Lobo, Email: marlene.lobo@ucl.ac.uk.
Nick Weir, Email: Nick.weir@ntu.ac.uk.
Lydia Hardowar, Email: lydia.hardowar@ntu.ac.uk.
Yara Al Ojaimi, Email: yara.alojaimi@ntu.ac.uk.
Ryan Madden, Email: Ryan.madden@nottingham.ac.uk.
Alex Gibson, Email: Alex.Gibson@nottingham.ac.uk.
Samuel M. Bestall, Email: Samuel.Bestall@nottingham.ac.uk.
Masanori Hirashima, Email: masonarihirashima@niigata.ac.jp.
Chris B. Schaffer, Email: Chrisschaffer@cornell.edu.
Lucy F. Donaldson, Email: Lucy.Donaldson@nottingham.ac.uk.
David O. Bates, Email: David.bates@nottingham.ac.uk.
References
- [1].Alam U, Sloan G, Tesfaye S. Treating pain in diabetic neuropathy: current and developmental drugs. Drugs 2020;80:363–84. [DOI] [PubMed] [Google Scholar]
- [2].Albuquerque RJ, Hayashi T, Cho WG, Kleinman ME, Dridi S, Takeda A, Baffi JZ, Yamada K, Kaneko H, Green MG, Chappell J, Wilting J, Weich HA, Yamagami S, Amano S, Mizuki N, Alexander JS, Peterson ML, Brekken RA, Hirashima M, Capoor S, Usui T, Ambati BK, Ambati J. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat Med 2009;15:1023–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Allen CL, Malhi NK, Whatmore JL, Bates DO, Arkill KP. Non-invasive measurement of retinal permeability in a diabetic rat model. Microcirculation 2020;27:e12623. [DOI] [PubMed] [Google Scholar]
- [4].Alles SRA, Smith PA. Etiology and pharmacology of neuropathic pain. Pharmacol Rev 2018;70:315–47. [DOI] [PubMed] [Google Scholar]
- [5].Asiedu MN, Mejia GL, Hübner CA, Kaila K, Price TJ. Inhibition of carbonic anhydrase augments GABAA receptor-mediated analgesia via a spinal mechanism of action. J Pain 2014;15:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bannister K, Sachau J, Baron R, Dickenson AH. Neuropathic pain: mechanism-based therapeutics. Annu Rev Pharmacol Toxicol 2020;60:257–74. [DOI] [PubMed] [Google Scholar]
- [7].Beazley-Long N, Moss CE, Ashby WR, Bestall SM, Almahasneh F, Durrant AM, Benest AV, Blackley Z, Ballmer-Hofer K, Hirashima M, Hulse RP, Bates DO, Donaldson LF. VEGFR2 promotes central endothelial activation and the spread of pain in inflammatory arthritis. Brain Behav Immun 2018;74:49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bestall SM, Hulse RP, Blackley Z, Swift M, Ved N, Paton K, Beazley-Long N, Bates DO, Donaldson LF. Sensory neuronal sensitisation occurs through HMGB-1-RAGE and TRPV1 in high-glucose conditions. J Cell Sci 2018;131:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Betteridge KB, Arkill KP, Neal CR, Harper SJ, Foster RR, Satchell SC, Bates DO, Salmon AHJ. Sialic acids regulate microvessel permeability, revealed by novel in vivo studies of endothelial glycocalyx structure and function. J Physiol 2017;595:5015–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nat Protoc 2007;2:1490–8. [DOI] [PubMed] [Google Scholar]
- [11].Chamessian A, Young M, Qadri Y, Berta T, Ji RR, Van De Ven T. Transcriptional profiling of somatostatin interneurons in the spinal dorsal horn. Sci Rep 2018;8:6809–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cobos EJ, Nickerson CA, Gao F, Chandran V, Bravo-Caparrós I, González-Cano R, Riva P, Andrews NA, Latremoliere A, Seehus CR, Perazzoli G, Nieto FR, Joller N, Painter MW, Ma CHE, Omura T, Chesler EJ, Geschwind DH, Coppola G, Rangachari M, Woolf CJ, Costigan M. Mechanistic differences in neuropathic pain modalities revealed by correlating behavior with global expression profiling. Cell Rep 2018;22:1301–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, Barrett L, Brenner GJ, Vardeh D, Woolf CJ, Fitzgerald M. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci 2009;29:14415–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Daneman R, Prat A. The blood–brain barrier. Cold Spring Harb Perspect Biol 2015;5:a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Erecińska M, Silver IA. Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol 2001;128:263–76. [DOI] [PubMed] [Google Scholar]
- [16].Farrar MJ, Bernstein IM, Schlafer DH, Cleland TA, Fetcho JR, Schaffer CB. Chronic in vivo imaging in the mouse spinal cord using an implanted chamber. Nat Methods 2012;9:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fayaz A, Croft P, Langford RM, Donaldson LJ, Jones GT. Prevalence of chronic pain in the UK: a systematic review and meta-analysis of population studies. BMJ Open 2016;6:e010364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fuchs D, Birklein F, Reeh PW, Sauer SK. Sensitized peripheral nociception in experimental diabetes of the rat. PAIN 2010;151:496–505. [DOI] [PubMed] [Google Scholar]
- [19].Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 2003;161:1163–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Goldberg DS, McGee SJ. Pain as a global public health priority. BMC Public Health 2011;11:770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. PAIN 1988;32:77–88. [DOI] [PubMed] [Google Scholar]
- [22].Hsiao HT, Lin YC, Wang JC, Tsai YC, Liu YC. Hypoxia inducible factor-1α inhibition produced anti-allodynia effect and suppressed inflammatory cytokine production in early stage of mouse complex regional pain syndrome model. Clin Exp Pharmacol Physiol 2016;43:355–9. [DOI] [PubMed] [Google Scholar]
- [23].Hsieh MT, Donaldson LF, Lumb BM. Differential contributions of A- and C-nociceptors to primary and secondary inflammatory hypersensitivity in the rat. PAIN 2015;156:1074–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hultgren NW, Fang JS, Ziegler ME, Ramirez RN, Phan DTT, Hatch MMS, Welch-Reardon KM, Paniagua AE, Kim LS, Shon NN, Williams DS, Mortazavi A, Hughes CCW. Slug regulates the Dll4-Notch-VEGFR2 axis to control endothelial cell activation and angiogenesis. Nat Commun 2020;11:5400–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jere M, Cassidy RM. VEGF-A165 b to the rescue: vascular integrity and pain sensitization. J Physiol 2018;596:5077–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jolivalt CG, Lee CA, Ramos KM, Calcutt NA. Allodynia and hyperalgesia in diabetic rats are mediated by GABA and depletion of spinal potassium-chloride co-transporters. PAIN 2008;140:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O'Brien SM, Davis RB, Gowen LC, Anderson KD, Thurston G, Joho S, Springer ML, Kuo CJ, McDonald DM. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol 2006;290:H560–76. [DOI] [PubMed] [Google Scholar]
- [28].Kanngiesser M, Mair N, Lim HY, Zschiebsch K, Blees J, Häussler A, Brüne B, Ferreiròs N, Kress M, Tegeder I. Hypoxia-inducible factor 1 regulates heat and cold pain sensitivity and persistence. Antioxid Redox Signal 2014;20:2555–71. [DOI] [PubMed] [Google Scholar]
- [29].Khacho M, Tarabay M, Patten D, Khacho P, MacLaurin JG, Guadagno J, Bergeron R, Cregan SP, Harper ME, Park DS, Slack RS. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat Commun 2014;5:3550–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci 2009;12:1364–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Korn C, Augustin HG. Mechanisms of vessel pruning and regression. Dev Cell 2015;34:5–17. [DOI] [PubMed] [Google Scholar]
- [32].Alban L, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 2009;10:895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee KY, Prescott SA. Chloride dysregulation and inhibitory receptor blockade yield equivalent disinhibition of spinal neurons yet are differentially reversed by carbonic anhydrase blockade. PAIN 2015;156:2431–7. [DOI] [PubMed] [Google Scholar]
- [34].Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell 2007;130:691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee-Kubli ACG, Calcutt NA. Altered rate-dependent depression of the spinal H-reflex as an indicator of spinal disinhibition in models of neuropathic pain. PAIN 2013;155:250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li Y, Lucas-Osma AM, Black S, Bandet MV, Stephens MJ, Vavrek R, Sanelli L, Fenrich KK, Di Narzo AF, Dracheva S, Winship IR, Fouad K, Bennett DJ. Pericytes impair capillary blood flow and motor function after chronic spinal cord injury. Nat Med 2017;23:733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lorenzo LE, Godin AG, Ferrini F, Bachand K, Plasencia-Fernandez I, Labrecque S, Girard AA, Boudreau D, Kianicka I, Gagnon M, Doyon N, Ribeiro-da-Silva A, De Koninck Y. Enhancing neuronal chloride extrusion rescues α2/α3 GABAA-mediated analgesia in neuropathic pain. Nat Commun 2020;11:869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ludwig J, Gorodetskaya N, Schattschneider J, Jänig W, Baron R. Behavioral and sensory changes after direct ischemia-reperfusion injury in rats. Eur J Pain 2007;11:677–84. [DOI] [PubMed] [Google Scholar]
- [39].Luo C, Kuner T, Kuner R. Synaptic plasticity in pathological pain. Trends Neurosci 2014;37:343–55. [DOI] [PubMed] [Google Scholar]
- [40].Mapplebeck JCS, Lorenzo LE, Lee KY, Gauthier C, Muley MM, De Koninck Y, Prescott SA, Salter MW. Chloride dysregulation through downregulation of KCC2 mediates neuropathic pain in both sexes. Cell Rep 2019;28:590–6.e4. [DOI] [PubMed] [Google Scholar]
- [41].Medrano MC, Dhanasobhon D, Yalcin I, Schlichter R, Cordero-Erausquin M. Loss of inhibitory tone on spinal cord dorsal horn spontaneously and nonspontaneously active neurons in a mouse model of neuropathic pain. PAIN 2016;157:1432–42. [DOI] [PubMed] [Google Scholar]
- [42].Montague-Cardoso K, Pitcher T, Chisolm K, Salera G, Lindstrom E, Hewitt E, Solito E, Malcangio M. Changes in vascular permeability in the spinal cord contribute to chemotherapy-induced neuropathic pain. Brain Behav Immun 2020;83:248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nalivaeva NN, Rybnikova EA. Brain hypoxia and ischemia: new insights into neurodegeneration and neuroprotection. Front Neurosci 2019;13:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, Kyrargyri V, Pfeiffer T, Khennouf L, Madry C, Gong H, Richard-Loendt A, Huang W, Saito T, Saido TC, Brandner S, Sethi H, Attwell D. Amyloid beta oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science 2019;365:eaav9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Paice JA. Chronic treatment-related pain in cancer survivors. PAIN 2011;152:S84–9. [DOI] [PubMed] [Google Scholar]
- [46].Pang V, Bates DO, Leach L. Regulation of human feto-placental endothelial barrier integrity by vascular endothelial growth factors: competitive interplay between VEGF-A165a, VEGF-A165b, PIGF and VE-cadherin. Clin Sci (Lond) 2017;131:2763–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Patel TP, Gullotti DM, Hernandez P, O'Brien WT, Capehart BP, Morrison B, Bass C, Eberwine JE, Abel T, Meaney DF. An open-source toolbox for automated phenotyping of mice in behavioral tasks. Front Behav Neurosci 2014;8:349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bickler PE, Donohoe PH. Adaptive responses of vertebrate neurons to hypoxia. J Exp Biol 2002;205:3579–86. [DOI] [PubMed] [Google Scholar]
- [49].Price TJ, Cervero F, de Koninck Y. Role of cation-chloride-cotransporters (CCC) in pain and hyperalgesia. Curr Top Med Chem 2005;5:547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Price TJ, Prescott SA. Inhibitory regulation of the pain gate and how its failure causes pathological pain. PAIN 2015;156:789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Reeson P, Choi K, Brown CE. VEGF signaling regulates the fate of obstructed capillaries in mouse cortex. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Reeson P, Tennant KA, Gerrow K, Wang J, Weiser Novak S, Thompson K, Lockhart KL, Holmes A, Nahirney PC, Brown CE. Delayed inhibition of VEGF signaling after stroke attenuates blood-brain barrier breakdown and improves functional recovery in a comorbidity-dependent manner. J Neurosci 2015;35:5128–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Richner M, Ferreira N, Dudele A, Jensen TS, Vaegter CB, Gonçalves NP. Functional and structural changes of the blood-nerve-barrier in diabetic neuropathy. Front Neurosci 2019;13:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ristoiu V, Shibasaki K, Uchida K, Zhou Y, Ton BH, Flonta ML, Tominaga M. Hypoxia-induced sensitization of transient receptor potential vanilloid 1 involves activation of hypoxia-inducible factor-1 alpha and PKC. PAIN 2011;152:936–45. [DOI] [PubMed] [Google Scholar]
- [55].Ross JL, Queme LF, Lamb JE, Green KJ, Jankowski MP. Sex differences in primary muscle afferent sensitization following ischemia and reperfusion injury. Biol Sex Differ 2018;9:2–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ruitenberg A, den Heijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MM. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol 2005;57:789–94. [DOI] [PubMed] [Google Scholar]
- [57].Ruscetti M, Iv JPM, Mezzadra R, Russell J, Romesser PB, Simon J, Kulick A, Ho Y, Fennell M, Li J, Norgard RJ, Wilkinson JE, Alonso-curbelo D, Heller DA, Stanchina E De, Stanger BZ, Sherr CJ, Lowe SW. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 2020;181:424–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ruusuvuori E, Huebner AK, Kirilkin I, Yukin AY, Blaesse P, Helmy M, Kang HJ, El Muayed M, Hennings JC, Voipio J, Šestan N, Hübner CA, Kaila K. Neuronal carbonic anhydrase VII provides GABAergic excitatory drive to exacerbate febrile seizures. EMBO J 2013;32:2275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Salmon AH, Neal CR, Bates DO, Harper SJ. Vascular endothelial growth factor increases the ultrafiltration coefficient in isolated intact Wistar rat glomeruli. J Physiol 2006;570:141–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sathyamurthy A, Johnson KR, Matson KJE, Dobrott CI, Li L, Ryba AR, Bergman TB, Kelly MC, Kelley MW, Levine AJ. Massively parallel single nucleus transcriptional profiling defines spinal cord neurons and their activity during behavior. Cell Rep 2018;22:2216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].SelvarajahEmery DCJ, Harris ND, Shaw PJ, Witte DR, Griffiths PD, Tesfaye SWID. Early involvement of the spinal cord in diabetic peripheral neuropathy. Diabetes Care 2006;29:2664–9. [DOI] [PubMed] [Google Scholar]
- [62].Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, Quaggin SE. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol 2010;21:1691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].So K, Tei Y, Zhao M, Miyake T, Hiyama H, Shirakawa H, Imai S, Mori Y, Nakagawa T, Matsubara K, Kaneko S. Hypoxia-induced sensitisation of TRPA1 in painful dysesthesia evoked by transient hindlimb ischemia/reperfusion in mice. Sci Rep 2016;6:23261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Suchting S, Freitas C, Toro R del, Noble F, Benedito R, Breant C, Duarte A, Eichmann A. The Notch ligand Delta‐like 4 (Dll4) negatively regulates endothelial tip cell formation and vessel branching. FASEB J 2007;21:3225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tan AM, Samad OA, Fischer TZ, Zhao P, Persson AK, Waxman SG. Maladaptive dendritic spine remodeling contributes to diabetic neuropathic pain. J Neurosci 2012;32:6795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Taylor SL, Trudeau D, Arnold B, Wang J, Gerrow K, Summerfeldt K, Holmes A, Zamani A, Brocardo PS, Brown CE. VEGF can protect against blood brain barrier dysfunction, dendritic spine loss and spatial memory impairment in an experimental model of diabetes. Neurobiol Dis 2015;78:1–11. [DOI] [PubMed] [Google Scholar]
- [67].Tesfaye S, Vileikyte L, Rayman G, Sindrup SH, Perkins BA, Baconja M, Vinik AI, Boulton AJ. Painful diabetic peripheral neuropathy: consensus recommendations on diagnosis, assessment and management. Diabetes Metab Res Rev 2011;27:629–38. [DOI] [PubMed] [Google Scholar]
- [68].Tichet M, Prod'Homme V, Fenouille N, Ambrosetti D, Mallavialle A, Cerezo M, Ohanna M, Audebert S, Rocchi S, Giacchero D, Boukari F, Allegra M, Chambard JC, Lacour JP, Michiels JF, Borg JP, Deckert M, Tartare-Deckert S. Tumour-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun 2015;6:6993. [DOI] [PubMed] [Google Scholar]
- [69].Ved N, Lobo MEDV, Bestall SM, Vidueira CL, Beazley-Long N, Ballmer-Hofer K, Hirashima M, Bates DO, Donaldson LF, Hulse RP. Diabetes-induced microvascular complications at the level of the spinal cord; a contributing factor in diabetic neuropathic pain. J Physiol 2018;596:3675–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Viitanen T, Ruusuvuori E, Kaila K, Voipio J. The K+-Cl- cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol 2010;588:1527–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Da Vitorio Lobo ME, Bates DO, Arkill KP, Hulse RP. Quantifying spinal cord vascular permeability in the mouse using intravital imaging. BioRxiv 2021. Available at: 10.1101/2021.06.09.447701. [DOI] [Google Scholar]
- [72].Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol 2019;18:684–96. [DOI] [PubMed] [Google Scholar]
- [73].Whittles CE, Pocock TM, Wedge SR, Kendrew J, Hennequin LF, Harper SJ, Bates DO. ZM323881, a novel inhibitor of vascular endothelial growth factor-receptor-2 tyrosine kinase activity. Microcirculation 2002;9:513–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B597.