
Figure 5: Synergy across pathogenic mechanisms in repeat expansion diseases.
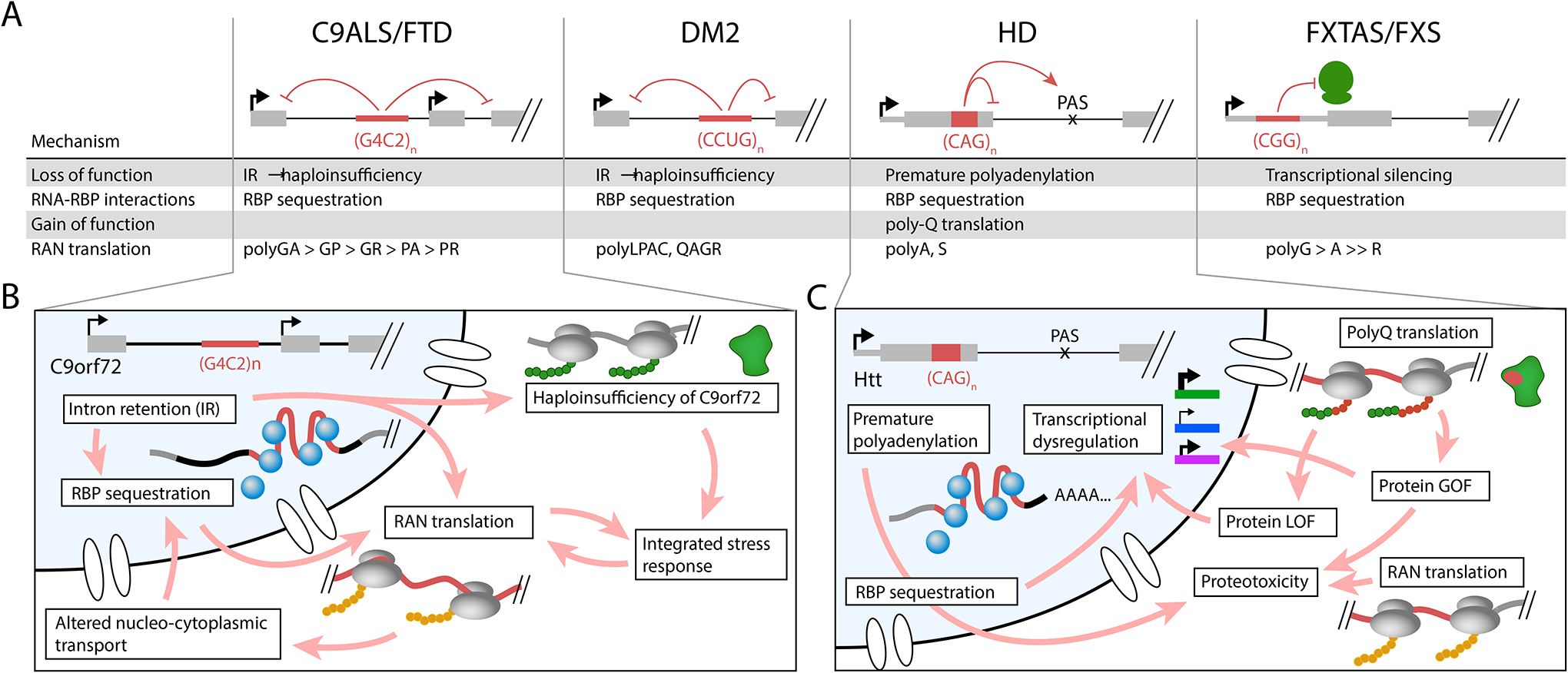
(A) In multiple diseases, the four major mechanisms detailed in this review can co-exist and/or synergize to drive complex pathology. For example, in C9 ALS/FTD, expanded GGGGCC repeats can induce intron retention, which leads to haploinsufficiency of C9ORF72, as well as exacerbates RBP sequestration by increasing the half-life of the repeat RNA. In addition, intron retention may increase the production of dipeptide repeats that activate numerous downstream pathogenic pathways. In DM2, expanded CCTG repeats lead to intron retention, which also results in reduction of mRNA available to generate full-length CNBP protein. RAN translation products can be generated from the intron-retained mRNA. In Huntington disease (HD), expanded CAG repeats alter RNA processing to impair recognition of the exon 1 donor splice site; this results in the formation of a truncated polyQ-containing HTT protein that is more toxic than full-length polyQ-containing HTT. RAN translation can also occur across the CAG repeat. In FXTAS/FXS, the CGG repeat can not only sequester RBPs, but can also enhance RAN translation of the uORF such that translation initiation for FMRP is reduced. (B) A more detailed view of pathways activated in C9 ALS/FTD shows that some pathogenic mechanisms can exacerbate or feed into other mechanisms. A complex network of cause and effect, including feed-forward loops, may synergize to drive disease pathology. (C) A more detailed view of pathways activated in HD also similarly reveals feedback loops in both the nucleus and cytoplasm.