

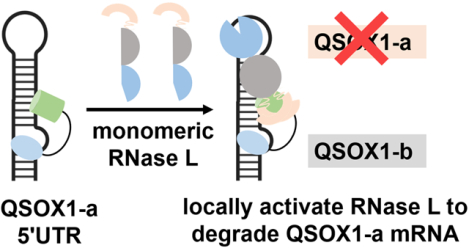
Abstract
The interactions between cellular RNAs in MDA-MB-231 triple negative breast cancer cells and a panel of small molecules appended with a diazirine cross-linking moiety and an alkyne tag were probed transcriptome-wide in live cells. The alkyne tag allows for facile pull-down of cellular RNAs bound by each small molecule, and the enrichment of each RNA target defines the compound’s molecular footprint. Among the 34 chemically diverse small molecules studied, six bound and enriched cellular RNAs. The most highly enriched interaction occurs between the novel RNA binding compound F1 and a structured region in the 5′ untranslated region of quiescin sulfhydryl oxidase 1 isoform a (QSOX1-a), not present in isoform b. Additional studies show that F1 specifically bound RNA over DNA and protein; that is, we studied the entire DNA, RNA, and protein interactome. This interaction was used to design a ribonuclease targeting chimera (RIBOTAC) to locally recruit Ribonuclease L to degrade QSOX1 mRNA in an isoform-specific manner, as QSOX1-a, but not QSOX1-b, mRNA and protein levels were reduced. The RIBOTAC alleviated QSOX1-mediated phenotypes in cancer cells. This approach can be broadly applied to discover ligands that bind RNA in cells, which could be bioactive themselves or augmented with functionality such as targeted degradation.
Graphical Abstract
INTRODUCTION
The importance of RNA in all aspects of biology is well-established, with its function dependent on its structure. (1,2) One way to model RNA structure is to use free energy minimization restrained by chemical probing data. (3,4) Alternatively, defining the binding sites of small molecules could allow for direct inference of the RNA structure in cells and could be particularly important if binding stabilizes dynamic structures, enhancing their detectability. Molecular recognition could be mediated by the RNA’s structure, an RNA-protein interface, or other factors. (5,6)
Covalent chemistry has been used to define RNAs bound by small molecules and their target sites in cells by using chemical cross-linking and isolation by pull-down (Chem-CLIP). (7–9) In this study, we describe how covalent chemistry and RNA profiling in live mammalian cells can define the RNA targets of low-molecular-weight small molecules. As such, this approach is the RNA parallel of profiling small molecules for protein and DNA targets. (10–13)
RESULTS AND DISCUSSION
A panel of 34 structurally diverse compounds (Figure S1) was appended with a diazirine moiety, which reacts with RNA upon photoactivation, and an alkyne tag (Figure 1A). The alkyne tag can be clicked to an azide-functionalized fluorescent dye for imaging (Figure 1B) or to azide-functionalized agarose beads for pull-down. A subset of the molecules was selected for their similarities to known RNA binders, as revealed by a two-dimensional uniform manifold approximation and projection (UMAP) analysis (Figure S2A). (14) The others were selected to include novel chemotypes not known to bind RNAs, such as thieno-pyridazines and phenyl-pyrazoles (Figures S1 and S2).
Figure 1.
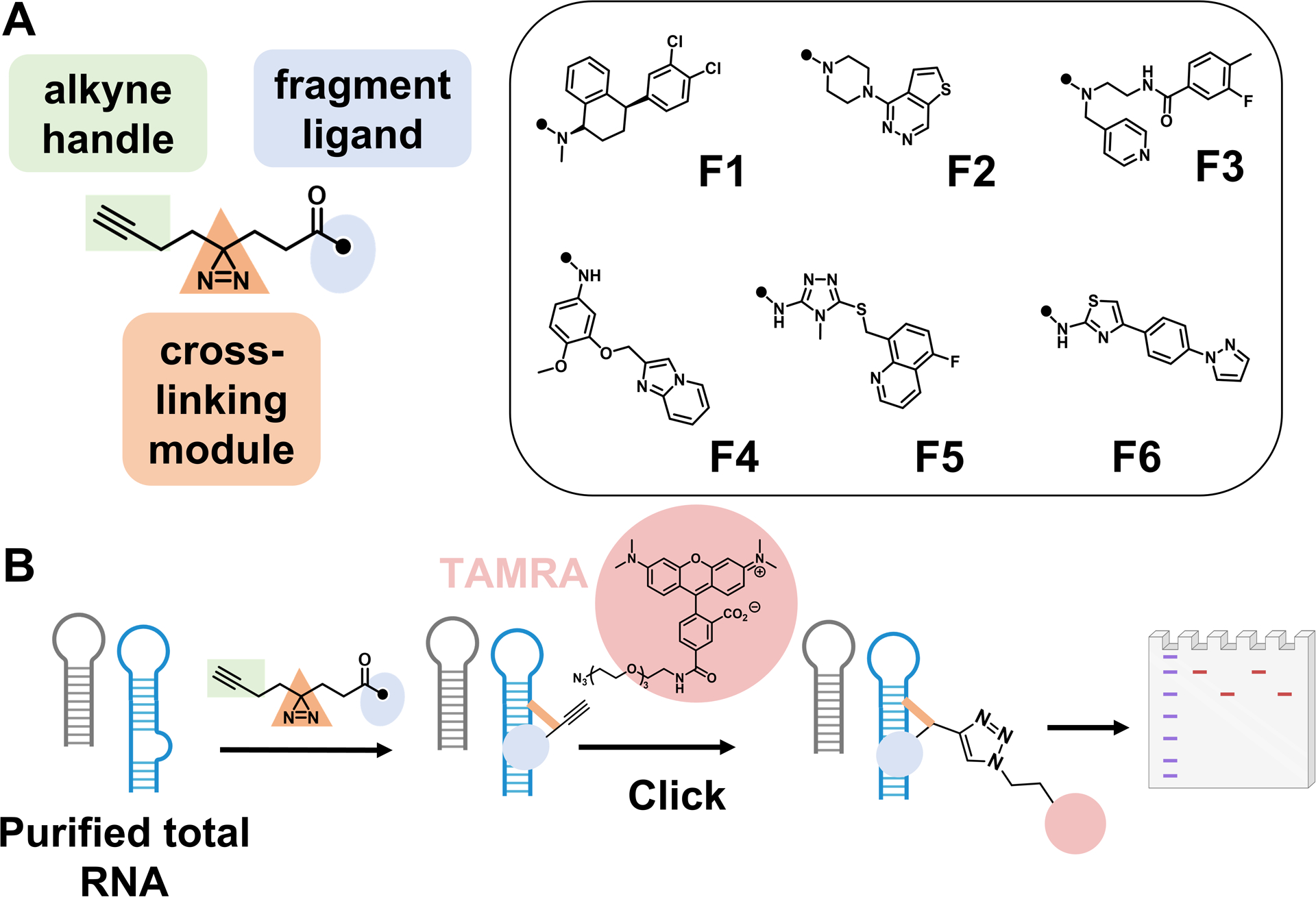
In vitro screening identifies low-molecular-weight ligands that preferentially bind RNA. (A) Structures of compounds identified as hits with fluorescence at least three-fold above the background. (B) Schematic depiction of in vitro screening of low-molecular-weight compounds binding to total human RNA from MDA-MB-231 TNBC cells.
Compounds were first evaluated in vitro with total RNA harvested from MDA-MB-231 triple negative breast cancer (TNBC) cells. Following irradiation, the cross-linked RNAs, which now contained an alkyne handle, were clicked to the fluorescent dye tetramethylrhodamine (TAMRA) azide (Figure 1B). Analysis of the samples by gel electrophoresis and fluorescence imaging identified six compounds (F1–F6; Figure 1B) with reproducible cross-linking to RNA targets, as defined by signal at least three-fold above background. Based on the size of the bands, many of the cross-linked targets included mRNAs (Figure S3).
All six compounds contain aromatic rings, including nitrogen-rich aromatic heterocycles, such as pyridine, triazole, and N-methyl piperazine that are known to bind RNA. (15–17) F1, however, is unique as it has no aromatic nitrogen. When compared to known RNA-binding molecules, (18,19) F1 is the most dissimilar, with an average Tanimoto coefficient of 0.27 ± 0.09 (Figure S2B). The average Tanimoto coefficient for the other molecules indicates that they, too, are chemically dissimilar (0.32–0.46), although the range of four compounds include coefficients ≥0.7. F1 also has the lowest topological polar surface area (TPSA); the TPSA for all six fragments is significantly lower than known RNA binders (Figure S2B,C). In contrast, when comparing atomic logP, only F1 is significantly different than known RNA binders (Figure S2D). When compared to molecules previously reported to target protein in chemoproteomics studies, (10,20) RNA-targeting compounds herein have a higher degree of aromaticity and more H-bond acceptors (Figure S4).
We next advanced our studies live and intact MDA-MB-231 cells to profile the cellular targets as well as the small molecule binding site within them, via Chem-CLIP-Map-Seq, (21) which had not previously been applied transcriptome-wide. After treatment (20 μM; 16 h) and cross-linking, total RNA was harvested and fragmented prior to pull-down (Figure 2A). Random fragmentation was completed first as it reduces: (1) sequence bias in library preparation; and (2) the background signal in the RNA-seq analysis. Most importantly, only the regions bound by the small molecules are pulled down rather than the entire transcript, identifying both the target and the binding site within it. In parallel, a control diazirine probe that lacks an RNA-binding functionality (Figure 2B) was also studied to identify targets that nonspecifically react with the cross-linking moiety, where cross-linking is not driven by the RNA-binding elements.
Figure 2.
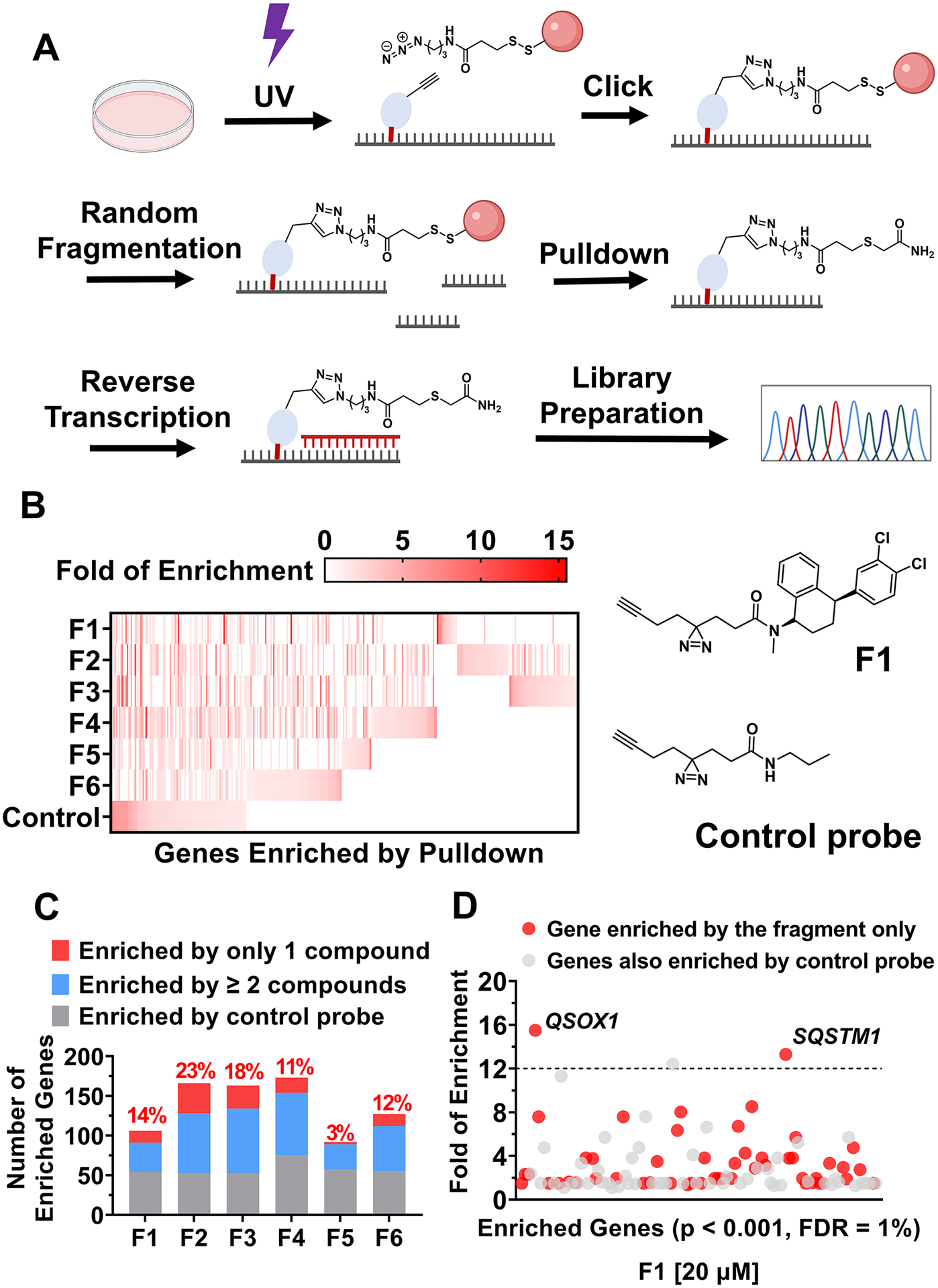
Chem-CLIP-Map-Seq profiles the targets of compounds across the human transcriptome in cellulis. (A) Schematic depiction of the Chem-CLIP-Seq workflow. (B) Heatmap representation of genes enriched by each compound. (C) Comparisons of genes enriched by each compound. (D) Genes that are significantly enriched by F1 identified by Chem-CLIP-Seq in MDA-MB-231 cells.
Significantly enriched regions across the human transcriptome (p < 0.001) were identified by using a publicly available package, Genrich (22) (Figures 2B, S5, and S6). Genrich uses a null model with a log-normal distribution to calculate p values by comparing sequencing runs of the same RNA sample before and after pull-down. Additional filters were applied to these significantly enriched regions, requiring a minimum read count of 5 and a minimum enrichment of 1.5, thereby eliminating low-confidence peaks. The distribution of the enrichment of RNAs bound by each molecule and the control probe is shown in Figures 2B, S4, and S5, and the complete dataset is publicly available in Mendeley Data.
Distinct patterns of target enrichment were observed for each compound when compared to each other and to the control probe (Figure 2B). Indeed, unique transcripts were identified for each small molecule [n = 3 (F5) to 38 (F2)] (Figure 2C). After eliminating transcripts that were also bound by the control diazirine probe, that is only bona fide targets remain, the number of transcripts enriched by F1 (n = 51) and F5 (n = 35) shows superior selectivity compared to other compounds (n > 70) (Figure S5). Notably, the largest enrichments were observed for F1, with two transcripts enriched >12-fold (Figure 2D). The compounds also appear to have different distributions across transcript regions, i.e., 5′ and 3′ untranslated regions (UTRs), coding regions (CDS), or introns, as well as noncoding RNAs (Figure S7).
Next, we sought to map the exact binding site within each transcript for all six molecules. Small molecules cross-linked to RNA can inhibit reverse transcriptase (RT) from proceeding near the cross-linked site, inducing an “RT stop” and resulting in a truncated cDNA strand. (21) Thus, we used the termini of enriched sequences to map the binding site of compounds to each RNA target. Some enriched regions have no discernable “RT stop” sites, suggesting that these regions may form flexible structures and lack a defined binding site. (23) Although mutations have also been observed in RNA-protein cross-linking studies, (24,25) no significant enrichment of mutations were observed in these studies. This is presumably due to the bulky size of cross-linked compounds compared to that of an amino acid residue after proteolysis in RNA-protein cross-linking studies.
As aforementioned, identifying the sites occupied by a small molecule could allow direct experimental inference of the RNA structure in cells. We therefore modeled the structure of each transcript at the small molecule binding site by using the state-of-the-art program ScanFold, which calculates the minimum free energy of the native sequence and compares it to that of random sequences to identify unusually stable structures within an RNA. (26) Over 60% of enriched regions are predicted to form stable, ordered structures (Figure S8).
We further investigated F1 as it is a novel RNA-binding chemotype, binds the second fewest number of transcripts, and has the highest fold of enrichment of a transcript–12-fold for two targets that are also unique to F1, QSOX1, and SQSTM1 (Sequestosome 1) (Figures 2D and S6). We first analyzed whether other biomolecules were pulled down by F1 as the diazirine could also cross-link to DNA or protein. Notably, no significant enrichment of protein or DNA targets was observed for F1 when compared to the control probe, suggesting that it preferentially engages RNAs in cells (Figure S9). In agreement with these data, we also completed full proteomics analysis of proteins cross-linked to F1. Only 79 proteins of the 3158 proteins detectable before pull-down were specifically pulled down by F1, that is, they were not pulled down by the control probe (Mendeley Data). Importantly, none of these proteins is known to form mRNA-protein complexes nor is QSOX1 or SQSTM1 pulled down.
The time and dose dependence of the pull down of QSOX1 mRNA by F1 was also studied by using real-time quantitative polymerase chain reaction (RT-qPCR) (see Figures S10 and S11 for primer validation). Compound F1 dose-dependently enriched QSOX1 mRNA, at 5 μM (~6-fold) and 20 μM (~12-fold) (16 h incubation; Figure S12A). The pull-down of QSOX1 mRNA was also time-dependent. After 1 h incubation, no enrichment of the QSOX1 was observed. An ~4-fold enrichment was observed after 4 h, with enrichment plateauing at the 8 and 16 h time points (Figure S12B). The time required to engage and cross-link to the target is likely due to a composite of factors and could include dissociation of proteins bound to the RNA, the association rate of the small molecule, the rate of the RNA’s folding, and/or structural dynamics.
The analysis of the RNA-seq data coupled with ScanFold structure prediction enabled mapping of the F1 binding site within the QSOX1 and SQSTM1, the 5’ UTR of the former and the coding region of the latter. The putative binding site in QSOX1 is predicted to form a stable hairpin structure containing a single bulged uridine (Figure 3A, B), the site of an “RT Stop” in the RNA-seq data. Notably, the sequence that forms this structure is present only in the QSOX1-a isoform and not in QSOX1-b (Figure S13). The SQSTM binding site also forms a structure, a hairpin with single bulged cytosine and two asymmetric internal loops. Here, the “RT Stop” occurs at a uridine residue in the hairpin loop (Figure S14).
Figure 3.
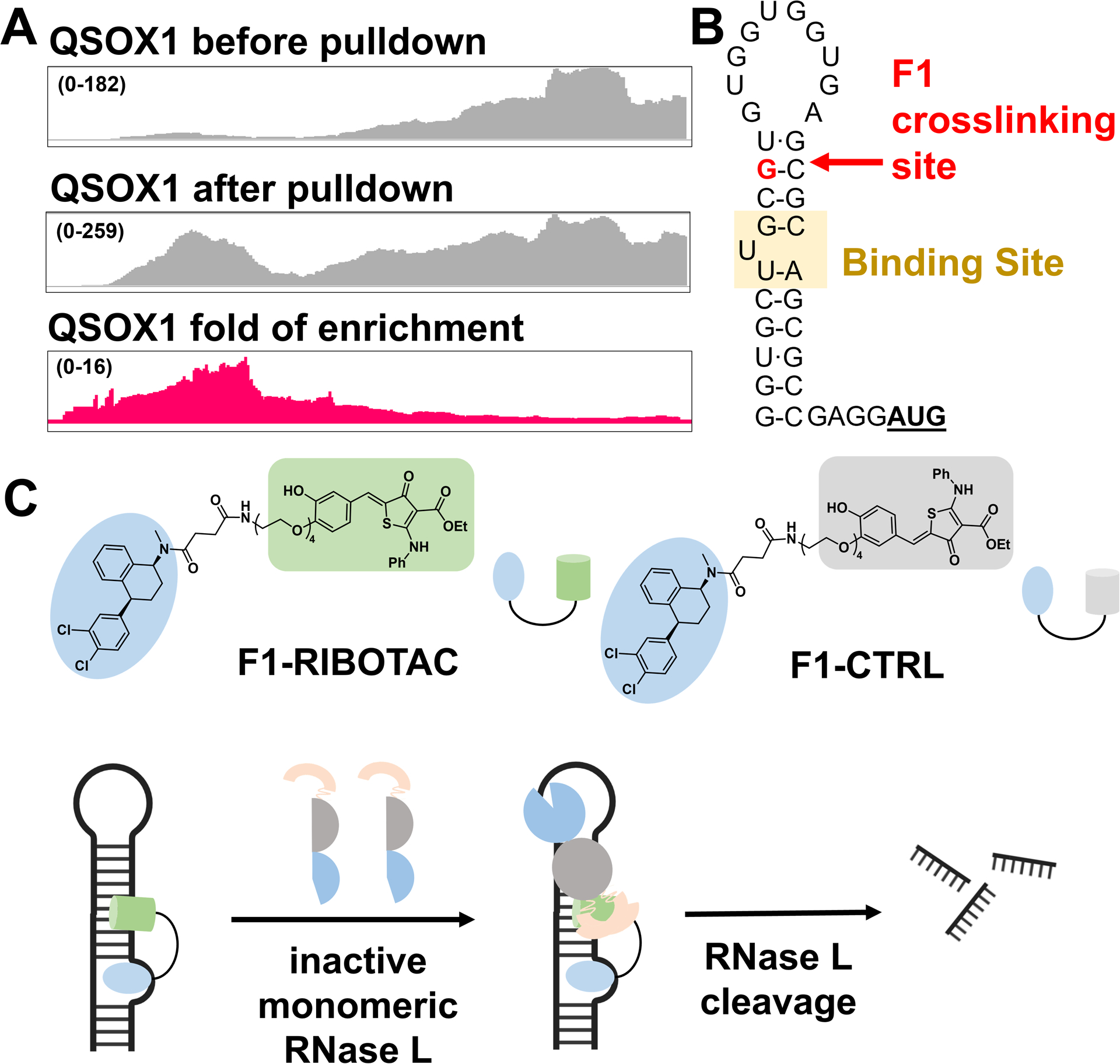
The F1 binding site was mapped to QSOX1 5’ UTR and F1 was subsequently converted to an RNA degrader. (A) RNA-seq tracks showing regions of the QSOX1 transcript enriched by F1 in cells. (B) The RNA sequence and structure nearby the mapped F1 cross-linking site. (C) Structures of F1-RIBOTAC that recruits RNase L, and F1-CTRL that is >20-fold less active in recruiting RNase L.
Because it is unknown whether these targetable structures in QSOX1-a or SQSTM1 are functional, we next examined the regions surrounding the binding site for potential sites sensitive to RNase L cleavage to enact a targeted degradation strategy. This strategy, which uses ribonuclease-targeting chimeras (RIBOTACs) comprising an RNA-binding module and an RNase L-recruiting module, has been advantageous for other RNA targets as they are catalytic, substoichiometric, more selective, and more potent. (27) RNase L, mainly localized in the cytoplasm, preferentially cleaves RNA with a UNN pattern (unpaired uridines). (28–30) Interestingly, three such sites are embedded in a hairpin adjacent to the F1 binding site in the QSOX1-a mRNA (Figure 3B). Only one such site is nearby the SQSTM binding site and at greater than twice the distance (6 vs 13 nucleotides; Figure S14). We therefore hypothesized that functional activity and selectivity could be engineered into F1 toward QSOX1 (vs SQSTM1) by converting it into a RIBOTAC.
We first studied the binding affinity of four molecules to a model of the QSOX1-a binding site by fluorescence polarization with Cy5-labeled RNA: F1, F1-Amide, F1-RIBOTAC, and F1-CTRL (Figures 3C and S15). F1-Amide mimics the conjugation of the RNase L recruiting module while F1-CTRL is a control RIBOTAC that uses an RNase L recruiting module with >20-fold reduced activity. (31) That is, F1-Amide and F1-CTRL are expected to bind with similar affinity and have similar cellular activity and the same mode of action–binding–as F1 does. F1 binds to the bulged U nucleotide in the QSOX1 mRNA with a Kd of 16 ± 6 μM; importantly no detectable binding to a fully paired RNA in which the U bulge was converted to an AU base pair was observed (Figure S15). As expected, F1-Amide (Kd = 26 ± 9 μM), F1-RIBOTAC (Kd = 11 ± 5 μM), and F1-CTRL (Kd = 10 ± 3 μM) bind with similar affinity as F1 (Figure S15). Additionally, no measurable binding was observed for all three compounds to a mutant RNA lacking the U-bulge (Kd > 100 μM) (Figure S15).
Two additional experiments were used to verify these binding data. We studied whether the binding of F1-Amide stabilized the folding of the RNA by optical melting. Indeed, the melting temperature (Tm) increased by 1.9 °C and the ΔG037 decreased by 0.3 kcal/mol upon the addition of a small molecule (Figure S16). No changes in the Tm or ΔG037 were observed when the mutant RNA was melted in the presence or absence of F1 (Figure S16). Binding was also studied by using in vitro Chem-CLIP, where F1 selectively cross-linked to a model of the QSOX1-a 5’ UTR hairpin. As expected, cross-linking of F1 was ablated by mutating the U-bulge into an AU base pair, supporting that F1 binds to the U-bulge that is three nucleotides upstream of the mapped cross-linking site (Figures 3B and S17A). Competitive Chem-CLIP further confirmed the binding of F1-Amide and F1-RIBOTAC on the same site with F1, as they dose-dependently competed off the cross-linking of F1 to the QSOX1-a target site in vitro (Figure S17B).
QSOX1 is an oncogene that promotes invasion and proliferation of breast cancer cells. (32,33) In MDA-MB-231 TBNC cells, F1-RIBOTAC dose-dependently decreased QSOX1-a mRNA levels up to ~35% at 10 μM, the highest dose with no effect on viability (Figure 4A). Neither F1-Amide nor F1-CTRL affected QSOX1-a mRNA levels (Figure S18A), suggesting that F1-RIBOTAC cleaves the target RNA by recruiting and activating RNase L. We further validated this mode of action by treating F1-RIBOTAC in CRISPR-edited cells with RNase L knockdown. As expected, the effect of F1-RIBOTAC on QSOX1-a mRNA was ablated in RNase L-knockdown cells but not in control cells, confirming that F1-RIBOTAC indeed decreases QSOX1 mRNA in an RNase L-dependent manner (Figure 4B).
Figure 4.
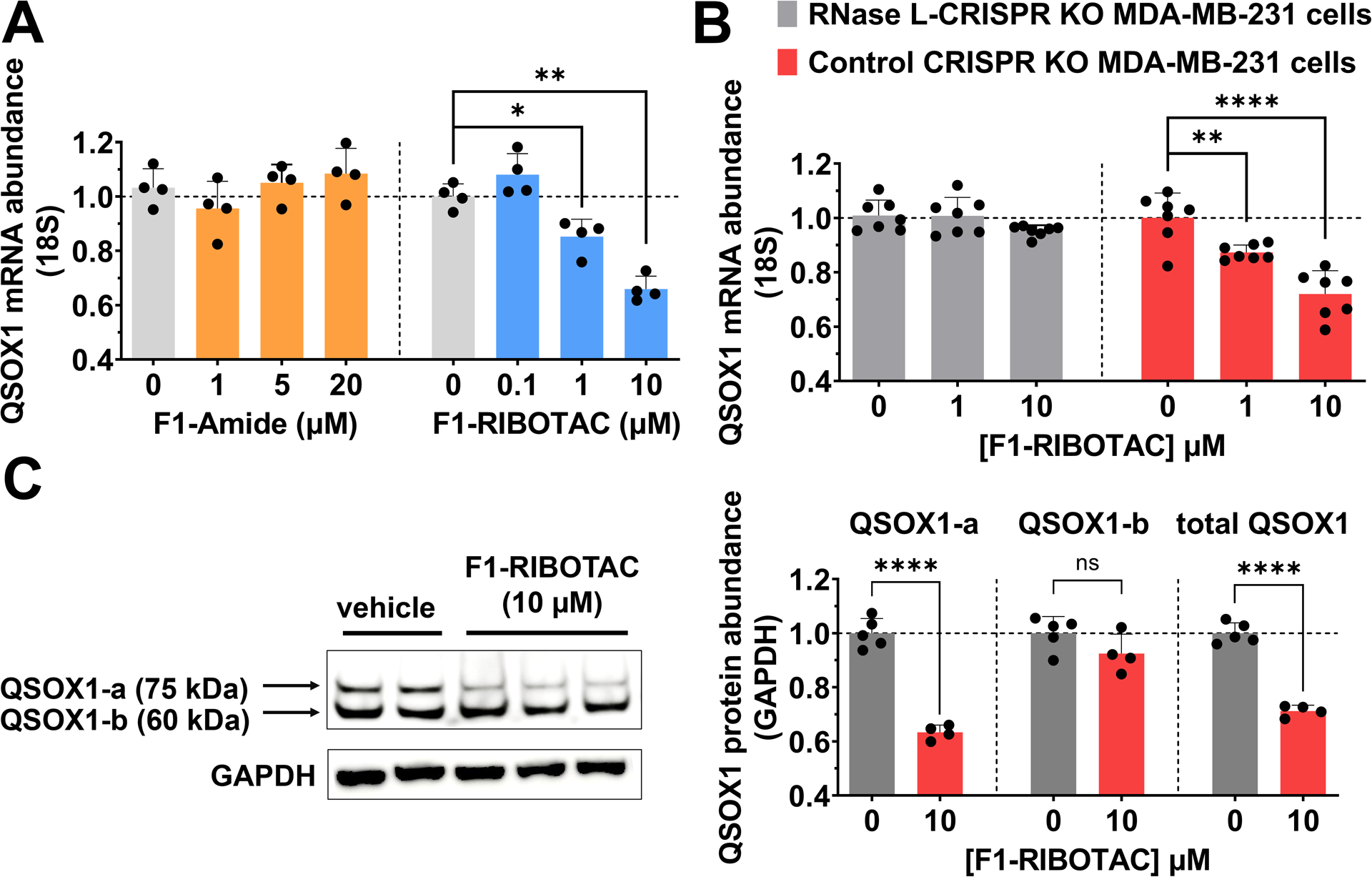
F1-RIBOTAC decreases QSOX1 mRNA and protein levels. (A) Effect of F1-Amide and F1-RIBOTAC on QSOX1 mRNA levels (n = 4). (B) Effect of F1-RIBOTAC on QSOX1 mRNA levels in RNase L knockout MDA-MB-231 cells and control CRISPR cells (n = 7). (C) F1-RIBOTAC on the expression of QSOX1 protein (n = 4). *p < 0.05, **p < 0.01, ****p < 0.0001, as determined by at two-tailed Student’s t-test. All data are reported as the mean ± SD.
Importantly, no significant effect was observed on SQSTM1 mRNA levels upon treatment with F1-RIBOTAC (Figure S19A). As F1 enriched QSOX1 and SQSTM1 similarly, this difference in the RIBOTAC’s activity is likely due to differences in the structure and sequence around the binding site, that is, a single distal unpaired U in SQSTM1, rendering it a poor substrate for RNase L. Interestingly, at the protein level, only QSOX1-a abundance was reduced by F1 RIBOTAC treatment, by ~35% at 10 μM (Figure 4C). No effect was observed on QSOX-1b levels, indicating that F1 RIBOTAC is isoform-specific. Previously, a small molecule was developed that targets both QSOX1 protein isoforms.(33) Therefore, F1-RIBOTAC could be useful to study the individual biological roles of each isoform by exploiting the structural differences in their encoding mRNAs.
In contrast to the results described above for F1 RIBOTAC, only an ~15% reduction of QSOX1-a protein was observed by treatment with 20 μM F1-Amide or F1-CTRL (Figure S18B, C). These results in conjunction with the lack of effect on mRNA abundance suggest that (i) F1-Amide and FI-CTRL bind the QSOX1-a 5’ UTR and impede translation, presumably by blocking ribosomal assembly or processivity, as has been described for other compounds targeting RNA; (34) and (ii) the QSOX1-a binding site is functional. Notably, neither F1-Amide nor F1-RIBOTAC had any effect on SQSTM1 protein levels (Figure S19B,C), the former suggesting that F1’s binding within SQSTM1 is biologically silent.
We next assessed whether the isoform-specific reduction of QSOX1-a protein by F1-RIBOTAC was sufficient to inhibit QSOX1-mediated phenotypes. Indeed, 10 μM F1-RIBOTAC significantly reduced the number of invasive MDA-MB-231 cells, by ~40% (p < 0.01; Figure 5A, B). In contrast, no significant effect was exerted by F1-Amide or F1-CTRL (Figure S20). In the proliferation assay, both F1-Amide and F1-RIBOTAC dose-dependently reduced proliferation of MDA-MB-231 cells, with F1-RIBOTAC showing enhanced activity compared to F1-Amide delivered at twice the dose (Figure 5C). Collectively, these results suggest that converting F1-Amide to F1-RIBOTAC improved its potency in reverting disease-related phenotypes.
Figure 5.
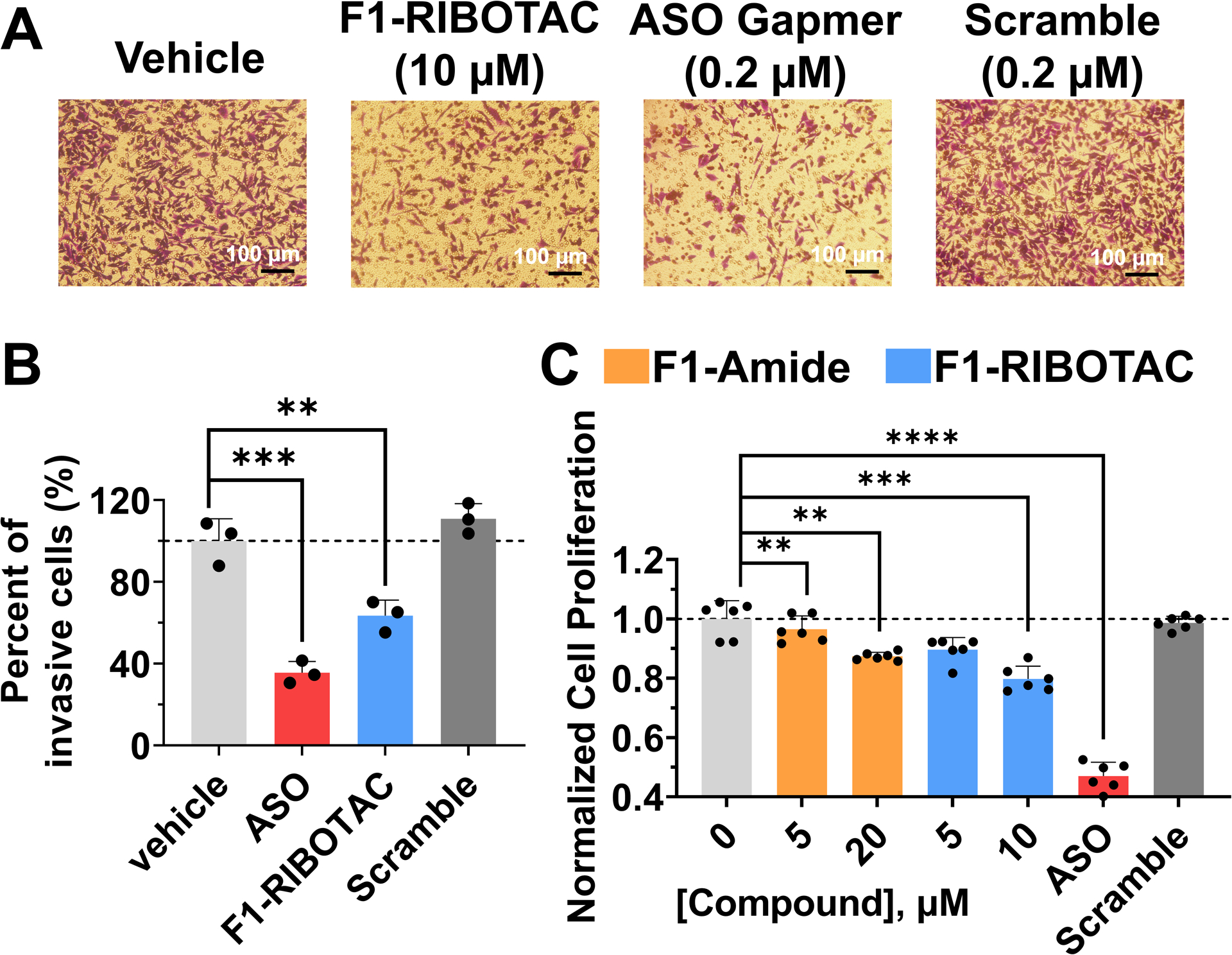
F1-RIBOTAC inhibits invasion and proliferation in MDA-MB-231 cells. (A) Images of invasive MDA-MB-231 with or without treatment. (B) Quantification of invasive cells (n = 3). (C) Effect of F1-RIBOTAC and F1-Amide on the proliferation of MDA-MB-231 cells after 48 h treatment (n = 6). **p < 0.01, ***p < 0.001, ****p < 0.0001, as determined by at two-tailed Student’s t-test. All data are reported as the mean ± SD.
CONCLUSIONS
In summary, we presented a method to map small-molecule binding sites across the human transcriptome in live cells. We demonstrated this method by targeting a structure in the 5’ UTR of oncogenic QSOX1-a mRNA and improved the potency and specificity of the initial hit compound by its conversion into a RIBOTAC. Interestingly, these studies afforded an isoform-specific degrader of QSOX1. Broadly, this method can be used to study the molecular recognition patterns between RNA and ligands in an unbiased way in intact biological systems. Additionally, it can also infer RNA structures mediated by RNA alone or by RNA-protein complexes that would be otherwise difficult to detect.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health R01 grant R01 CA249180 (to M.D.D.), R01 GM133810 (to W.N.M.), and F31 CA257090 (to W.B.R.). We thank Christopher C. Williams and Dr. Haruo Aikawa for aiding in chemical synthesis.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c01929.
The authors declare the following competing financial interest(s): M.D.D. is a founder of Expansion Therapeutics.
The results of Chem-CLIP-Seq analysis were deposited in Mendeley Data (DOI: 10.17632/56r9zmjps2.1).
The results of proteomic studies were deposited in Mendeley Data (DOI: 10.17632/5z3k487xmw.2).
Contributor Information
Yuquan Tong, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Quentin M. R. Gibaut, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States
Warren Rouse, Roy J. Carver Department of Biochemistry, Biophysics, and Molecular Biology, Iowa State University, Ames, Iowa 50011, United States.
Jessica Childs-Disney, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Blessy M. Suresh, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States
Daniel Abegg, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Shruti Choudhary, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Yoshihiro Akahori, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Alexander Adibekian, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Walter N. Moss, Roy J. Carver Department of Biochemistry, Biophysics, and Molecular Biology, Iowa State University, Ames, Iowa 50011, United States
Matthew D. Disney, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States
References
- 1.Ding Y; Tang Y; Kwok CK; Zhang Y; Bevilacqua PC; Assmann SM In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700, DOI: 10.1038/nature12756 [DOI] [PubMed] [Google Scholar]
- 2.Wan Y; Kertesz M; Spitale RC; Segal E; Chang HY Understanding the transcriptome through RNA structure. Nat. Rev. Genet 2011, 12, 641–655, DOI: 10.1038/nrg3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strobel EJ; Yu AM; Lucks JB High-throughput determination of RNA structures. Nat. Rev. Genet 2018, 19, 615–634, DOI: 10.1038/s41576-018-0034-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spitale RC; Flynn RA; Zhang QC; Crisalli P; Lee B; Jung JW; Kuchelmeister HY; Batista PJ; Torre EA; Kool ET; Chang HY Structural imprints in vivo decode RNA regulatory mechanisms. Nature 2015, 519, 486–490, DOI: 10.1038/nature14263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ratni H; Ebeling M; Baird J; Bendels S; Bylund J; Chen KS; Denk N; Feng Z; Green L; Guerard M; Jablonski P; Jacobsen B; Khwaja O; Kletzl H; Ko CP; Kustermann S; Marquet A; Metzger F; Mueller B; Naryshkin NA; Paushkin SV; Pinard E; Poirier A; Reutlinger M; Weetall M; Zeller A; Zhao X; Mueller L Discovery of Risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J. Med. Chem 2018, 61, 6501–6517, DOI: 10.1021/acs.jmedchem.8b00741 [DOI] [PubMed] [Google Scholar]
- 6.Disney MD Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J. Am. Chem. Soc 2019, 141, 6776–6790, DOI: 10.1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guan L; Disney MD Covalent small molecule-RNA complex formation enables cellular profiling of small molecule-RNA interactions. Angew. Chem. Int. Ed 2013, 52, 10010–10013, DOI: 10.1002/anie.201301639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balaratnam S; Rhodes C; Bume DD; Connelly C; Lai CC; Kelley JA; Yazdani K; Homan PJ; Incarnato D; Numata T; Schneekloth JS Jr A chemical probe based on the PreQ1 metabolite enables transcriptome-wide mapping of binding sites. Nat. Commun 2021, 12, 5856, DOI: 10.1038/s41467-021-25973-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J; Schultz PG; Johnson KA Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E4604–e4612, DOI: 10.1073/pnas.180026011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parker CG; Galmozzi A; Wang Y; Correia BE; Sasaki K; Joslyn CM; Kim AS; Cavallaro CL; Lawrence RM; Johnson SR; Narvaiza I; Saez E; Cravatt BF Ligand and target discovery by fragment-based screening in human cells. Cell 2017, 168, 527–541, DOI: 10.1016/j.cell.2016.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spradlin JN; Zhang E; Nomura DK Reimagining druggability using chemoproteomic platforms. Acc. Chem. Res 2021, 54, 1801–1813, DOI: 10.1021/acs.accounts.1c00065 [DOI] [PubMed] [Google Scholar]
- 12.Zhang X; Spiegel J; Martínez Cuesta S; Adhikari S; Balasubramanian S Chemical profiling of DNA G-quadruplex-interacting proteins in live cells. Nat. Chem 2021, 13, 626–633, DOI: 10.1038/s41557-021-00736-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.di Antonio M; McLuckie KI; Balasubramanian S Reprogramming the mechanism of action of chlorambucil by coupling to a G-quadruplex ligand. J. Am. Chem. Soc 2014, 136, 5860–5863, DOI: 10.1021/ja5014344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McInnes L; Healy J; Melville J Umap: Uniform manifold approximation and projection for dimension reduction. Submitted on 2018–02-09. arXiv preprint:1802.03426 DOI: 10.48550/arXiv.1802.03426. (accessed 2022-01-09). [DOI] [Google Scholar]
- 15.Carnevali M; Parsons J; Wyles DL; Hermann T A modular approach to synthetic RNA binders of the hepatitis C virus internal ribosome entry site. ChemBioChem 2010, 11, 1364–1367, DOI: 10.1002/cbic.201000177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilimire TA; Chamberlain JM; Anokhina V; Bennett RP; Swart O; Myers JR; Ashton JM; Stewart RA; Featherston AL; Gates K; Helms ED; Smith HC; Dewhurst S; Miller BL HIV-1 frameshift RNA-targeted triazoles inhibit propagation of replication-competent and multi-drug-resistant HIV in human cells. ACS Chem. Biol 2017, 12, 1674–1682, DOI: 10.1021/acschembio.7b00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Giorgio A; Tran TP; Duca M Small-molecule approaches toward the targeting of oncogenic miRNAs: roadmap for the discovery of RNA modulators. Future Med. Chem 2016, 8, 803–816, DOI: 10.4155/fmc-2016-0018 [DOI] [PubMed] [Google Scholar]
- 18.Disney MD; Winkelsas AM; Velagapudi SP; Southern M; Fallahi M; Childs-Disney JL Inforna 2.0: A platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem. Biol 2016, 11, 1720–1728, DOI: 10.1021/acschembio.6b00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan BS; Sanaba BG; Donlic A; Karloff DB; Forte JE; Zhang Y; Hargrove AE R-BIND: an interactive database for exploring and developing RNA-targeted chemical probes. ACS Chem. Biol 2019, 14, 2691–2700, DOI: 10.1021/acschembio.9b00631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang YJ; Dix MM; Bianco G; Remsberg JR; Lee HY; Kalocsay M; Gygi SP; Forli S; Vite G; Lawrence RM; Parker CG; Cravatt BF Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat. Chem 2019, 11, 1113–1123, DOI: 10.1038/s41557-019-0351-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velagapudi SP; Li Y; Disney MD A cross-linking approach to map small molecule-RNA binding sites in cells. Bioorg. Med. Chem. Lett 2019, 29, 1532–1536, DOI: 10.1016/j.bmcl.2019.04.00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.https://github.com/jsh58/Genrich [acesssed 2022-01-09].
- 23.Mukherjee H; Blain JC; Vandivier LE; Chin DN; Friedman JE; Liu F; Maillet A; Fang C; Kaplan JB; Li J; Chenoweth DM; Christensen AB; Petersen LK; Hansen NJV; Barrera L; Kubica N; Kumaravel G; Petter JC PEARL-seq: a photoaffinity platform for the analysis of small molecule-RNA interactions. ACS Chem. Biol 2020, 15, 2374–2381, DOI: 10.1021/acschembio.0c00357 [DOI] [PubMed] [Google Scholar]
- 24.Linder B; Grozhik AV; Olarerin-George AO; Meydan C; Mason CE; Jaffrey SR Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772, DOI: 10.1038/nmeth.3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hafner M; Katsantoni M; Köster T; Marks J; Mukherjee J; Staiger D; Ule J; Zavolan M CLIP and complementary methods. Nat. Rev. Dis. Primers 2021, 1, 20, DOI: 10.1038/s43586-021-00018-1 [DOI] [Google Scholar]
- 26.Andrews RJ; Roche J; Moss WN ScanFold: an approach for genome-wide discovery of local RNA structural elements-applications to Zika virus and HIV. PeerJ 2018, 6, e6136 DOI: 10.7717/peerj.6136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang P; Liu X; Abegg D; Tanaka T; Tong Y; Benhamou RI; Baisden J; Crynen G; Meyer SM; Cameron MD; Chatterjee AK; Adibekian A; Childs-Disney JL; Disney MD Reprogramming of protein-targeted small-molecule medicines to RNA by ribonuclease recruitment. J. Am. Chem. Soc 2021, 143, 13044–13055, DOI: 10.1021/jacs.1c02248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han Y; Donovan J; Rath S; Whitney G; Chitrakar A; Korennykh A Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 2014, 343, 1244–1248, DOI: 10.1126/science [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wreschner DH; McCauley JW; Skehel JJ; Kerr IM Interferon action--sequence specificity of the ppp(A2’p)nA-dependent ribonuclease. Nature 1981, 289, 414–417, DOI: 10.1038/289414a0 [DOI] [PubMed] [Google Scholar]
- 30.Floyd-Smith G; Slattery E; Lengyel P Interferon action: RNA cleavage pattern of a (2′−5′)oligoadenylate--dependent endonuclease. Science 1981, 212, 1030–1032, DOI: 10.1126/science.6165080 [DOI] [PubMed] [Google Scholar]
- 31.Costales MG; Aikawa H; Li Y; Childs-Disney JL; Abegg D; Hoch DG; Pradeep Velagapudi S; Nakai Y; Khan T; Wang KW; Yildirim I; Adibekian A; Wang ET; Disney MD Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 2406–2411, DOI: 10.1073/pnas.1914286117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baek JA; Song PH; Ko Y; Gu MJ High expression of QSOX1 is associated with tumor invasiveness and high grades groups in prostate cancer. Pathol. Res. Pract 2018, 214, 964–967, DOI: 10.1016/j.prp.2018.05.019 [DOI] [PubMed] [Google Scholar]
- 33.Fifield AL; Hanavan PD; Faigel DO; Sergienko E; Bobkov A; Meurice N; Petit JL; Polito A; Caulfield TR; Castle EP; Copland JA; Mukhopadhyay D; Pal K; Dutta SK; Luo H; Ho TH; Lake DF Molecular inhibitor of QSOX1 suppresses tumor growth in vivo. Mol. Cancer Ther 2020, 19, 112–122, DOI: 10.1158/1535-7163.MCT-19-0233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang PY; Park HJ; Zhang J; Junn E; Andrews RJ; Velagapudi SP; Abegg D; Vishnu K; Costales MG; Childs-Disney JL; Adibekian A; Moss WN; Mouradian MM; Disney MD Translation of the intrinsically disordered protein alpha-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 1457–1467, DOI: 10.1073/pnas.190505711 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.