

The transforming growth factor beta (TGFβ) pathway exhibits pro-tumorigenic effects across multiple malignancies, via its impact on tumor cells, microenvironment, and/or the immune system1, 2. There is therefore a growing impetus to target the TGFβ signaling axis across malignancies1, 2. While early-phase/dose-defining trials with TGFβR1 (ALK5) inhibitor in glioma3 and TGFβ2 oligo therapy in anaplastic astrocytoma4 do show improved outcomes, albeit in a small subset of patients, the molecular determinants that dictate response to TGFβ-pathway targeted therapies however remain elusive; underscoring a broader unmet need in precision oncology. Furthermore, lack of such robust response biomarkers will continue to pose challenges, undermining ongoing/future clinical trials with TGFβ-pathway targeted therapies2. Identifying pertinent molecular- and tumor-contexts where such treatments could be most beneficial will not only improve treatment paradigms and clinical outcomes, but also minimize unwarranted therapy-associated burdens.
We recently reported that esophageal adenocarcinomas (EAC) are characterized by hyperactivation of the TGFβ pathway early on in their development, unlike other gastrointestinal (GI) malignancies5. These distal-esophageal tumors in particular are alarmingly increasing in incidence, even among younger individuals under 50 years of age6. Current treatment options (targeted or otherwise) for EAC are extremely limited and/or ineffective, with vast majority becoming refractory to standard chemoradiation therapies; overall leading to a dismal 5-year survival7. Consequently, we posited whether TGFβ-pathway targeted therapies could be beneficial in EACs, and accordingly evaluated the anti-tumor efficacy of a new generation ALK5 inhibitor (Vactosertib)8 in multiple in-vivo preclinical models of EAC.
Of the EAC models tested, we observed ALK5i to effectively suppress or even abrogate the growth of a subset of EAC tumors (Figure 1A). ALK5i treatment nonetheless effectively abrogated Smad2/3 phosphorylation (p-Smad), a critical mediator of TGFβ signaling, across all the EAC tumor models in-vivo (Figure 1A); suggesting that sensitivity to TGFβ pathway inhibition is not likely due to differences in ALK5i-pharmacodynamics or in TGFβ receptor activities amongst the EAC models.
Figure 1: HNF4A mediates ALK5i resistance in EAC and molecular subtypes of EAC.
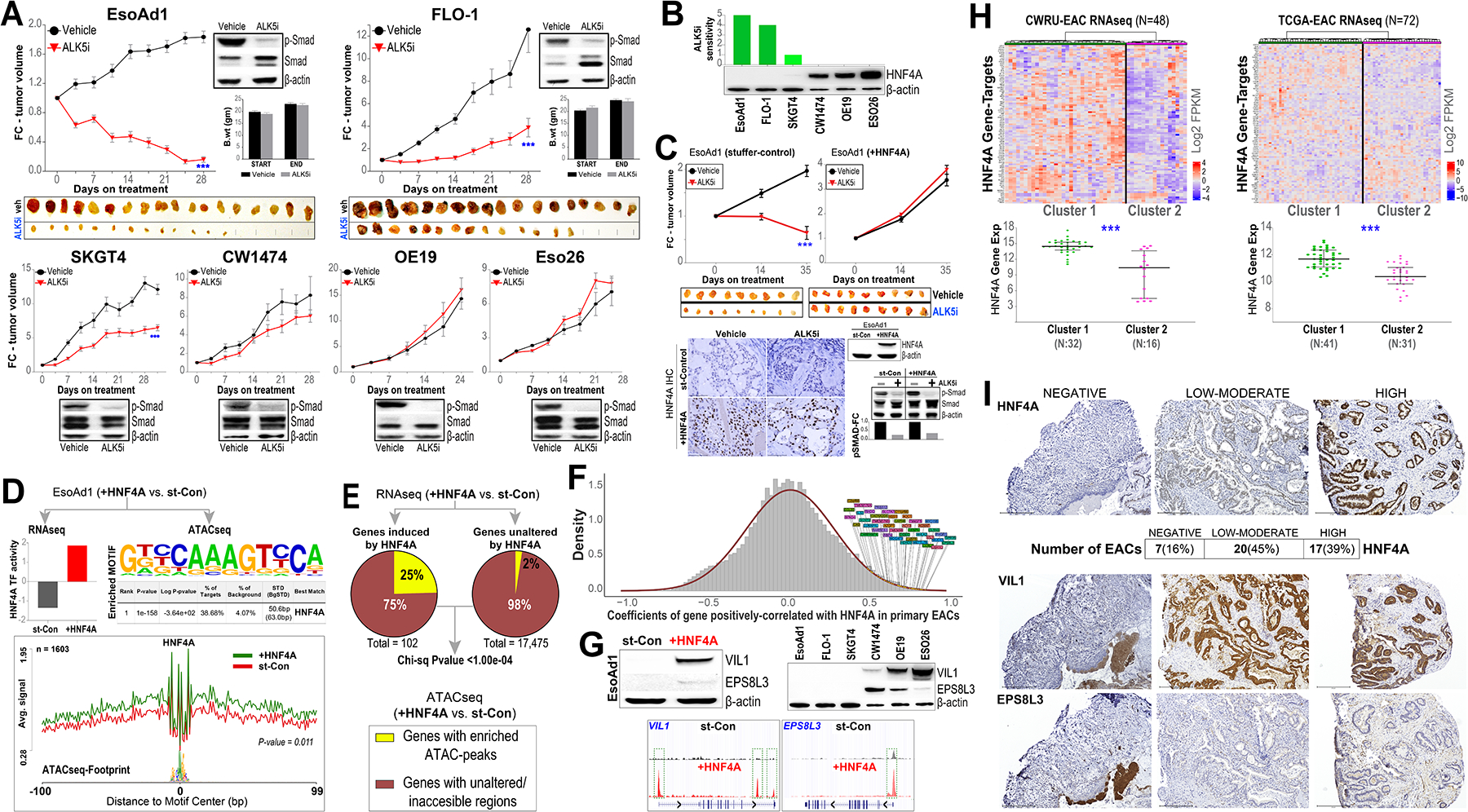
(A) Tumor xenografts from indicated EAC cell lines were established subcutaneously in immune-compromised mice, and randomized to Vactosertib (ALK5i;100 mg/kg) or vehicle treatment via oral gavage, once daily for 5 consecutive days and 2-day off-treatment. Y-axis in the line graph depicts the average fold-change (FC) in tumor volume over time (x-axis) in respective treatment groups, normalized to the first day of treatment (time point 0 on x-axis). Data are plotted as mean ± SEM, obtained from at least 8 tumors per ALK5i or vehicle arm. *** (P<0.0005) indicates significant differences in tumor volume at the final time point between ALK5i- vs vehicle-treatment groups, estimated using a Student’s t-test. Western blot images depict the levels of total and p-Smad proteins in representative tumor xenografts, obtained 1-week after treatment initiation, from respective EAC models. β-actin was used as an endogenous loading control in the Western blots. EACs (EsoAd1, FLO-1) that were highly sensitive to ALK5i-mediated growth inhibition were shown in the top row, along with photographs of excised tumors from ALK5i and vehicle treated mice at the end of the study. SKGT4 showed modest sensitivity to ALK5i, whereas CW1474, OE19, and Eso26 were resistant to ALK5i (bottom row). Average body weights (B. wt) of mice at the start and end of treatments were provided for the sensitive EsoAd1 and FLO-1 xenografts to demonstrate no apparent adverse effects from ALK5i treatment in-vivo. (B) Western blot analyses demonstrating relative HNF4A protein expression, and the green bars above depict the degree of sensitivity (on an arbitrary 1–5 scale) to ALK5i-induced growth suppression across EAC models. Note the striking correlation of HNF4A RNA/protein expression with ALK5i sensitivity. (C) Tumor xenografts from EsoAd1 EAC cells, stably-reconstituted with HNF4A (+HNF4A) or isogenic stuffer control (st-Control), were established in immune-deficient mice and treated with Vactosertib (ALK5i) or vehicle, and growth kinetics measured over time as in Figure 1A. Data are plotted as mean ± SEM, obtained from at least 8 tumors per ALK5i or vehicle arm. *** (P<0.0005) indicates significant differences in tumor volume at the final time point between ALK5i- vs vehicle-treatment groups, estimated using a Student’s t-test. Also shown are photographs of excised xenograft tumors obtained at the end of the study. Note that restoration of HNF4A completely abrogates sensitivity to ALK5i in EsoAd1 cells. (Bottom) representative images from IHC analysis depicting HNF4A protein levels in respective xenograft tumors, obtained 1-week after treatment initiation. Scale bar, 75 μm. Western blot images to the right show protein levels of HNF4A in control (st-Con) and in HNF4A-reconstituted (+HNF4A) EsoAd1 tumor-xenografts derived from immune-deficient mice treated with ALK5i or vehicle. Bar graphs below show relative fold-change (FC) in p-Smad protein levels, normalized to vehicle control treatments, in respective control and +HNF4A groups. (D) (Top) HNF4A-reconstituted EsoAd1 cells showing significant increase in HNF4A transcription-factor (TF) activity, and (bottom) enrichment of HNF4A motif and footprint in accessible chromatin regions (bp; basepair), when compared to isogenic control (st-Con) EsoAd1 cells. (E) Pie charts demonstrating that 25% of HNF4A-induced genes (≥2-fold increased expression in HNF4A-reconstituted vs. st-Con EsoAd1 cells) are associated with regions exhibiting enhanced chromatin accessibility upon HNF4A induction; significantly different from the 2% increase in chromatin accessibility for genes unaltered by HNF4A. (F) RNAseq-based analysis showing genes that are significantly and positively correlated with HNF4A expression in primary EAC tumors. (G) (Top row) Western blot analysis of representative HNF4A-induced genes (VIL1, EPS8L3) showing significant induction of respective proteins in HNF4A-reconstituted EsoAd1, and in ALK5i-resistant parental EAC cells (CW1474, OE19, ESO26). β-actin was included as a loading control in the Western blots. (Bottom row) ATACseq-based Tag density plots (UCSC Genome Browser) show enhanced peaks in respective VIL1 and EPS8L3 gene promoters (green boxes), upon HNF4A induction. (H) Heatmaps from unsupervised hierarchical clustering of HNF4A gene-targets in primary EACs, derived from in-house (CWRU) and TCGA RNAseq datasets, revealing two major molecular subclasses/clusters of EAC. Note the significant (***; P<0.0005) differences in HNF4A gene expression (Y-axis; log2 FPKM values) between the two EAC clusters in CWRU and TCGA RNAseq datasets (see Methods). (I) (Top) IHC images demonstrating nuclear HNF4A protein (brown staining) in representative HNF4A-negative, -low/moderate, and -high EACs. Shown below are the number (percentage) of EAC samples with negative, low-moderate, and high protein expression of HNF4A by IHC. (Below) IHC analysis depicting protein expression of HNF4A-targets, VIL1 and EPS8L3, in the respective tumor sections from above. Scale bar, 275 μm.
Our further assessments of aberrations in specific EAC-associated driver genes, such as P53, SMAD4, ERBB2, or our RNAseq-based assessments of TGFβ response signatures showed no apparent associations with ALK5i responses in EACs (Supplementary Figure 1). We therefore used an agnostic approach to identify molecular factors associated with ALK5i response. Intriguingly, HNF4A, a GI lineage defining transcription factor implicated in BE and EAC9, 10, emerged as the top candidate with markedly increased expression in ALK5i-resistant compared to ALK5i-sensitive EACs (Figure 1B, Supplementary Figure 1). We next asked whether HNF4A could also functionally regulate ALK5i response in EACs. Since cancer cells with high baseline HNF4A expression are highly sensitive to HNF4A depletion9 (and our own observations), we conversely assessed whether induction of HNF4A would impart resistance to ALK5i treatment in otherwise HNF4A-low/null (ALK5i-sensitive) EAC xenografts. Indeed, stable reconstitution of HNF4A completely abrogated the anti-tumor efficacy of ALK5i in-vivo, again with no differences in p-Smad levels between control and HNF4A-reconstituted EAC tumors (Figure 1C). These findings provocatively suggest HNF4A as playing a functional role in conferring inherent resistance to ALK5i therapy.
Although HNF4A is a well-recognized GI lineage transcription factor, its molecular effects in the EAC context remains unknown. We therefore sought to understand if/how HNF4A impacts the molecular programs in EAC, as this could provide insights into its function in this malignancy. We performed integrative RNAseq and ATACseq analyses in HNF4A-low/negative EAC (EsoAd1) cells, with or without stable reconstitution of HNF4A. Ensuring that ectopically reconstituted HNF4A is functionally active, we observed a significant enrichment in HNF4A transcriptional activity and genomic footprint in EAC cells (Figure 1D). Notably, we found HNF4A-induced genes were much more likely to be within HNF4A-induced open chromatin regions, compared to genes that were unaltered by HNF4A (P=0.0001) (Figure 1E). Gene Ontology analysis of HNF4A-induced genes revealed a significant enrichment of digestion-associated biologic process (P=8.65E-08), consistent with its role in promoting GI cell fate. Furthermore, ~40% of HNF4A-induced genes showed significant positive correlation with HNF4A expression in primary EAC tumors (Figure 1F, and Supplementary Figure 2). Subsequent Western blot analyses of representative HNF4A gene-targets, VIL1 and EPS8L3 (intestinal epithelial/differentiation markers), confirmed a marked increase in protein expression along with enriched ATAC peaks in respective gene-promoters in EAC cells (Figure 1G). In line with this, we found both VIL1 and EPS8L3 to be expressed selectively in HNF4A-high (ALK5i-resistant) parental EAC lines (Figure 1G). Collectively, these findings strongly suggest that baseline HNF4A expression confers a digestive/intestinal-type molecular phenotype in EACs with potential implications in disease pathogenesis.
To further understand how HNF4A could be impacting ALK5i response on a molecular level, we performed RNAseq in HNF4A-reconstituted vs. isogenic control EAC cells treated with ALK5i. Pathway and protein-protein interaction enrichment analyses revealed significant (P<0.05) alterations in interleukin-18/matrix-metalloprotein/inflammatory sub-network between HNF4A-positive vs. -negative isogenic EAC cells treated with ALK5i (Supplementary Figure 2). Orthogonal qPCR-based assessment of a representative gene of this network, TNFAIP3 (a TNF alpha induced protein and an upstream regulator of IL-18), showed HNF4A to abrogate ALK5i-mediated suppression of TNFAIP3 in isogenic, as well as in HNF4A-high parental EAC cells (Supplementary Figure 2). These studies besides revealing ALK5i response pathways, also offer potential mechanisms by which HNF4A impedes response to ALK5i therapy.
As our findings above implicate HNF4A as a candidate biomarker of ALK5i response in EACs, it is imperative to understand the extent to which primary EAC tumors differ in HNF4A status. Unsupervised clustering using an in-house RNAseq dataset of treatment-naïve EACs (n=48)5 revealed two broad and distinct clusters of EACs stratified by differences in the expression of HNF4A target-genes (Figure 1H). Subsequent supervised analyses showed a significant (P<0.0005) difference in HNF4A expression between the respective sample clusters (Figure 1H); findings that were similarly observed in an independent TCGA EAC RNAseq dataset (n=72) (Figure 1H). Additional evaluation of HNF4A protein status using IHC in a third independent in-house cohort of treatment-naïve EAC tumor biopsy tissues (n=44) showed markedly distinct HNF4A protein expression, as well as HNF4A targets (VIL1, EPS8L3), in EAC tumors (Figure 1I). Taken together the RNAseq and protein analyses, we estimate the majority of primary EAC tumors (60%−80%) to exhibit low/moderate or high HNF4A expression, with ~20% of tumors lacking HNF4A. Overall survival probability based on HNF4A expression status however did not significantly differ in any of the three patient cohorts (data not shown). Nonetheless, intriguingly, majority (70%) of HNF4A-negative tumors (based on histologically-defined IHC cohort) were poorly differentiated, as opposed to only 5% of HNF4A-high tumors (Fisher’s Exact P=0.0027), suggesting that HNF4A may further define a histologic subset of EACs. Collectively, these findings underscore the inherent heterogeneity of EAC tumors, defined by HNF4A, and their associated implications for TGFβ-pathway targeted therapies in this disease.
In summary, we identify TGFβ pathway as a therapeutic vulnerability in a highly aggressive GI malignancy. Our findings are timely given the unmet need in identifying effective targeted therapies for these highly-refractory upper GI malignancies1, and the growing efforts in exploiting this pathway for cancer treatment1, 2. Also, our identification of HNF4A as an intrinsic modulator of ALK5i response has broader clinical implications as a predictive biomarker that can be evaluated in ongoing/future clinical trials, such that evidence-based strategies can be implemented to aid in selecting appropriate tumor contexts and patient cohorts who may derive most benefit from TGFβ-pathway targeted therapies.
Supplementary Material
Data Transparency Statement:
Pertinent high-throughput sequencing and integrative analytics in cell line models, where applicable, will be deposited in SRA and made freely available to general public following acceptance of the manuscript.
ACKNOWLEDGEMENTS:
We thank Dr. Jason Mills and Dr. Ramon Jin for sharing their technical expertise in the culturing of patient derived samples. This work was also supported by the Tissue Resources Core and Animal Resource Center at Case Western Reserve University. We acknowledge Wendy Brock, Rajesh Gupta, and Komal Keerthy for their assistance with patient samples. We Acknowledge Sarada Chadalawada and Maeve Slife for their technical contributions. We acknowledge the Barrett’s Esophagus Translational Research Network (BETRNet) for contributions to this manuscript and ongoing efforts to speed the translation of important research findings from laboratory and clinical studies into useful medical applications; BETRNet contributors to this manuscript are listed on the title page of the manuscript. This work was supported in part by Career Development Award, Award #1IK2CX001831 from the United States (U.S.) Department of Veterans Affairs (Biomedical Laboratory Research and Development Service).
Grant Support:
This research was supported by PHS awards: R01 CA204549 (K. Guda), U01 CA152756 (K. Guda), Case BETRNet U54 CA163060 (A. Chak, K. Guda), Case GI SPORE P50 CA150964 (A. Chak, K. Guda, A. Blum), K25 DK115904 (V. Varadan), P30 CA043703 (V. Varadan, K. Guda); CDA-2 1IK2CX001831 United States (U.S.) Department of Veterans Affairs, Biomedical Laboratory Research and Development Service (A. Blum); and the DeGregorio Family Foundation, the Savone Family, and the Esophageal Cancer Awareness Association (K. Guda), and Torrey Coast Foundation GEMINI Network (K. Guda)
Abbreviations:
- EAC
Esophageal adenocarcinoma
- ALK5i
ALK5 inhibitor
- IHC
Immunohistochemistry
- ATACseq
Assay for Transposase-Accessible Chromatin using sequencing
- GI
gastrointestinal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have no potential conflicts of interest to disclose.
Disclaimer: The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Contributor Information
BETRNet Consortium:
Salendra Singh, Yanling Miao, Adam M. Kresak, Joseph E. Willis, Marcia I. Canto, Jean S. Wang, Nicholas J. Shaheen, and Amitabh Chak
REFERENCES
- 1.Veen LM, Skrabanja TLP, Derks S, et al. The role of transforming growth factor beta in upper gastrointestinal cancers: A systematic review. Cancer Treat Rev 2021;100:102285. [DOI] [PubMed] [Google Scholar]
- 2.Tauriello DVF, Sancho E, Batlle E. Overcoming TGFbeta-mediated immune evasion in cancer. Nat Rev Cancer 2022;22:25–44. [DOI] [PubMed] [Google Scholar]
- 3.Rodon J, Carducci MA, Sepulveda-Sanchez JM, et al. First-in-human dose study of the novel transforming growth factor-beta receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res 2015;21:553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bogdahn U, Hau P, Stockhammer G, et al. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol 2011;13:132–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blum AE, Venkitachalam S, Ravillah D, et al. Systems Biology Analyses Show Hyperactivation of Transforming Growth Factor-beta and JNK Signaling Pathways in Esophageal Cancer. Gastroenterology 2019;156:1761–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Codipilly DC, Sawas T, Dhaliwal L, et al. Epidemiology and Outcomes of Young-Onset Esophageal Adenocarcinoma: An Analysis from a Population-Based Database. Cancer Epidemiol Biomarkers Prev 2021;30:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smyth EC, Lagergren J, Fitzgerald RC, et al. Oesophageal cancer. Nat Rev Dis Primers 2017;3:17048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin CH, Krishnaiah M, Sreenu D, et al. Discovery of N-((4-([1,2,4]triazolo[1,5-a]pyridin-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2 -yl)methyl)-2-fluoroaniline (EW-7197): a highly potent, selective, and orally bioavailable inhibitor of TGF-beta type I receptor kinase as cancer immunotherapeutic/antifibrotic agent. J Med Chem 2014;57:4213–38. [DOI] [PubMed] [Google Scholar]
- 9.Pan J, Silva TC, Gull N, et al. Lineage-Specific Epigenomic and Genomic Activation of Oncogene HNF4A Promotes Gastrointestinal Adenocarcinomas. Cancer Res 2020;80:2722–2736. [DOI] [PubMed] [Google Scholar]
- 10.Rogerson C, Britton E, Withey S, et al. Identification of a primitive intestinal transcription factor network shared between esophageal adenocarcinoma and its precancerous precursor state. Genome Res 2019;29:723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.