
Figure 3.
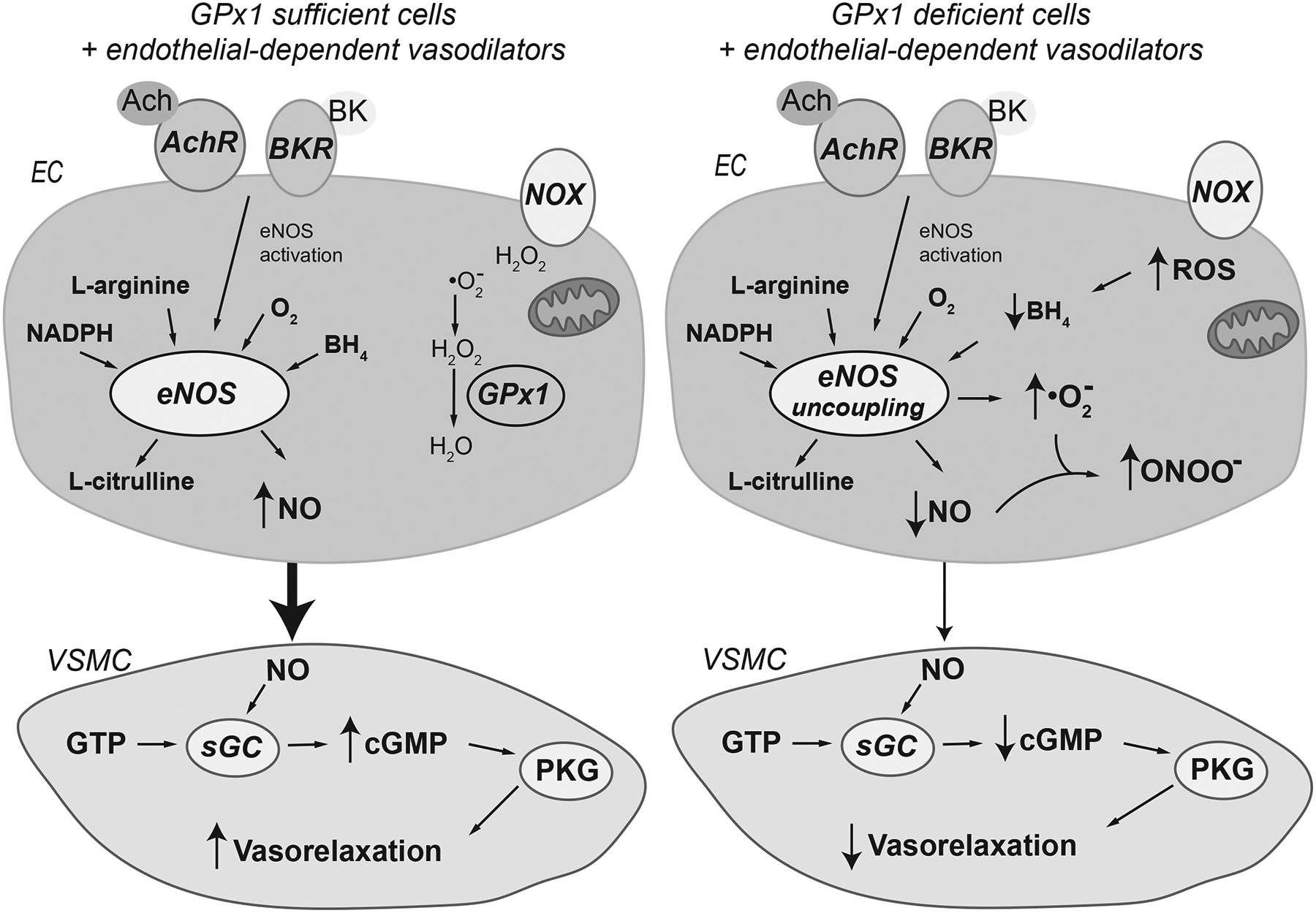
Endothelial dysfunction caused by GPx1 deficiency. The binding of the vasodilators acetylcholine (Ach) and bradykinin (BK) to their receptors, ACHR (muscarinic receptor) and BKR (bradykinin receptor), on endothelial cells (EC) activates endothelial nitric oxide synthase (eNOS) to promote the production of nitric oxide. Nitric oxide (NO) produced in endothelial cells (EC) promotes the relaxation of vascular smooth muscle cells (VSMC) via the cGMP-mediated activation of protein kinase G (PKG) to dilate blood vessels. Nitric oxide binding to the soluble guanylyl cyclase (sGC) stimulates the conversion of GTP to cGMP. In normal EC cells, basal NADPH oxidase (NOX) activity (primarily the hydrogen peroxide producing NOX4) and mitochondrial respiration provide most of the cellular ROS. GPx1 deficiency is associated with increased levels of ROS including increases in hydrogen peroxide and superoxide. Loss of GPx1 can promote the upregulation of superoxide generators, such as NOX2, and increased production and release of ROS from mitochondria, in part, due to ROS-mediated ROS production. ROS, including hydrogen peroxide, can oxidize the eNOS cofactor tetrahydrobiopterin (BH4) to uncouple eNOS. Uncoupling of eNOS results in substantially lower production of nitric oxide by eNOS and greater production of superoxide. The increased production of superoxide by eNOS promotes the formation of peroxynitrite, an oxidant that promotes additional oxidation of BH4, as well as the modification of protein tyrosine residues (3-nitrotyrosine modifications, not illustrated) and lipids. Note that decreased production of NO by EC results in diminished production of cGMP by sGC and a decrease in vasorelaxation and, in some cases, paradoxical vasoconstriction in response to agonist stimulation. GPx1 deficiency, however, does not affect the ability of VSMC to relax in response to nitric oxide donors, consistent with endothelial dysfunction.