
Fig. 3.
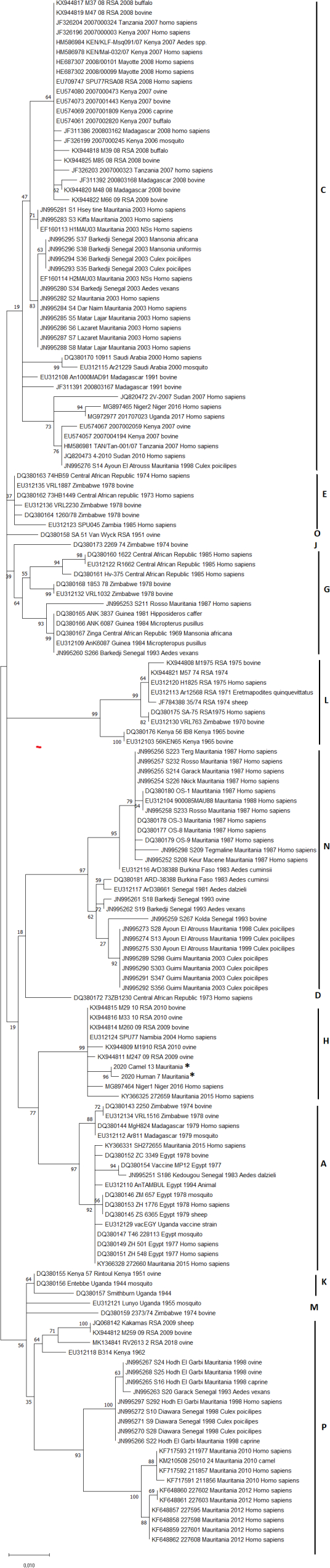
Phylogenetic tree derived from nucleotide sequence data of the S segment, partial NSs gene. The tree with the highest log likelihood (-2460.73) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heu-ristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites. (5 categories (+G, parameter = 0.3199)). The rate variation model allowed for some sites to be evolutionari-ly invariable ([+I], 35.26% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Classification of the isolates followed the lineage terminology of Grobbelaar et al., 2011 [46]. The GenBank accession numbers for the NSs gene of the virus S segment are ON052829(2020 Camel 13 Mauritania) and ON052830 (2020 Human 7 Mauritania).