

Abstract
Background:
Biochemical reaction networks are self-regulated in part due to feedback activation mechanisms. The tissue factor (TF) pathway of blood coagulation is a complex reaction network controlled by multiple feedback loops that coalesce around the serine protease thrombin.
Objectives:
Our goal was to evaluate the relative contribution of the feedback activation of coagulation factor XI (FXI) in TF-mediated thrombin generation using a comprehensive systems-based analysis.
Materials and Methods:
We developed a systems biology model that improves the existing Hockin-Mann (HM) model through an integrative approach of mathematical modeling and in vitro experiments. Thrombin generation measured using in vitro assays revealed that the feedback activation of FXI contributes to the propagation of thrombin generation based on the initial concentrations of TF or activated coagulation factor X (FXa). We utilized experimental data to improve the robustness of the HM model to capture thrombin generation kinetics without a role for FXI before including the feedback activation of FXI by thrombin to construct the extended (ext.) HM model.
Results and Conclusions:
Using the ext.HM model, we predicted that the contribution of positive feedback of FXI activation by thrombin can be abolished by selectively eliminating the inhibitory function of tissue factor pathway inhibitor (TFPI), a serine protease inhibitor of FXa and TF-activated factor VII (FVIIa) complex. This prediction from the ext.HM model was experimentally validated using thrombin generation assays with function blocking antibodies against TFPI and plasmas depleted of FXI. Together, our results demonstrate the applications of combining experimental and modeling techniques in predicting complex biochemical reaction systems.
Keywords: coagulation factor XI, feedback regulation, systems biology, thrombin, tissue factor pathway inhibitor
1 |. INTRODUCTION
Blood coagulation is a network of complex reactions dependent upon the conversion of inactive species (zymogens) of coagulation factors to active serine proteases. Often portrayed as a waterfall, the plasma coagulation was originally described as a series of enzymatic events initiated by two sources, tissue factor (TF) and coagulation factor XII (FXII), converging in the activation of coagulation factor X (FX). TF-initiated coagulation, referred to here as the TF pathway, starts when blood is exposed to extravascular TF that forms a complex with coagulation factor VII (FVII) to catalyze the conversion of FVII to its activated version FVIIa.1,2 The TF–FVIIa complex then activates coagulation factors X and IX (FIX) leading to the generation of thrombin. Thrombin is the central serine protease that serves as a master regulator of coagulation by activating both procoagulant and anticoagulants species. For instance, thrombin amplifies its generation by direct activation of the cofactors factor V (FV) and factor VIII (FVIII).3,4 Moreover, thrombin activates coagulation factor XI (FXI), a member of the contact activation pathway, which in turn activates FIX to create a feedback loop of thrombin generation.5
While experimental studies utilizing purified components or selective inhibitors have been useful for delineating the contribution of each individual component of the coagulation system, an integrative systems biology approach is required to enable data-driven knowledge discovery and predict yet unknown pathways for thrombin generation. With this as a goal, Hockin et al. built one of the first mathematical models of the TF pathway of thrombin generation by assembling 42 individual coagulation factor activation or inhibition reactions into ordinary differential equations (ODEs).6 The Hockin-Mann (HM) model enabled an understanding of the temporal variation of individual species involved in the TF pathway and their sensitivity to variable initial conditions.7–10 Naturally, the HM model was quickly adapted into several models of coagulation that included contributions of flow, diffusion, fibrin formation, and platelets to thrombin generation.11–14 While the HM model has proven extremely useful in studying the TF pathway, it was built specifically to exclude coagulation FXI, in part due to an incomplete mechanistic understanding of the role of FXI in thrombin generation. Moreover, at that time, the physiological relevance of FXI to TF-mediated thrombin generation was in question in part due to in vitro studies that utilized supraphysiological concentrations of TF as initiators.15 Therefore, by definition, the utility of the HM model is limited to conditions in which FXI does not play a role in thrombin generation.
In this work, we aim to extend the applications of the HM model for studying the TF pathway of thrombin generation by including the feedback activation of FXI by thrombin. Toward this goal, we used an integrative approach of thrombin generation experiments and numerical analysis to extend the capacity of the HM model to predict the effect of FXI on TF-mediated thrombin generation. We added the mechanisms of feedback activation of FXI by thrombin, activation of FIX by FXIa, and inhibition of FXIa by plasma proteins. We utilized in vitro thrombin generation assays to support the development of our model. Finally, we tested the robustness of our model by predicting and experimentally validating the role of FXI in the TF pathway with respect to tissue factor pathway inhibitor (TFPI), an endogenous inhibitor circulating in plasma. Our model and experiments suggest the hypothesis that the effect of FXI in TF- mediated thrombin generation can be modified by neutralizing the function of TFPI in plasma. Our work is a demonstration of how mathematical models can be a useful method for testing and deriving novel mechanistic ideas that can be experimentally verified.
2 |. MATERIALS AND METHODS
2.1 |. Reagents
FXI-depleted plasma was from Affinity Biologicals Inc. Dade™ Innovin™ from Siemens Healthineers wasthe source of TF. Neutralizing monoclonal mouse anti-human TFPI antibodies, specific for the Kunitz 1 or 2 domains, was from MyBioSource Inc. Corn trypsin inhibitor (CTI) was obtained from Enzyme Research Laboratories. Plasma-derived FXa and FXI were from Haematologic Technologies, Inc. The anti-factor XI antibody 1A6, which blocks the activation of FIX by FXIa, was generated as previously described.16 The fluorogenic substrate and calibrator for thrombin generation assay was from TECHNOTHROMBIN® TGA kit (technoclone, GmbH). The TGA kit consists of the fluorogenic substrate, 1 mM Z-G-G-Ar-AMC in 15 mM CaCl2, and the calibrator of 1 nM thrombin. Black 96-well plates were from Corning. HEPES buffered saline (HBS) containing 25 mM HEPES and 150 mM NaCl (pH7.4) was used as the buffer for all dilutions unless stated otherwise.
2.2 |. Thrombin generation—Experiments
Thrombin generation in plasma was measured by following the cleavage of fluorogenic substrate Z-G-G-Ar-AMC at 37°C on a Tecan’s Infinite M200 microplate reader. Experiments were performed in 96-well black plates coated with polyethylene glycol 20000 (2%). FXI-depleted plasma was supplemented with 30nM FXI or vehicle and with 40 μg/ml CTI. In select experiments, plasma was additionally supplemented with 30 μg/ml 1A6 or vehicle or anti-TFPI antibodies (6 μg/ml) for 30 mins before use at room temperature. HBS with 15mM CaCl2 was used as the buffer for diluting TF (0.125–1 pM) or FXa (0.1–0.5 nM) seeing the fluorogenic substrate contained 15 mM CaCl2.
Thrombin generation was initiated by mixing 40 μl of supplemented plasma with 60 μl of reagent substrate mixture, containing 50 μl of fluorogenic substrate and 10 μl of TF or FXa. The fluorescence was recorded at 60 s intervals with an excitation wavelength of 360 nm and emission wavelength of 460 nm for 60 min. Thrombin generation was calculated by using TECHNOTHROMBIN TGA evaluation software. The resulting thrombin generation curves from the software were analyzed to estimate the kinetics of thrombin generation.
2.3 |. Thrombin generation—Simulations
The mathematical models of tissue factor pathway of thrombin generation were set up as systems of ODEs as described in Hockin et al.6 and.11 and solved using odeint from SciPy in Python with a tolerance of 1.5 × 10−8. Odeint uses a Livermore Solver for Ordinary Differential Equations (LSODA) algorithm with dynamic step sizing to solve ODEs and a classic example of the algorithm’s usage for a similar purpose is the original implementation of the HM model.6 Initially, simulations were performed using just SciPy, which led to an average run time of 5 min making it computationally very expensive to run further studies using the model. Therefore, to improve the speed of our simulations, we utilized Numba, which translates Python functions to machine code. This led to a decrease in average run time from 204 to 30 s. For this work, all computations were implemented in Spyder 4.2.5 IDE running on an Intel i7 core system with 16 GB RAM.
Both the models assume the plasma is well mixed without any spatial variations in enzyme levels and transport limitations such as diffusion. The models assume that all reactions occur in the fluid phase and do not explicitly model the surface reactions. As such, our models do not include contact activation of FXII or activation of FXI by FXIIa; experimentally, we included CTI to pharmacologically block the contribution of FXII to thrombin generation. The HM model also does not include fibrin-related reactions and these conditions were unchanged when we improved the model. In all cases, 60 min of thrombin generation from the addition of initiator was simulated.
The HM model is taken from the work of Hockin et al.6 without any modifications. The initial concentrations of enzymes used in all simulations are normal plasma concentrations as provided in the supporting information, and all these concentrations were diluted 2/5th to match the experimental conditions used in the study. To simulate conditions of FXI-depleted plasma or TFPI-neutralized plasma, the initial concentrations of FXI or TFPI were set to “0.”
The following output parameters were determined from the simulations as described below:
Endogenous thrombin potential (ETP, nM.min)—area under the thrombin generation curve.
Lag time (min)—time taken to generate 2 nM thrombin; if less than 2 nM thrombin was generated within the simulated 60 mins (maximum timespan of simulation), lag time is counted as 60 min.
Peak thrombin (nM)—maximum thrombin concentration observed within the simulated 60 min of thrombin generation.
Peak time (min)—time taken to reach peak thrombin.
2.4 |. Estimation of kinetic constants
Kinetic constants of the mod.HM model were estimated by simultaneously fitting all four outputs of thrombin generation, ETP, lag time, peak thrombin, and peak time to the experimental data by using the Sequential Least Squares Programming (SLSQP) algorithm available in SciPy package. Values of kinetic constants from the HM model were used as initial guesses to estimate the kinetic constants for mod.HM model. For estimating the kinetics of FXI-related reactions to use in the ext.HM model, the values of kinetic constants obtained from the literature were used as starting points.11 The supporting information includes details on the initial plasma enzyme concentrations and the values of the estimated kinetic constants used in the model.
2.5 |. Statistical analysis
Experimental data were analyzed using GraphPad Prism 9 software. Data are shown as means ± standard error of means (SEMs) of three independent experiments. Illustrated curves from experiments are representative of three independent trials performed in duplicates. Single pair comparisons were analyzed by 2-tailed Student’s t test. Dunnett’s multiple comparisons tests with two-way analysis of variance (ANOVA) were performed to compare vehicle to treatments for more than two groups.
3 |. RESULTS
3.1 |. FXI increases TF-initiated thrombin generation in a TF concentration-dependent manner
First, we experimentally analyzed the contribution of FXI to thrombin generation initiated via TF in vitro to understand conditions for which the HM model required extension to include the contribution of FXI. To achieve this, we measured thrombin generation in FXI-depleted human plasma supplemented with either a physiological concentration of human FXI (30 nM) or vehicle. We used a fluorogenic substrate assay to measure thrombin generation.17 In this assay, the thrombin concentration was measured every minute by recording the fluorescent signal resulting from the cleavage of the fluorogenic substrate, Z-G-G-Ar-AMC. The thrombin concentration at each time point was computed and used to prepare a thrombin generation curve. The resulting curves were used to derive the following four output parameters to assess different aspects of thrombin generation: ETP—area under the curve (nM.min); peak thrombin—maximum thrombin concentration measured during the observation period (nM); lag time—time to generate 2 nM of thrombin (min); and peak time—time to reach peak thrombin (min). Increases in ETP and/or peak thrombin or decreases in lag time and/ or peak time is indicative of enhanced thrombin generation. To restrict our study to the TF-initiated FXI feedback pathway for thrombin generation, we pretreated all plasmas with the FXIIa inhibitor CTI (40 μg/ml).18 This eliminates potential contribution from FXI activation by FXIIa within the experimental time course of 60 min (Figure S1 in supporting information).
When thrombin generation was initiated in FXI-depleted plasma with 0.125 pM TF, the inclusion of FXI dramatically increased the rate and extent, but not the peak time of thrombin generation (Figure 1A). This was exemplified by a 145% increase in ETP (1166 ± 90 to 2859 ± 121 nM/min; mean ± SEM; Figure 1E) and a 140% increase in peak thrombin (49.7 ± 1.2 to 119.9 ± 3.1 nM Figure 1F) coupled with a 50% decrease in lag time observed in the presence of FXI (Figure 1G, H). Similar trends were observed for 0.25 pM TF-initiated thrombin generation (Figure 1B, E–H). As the initiating concentration of TF increased up to 1 pM, the relative contribution of FXI to the overall rate and extent of thrombin generation fell below 10% (Figure 1A–H). As expected, no thrombin generation was observed in the absence of TF (Figure S2A, B in supporting information). FXIa activates FIX to promote thrombin generation. To determine if the observed increase in thrombin generation in the presence of FXI was due to the activation of FIX by FXIa, we pretreated plasmas with an anti-FXI mAb 1A6, to selectively eliminate the activation of FIX by FXIa.16,19,20 Note that 1A6 does not inhibit the activation of FXI by thrombin.21 Indeed, pretreatment with 1A6 reversed the FXI-dependent increase in thrombin generation in plasma induced to clot with 0.125 or 0.25 pM TF but did not produce a significant change with 0.5 or 1 pM TF (Figure 1A–H). To address the possibility that the purified FXI used in our experiments could be contaminated with FXIa, we performed thrombin generation experiments in which we self-contaminated with known concentrations of FXIa (0.3–3000 pM). As confirmed in Figure S2, FXIa contamination would need to exceed 300 pM to contribute to the signal of thrombin generation in our system.
FIGURE 1.
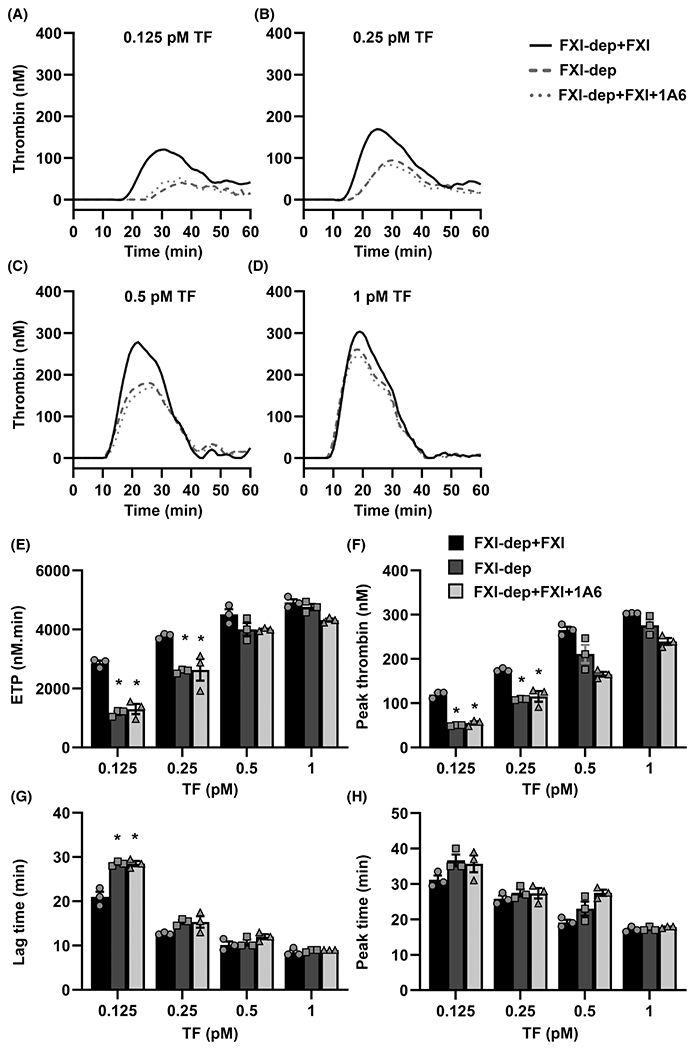
Sensitivity of tissue factor (TF)-initiated thrombin generation to factor XI (FXI). A–D, Thrombin generation in FXI-depleted plasma (FXI-dep) supplemented with 30 nM FXI or vehicle was initiated by the addition of TF (0.125–1 pM) and Ca2+. Experiments were run in the presence or absence of the anti-FXI mAb, 1A6 (30 μg/ml). E, Endogenous thrombin generation potential (ETP), (F) peak thrombin concentrations, (G) lag time, and (H) peak time were estimated from the thrombin generation curves for each condition. Data are means ± standard error of the mean (n = 3). *P < .05 with respect to FXI-dep+FXI
These experimental results established a range of TF concentrations for which FXI makes substantial contribution to the rate and extent of thrombin generation. Based on these results, the inclusion of the feedback loop of FXI activation to the HM model of thrombin generation at low concentrations of TF is warranted.
3.2 |. Effect of FXI on FXa-initiated thrombin generation
As the TF–FVIIa complex drives thrombin generation by activating FX, we sought to identify the range of FXa concentrations at which FXI contributes to the rate and extent of FXa-mediated thrombin generation. When thrombin generation was initiated with 0.1 nM FXa, similar to our observations with low TF, the addition of FXI to FXI-depleted plasma dramatically increased the rate and extent of thrombin generation (Figure 1A). Under these conditions, both the FXa-generated ETP (259 ± 149 to 3722 ± 400 nM/min; Figure 2C) and the peak thrombin (13.0 ± 6.0 to 154.8 ± 19.5 nM; Figure 2D) increased more than 1000% in the presence of FXI compared to the presence of vehicle. We also observed a decrease in lag time (Figure 2E) and peak time (Figure 2F) in the presence of FXI. As with TF-initiated thrombin generation, the selective blockade of FIX activation by FXIa with 1A6 eliminated the contribution of FXI to FXa-initiated thrombin generation at low FXa levels (Figure 2A, C). As the initiating concentration of FXa increased up to 0.5 nM, the relative contribution of FXI to the overall rate and extent of thrombin generation fell below 10% (Figure 2B–F).
FIGURE 2.
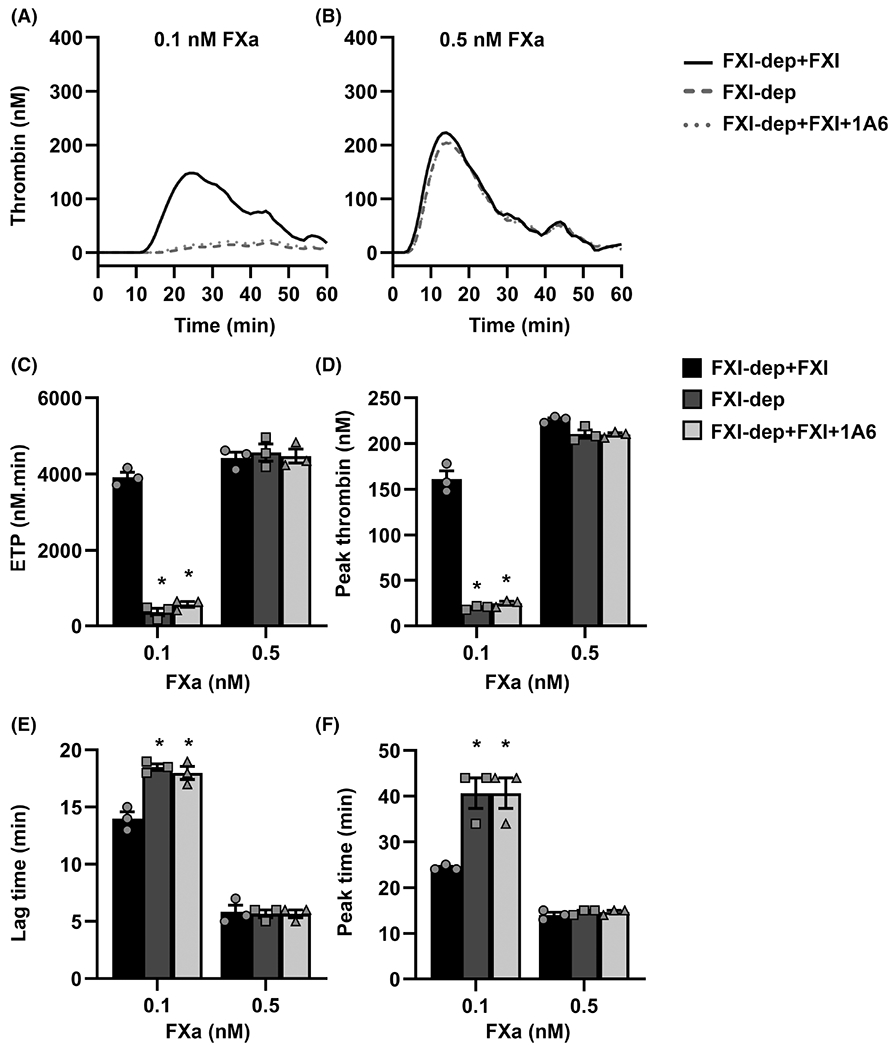
Sensitivity of activated factor X (FXa)-initiated thrombin generation to factor XI (FXI). A–B, FXa (0.1 or 0.5 nM) was used to initiate thrombin generation in FXI-depleted plasma (FXI-dep) supplemented with 30 nM FXI or vehicle in the presence or absence of the anti-FXI mAb, 1A6 (30 μg/ml). C, Endogenous thrombin generation potential (ETP), (D) peak thrombin concentration, (E) lag time, and (F) peak time were estimated from the thrombin generation curves recorded for each condition. Data are means ± standard error of the mean (n = 3). *P < .05 with respect to FXI-dep+FXI
Together, these results demonstrate that the contribution of FXI to thrombin generation is dependent on the initial concentration of FXa or TF used to trigger coagulation.
3.3 |. A systems biology model of TF pathway of thrombin generation
The HM model was one of the first mathematical models to describe the TF pathway of thrombin generation. The model does not include FXI as the mechanistic role of FXI in thrombin generation was unclear at the time the HM model was developed.5,22 The HM model was built for simulating in vitro thrombin generation and was originally validated with measurements of thrombin concentrations formed in a mixture of purified plasma zymogens at their physiologic concentrations rather than in plasma itself.4,23 It is noteworthy that this “synthetic” plasma did not include FXI and was studied at TF-initiating concentrations of more than 1 pM. Thus, we suspect that the original HM model would be unable to predict the thrombin generation dynamics observed in our studies with low concentrations of TF where thrombin generation is most dependent on FXI.
To test this hypothesis, we compared our experimental results for thrombin generation initiated by 0.125 and 0.25 pM TF in FXI-depleted plasma. The initial concentrations of the plasma species were computationally set to the physiological levels of zymogens and enzymes normally found in plasma (values listed in Table S1 in supporting information). The initial concentration of FVIIa was set to 100 pM based on reports in literature that measured the circulating concentration of FVIIa to be in the range of 10–160 pM.24 The computed thrombin curves from the simulations were used to derive ETP, peak thrombin, lag time, and peak time. As shown in Figure 3A–H, the HM model was unable to predict the thrombin generation curve or the four kinetic outputs to match the experimentally derived data for either 0.125 or 0.25 pM TF. This was particularly evident for the condition using 0.125 pM TF, where the HM model predicted an ETP <1 nM/min compared to an experimental average of 1150 nM/min ETP (Figure 3E). The inability of the HM model to capture the kinetics of thrombin generation in a system depleted of FXI provided the rationale for making the following modifications:
FIGURE 3.
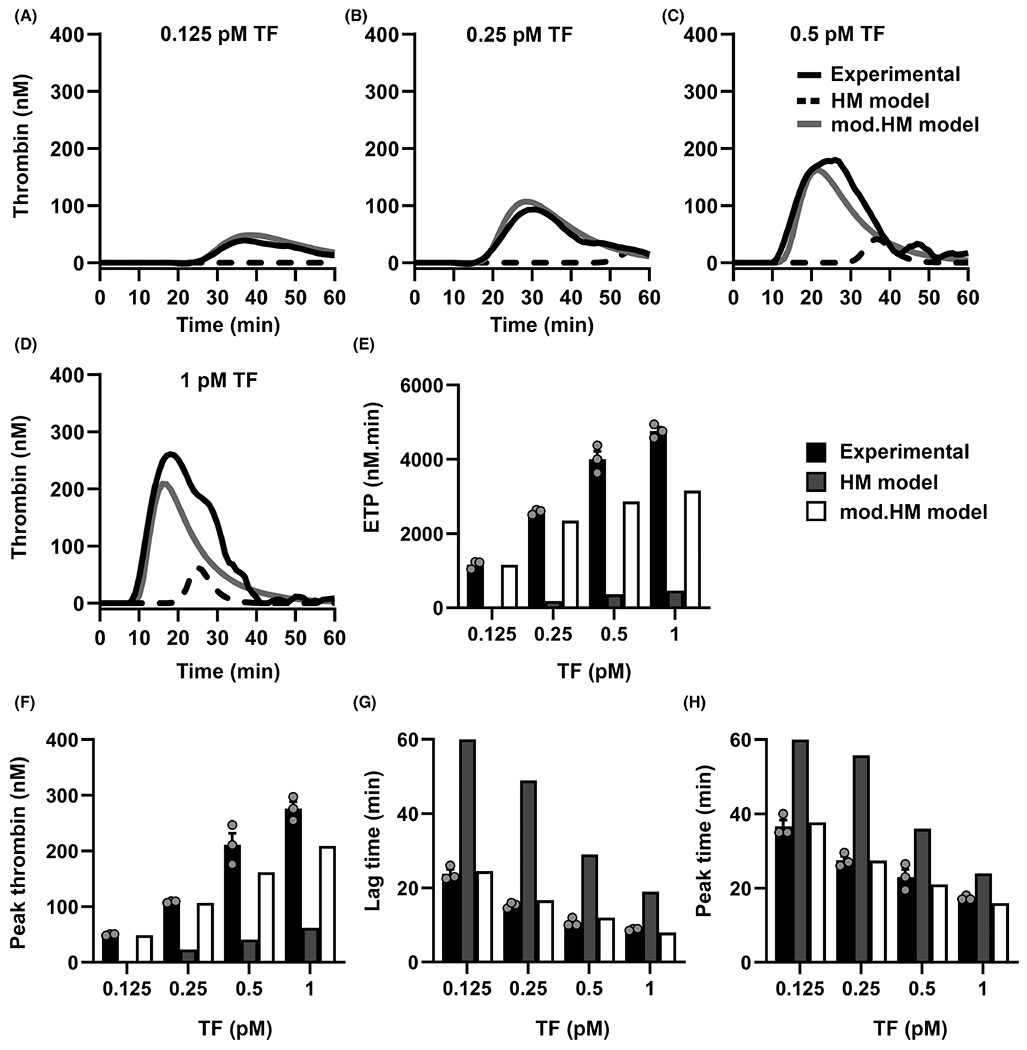
Comparison of in vitro and in silico tissue factor (TF)-initiated thrombin generation in factor XI (FXI)-depleted plasma. Thrombin generation in FXI-depleted plasma was simulated using the Hockin-Mann (HM) model (black dotted line) or the modified (mod.)-Hockin-Mann (mod.HM) model (gray line) with physiological concentrations of enzymes as described in Methods, 0 nM FXI and (A) 0.125 or (B) 0.25 or (C) 0.5 or (D) 1 pM TF and (A-D) plotted alongside the curves obtained from experiments in FXI-depleted plasma with vehicle (black solid line). E, Endogenous thrombin generation potential (ETP), (F) peak thrombin concentration, (G) lag time, and (H) peak time were estimated from HM model (gray bars), mod.HM model (white bars), and experiments in FXI-depleted plasma (black bars). Lag time and peak time were recorded as 60 min if <2 nM thrombin was generated within 60 min. No bars in (E) and (F) for HM model indicate that <2 nM of thrombin generation was recorded within 60 min of simulation. Experimental data are shown as means ± standard error of the mean (n = 3)
Include reactions describing the activation of FVII in the TF–FVII complex to FVIIa by FXa, thrombin, and the TF–FVIIa complex itself. We assumed that the kinetics of TF–FVII activation is similar to the activation of FVII by FXa, thrombin, and TF–FVIIa complex allowing us to use the same kinetic parameters for both sets of reactions. This is required in light of recent studies suggesting that FVII can be activated by several proteases while in a complex with TF.12,25 We also included the activation of FX by FIXa.26
The kinetic constants of the reactions were updated by fitting them to the experimental data derived from 0.125 and 0.25 pM TF-initiated thrombin generation in FXI-depleted plasma. The error between the experimental data and the predicted values from the model simulations was minimized according to an objective function as described below. The objective function consists of experimentally estimated average values of ETP, peak thrombin, lag time, and peak time and the corresponding predictions from HM model simulations. We used these outputs rather than time-dependent thrombin concentration alone in order to avoid noise associated with single variable measurements.27
Hereafter, we refer to the HM model after modifications 1 and 2 as the modified (mod.) HM model. The kinetic constants of the mod.HM model are shown in Table S2 in supporting information. We repeated the simulations using the mod.HM model using TF concentrations from 0.125 to 1 pM. While experimental data obtained using 0.125 and 0.25 pM TF were used in estimating the kinetic parameters, the simulations for 0.5 and 1 pM are a test for the model’s predictive capabilities (Figure 3C–H). As shown in Figure 3A–D, the predictions derived from the mod.HM model showed excellent agreement with the experimental data using FXI-depleted plasma. Thus, our modifications improved the robustness of the HM model of TF-mediated thrombin generation under conditions in which plasma lacks FXI activity.
3.4 |. Extending the mod.HM model to include FXI
As the mod.HM model does not include FXI, it cannot capture effects of FXI on thrombin generation. To address this limitation, we included the following mechanisms related to FXI activation and activity in the mod.HM model (herein referred to as the “extended HM model,” or ext.HM model):
FXI activation by thrombin.
FIX activation by FXIa.
FXIa inhibition by antithrombin (ATIII) and C1-inhibitor (C1inh).
We took parallel approaches to extend the mod.HM model. First, we incorporated the literature values for kinetic constants of the activation of FXI by thrombin. In the second approach, we estimated the kinetic constants based on fitting curves to our experimental data. The caveat for the literature values was that they were measured in the presence of dextran sulfate—an exogenous cofactor of FXI activation by thrombin—and thus may overestimate the kinetics of endogenous FXI activation by thrombin.11 Our experimental data were derived from conditions under which thrombin generation was most sensitive to FXI: 0.125 and 0.25pM TF-initiated thrombin generation in FXI-depleted plasma supplemented with 30nM FXI.
Both simulation approaches were performed for initial TF concentrations of 0.125 or 0.25pM TF and the initial concentration of FXI computationally set to 30nM. Simulated thrombin generation curves (Figure 4A–D) and the outputs derived from the curves were then compared to the experimental data (Figure 4E–H).
FIGURE 4.
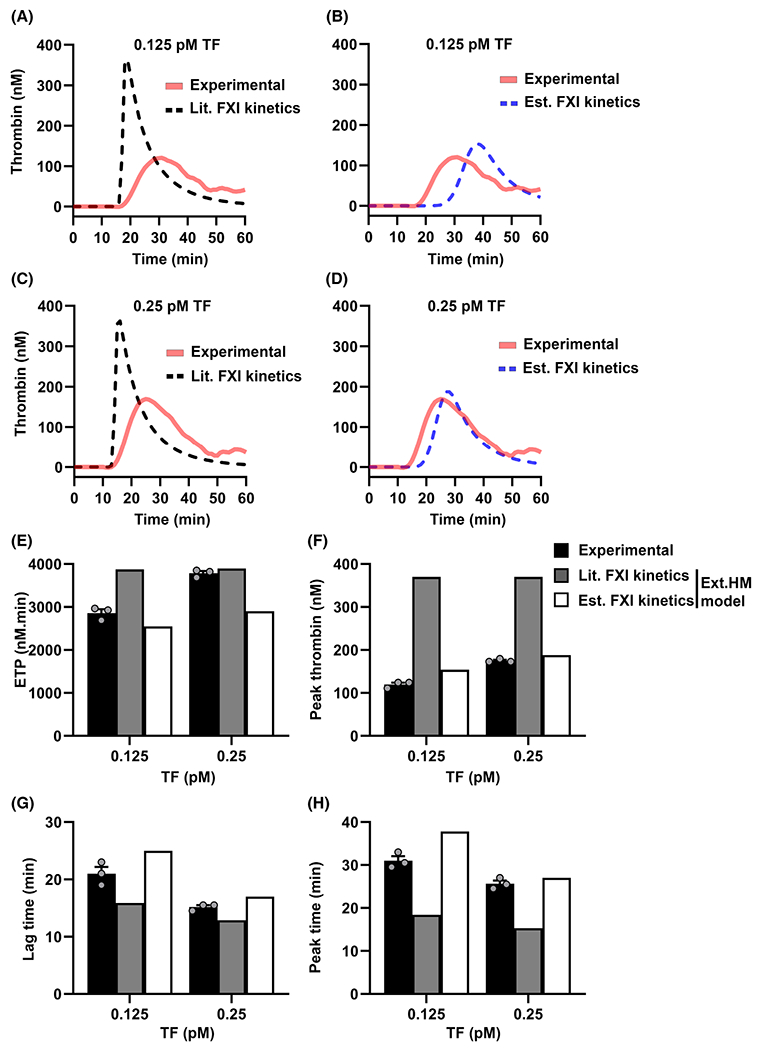
Comparison of in vitro and in silico tissue factor (TF)-initiated thrombin generation in plasma with factor XI (FXI). Experimental curves are from experiments in FXI-depleted plasma supplemented with 30 nM FXI as described in Figure 1. A, Thrombin generation curves from simulations of modified (mod.)-Hockin–Mann model with FXI-related kinetics from literature (Lit. FXI kinetics; black broken line) and experiments (red solid line). B, Thrombin generation from simulation using mod.HM model with FXI-related kinetic parameters estimated utilizing experimental data (Est. FXI kinetics; blue broken line) and experiments (red solid line). (E) Endogenous thrombin generation potential (ETP), (F) peak thrombin, (G) lag time, and (H) peak time were estimated from the experiments (black bars) and the simulations of mod.HM model with literature kinetic parameters (dark gray bars) and estimated kinetic parameters (white bars) as described in Methods. Experimental data are expressed as means ± standard error of the mean (n = 3)
The ext.HM model run with kinetic parameters from the literature predicted peak thrombin values that were >2-fold higher than the experimentally derived values (Figure 4A, C, F). Moreover, the literature-based model predictions were insensitive to changes in TF concentration (Figure 4A, C). For example, the model predicted a <1% decrease in ETP and peak thrombin when the TF concentration fell from 0.25 to 0.125 pM, whereas a >33% decrease was observed experimentally (Figure 4E, F). Similarly, both the lag time and peak time were insensitive to TF concentration when ext.HM model was run with kinetic parameters from the literature (Figure 4G, H). However, the predicted thrombin generation curves were sensitive to TF concentration in a manner consistent with experimental observation when the ext.HM model was simulated with estimated kinetic constants (values listed in supporting information Table S2). ext.HM model simulations with estimated kinetic parameters predicted a decrease in ETP and peak thrombin consistent with the experimental observation when the TF concentration was decreased from 0.25 pM to 0.125 pM (Figure 4E, F). Therefore, we chose to use experimentally derived estimated kinetics for extending the mod. HM model to predict the role of the FXI feedback mechanism in thrombin generation. The extended HM model based on estimated kinetics for FXI-related reactions is referred to hereafter as “ext.HM model.” As a first step, we validated the predictive capability of the ext.HM model by experimentally initiating coagulation with increasing concentrations of exogenously added FXIa (Figure S5 in supporting information); our results show good agreement between experimental and simulated results, verifying the sensitivity of the ext.HM model to FXIa.
3.5 |. Validating the ext.HM model
We tested the predictive capabilities of the ext.HM model using a novel set of conditions not previously used during kinetic parameter estimation. We chose to predict and experimentally validate the role of FXI in TF-mediated thrombin generation in the presence or absence of an endogenous inhibitor of the TF/FVIIa/FXa complex, TFPI. The ability of TFPI to reduce thrombin generation becomes more pronounced as TF concentrations decrease6,28 and for the purpose of our study, we predicted that this effect would be maximal for conditions of low TF where FXI makes its most substantial contribution to thrombin generation. Simulations were performed for TF concentrations of 0.125 or 0.25 pM with or without physiological levels of FXI (30 nM) or TFPI (2.5 nM).
The simulations indicate that removing TFPI from the in silico system eliminates the contribution of FXI to thrombin generation (Figure 5A, E). In particular, our model predicts that for the initiating condition of 0.125 pM TF, removing TFPI from the system caused a 10-fold reduction in the sensitivity of ETP to FXI (Figure 5E, F). This suggests that the inhibitory function of TFPI is necessary to observe the effect of FXI on thrombin generation under conditions of low TF.
FIGURE 5.
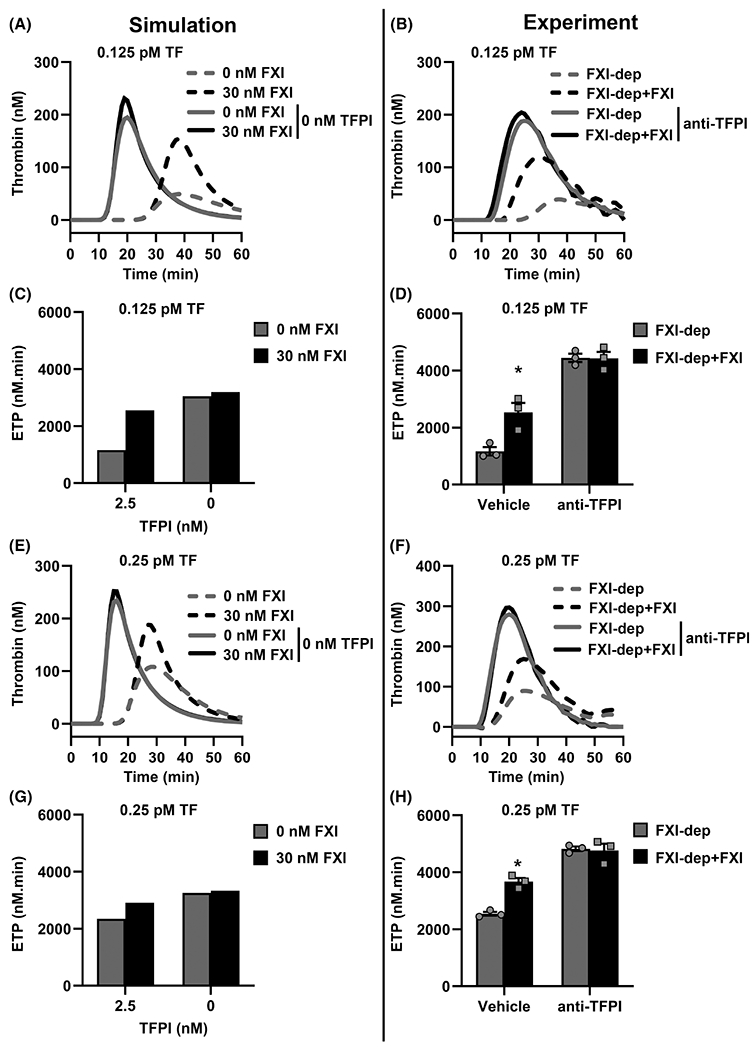
Validation of extended-Hockin-Mann model. The effect of tissue factor pathway inhibitor (TFPI) on tissue factor (TF)-initiated thrombin generation in the presence or absence of factor XI (FXI) is predicted using the extended (ext.)-Hockin–Mann model with fitted FXI-related kinetic parameters. Experiments were conducted in FXI-depleted plasma (FXI-dep) supplemented with 30 nM FXI or vehicle in the presence or absence of TFPI blocking antibodies (5 μg/ml). All plasmas were incubated with corn trypsin inhibitor to block activated factor XII (FXIIa) activity. Thrombin generation curves from simulations initiated with (A) 0.125 or (E) 0.25 pM TF and 0 or 30 nM FXI with 0 or 2.5 nM TFPI. Thrombin generation was initiated with (B) 0.125 or (F) 0.25 pM TF in FXI-depleted plasma supplemented with 30 nM FXI or vehicle. In select experiments, plasma was pretreated with 5 μg/ml blocking antibodies against K-1 and K-2 domains of TFPI; (C) and (D) show the ETP for 0.125 pM TF-initiated thrombin generation as estimated from simulations and experiments for each condition, respectively; (G) and (H) show the ETP of 0.25 pM TF-initiated thrombin generation from simulations and experiments, respectively. Data from experiments are shown as means ± standard error of the mean (n = 3). *P < .05 with respect to FXI-dep
Next, we took an integrative approach to validate the effect of TFPI on thrombin generation predicted by the ext.HM model under conditions of low TF. We measured thrombin generation initiated with 0.125 or 0.25 pM TF, in FXI-depleted plasma supplemented with vehicle or 30 nM FXI. We neutralized the function of plasma TFPI with anti-TFPI antibodies. Any potential effects of the FXII-mediated contact activation pathway to thrombin generation were abrogated by inclusion of CTI (40 μg/ml) in plasma.
In agreement with the effect predicted by the ext.HM model for TFPI on FXI-mediated thrombin generation, we observed that blocking plasma TFPI abrogated the potentiating effect of FXI on the total extent of thrombin generated, ETP (Figure 5D, H), as well as the kinetics of the thrombin generation curve (Figure 5B, F). This integrative approach demonstrates the robustness and utility of the ext.HM model to predict the influence of FXI on thrombin generation.
4 |. DISCUSSION
Feedback activation mechanisms are central to the self-regulation of biological processes ranging from cellular signaling to protein synthesis.29–33 For instance, an organized set of positive and negative feedback mechanisms control the complex biochemical reaction networks observed in metabolic pathways, complement pathways of the immune system, and the pathways of blood coagulation. Due to the inherent complexity associated with the presence of several series and parallel reactions in biochemical reaction networks, intuitive analysis alone is insufficient to assess the importance of individual reactions, necessitating a systems-based approach. In this work, we present a case example of the TF pathway of thrombin generation to demonstrate the utility of synchronizing experimental and computational systems biology models to map the feedback mechanisms of a complex reaction network. We believe our approach is versatile and simple and can be extended to the study of any complex reaction system with feedback loops.
In the initiation phase of coagulation, formation of a complex of TF and FVIIa catalyzes a series of enzymatic reactions that culminate in the generation of thrombin.34 In the propagation phase, thrombin self-regulates its production through feedback of activation of FVII, FVIII, FV, and FXI.35 The mean plasma concentration of FXI (30 nM) is higher than the other zymogens FVII (10 nM), and cofactors FVIII (0.3 nM) and FV (20 nM), yet the catalytic efficiency (kcat/Km) of FXI activation by thrombin is at least 100-fold lower than those for activation of the other coagulation factors.3,5,36 Moreover, the primary substrate for FXIa is FIX, which can also be directly activated by the TF-FVIIa complex. Thus, robust thrombin generation driven by relatively high concentrations of TF or FXa would be predicted to be insensitive to FXI. This has led some to question the physiological relevance of FXI activation by thrombin.15,27 However, we and others hypothesize that FXI activation by thrombin affects the propagation of thrombin generation when the initial thrombin generation rate through the main reaction pathways is initiated by relatively low initial concentrations of TF or FXa.16,19 By incorporating the FXI feedback activation mechanism, we have improved a robust network model that encompassed not only the major reaction pathways but also the role of regulatory mechanisms in the generation of thrombin.
Our network model predicts that manipulating the initial rates of thrombin generation by selectively neutralizing a specific inhibitor also changes the sensitivity of thrombin generation to FXI. TFPI is an endogenous inhibitor of FXa and the TF–FVIIa complex. In simulations of thrombin generation initiated with a low TF concentration, the model predicted that eliminating TFPI will increase the initial rate of thrombin generation and abolish the effect of FXI on TF-mediated thrombin generation. Our experimental validation of this computationally derived hypothesis provides credence to the theory that amplifying the propagation phase of thrombin generation through feedback activation of FXI is important in low TF environments. Interestingly, we observed an effect of FXI on the initiation phase of thrombin generation under low TF conditions (0.125 pM). The relevance of FXI to thrombin generation under low TF was validated using fibrin generation experiments; indeed, we found that FXI decreased the lag phase of fibrin generation under low TF conditions (Figure S4 in supporting information).
Applied to physiology, the feedback activation pathway could underlie a spatial role for FXI in propagating thrombin generation during the growth and stabilization phase of hemostatic plug development beyond the initial burst of thrombin generated by surface-expressed TF at the site of a vascular breach.20,37–40 Furthermore, this mechanism could propagate the growth of pathologic occlusive thrombi, suggesting it may be a useful target for antithrombotic therapy. For instance, several biological substances that are likely to be present in a growing thrombus, including polyphosphates released from platelets,41 the platelet surface itself,42 and neutrophil extracellular traps43,44 have been shown to affect the activation rate of FXI or the kinetics of FXI activity.
Similar to the HM model we approximate surface and bulk reactions together as bulk reactions but this did not affect the capacity of ext.HM model to predict qualitative changes in thrombin generation profiles in response to changes in stimulus as observed in vitro. Potential roles for the surface-based FXI activation or inhibition are not included in our model. It is noteworthy that the kinetic constants for FXI activation used in the systems biology model developed by Chatterjee et al. were derived from experiments performed in the presence of the cofactor dextran sulfate.11 We based our search for a kinetic constant for the feedback activation of FXI in the absence of any exogenous cofactors based on our experimental thrombin generation data measured under such conditions. Future work focused on modifying the kinetics of FXI feedback activation by cofactors and biologically relevant surfaces can establish the effect of this feedback mechanism on thrombin generation and thrombus formation differentially in a disease setting.
The fluorogenic substrate-based assay allows for the continuous measurement of the amount of thrombin generated during coagulation. While Z-G-G-Ar-AMC is highly specific to thrombin, at higher concentrations it can also be cleaved to a minor extent by other enzymes in plasma.45 While our model does not explicitly include the kinetics of the substrate cleavage by thrombin, the kinetic parameters in our model were estimated to reflect the thrombin generation curve features that should include the effect of the fluorogenic substrate on thrombin generation. In addition, it is to be noted that the plasma in our system is diluted 2.5 times by the fluorogenic substrate to measure thrombin generation. This is still below the threshold dilution factor of 12 that has been reported to introduce measurable errors in plasma-based coagulation assays.46
It is noteworthy that the original HM model used synthetic plasma that lacked any natural cofactors such as microparticles and lipids that have been shown to accelerate several reactions that are part of the TF-initiated thrombin generation. Moreover, the kinetics of enzymatic reactions were measured in purified systems and often vary several fold in values between laboratories. Therefore, all the kinetic parameters of the HM model were tuned by the SLSQP algorithm. But it is the kinetic constants of TF-related reactions that varied the most from their values used in the original HM model. The kinetic constants of reactions involving TF and FVIIa were estimated to be at least 10-fold different from the original HM model. These reactions are either procoagulant, if they lead to an increase in thrombin generation directly or indirectly, or anticoagulant if they slow down the rate of thrombin generation, that is, inhibition of FXa or TF–FVIIa complex by TFPI. Most importantly, the kinetics of TF and FVIIa complex formation was estimated to be 100-fold more than values used in the HM model. This is expected as these reactions control the initiation phase of the thrombin generation, which was five times faster than was predicted by the original HM model (Figure 3A–D). The fact that our ext.HM model incorporates the physiological concentrations of all blood enzymes including FXI allowed us to compare our predicted values to experimental data in the literature; as a case example, our simulations show good qualitative agreement with the experimental data published by Kravtsov et al., as shown in Figure S6 in supporting information.16
In addition to activating FIX, FXIa can contribute to thrombin generation by activating FX, FV, and FVIII, and by inhibiting TFPI.47–49 Further refinements to the model will be required to include these mechanisms to the reaction topology of the current model. However, even in the absence of explicit consideration of surface reactions and additional mechanisms of FXI, the ext.HM model can be useful in predicting qualitative changes on thrombin generation profiles in response to changes induced by stimulus as observed in vitro.
In conclusion, we observed a threshold behavior for the involvement of FXI in thrombin generation based on the initial rate of thrombin generation determined by the concentrations of TF or FXa used to initiate coagulation. Experimental data from thrombin generation assays were first used to improve the robustness of the systems biology model of TF pathway, the HM model, followed by extension of the model to include FXI. The ext.HM model was subsequently used to predict the ability of TFPI to influence the importance of FXI on thrombin generation, which was experimentally validated using thrombin generation assays. This demonstrates the utility of computational systems biology approaches to evaluate complex reaction systems with feedback loops (Figure 6).
FIGURE 6.
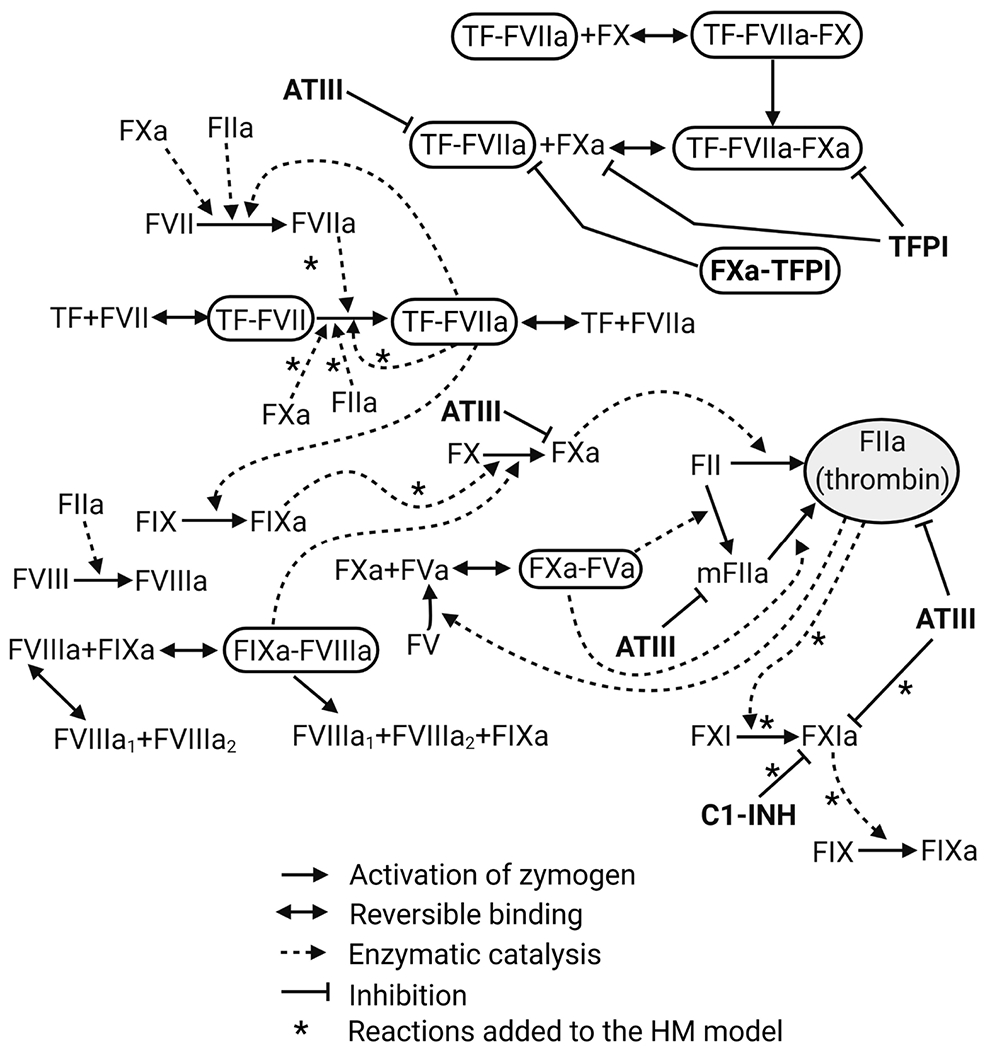
Extended Hockin-Mann model. Reactions in the extended (ext.) HM model that describes the TF pathway of thrombin generation. Arrows with asterisks (*) indicate reactions that are added to the original HM model. ATIII, antithrombin; C1-INH, C1 inhibitor; FII, prothrombin; FIX, coagulation factor IX; FV, coagulation factor V; FVII, coagulation factor VII; FVIII, coagulation factor VIII; FVIIIa1 and FVIIIa2, trimeric forms of FVIIIa; FX, coagulation factor X; FXI, coagulation factor XI; HM, Hockin–Mann model; mFIIa, meizothrombin; TF, tissue factor; TFPI, tissue factor pathway inhibitor; the activated forms of the coagulation factor are represented with “a” added as the suffix, such as FIIa, FVIIa, FVIIIa, FIXa, FXa, and FVa
Supplementary Material
ESSENTIALS.
A systems-based approach is vital for analyzing the role of feedback activation of coagulation factor XI (FXI) in thrombin generation.
A mathematical model of thrombin generation was developed using experimental data.
We computationally predicted and experimentally validated that tissue factor pathway inhibitor (TFPI) modifies the role of FXI in thrombin generation.
Our study demonstrates the utility of integrating mathematical models and experiments.
ACKNOWLEDGMENTS
We thank Daniel M. Zuckerman for his stimulating comments and feedback on this study. O.J.T. McCarty is an American Heart Association Established Investigator.
Funding Information
This work was supported by the National Institutes of Health (R01HL101972 and R01HL144113 to O.J.T.M., R35HL140025 to D.G., and F30HL158079 to T.J.Z.) and the American Heart Association (828839 to S.E.R.).
Footnotes
CONFLICTS OF INTEREST
E.T. is an employee of Aronora, Inc. E.T. and Oregon Health and Science University have a significant financial interest in Aronora, Inc., a company that may have a commercial interest in the results of this research. This potential conflict of interest has been reviewed and managed by the Oregon Health & Science University Conflict of Interest in Research Committee. The other authors declare that they have no conflicts of interest with the contents of this article.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY
Data for all figures are contained within the article.
REFERENCES
- 1.Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93(1):327–358. [DOI] [PubMed] [Google Scholar]
- 2.Yamamoto M, Nakagaki T, Kisiel W. Tissue factor-dependent autoactivation of human blood coagulation factor VII. J Biol Chem. 1992;267(27):19089–19094. [PubMed] [Google Scholar]
- 3.Giddings JC, Bloom AL. Factor-V activation by thrombin and its role in prothrombin conversion. Br J Haematol. 1975;29(2):349–364. [DOI] [PubMed] [Google Scholar]
- 4.Lawson JH, Kalafatis M, Stram S, Mann KG. A model for the tissue factor pathway to thrombin. I. An empirical study. J Biol Chem. 1994;269(37):23357–23366. [PubMed] [Google Scholar]
- 5.Gailani D, Broze GJ Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253(5022):909–912. [DOI] [PubMed] [Google Scholar]
- 6.Hockin MF, Jones KC, Everse SJ, Mann KG. A model for the stoichiometric regulation of blood coagulation. J Biol Chem. 2002;277(21):18322–18333. [DOI] [PubMed] [Google Scholar]
- 7.Chelle P, Morin C, Montmartin A, Piot M, Cournil M, Tardy-Poncet B. Evaluation and calibration of in silico models of thrombin generation using experimental data from healthy and haemophilic subjects. Bull Math Biol. 2018;80(8):1989–2025. [DOI] [PubMed] [Google Scholar]
- 8.Chua HN, Roth FP. Discovering the targets of drugs via computational systems biology. J Biol Chem. 2011;286(27):23653–23658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iyengar R. Computational biochemistry: systems biology minireview series. J Biol Chem. 2009;284(9):5425–5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu B, Zhang J, Tan PY, et al. A computational and experimental study of the regulatory mechanisms of the complement system. PLoS Comput Biol. 2011;7(1):e1001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chatterjee MS, Denney WS, Jing H, Diamond SL. Systems biology of coagulation initiation: kinetics of thrombin generation in resting and activated human blood. PLoS Comput Biol. 2010;6(9):e1000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuharsky AL, Fogelson AL. Surface-mediated control of blood coagulation: the role of binding site densities and platelet deposition. Biophys J. 2001;80(3):1050–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dydek EV, Chaikof EL. Simulated thrombin generation in the presence of surface-bound heparin and circulating tissue factor. Ann Biomed Eng. 2016;44(4):1072–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jordan SW, Chaikof EL. Simulated surface-induced thrombin generation in a flow field. Biophys J. 2011;101(2):276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pedicord DL, Seiffert D, Blat Y. Feedback activation of factor XI by thrombin does not occur in plasma. Proc Natl Acad Sci USA. 2007;104(31):12855–12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kravtsov DV, Matafonov A, Tucker EI, et al. Factor XI contributes to thrombin generation in the absence of factor XII. Blood. 2009;114(2):452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ay L, Thaler J, Brix JM, et al. Decrease in microvesicle-associated tissue factor activity in morbidly obese patients after bariatric surgery. Int J Obes. 2016;40(5):768–772. [DOI] [PubMed] [Google Scholar]
- 18.Puy C, Ngo ATP, Pang J, et al. Endothelial PAI-1 (plasminogen activator inhibitor-1) blocks the intrinsic pathway of coagulation, inducing the clearance and degradation of FXIa (activated factor XI). Arterioscler Thromb Vasc Biol. 2019;39(7):1390–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tucker EI, Marzec UM, White TC, et al. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood. 2009;113(4):936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zilberman-Rudenko J, Itakura A, Wiesenekker CP, et al. Coagulation factor XI promotes distal platelet activation and single platelet consumption in the bloodstream under shear flow. Arterioscler Thromb Vasc Biol. 2016;36(3):510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng Q, Tucker EI, Pine MS, et al. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116(19):3981–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brunnee T, La Porta C, Reddigari SR, Salerno VM, Kaplan AP, Silverberg M. Activation of factor XI in plasma is dependent on factor XII. Blood. 1993;81(3):580–586. [PubMed] [Google Scholar]
- 23.Jones KC, Mann KG. A model for the tissue factor pathway to thrombin. II. A mathematical simulation. J Biol Chem. 1994;269(37):23367–23373. [PubMed] [Google Scholar]
- 24.Morrissey JH, Macik BG, Neuenschwander PF, Comp PC. Quantitation of activated factor VII levels in plasma using a tissue factor mutant selectively deficient in promoting factor VII activation. Blood. 1993;81(3):734–744. [PubMed] [Google Scholar]
- 25.Link KG, Stobb MT, Sorrells MG, et al. A mathematical model of coagulation under flow identifies factor V as a modifier of thrombin generation in hemophilia A. J Thromb Haemost. 2020;18(2):306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duffy EJ, Lollar P. Intrinsic pathway activation of factor X and its activation peptide-deficient derivative, factor Xdes-143-191. J Biol Chem. 1992;267(11):7821–7827. [PubMed] [Google Scholar]
- 27.Nayak S, Lee D, Patel-Hett S, et al. Using a systems pharmacology model of the blood coagulation network to predict the effects of various therapies on biomarkers. CPT Pharmacometrics Syst Pharmacol. 2015;4(7):396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van’t Veer C, Mann KG. Regulation of tissue factor initiated thrombin generation by the stoichiometric inhibitors tissue factor pathway inhibitor, antithrombin-III, and heparin cofactor-II. J Biol Chem. 1997;272(7):4367–4377. [DOI] [PubMed] [Google Scholar]
- 29.Tucker EI, Verbout NG, Markway BD, et al. The protein C activator AB002 rapidly interrupts thrombus development in baboons. Blood. 2020;135(9):689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hetmanski JH, Zindy E, Schwartz JM, Caswell PT. A MAPK-driven feedback loop suppresses Rac activity to promote RhoA-driven cancer cell invasion. PLoS Comput Biol. 2016;12(5):e1004909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang S, Hartman IZ, Calhoun LN. et al. Contribution of accelerated degradation to feedback regulation of 3-Hydroxy-3-methylglutaryl coenzyme A reductase and cholesterol metabolism in the liver. J Biol Chem. 2016;291(26):13479–13494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mao XM, Sun N, Wang F, et al. Dual positive feedback regulation of protein degradation of an extra-cytoplasmic function sigma factor for cell differentiation in Streptomyces coelicolor. J Biol Chem. 2013;288(43):31217–31228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nemeth T, Mocsai A. Feedback amplification of neutrophil function. Trends Immunol. 2016;37(6):412–424. [DOI] [PubMed] [Google Scholar]
- 34.Mann KG, Butenas S, Brummel K. The dynamics of thrombin formation. Arterioscler Thromb Vasc Biol. 2003;23(1):17–25. [DOI] [PubMed] [Google Scholar]
- 35.Jesty J, Beltrami E. Positive feedbacks of coagulation: their role in threshold regulation. Arterioscler Thromb Vasc Biol. 2005;25(12):2463–2469. [DOI] [PubMed] [Google Scholar]
- 36.Mann KG, Brummel K, Butenas S. What is all that thrombin for? J Thromb Haemost. 2003;1(7):1504–1514. [DOI] [PubMed] [Google Scholar]
- 37.Crosby JR, Marzec U, Revenko AS, et al. Antithrombotic effect of antisense factor XI oligonucleotide treatment in primates. Arterioscler Thromb Vasc Biol. 2013;33(7):1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu C, Hutt E, Bloomfield DM, Gailani D, Weitz JI. Factor XI inhibition to uncouple thrombosis from hemostasis: JACC review topic of the week. J Am Coll Cardiol. 2021;78(6):625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J, Diamond SL. Reduced model to predict thrombin and fibrin during thrombosis on collagen/tissue factor under venous flow: roles of gamma′-fibrin and factor XIa. PLoS Comput Biol. 2019;15(8):e1007266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeCortin ME, Brass LF, Diamond SL. Core and shell platelets of a thrombus: a new microfluidic assay to study mechanics and biochemistry. Res Pract Thromb Haemost. 2020;4(7):1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puy C, Rigg RA, McCarty OJ. The hemostatic role of factor XI. Thromb Res. 2016;141(Suppl 2):S8–S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baglia FA, Walsh PN. Thrombin-mediated feedback activation of factor XI on the activated platelet surface is preferred over contact activation by factor XIIa or factor XIa. J Biol Chem. 2007;282(39):29067. [DOI] [PubMed] [Google Scholar]
- 43.Ivanov I, Shakhawat R, Sun MF, et al. Nucleic acids as cofactors for factor XI and prekallikrein activation: different roles for high-molecular-weight kininogen. Thromb Haemost. 2017;117(4):671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohammed BM, Matafonov A, Ivanov I, et al. An update on factor XI structure and function. Thromb Res. 2018;161:94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Berkel SS, van der Lee B, van Delft FL, Wagenvoord R, Hemker HC, Rutjes FP. Fluorogenic peptide-based substrates for monitoring thrombin activity. ChemMedChem. 2012;7(4):606–617. [DOI] [PubMed] [Google Scholar]
- 46.De Smedt E, Wagenvoord R, Coen HH. The technique of measuring thrombin generation with fluorogenic substrates: 3. The effects of sample dilution. Thromb Haemost. 2009;101(1):165–170. [PubMed] [Google Scholar]
- 47.Puy C, Tucker EI, Matafonov A, et al. Activated factor XI increases the procoagulant activity of the extrinsic pathway by inactivating tissue factor pathway inhibitor. Blood. 2015;125(9):1488–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matafonov A, Cheng Q, Geng Y, et al. Evidence for factor IX-independent roles for factor XIa in blood coagulation. J Thromb Haemost. 2013;11(12):2118–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whelihan MF, Orfeo T, Gissel MT, Mann KG. Coagulation procofactor activation by factor XIa. J Thromb Haemost. 2010;8(7):1532–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for all figures are contained within the article.