

Abstract
Oxidoreductases have evolved over millions of years to perform a variety of metabolic tasks crucial for life. Understanding how these tasks are engineered relies on delivering external electron donors or acceptors to initiate electron transfer reactions. This is a challenge. Small-molecule redox reagents can act indiscriminately, poisoning the cell. Natural redox proteins are more selective, but finding the right partner can be difficult due to the limited number of redox potentials and difficulty tuning them. De novo proteins offer an alternative path. They are robust and can withstand mutations that allow for tailorable changes. They are also devoid of evolutionary artifacts and readily bind redox cofactors. However, no reliable set of engineering principles have been developed that allow for these proteins to be fine-tuned so their redox midpoint potential (Em) can form donor/acceptor pairs with any natural oxidoreductase. This work dissects protein-cofactor interactions that can be tuned to modulate redox potentials of acceptors and donors using a mutable de novo designed tetrahelical protein platform with iron tetrapyrrole cofactors as a test case. We show a series of engineered heme b-binding de novo proteins and quantify their resulting effect on Em. By focusing on the surface charge and buried charges, as well as cofactor placement, chemical modification, and ligation of cofactors, we are able to achieve a broad range of Em values spanning a range of 330 mV. We anticipate this work will guide the design of proteinaceous tools that can interface with natural oxidoreductases inside and outside the cell while shedding light on how natural proteins modulate Em values of bound cofactors.
Introduction
Natural oxidoreductases mediate a large number of metabolic reactions involving chemical catalysis and DNA binding.1−3 Unraveling their functional design has been hampered by the high levels of complexity brought on by evolution. Even minor sequence changes can result in defective natural proteins or lowered reactivity.4,5 Unfortunately, small chemical oxidants and reductants have proven to be imperfect tools.6−10 They are frequently toxic impeding in vivo experiments and are often not biocompatible. An alternative approach employs a separate protein at a specific redox potential to interface with the oxidoreductase.11 Yet, connecting with the large number of different redox proteins, all with their own unique behavior, will be quite a challenge.12,13 Even seemingly benign mutations can have unintended consequences on the redox midpoint potential (Em) and generate misleading data. We need a reliable toolkit to probe oxidoreductase function in a neutral fashion. Ideally, this will take the form of a single protein platform with various tunable properties.
De novo proteins can function as redox tools that do not require significant genetic modifications, stumble over evolutionary artifacts, or poison the system with harsh chemicals.14−20 To date, there have been an impressive number of these systems and more are being developed every day.4,21−26 However, we are missing a set of fundamental engineering guidelines that allow for the reliable tuning of Em values impeding their utility.27,28 Currently, designed proteins are characterized as they are made and used where appropriate.25 More broadly, our knowledge of how natural proteins modify Em values comes from a limited set of data, which may be complicated by evolutionary artifacts like misfolding or compensating amino acids we have not identified. Zheng and Gunner have highlighted this fact concluding that no single type of protein-cofactor interaction can be pinpointed as the single most important and that different proteins use different structural and chemical features to tune the Em values of heme cofactors.29 However, it has been shown that Em is integral to function and even minor sequence changes can result in defective proteins. For de novo designed proteins to reach the next level of integration with natural proteins and be a true set of tools with tunable potentials, we first need to understand how to modulate the Em value with precision.
Here, we use de novo design to investigate the protein-engineering requirements for fine-tuning Em to a desired value for a specific redox function, principles which will help other groups develop effective tools that can interface with natural oxidoreductases over a wide range of donor/acceptor potentials and act as tunable ET partners. The proteins in this paper have been intentionally developed from first principles bearing no resemblance to any natural sequence. This lack of evolutionary baggage allows us to extract fundamental biophysical parameters from the work, leading to new biotechnological possibilities. Similarly designed proteins have been shown to interact with a range of natural proteins without altering their own structure or damaging the natural protein.30−35 Their simple designed structure allows them to be altered easily without the risk of unforeseen side effects. These de novo designed proteins are practical systems for studying biological redox reactions.19,36
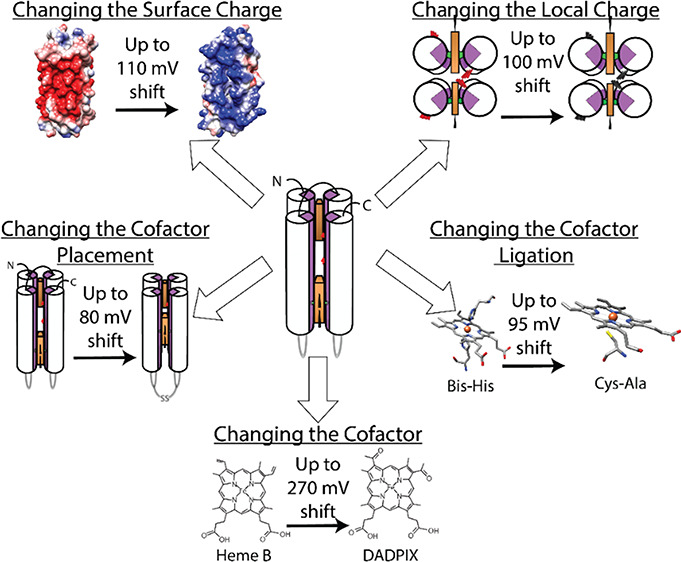
In this work, we explore new dimensions of protein design and cofactor modifications to identify four different ways of modulating Em: surface charge, different cofactors, cofactor placement, and cofactor ligations. These parameters all take advantage of the added versatility of de novo designed proteins. The principles in this paper can be used to tailor proteins to function as ET donors and acceptors, guiding the design process from the selection of cofactors, their placement, and, importantly, Em control.
Materials and Methods
Materials
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. Solvents were purchased from Fisher Scientific.
Porphyrins
All porphyrins were purchased from Frontier Scientific unless otherwise noted. They were dissolved in DMSO, and these stock solution concentrations weree determined by mass. The exceptions are heme b, Fe-mesoporphyrin, and Fe-deuteroporphyrin, which had their concentrations measured by the pyridine hemochrome assay, utilizing the extinction coefficient at 556 nm as described here.37
Protein Preparation
The proteins used in this paper were obtained as follows. The gene for the protein was ordered from DNA2.0. The construct contained a His-tag, a TEV cleavage site containing linker, and the protein of interest. Upon delivery, the DNA was transformed into BL-21 strain Escherichia coli (E. coli) cells. For expression, the BL-21 cells were grown in TB media until the OD600 was at 0.6 AU. Overexpression was induced by addition of 1 mM IPTG. The cells overexpressed the protein for 4.5 h and then were spun down to a pellet. The pellet was then resuspended in resuspection buffer (300 mM NaCl, 50 mM NaH2PO4, 10 mM Imidazole, pH 7) and sonicated to lyse the cells. The lysate was spun down for 1 h at 20,000g. The supernatant was then run through a GE-Histrap column. A linear gradient from resuspension buffer to elution buffer (300 mM NaCl, 50 mM NaH2PO4, 500 mM Imidazole, pH 7) over 1 h was used to elute the protein. The eluted protein was mixed with TEV protease to cleave the His-tag off and rerun. The flow-through of this second column was dialyzed into CHES buffer (20 mM CHES, 150 mM KCl, pH 9). Its concentration was determined by the absorbance at 280 nm using an extinction coefficient of 22,500 M–1 cm–1. The extinction coefficient was determined with the ProtParam software on the ExPASy website (http://web.expasy.org/protparam/). Where mutagenesis was required, primers were ordered from Integrated DNA Technologies (IDT) and dissolved to 10 uM. Using this stock, 1 μL of this primer was mixed with 5 ng of template DNA, Phusion Hot Start DNA polymerase mix to 1× (2× master mix: 4 μL of 5× Phusion buffer, 0.5 μL of 10 mM dNTP, 0.2 μL of Phusion Hot Start, 0.2 μL of 50 mM MgCl2, 5.1 μL of Milli-Q water), and Milli-Q water to obtain a 10 μL final volume. This was subject to a PCR cycle of 98 °C for 30 s, followed by 98 °C for 10 s, annealing temperature for 30 s, 72 °C for the extension time (repeat for 15 cycles), and followed by a final extension step at 72 °C. This DNA was then transformed as discussed above, and the protein was purified as discussed above.
Certain proteins were unable to be made in E. coli. These were prepared at a 0.1 mmol scale on a CEM Liberty microwave peptide synthesizer using standard FMOC/tBu protection protocols.38 Amino acids were purchased from Nova Biochem. After synthesis, the protein was cleaved from the resin by incubating it with a mixture of trifluoroacetic acid (TFA), ethanedithiol, anisole, and thioanisole in a 9:0.2:0.5:0.3 ratio for two and a half hours protected from light. The crude product then had an excess reagent removed via a rotovap and was subsequently precipitated with 4 ° C methyl, t-butyl ether. The protein was then purified by reverse-phase HPLC using a C18 column and a linear gradient of acetonitrile with 0.1% (v/v) TFA in water with 0.1% (v/v) TFA. The products were verified with MALDI mass spectroscopy using a 2-(4′-hydroxybenzeneazo) benzoic acid (HABA) matrix. All proteins have Trp in the sequence allowing for quantification of the concentration using UV–vis spectroscopy.
Em Titrations
Preparation of all porphyrin-protein samples for redox titration experiments was carried out using the following protocol. Three milliliters of each protein were mixed with 0.9 equiv of porphyrin and allowed to equilibrate for approximately 10 min. Samples were then run through a PD-10 desalting column to remove any residual free porphyrin. In the case of the offset protein variant, the protein was mixed with 2.2 equiv of heme and allowed to equilibrate for approximately 15 min. After this time, the protein was put through a PD-10 column to remove unbound heme. In all cases, the UV–vis spectra were taken to confirm that no free porphyrin was present.
Redox titrations were performed in combination with UV/vis monitoring, adapted from Dutton.39 Samples of typically 5–25 μM were monitored electrochemically by a calomel electrode purchased from Radiometer Analytical. The change from oxidized to reduced was monitored by the change in absorbance at the q-band region of the Bis-His ligated porphyrin. These titrations were done anaerobically under a constant stream of Ar. All redox titrations were performed in CHES buffer pH 9. The environmental potential was modulated by 1–3 μL injections of a freshly prepared sodium dithionite solution when trying to lower the potential or a potassium ferricyanide solution when trying to increase the potential. The following redox mediators were used: anthraquinone-2-sulfonate (20 μM), benzly viologen (10 μM), methyl viologen (10 μM), sulfanilamide (10 μM), indigo trisulfonate (10 μM), phenazine (10 μM), pyocyanin (10 μM), and hydroxynapthquinone (10 μM).
The data was analyzed the same as Gibney et al.38 The absorbance value of the reduced peak (corrected for baseline shifts) was plotted against voltage and fit to the Nernst equation described in the introduction and displayed below:
| 1 |
where Ecell is the reduction potential and E°cell is the standard potential (also denoted as Em and Eh, respectively), R is the gas constant, T is the temperature in kelvin, z is the number of electrons involved in the half-cell reaction, F is Faraday’s constant (96,485 J V–1 mol–1), [red] is the concentration of reduced components, and [ox] is the concentration of oxidized components. This equation is converted to the following when used for fitting:
| 2 |
When two midpoint potentials are present, the equation can be expanded to account for that.
| 3 |
Where the fraction reduced is a combination of the sum of the concentrations of each species. All values from these experiments are reported versus the standard hydrogen electrode (SHE).
Spectral-Electrochemistry Titrations
Spectroelectrochemical titrations were carried out with a platinum working electrode, gold counter wire, and a Ag/AgCl reference electrode. A CH Instruments (Austin Texas USA) Electrochemical Workstation was used with the CH Instruments interface program version 9.07. This experiment was performed in a cuvette with a 0.1 cm path length, 200 μM protein with one porphyrin bound. The reagents were dissolved in 20 mM CHES 150 mM KCl buffer pH 9. Mediators used were: anthraquinone-2-sulfonate (100 μM), benzly viologen (50 μM), methyl viologen (50 μM), sulfanilamide (50 μM), indigo trisulfonate (50 μM), phenazine (50 μM), pyocyanin (50 μM), and hydroxynapthquinone (50 μM). Eh was set by the computer, and the current was monitored. When the current reached zero, a spectrum was taken. This was repeated in 20 mV steps from −450 mV up to −150 mV. This was done multiple times (two oxidative, and two reductive) to see if there was any hysteresis. Afterward, the absorbance value of 535–517 nm was plotted against voltage and fit to the Nernst equation as above yielding an Em value. All values from these experiments are reported versus SHE.
Computational Characterization
Proteins and heme-coordination states were modeled using PyMol version 4.1 and were adapted from the crystal structure of a progenitor 4-helix bundle protein termed “BB”.40 Mutations were made to convert the sequence into the protein variants seen in this paper, but the backbone confirmation was held constant. Electrostatic maps were also created in PyMol using the ABPS and surface display tools.
Data Analysis and Coding
Data were analyzed in a Jupyter notebook.41 The spectra were imported and converted to dataframes using the Pandas package. The data was plotted as a scatterplot, and the best fit line found with the scipy.optimize.curve_fit function. Both midpoints and the n value were the variable parameters. The scatter plot data was converted into a linear space before being fit, to avoid biasing from outlier datapoints. The error in the measurements is denoted as a “±” value whose units are mV.
Results
After a brief introduction to the proteins we are using, we demonstrate a series of changes showing how individual effects to the protein scaffold or porphyrin modulate the Em value. We then show the additive effects of the bundle system.
De Novo Protein Morphologies Used in This Work
Table 1 displays the three different morphologies of de novo designed proteins, all of which assume a 4-α-helix bundle structure in solution. For much of this work, we will be concerned with the single chain (SC) morphology as its lack of repeating units allows for individual amino acids to be changed without affecting other helices. Sequences were also developed using the helix-loop-helix (HLH) motif. Both the SC and HLH variants shown in Table 1 have been previously characterized and display midpoint potentials of −260 mV, providing a baseline for comparison in this study.34 All sequences have glutamate residues that become buried upon heme b binding (depicted as red wedges in the cartoons). These amino acids were added to provide strain on the His-heme b bond and foster oxygen binding activity as observed here.16
Table 1. De Novo Protein Morphologies.

Environmental Changes
We first chose to measure the effect of surface charge on the Em values of bound heme b. We developed three different SC variants: a negatively charged protein, a positively charged variant, and a mixed charged bundle—termed binary variant. Table 2 shows a comparison of these three proteins with different surface charge patterning, which were generated in PyMol using the APBS plugin. Starting with the single chain protein that has been previously characterized and whose net charge is −16;34 the charge pattern along the outside was altered such that the net charges become −2 for the binary protein and +11 for the positively charged variant (+2 charge in each helix, and an additional +1 from lysine residues in each loop). For the binary protein, the minus-two charge is a sum over the entire protein, including lysine residues in two of the loops connecting the helices. This protein has two domains, one positively charged domain of +6 and a negatively charged domain of −8. These two environments are created by arranging two negatively charged helices and two positively charged helices such that each pair binds one heme b. We observe that the charge patterning has a significant effect on Em. The wholly negatively charged protein (−16) has an Em value of −261 (± 4) mV. Increasing the charge of the outside of the protein shifts this value up to −151 (± 6) mV difference (Figure 1). The binary protein, due to its individual environments, generates a split in potentials at −225 (± 9) mV and −151 (± 6) mV, a ∼74 mV split. In this variant, the core sequence for all four helices is identical, and the only changes made are to the surface.
Table 2. Comparison of the Three Charge Variants Used to Modulate Ema.

Positive and binary variants contain additional lysine residues in the loops connecting the helices and are not shown here.
Figure 1.

Nernst curves showing the shift in Em as a function of surface charge. Protein sequences and structures depicted in Table 2. Filled circles are reductive, and open circles are oxidative titration points.
We further wanted to test the effect of buried negative charges. We generated a series of HLH proteins that replaced certain buried Glu residues with Ala maintaining the surface charge of the negatively charged variant. Single residue charge modifications do not sufficiently change Em (Figure 2). We mutated targeted Glu residues into Alanine in the vicinity of the heme b cofactor. We chose E8 and E57, one Glu near each heme b binding site. Given the 2-fold symmetry of the HLH motif, this yielded two changes to the protein. Neither of the two HLH proteins E8A and E57A shows a substantial shift in potential from the original HLH protein remaining at approximately −280 mV (Figure 2). We next mutated all of the buried Glu residues to alanine generating the protein A6 in the HLH motif. This protein only had one midpoint potential of −169 (± 3) mV, a ∼100 mV shift compared to the single chain negatively charged variant, and the related single mutants E8A and E57A. Removing single negative charges from the vicinity of the heme b is not enough; more global changes need to be made to exert a noticeable effect on the cofactor.
Figure 2.

Replacement of Glu residues with Ala causes a shift in Em. (A–D) Cartoons showing the specific Ala residues replaced. (E) Nernst curves of the various Ala substitutions maquettes. Replacement of single Glu residues with Ala does not cause a significant shift, while replacement of all six increases the midpoint potential by 100 mV. Filled circles are reductive, and open circles are oxidative titration points.
Ligation Changes
Heme b ligation bonds are another means of shifting Em. Figure 3 demonstrates the change in Em when Bis-His ligation—the standard ligation in both the SC and HLH proteins—is mutated to His-Cys and Cys-Ala. All of these proteins are variants of the negatively charged SC morphology. The His-Cys ligation changes Em to change from −261 (± 4) mV in Bis-His to −280 (± 3) mV. Although slight, this change also opens up the possibilities for other functions as the Cys residue is used as a ligand in many P450 enzymes. Changing the ligation to Cys-Ala yields spectra that correspond to a 5-coordinate state typically seen in P450s (Figure S8, Das, Sligar42). This ligation change has a more profound effect on Em, raising it to −165 (± 9) mV, a difference of ∼95 mV. This is in line with natural P450 enzymes43 and represents a major increase in the reduction potential of the heme b.
Figure 3.

(A–C) Various heme-ligations that are possible in the single chain maquette protein. These ligations allow for changes to the midpoint potential as seen in (D): Nernst curves of the single chain Bis-His (seen in Figures 1 and 2), along with His-Cys, and Cys-Ala. Filled circles are reductive, and open circles are oxidative titration points.
While cysteine residues are tolerated, other ligations tend to not have a comparable affinity. A single His site is unable to bind heme b at a measurable level. Met residues, like those seen in the natural protein b562, do not bind heme b in our de novo protein system. This is comparable to natural systems. Changing the Met residue of b562 to a histidine shifts the midpoint potential down approximately ∼262 mV; however, it decreases the affinity by at least 13-fold.44
Using Cofactors to Shift the Potential
We next attempted to move two heme b-cofactors within van der Waals distance of one another so that they would be electronically coupled in an effort explore new methods to change the midpoint potential. We moved the ligating histidine amino acids in the HLH maquette such that the two heme b cofactors would be adjacent to one another. However, the adjacent HLH was unable to bind more than 1 equiv of heme b. To address this, the next variant moved one heme b down one heptad such that the two cofactors were now “offset” to one another. Figure 4 shows redox titration of the offset-variant with Em values of −180 (± 3) mV versus the single chain protein with heme b at −261 (± 4) mV, a split of 80 mV. This effect stems from either the positive charge on one heme b affecting the other or destabilization of the core. To verify that this effect is not due to the placement of the site itself, we performed a redox titration with less than 1 equiv of heme (Figure S10). In these data, we only see one potential at ∼−271 (± 3) mV. We are unable to identify the predominant factor from these data.
Figure 4.

(A, B) HLH proteins with heme cofactors placed in distant or offset positions relative to one another (C): Nernst curves showing the effect heme molecules have on one another when placed close together. Filled circles are reductive, and open circles are oxidative titration points.
Using Alternative Porphyrins
The final method we used to change the midpoint potential of the cofactor was to replace heme b in the SC variant with a different Fe-centered porphyrin. We obtained Em values for a variety of synthetic Fe-centered porphyrins bound to the single chain (negatively charged) protein variant (Figures S11–S15). For these studies, one porphyrin was bound to the SC variant; all proteins have similar KD values.45 Using modified porphyrins expands the potential range in both oxidizing and reducing directions (Supplemental Table 1, Figure 5). Mesoporphyrin despite differing by a minor structural change has a shift downward to −319 (± 1) mV (approximately 60 mV relative to heme b). This is not the case with deuteroporphyrin whose Em is −270 (± 1) mV, an approximate 10 mV shift downward, within error for this method. The last porphyrin with a more reducing potential is the Fe-tetracarboxyphenyl porphyrin (TCP), which has four electron donating carboxyphenyl substituents that drive the potential down to −320 (± 9) mV.
Figure 5.

Reduction potentials of the Fe-porphyrins able to bind to the single chain protein. All titrations done at 25 μM. All titrations done in CHES buffer pH 9 at room temperature. Nernst curves in Figures S10–S15.
The rest of this series of porphyrins have substituents that push the potential in the more oxidizing direction (Figure 5). Isohematoporphyrin shifts the potential up to −236 (± 5) mV, likely caused by the hydroxyl groups increasing the water concentration in the core. Hydroxyl groups are polar, which could bond to water inside the protein. It is also possible that the hydroxyl groups are interacting with the porphyrin skeleton stabilizing the reduced state. However, it is not possible to elucidate the main contributing factor from these data. The largest increase in midpoint potential was seen when highly electron withdrawing groups are attached to the porphyrin macrocycle. This destabilizes the reduced state by removing electron density.46 This is most clearly seen in DADPIX where the potential is −48 (± 5) mV. This porphyrin has the two vinyl groups of heme b replaced with highly electronegative acetyl groups, raising Em by approximately 210 mV.
Additivity in Our Maquette System
A key benefit of the maquette system is the ability to combine characteristics. Throughout this work, we have been identifying individual parameters of the protein or cofactor and modifying them to measure their effect onEm. We sought to take two of those parameters to measure the effect of additivity in our system. We utilized the positive mutant’s +11-charged surface that was able to elevate the heme b Em to −151 (± 6) mV and combined it with the DADPIX porphyrin whose electron withdrawing groups push Em up to −48 (± 5) mV. Figure 6 shows the Nernst curve for this combined maquette where we observe a 10 (± 6) mV Em, raising the DADPIX Em up ∼62 mV. Compared to the negatively charged protein with mesoporphyrin, we are able to cover a range of over 330 mV in our de novo protein system. We anticipate that exchange of the charged residues near the heme b with nonpolar Ala would extend this range even further, bringing these proteins in line with many electron transfer proteins in nature and allowing for our tunable system to interface with naturally occurring oxidoreductase and taking part in a slew of electron transfer reactions.
Figure 6.

Cartoon depictions of the SC protein with heme (A), DADPIX (B), and the positively charged protein with DADPIX (C). The combination of charge variation with a different porphyrin can raise the midpoint potential ∼200 mV. Filled circles are reductive, and open circles are oxidative titration points.
Discussion
Here, we demonstrate a simple straightforward set of engineering principles that can guide future designs and allow for a tunable Em: (1) surface charge, (2) altering buried charged groups near the heme b itself, (3) changing the ligation to the heme b toa Cys-Ala, and (4) placing a second cofactor near in the vicinity. A fifth engineering principle replacing the heme b with other porphyrins allowed us to drastically increase the Em of the holo-protein, but the effects of the protein itself did not contribute. We were able to establish that the Em of the holo-protein can be raised higher due to the partial additivity of these principles when we combined the positively charged helical sequence with the DADPIX cofactor.
This work represents the next steps in the findings of Shifman et al., Gibney et al., Koder et al., and others who have studied the mutational effects on protein electrochemistry in de novo designed 4-helix bundle proteins.38,47−50 Shifman et al., in particular, looked into many of the effects that we see studied here.48 However, the bundles in that work were exclusively homotetrameric, and therefore, the authors could not make targeted mutations in the same way we do here. This additional control led to a diversity of heme ligation modes, surface charge differences, and targeted alanine mutations that provide new insights into the functional roles of amino acids in the Heme binding sites of natural proteins. Proteins in this work can have separate redox domains that can be individually modified to drive electron transfer in one direction.
The base de novo protein in Shifman et al.’s work, named H10A24 had a different, electrostatically neutral sequence, and the bound Heme B had two Em values at −216 and −144 mV.48 One heme B bound had an Em of −156 mV. The splitting with two hemes bound occurred due to their adjacent placement to one another and electrostatic coupling. Replacement of one of those heme cofactors with the neutral Zn-protoporphyrin IX resulting in a single midpoint potential of −148 mV. This corroborates our results showing a large splitting when two hemes are placed offset to one another and electrostatically coupled. However, due to difference of the surface charges, the single-heme Em is lower in our SC variant (Figure 4 and Table 3). Similarly, Shifman et al. showed a large change in Em when the porphyrin itself is changed. They bound heme A, a known high potential porphyrin, to H10A24 and showed an increase in Em from −156 mV up to +18 mV, a difference of 174 mV. This was primarily due to the electron withdrawing effects of the aldehyde group directly attached to the tetrapyrrole macrocycle. In our work, we used a larger library of porphyrins with more variable modifications. We showed a greater range of −270 mV in this system purely though different porphyrins as well as additional additivity when these are combined with other changes depicted here (Figure 6 and Table 3).
Table 3. Proteins Used in This Work, Their Topologies, Sequences, and Respective Em Values.
| protein | topology | sequence | cofactor | Em (in mV) |
|---|---|---|---|---|
| negatively charged | SC | EIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQL | heme B | –261 (±4) |
| positively charged | SC | EIKRQHEDALRKFEEALKRFEDKKQKGGSGKGSGGEIWKRHEDALRKFEEALKRFEDKKQKGGSGKGSGGEIWKRHEDALRKFEEALKRFEDKKQKGGSGKGSGGEIKQRHEDALRKFEEALKRFEDKKQK | heme B | –151 (±6) |
| binary charged | SC | EIWKQHEDALQKFEEALNQFEDLKQLGGSGKGSGGEIKRQHEDALRKFEEALKRFEDKKQKGGSGSGSGGEIWKQHEDALQKFEEALNQFEDLKQLGGSGKGSGGEIKRQHEDALRKFEEALKRFEDKKQK | heme B | –151 (±6), −225 (±9) |
| E57A | HLH | EIWKQHEDALQKFEDALNQFEDLKQLGGSGCGSGGEIWKQHEDALQKFEDALNQFADLKQL | heme B | –280 (±10) |
| E8A | HLH | EIWKQHADALQKFEDALNQFEDLKQLGGSGCGSGGEIWKQHEDALQKFEDALNQFEDLKQL | heme B | –307 (±1) |
| A6 | HLH | EIWKQHADALQKFADALNQFADLKQLGGSGCGSGGEIWKQHADALQKFADALNQFADLKQL | heme B | –169 (±3) |
| His-Cys ligation | SC | EIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQAEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQCEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQAEDALQKFEDALNQFEDLKQL | heme B | –280 (±3) |
| Cys-Ala ligation | SC | EIWKQCEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQAEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQAEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQAEDALQKFEDALNQFEDLKQL | heme B | –165 (±9) |
| offset | HLH | EIWKQHEDALQKFEDALNQFEDLKQLGGSGCGSGGEIWKQFEDALQKHEDALNQFEDLKQL | heme B | –180 (±3), −261 (±4) |
| negatively charged | SC | EIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQL | mesoporphyrin | –319 (±1) |
| negatively charged | SC | EIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQL | tetracarboxyphenyl porphyrin | –320 (±9) |
| negatively charged | SC | EIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQL | deuteroporphyrin | –270 (±1) |
| negatively charged | SC | EIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQL | isohematoporphyrin | –236 (±5) |
| negatively charged | SC | EIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQLGGSGSGSGGEIWKQHEDALQKFEDALNQFEDLKQL | diacetyldeuteroporphyrin | –48 (±5) |
| positively charged | SC | EIKRQHEDALRKFEEALKRFEDKKQKGGSGKGSGGEIWKRHEDALRKFEEALKRFEDKKQKGGSGKGSGGEIWKRHEDALRKFEEALKRFEDKKQKGGSGKGSGGEIKQRHEDALRKFEEALKRFEDKKQK | diacetyldeuteroporphyrin | 10 (±6) |
This work studying the effect of protein structure on the Em of bound porphyrin cofactors improves our understanding of oxidoreductase function and the extent to which certain parameters can tune the potential. Our work generally agrees with much of the literature examining heme-protein interactions.51−53 However, we believe our work, due to its more direct characterization of individual parameters, can shed further light on the effects of protein structure. Zheng and Gunner computationally analyzed a library of heme-proteins and showed that combinations of the charge, ligand, dielectric, and other key parameters all contribute toEm.29 Incorporation of this work into their analysis may clarify individual roles and lead to an even deeper understanding the protein structure-Em relationship. Similarly, this work supports that of Bhagi-Damodaran et al. who examined the effect of a serine to alanine mutation in a myoglobin mutant.52 The authors attributed the Em change to a combination of the hydrogen bonding network and an increase in hydrophobicity in the heme binding site. Our work agrees that increasing hydrophobicity increases Em but the magnitude is different. The impact in their myoglobin mutant was ∼28 mV compared to ∼100 mV here. We attribute that difference to unknown compensating factors built into the myoglobin structure. This hypothesis is further supported by Zhang et al. who made similar alanine scanning mutations to a related de novo four-helix bundle protein.49 Although their primary focus was on the oxygen binding functions of the resulting protein, electrochemistry was a significant part of the work. In that paper, the authors measured a 65 mV split between their normal and alanine variants. It is unclear what is causing the ∼30 mV discrepancy in measured values, but the effect in both cases, Em is raised when all six Glu residues are replaced with Ala. Zhang et al. did not make individual mutations to identify which Ala residues contributed to the effect or search for the minimal number of hydrophobic amino acids needed to change Em. Here, we made two mutations near the heme itself. In this topology, the single residue changes were duplicated on the other side as well resulting in two changes per mutation. However, neither position yielded any noticeable difference compared to the SC variant (Figure 2 and Table 3). From this, we can conclude that removing single Glu residues does not materially affect the Em of heme B.
A boon for the tetrahelical bundle system is the malleability of the de novo scaffold. This is very clearly shown in the wide range of midpoint potentials achieved in this paper, and the variety of functions they have been shown to accomodate.15,34 Making surface mutations and being able to couple them to modified porphyrins is a significant advantage over natural proteins, allowing both large scale and fine-tuning control of Em values over approximately a 330 mV range.
This work also establishes principles needed to create subdomains within our simplified scaffold, which is important for directing electron transfer. Complex I of the mitochondrial respiratory chain uses different local environments to shift the Em of Fe-S clusters to direct electrons transfer.54 A small number of de novo protein model systems were designed to study Fe-S clusters.55−57 In those studies, the authors have one cluster per protein and therefore cannot show directionality in electron transfer reactions. In this work, we show tetrahelical bundles engineered to create different redox domains—with tailorable distances—that can drive electron transfer in one direction. This concept of protein modularity is an important aspect of the maquette platform. This also allows for de novo proteins to be inserted into more diverse electron transfer pathways, bridging multiple redox centers.
For design considerations, changing the porphyrin directly yields the most extensive range going from −311 mV up to −48 mV in the same protein scaffold without significant changes to the affinity or having to make mutations to the protein. Adding electron-withdrawing groups to the porphyrin increases the redox potential, an effect seen in nature between groups like heme b and heme a or in in vitro settings between heme a and synthetic mimics.46,52,58 However, there previously existed no neutral way to test their functional implications without potentially upsetting other unknown interactions. Inserting a new cofactor into an existing natural protein can be difficult due to steric constraints or misaligned contacts. In our robust tetrahelical bundle platform, cofactors can be swapped out easily—as can the ligating amino acids—providing a simplified system that can highlight the effect of Em changes on function. It has been shown that Em is a key factor, and this work provides an excellent system for these studies.5
Changing the charge pattern on the helix can allow for Em changes of approximately 150 mV. Recent literature has shown protein supercharging to be a novel way of controlling protein function.59,60 That work focused primarily on allosteric regulation and molecular switching. In this work, we expand on its potential by considering the implications this would have on the redox. First, the change in charge itself is shown here to be a major factor in Em. However, there is a second important element, the change in structure and oligomerization. The balancing of charge with high levels of salt allowed for Cyt b562 to adopt its native structure or the GFP variants to oligomerize. These morphological and structural changes could be used to develop new domains for redox cofactors that have different potentials—and therefore different functions—based entirely on the salt concentration. Furthermore, the interfaces between two separately supercharged dimers could form a neutralizing allosteric effector capable of switching an oxidoreductase function on or off.
Ligation changes result in a 95 mV shift in the heme Em in these tetrahelical bundles, which provide an ideal way to study its effects on function and other downstream properties. It has been shown that ligations are important to function and can even change a protein from a mono-oxygenase into a carbene transferase.61 Our system, with its robust tolerance of mutations, can be a new tool for these studies. Recent work has shown that the pocket of myoglobin can be affected by the heme itself causing ligand switches if the propionates are esterified.62 In our system, this porphyrin also binds but the ligation is unaffected.45 In this work, we have engineered tools that can further investigate function without any complications like competing ligands in the binding site. The tetrahelical bundles can be made with the peptide synthesizer, which can incorporate nonstandard amino acids with minimal structural perturbation.63 Recent work has also shown the need to expand the possible ligand set to Fe-centered porphyrins to help study and promote carbene transfer.64 These proteins can fill that niche and be an impactful tool for these studies.
Cofactor placement impacts Em by approximately 80 mV. In the offset-HLH, despite the low affinity in the reduced state, the two-heme b cofactors are electronically coupled as seen by their redox activity. The natural OmcS protein from Geobacter sulfurreducens has six closely stacked heme cofactors that all are used to transport electrons over long distances, but it is unclear how their relative orientation affects their electrochemical properties.65,66 This work can help explain how the directionality of electron transport is achieved and how the Em of the multiple cofactors is set and maintained at both the local-subunit and whole-fiber levels.
Conclusions
The engineering principles we uncovered here: surface charge, buried charge, cofactor ligation, cofactor coupling, and cofactor replacement, all have effect on the Em of bound porphyrins and can be combined to produce potent additive effects. This tunability can lead to a library of de novo proteins with tailored Em values that can accommodate a large range of ET partners in varying directions. All mutations were made in a similar four-helix bundle architecture, meaning that different folds are not required to achieve a difference in potential and that the same interface can be used at different levels. These principles can also be applied to natural proteins to better understand how they poise Em values and better diagnose redox-based metabolic diseases from a functional perspective. These de novo proteins can potentially lead to a variety of tools that make studying oxidoreductases far easier and provide a new way to study electron transfer. In the future, groups will choose the midpoint potential they want when studying oxidoreductases in vivo and in vitro. The modularity allows these tetrahelical bundles to interface with existing proteins as either electron donors or acceptors by changing Em without significantly changing the rest of the structure.
Acknowledgments
The authors would like to thank Joel Schnur for assistance editing this manuscript. This work was funded by the Johnson Research Foundation. This research was supported in part by the U.S. National Institutes of Health, General Medical Institutes (RO1GM 41048), which funded molecular biology supplies, expression, and purification from E. coli of the different heme binding proteins that included five structurally distinct maquettes, also the electrochemical characterization of these proteins. The research was also supported in part by U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering (DE-FG02-05ER46223), which funded materials, design, synthesis, characterization, and development of the family of structural and polarity variants of Fe-tetrapyrroles and the characterization and analysis of their binding rates in the proteins.
Glossary
Abbreviations
- TLC
thin-layer chromatography
- DADPIX
diacetyldeuteroporphyrin
- Em
redox midpoint potential
- CHES
N-cyclohexyl-2-aminoethanesulfonic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.2c05119.
Spectra corresponding to the Nernst curves in the manuscript, the Nernst curve, and UV–vis spectra for the offset mutation with one heme, the Nernst curves for the alternative porphyrins, and KD information for the alternative porphyrins binding to the SC protein (PDF)
Author Present Address
⊥ Department of Human Genetics, University of Pittsburg, Pittsburg, Pennsylvania 15213, United States (J.W.)
Author Present Address
# Molecular Templates, Austin, Texas 78729, United States (G.K.).
Author Contributions
The manuscript was written through contributions of all authors. L.A.S. performed the experiments and performed data analysis. J.W. assisted with data analysis and wrote the python code to clean and process the data. G.K., C.M., and P.L.D. assisted with the protein design and experimental interpretation and analysis. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Nelson D. L.; Cox M. M.; Lehninger A. L.. Lehninger Principles of Biochemistry; Seventh edition.; W.H. Freeman and Company; Macmillan Higher Education: New York, NY: Houndmills, Basingstoke, 2017. [Google Scholar]
- Franco R.; Vargas M. R. Redox Biology in Neurological Function, Dysfunction, and Aging. Antioxid. Redox Signaling 2018, 28, 1583–1586. 10.1089/ars.2018.7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tell G.; Scaloni A.; Pellizzari L.; Formisano S.; Pucillo C.; Damante G. Redox Potential Controls the Structure and DNA Binding Activity of the Paired Domain. J. Biol. Chem. 1998, 273, 25062–25072. 10.1074/jbc.273.39.25062. [DOI] [PubMed] [Google Scholar]
- Nastri F.; D’Alonzo D.; Leone L.; Zambrano G.; Pavone V.; Lombardi A. Engineering Metalloprotein Functions in Designed and Native Scaffolds. Trends Biochem. Sci. 2019, 44, 1022–1040. 10.1016/j.tibs.2019.06.006. [DOI] [PubMed] [Google Scholar]
- Bhagi-Damodaran A.; Reed J. H.; Zhu Q.; Shi Y.; Hosseinzadeh P.; Sandoval B. A.; Harnden K. A.; Wang S.; Sponholtz M. R.; Mirts E. N.; Dwaraknath S.; Zhang Y.; Moënne-Loccoz P.; Lu Y. Heme Redox Potentials Hold the Key to Reactivity Differences between Nitric Oxide Reductase and Heme-Copper Oxidase. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 6195–6200. 10.1073/pnas.1720298115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis V.; Cascione M.; Toma C.; Leporatti S. Silver Nanoparticles: Synthetic Routes, In Vitro Toxicity and Theranostic Applications for Cancer Disease. Nanomaterials 2018, 8, 319. 10.3390/nano8050319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day B. J. Antioxidant Therapeutics: Pandora′s Box. Free Radical Biol. Med. 2014, 66, 58–64. 10.1016/j.freeradbiomed.2013.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges R. S.; Carneiro A. S.; Barros T. G.; Barros C. A. L.; Neto A. M. J. C.; da Silva A. B. F. Understanding the Cytotoxicity or Cytoprotective Effects of Biological and Synthetic Quinone Derivatives by Redox Mechanism. J. Mol. Model. 2014, 20, 2541. 10.1007/s00894-014-2541-9. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C. The Challenges of Using Fluorescent Probes to Detect and Quantify Specific Reactive Oxygen Species in Living Cells. Biochim. Biophys. Acta, Gen. Subj. 2014, 1840, 730–738. 10.1016/j.bbagen.2013.05.004. [DOI] [PubMed] [Google Scholar]
- Arts M. J. T. J.; Haenen G. R. M. M.; Voss H.-P.; Bast A. Antioxidant Capacity of Reaction Products Limits the Applicability of the Trolox Equivalent Antioxidant Capacity (TEAC) Assay. Food Chem. Toxicol. 2004, 42, 45–49. 10.1016/j.fct.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Volkov A. N.; van Nuland N. A. J. Electron Transfer Interactome of Cytochrome c. PLoS Comput. Biol. 2012, 8, e1002807 10.1371/journal.pcbi.1002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser C. C.; Keske J. M.; Warncke K.; Farid R. S.; Dutton P. L. Nature of Biological Electron Transfer. Nature 1992, 355, 796–802. 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Electron Transfer in Proteins. Annu. Rev. Biochem. 1996, 65, 537–561. 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- Ferrando J.; Solomon L. A. Recent Progress Using De Novo Design to Study Protein Structure, Design and Binding Interactions. Life 2021, 11, 225. 10.3390/life11030225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korendovych I. V.; DeGrado W. F. De Novo Protein Design, a Retrospective. Quart. Rev. Biophys. 2020, 53, e3 10.1017/S0033583519000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koder R. L.; Anderson J. L. R.; Solomon L. A.; Reddy K. S.; Moser C. C.; Dutton P. L. Design and Engineering of an O2 Transport Protein. Nature 2009, 458, 305–309. 10.1038/nature07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun J. R.; Nastri F.; Maglio O.; Pavone V.; Lombardi A.; DeGrado W. F. Artificial Diiron Proteins: From Structure to Function. Biopolymers 2005, 80, 264–278. 10.1002/bip.20230. [DOI] [PubMed] [Google Scholar]
- Baker D. What Has de Novo Protein Design Taught Us about Protein Folding and Biophysics?. Protein Sci. 2019, 28, 678–683. 10.1002/pro.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins G. H.; Noble C. E. M.; Blackburn H.; Hardy B.; Landau C.; Parnell A. E.; Yadav S.; Williams C.; Race P. R.; Oliveira A. S. F.; et al. Precision Design of Single and Multi-Heme de Novo Proteins; preprint; Synthetic Biology, 2020. [Google Scholar]
- Klein A. S.; Zeymer C. Design and Engineering of Artificial Metalloproteins: From de Novo Metal Coordination to Catalysis. Protein Eng., Des. Sel. 2021, 34, gzab003. 10.1093/protein/gzab003. [DOI] [PubMed] [Google Scholar]
- Grayson K. J.; Anderson J. L. R. Designed for Life: Biocompatible de Novo Designed Proteins and Components. J. R. Soc., Interface 2018, 15, 20180472. 10.1098/rsif.2018.0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins D. W.; Jenkins J. M. X.; Grayson K. J.; Wood N.; Steventon J. W.; Le Vay K. K.; Goodwin M. I.; Mullen A. S.; Bailey H. J.; Crump M. P.; et al. Construction and in Vivo Assembly of a Catalytically Proficient and Hyperthermostable de Novo Enzyme. Nat. Commun. 2017, 8, 358. 10.1038/s41467-017-00541-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procko E.; Berguig G. Y.; Shen B. W.; Song Y.; Frayo S.; Convertine A. J.; Margineantu D.; Booth G.; Correia B. E.; Cheng Y.; Schief W. R.; Hockenbery D. M.; Press O. W.; Stoddard B. L.; Stayton P. S.; Baker D. A Computationally Designed Inhibitor of an Epstein-Barr Viral Bcl-2 Protein Induces Apoptosis in Infected Cells. Cell 2014, 157, 1644–1656. 10.1016/j.cell.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L.; Goreshnik I.; Coventry B.; Case J. B.; Miller L.; Kozodoy L.; Chen R. E.; Carter L.; Walls A. C.; Park Y.-J.; Strauch E. M.; Stewart L.; Diamond M. S.; Veesler D.; Baker D. De Novo Design of Picomolar SARS-CoV-2 Miniprotein Inhibitors. Science 2020, 370, 426–431. 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins D. W.; Armstrong C. T.; Beesley J. L.; Marsh J. E.; Jenkins J. M. X.; Sessions R. B.; Mann S.; Ross Anderson J. L. A Suite of de Novo c-Type Cytochromes for Functional Oxidoreductase Engineering. Biochim. Biophys. Acta, Rev. Bioenerg. 2016, 1857, 493–502. 10.1016/j.bbabio.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Hecht M. H.; Zarzhitsky S.; Karas C.; Chari S. Are Natural Proteins Special? Can We Do That?. Curr. Opin. Struct. Biol. 2018, 48, 124–132. 10.1016/j.sbi.2017.11.009. [DOI] [PubMed] [Google Scholar]
- Mirts E. N.; Bhagi-Damodaran A.; Lu Y. Understanding and Modulating Metalloenzymes with Unnatural Amino Acids, Non-Native Metal Ions, and Non-Native Metallocofactors. Acc. Chem. Res. 2019, 52, 935–944. 10.1021/acs.accounts.9b00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naowarojna N.; Cheng R.; Lopez J.; Wong C.; Qiao L.; Liu P. Chemical Modifications of Proteins and Their Applications in Metalloenzyme Studies. Synth. Syst. Biotechnol. 2021, 6, 32–49. 10.1016/j.synbio.2021.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z.; Gunner M. R. Analysis of the Electrochemistry of Hemes with Em s Spanning 800 MV. Proteins 2009, 75, 719–734. 10.1002/prot.22282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenner R.; Steventon J. W.; Seddon A.; Anderson J. L. R. A de Novo Peroxidase Is Also a Promiscuous yet Stereoselective Carbene Transferase. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 1419–1428. 10.1073/pnas.1915054117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutter A. C.; Tyryshkin A. M.; Campbell I. J.; Poudel S.; Bennett G. N.; Silberg J. J.; Nanda V.; De Falkowski P. G. Novo Design of Symmetric Ferredoxins That Shuttle Electrons in Vivo. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 14557–14562. 10.1073/pnas.1905643116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M. A.; McKinley K. L.; Bradley L. H.; Viola S. R.; De Hecht M. H. Novo Designed Proteins from a Library of Artificial Sequences Function in Escherichia Coli and Enable Cell Growth. PLoS One 2011, 6, e15364 10.1371/journal.pone.0015364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini J. A.; Sheehan M.; Kodali G.; Chow B. Y.; Bryant D. A.; Dutton P. L.; De Moser C. C. Novo Synthetic Biliprotein Design, Assembly and Excitation Energy Transfer. J. R. Soc., Interface 2018, 15, 20180021. 10.1098/rsif.2018.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farid T. A.; Kodali G.; Solomon L. A.; Lichtenstein B. R.; Sheehan M. M.; Fry B. A.; Bialas C.; Ennist N. M.; Siedlecki J. A.; Zhao Z.; Stetz M. A.; Valentine K. G.; Anderson J. L. R.; Wand A. J.; Discher B. M.; Moser C. C.; Dutton P. L. Elementary Tetrahelical Protein Design for Diverse Oxidoreductase Functions. Nat. Chem. Biol. 2013, 9, 826–833. 10.1038/nchembio.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland G. A.; Grayson K. J.; Adams N. B. P.; Mermans D. M. J.; Jones A. S.; Robertson A. J.; Auman D. B.; Brindley A. A.; Sterpone F.; Tuffery P.; Derreumaux P.; Dutton P. L.; Robinson C.; Hitchcock A.; Hunter C. N. Probing the Quality Control Mechanism of the Escherichia Coli Twin-Arginine Translocase with Folding Variants of a de Novo–Designed Heme Protein. J. Biol. Chem. 2018, 293, 6672–6681. 10.1074/jbc.RA117.000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson K. J.; Anderson J. R. The Ascent of Man(Made Oxidoreductases). Curr. Opin. Struct. Biol. 2018, 51, 149–155. 10.1016/j.sbi.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry E. A.; Trumpower B. L. Simultaneous Determination of Hemes a, b, and c from Pyridine Hemochrome Spectra. Anal. Biochem. 1987, 161, 1–15. 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- Gibney B. R.; Rabanal F.; Reddy K. S.; Dutton P. L. Effect of Four Helix Bundle Topology on Heme Binding and Redox Properties †. Biochemistry 1998, 37, 4635–4643. 10.1021/bi971856s. [DOI] [PubMed] [Google Scholar]
- Dutton P. L.[23] Redox Potentiometry: Determination of Midpoint Potentials of Oxidation-Reduction Components of Biological Electron-Transfer Systems. In Methods in Enzymology; Elsevier, 1978; Vol. 54, pp. 411–435. [DOI] [PubMed] [Google Scholar]
- Huang S. S.; Gibney B. R.; Stayrook S. E.; Leslie Dutton P.; Lewis M. X-Ray Structure of a Maquette Scaffold. J. Mol. Biol. 2003, 326, 1219–1225. 10.1016/S0022-2836(02)01441-9. [DOI] [PubMed] [Google Scholar]
- Kluyver T.; Ragan-Kelley B.; Perez F.; Granger B.; Bussonnier M.. Jupyter Notebooks – a Publishing Format for Reproducible Computational Workflows. In Loizides F.; Schmidt B, editors. Positioning and Power in Academic Publishing: Players, Agents and Agendas .; 2016; pp. 87–90.
- Das A.; Grinkova Y. V.; Sligar S. G. Redox Potential Control by Drug Binding to Cytochrome P450 3A4. J. Am. Chem. Soc. 2007, 129, 13778–13779. 10.1021/ja074864x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming B. D.; Tian Y.; Bell S. G.; Wong L.-L.; Urlacher V.; Hill H. A. O. Redox Properties of Cytochrome P450 BM3 Measured by Direct Methods: Redox Properties of Cytochrome P450 BM3. Eur. J. Biochem. 2003, 270, 4082–4088. 10.1046/j.1432-1033.2003.03799.x. [DOI] [PubMed] [Google Scholar]
- Hay S.; Wydrzynski T. Conversion of the Escherichia Coli Cytochrome b562 to an Archetype Cytochrome b: A Mutant with Bis-Histidine Ligation of Heme Iron †. Biochemistry 2005, 44, 431–439. 10.1021/bi0492298. [DOI] [PubMed] [Google Scholar]
- Solomon L. A.; Kodali G.; Moser C. C.; Dutton P. L. Engineering the Assembly of Heme Cofactors in Man-Made Proteins. J. Am. Chem. Soc. 2014, 136, 3192–3199. 10.1021/ja411845f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanullah S.; Saha P.; Saha R.; Dey A. Synthetic Iron Porphyrins for Probing the Differences in the Electronic Structures of Heme a3, Heme d, and Heme d1. Inorg. Chem. 2019, 58, 152–164. 10.1021/acs.inorgchem.8b02063. [DOI] [PubMed] [Google Scholar]
- Shifman J. M.; Moser C. C.; Kalsbeck W. A.; Bocian D. F.; Dutton P. L. Functionalized de Novo Designed Proteins: Mechanism of Proton Coupling to Oxidation/Reduction in Heme Protein Maquettes †. Biochemistry 1998, 37, 16815–16827. 10.1021/bi9816857. [DOI] [PubMed] [Google Scholar]
- Shifman J. M.; Gibney B. R.; Sharp R. E.; Dutton P. L. Heme Redox Potential Control in de Novo Designed Four-α-Helix Bundle Proteins †. Biochemistry 2000, 39, 14813–14821. 10.1021/bi000927b. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Anderson J. L. R.; Ahmed I.; Norman J. A.; Negron C.; Mutter A. C.; Dutton P. L.; Koder R. L. Manipulating Cofactor Binding Thermodynamics in an Artificial Oxygen Transport Protein. Biochemistry 2011, 50, 10254–10261. 10.1021/bi201242a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju G.; Capo J.; Lichtenstein B. R.; Cerda J. F.; Koder R. L. Manipulating Reduction Potentials in an Artificial Safranin Cofactor. Tetrahedron Lett. 2012, 53, 1201–1203. 10.1016/j.tetlet.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melin F.; Hellwig P. Redox Properties of the Membrane Proteins from the Respiratory Chain. Chem. Rev. 2020, 120, 10244–10297. 10.1021/acs.chemrev.0c00249. [DOI] [PubMed] [Google Scholar]
- Bhagi-Damodaran A.; Petrik I. D.; Marshall N. M.; Robinson H.; Lu Y. Systematic Tuning of Heme Redox Potentials and Its Effects on O2 Reduction Rates in a Designed Oxidase in Myoglobin. J. Am. Chem. Soc. 2014, 136, 11882–11885. 10.1021/ja5054863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseinzadeh P.; Lu Y. Design and Fine-Tuning Redox Potentials of Metalloproteins Involved in Electron Transfer in Bioenergetics. Biochim. Biophys. Acta, Rev. Bioenerg. 2016, 1857, 557–581. 10.1016/j.bbabio.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma L. K.; Lu J.; Bai Y. Mitochondrial Respiratory Complex I: Structure, Function and Implication in Human Diseases. Curr. Med. Chem. 2009, 16, 1266–1277. 10.2174/092986709787846578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda V.; Senn S.; Pike D. H.; Rodriguez-Granillo A.; Hansen W. A.; Khare S. D.; Noy D. Structural Principles for Computational and de Novo Design of 4Fe–4S Metalloproteins. Biochim. Biophys. Acta, Rev. Bioenerg. 2016, 1857, 531–538. 10.1016/j.bbabio.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzyb J.; Xu F.; Weiner L.; Reijerse E. J.; Lubitz W.; Nanda V.; De Noy D. Novo Design of a Non-Natural Fold for an Iron–Sulfur Protein: Alpha-Helical Coiled-Coil with a Four-Iron Four-Sulfur Cluster Binding Site in Its Central Core. Biochim. Biophys. Acta, Rev. Bioenerg. 2010, 1797, 406–413. 10.1016/j.bbabio.2009.12.012. [DOI] [PubMed] [Google Scholar]
- Mancini J. A.; Pike D. H.; Tyryshkin A. M.; Haramaty L.; Wang M. S.; Poudel S.; Hecht M.; Nanda V. Design of a Fe 4 S 4 Cluster into the Core of a de Novo Four-helix Bundle. Biotechnol. Appl. Biochem. 2020, 67, 574–585. 10.1002/bab.2003. [DOI] [PubMed] [Google Scholar]
- Gibney B. R.; Isogai Y.; Rabanal F.; Reddy K. S.; Grosset A. M.; Moser C. C.; Dutton P. L. Self-Assembly of Heme A and Heme B in a Designed Four-Helix Bundle: Implications for a Cytochrome c Oxidase Maquette †. Biochemistry 2000, 39, 11041–11049. 10.1021/bi000925r. [DOI] [PubMed] [Google Scholar]
- Schnatz P. J.; Brisendine J. M.; Laing C. C.; Everson B. H.; French C. A.; Molinaro P. M.; Koder R. L. Designing Heterotropically Activated Allosteric Conformational Switches Using Supercharging. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 5291–5297. 10.1073/pnas.1916046117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon A. J.; Zhou Y.; Ramasubramani V.; Glaser J.; Pothukuchy A.; Gollihar J.; Gerberich J. C.; Leggere J. C.; Morrow B. R.; Jung C.; Glotzer S. C.; Taylor D. W.; Ellington A. D. Supercharging Enables Organized Assembly of Synthetic Biomolecules. Nat. Chem. 2019, 11, 204–212. 10.1038/s41557-018-0196-3. [DOI] [PubMed] [Google Scholar]
- McIntosh J. A.; Heel T.; Buller A. R.; Chio L.; Arnold F. H. Structural Adaptability Facilitates Histidine Heme Ligation in a Cytochrome P450. J. Am. Chem. Soc. 2015, 137, 13861–13865. 10.1021/jacs.5b07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nye D. B.; Preimesberger M. R.; Majumdar A.; Lecomte J. T. J. Histidine–Lysine Axial Ligand Switching in a Hemoglobin: A Role for Heme Propionates. Biochemistry 2018, 57, 631–644. 10.1021/acs.biochem.7b01155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein B. R.; Bialas C.; Cerda J. F.; Fry B. A.; Dutton P. L.; Moser C. C. Designing Light-Activated Charge-Separating Proteins with a Naphthoquinone Amino Acid. Angew. Chem., Int. Ed. 2015, 54, 13626–13629. 10.1002/anie.201507094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott M.; Tinzl M.; Hayashi T.; Ota Y.; Dunkelmann D.; Mittl P. R. E.; Hilvert D. Noncanonical Heme Ligands Steer Carbene Transfer Reactivity in an Artificial Metalloenzyme**. Angew. Chem., Int. Ed. 2021, 60, 15063–15068. 10.1002/anie.202103437. [DOI] [PubMed] [Google Scholar]
- Qian X.; Mester T.; Morgado L.; Arakawa T.; Sharma M. L.; Inoue K.; Joseph C.; Salgueiro C. A.; Maroney M. J.; Lovley D. R. Biochemical Characterization of Purified OmcS, a c-Type Cytochrome Required for Insoluble Fe(III) Reduction in Geobacter Sulfurreducens. Biochim. Biophys. Acta, Rev. Bioenerg. 2011, 1807, 404–412. 10.1016/j.bbabio.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Wang F.; Gu Y.; O’Brien J. P.; Yi S. M.; Yalcin S. E.; Srikanth V.; Shen C.; Vu D.; Ing N. L.; Hochbaum A. I.; Egelman E. H.; Malvankar N. S. Structure of Microbial Nanowires Reveals Stacked Hemes That Transport Electrons over Micrometers. Cell 2019, 177, 361–369.e10. 10.1016/j.cell.2019.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.