

Abstract
Synaptic and cognitive deficits mediated by a severe reduction in excitatory neurotransmission caused by a disproportionate accumulation of the neuronal protein tau in dendritic spines is a fundamental mechanism that has been found repeatedly in models of tauopathies, including Alzheimer’s disease, Lewy body dementia, frontotemporal dementia, and traumatic brain injury. Synapses thus damaged may contribute to dementia, among the most feared cause of debilitation in the elderly, and currently there are no treatments to repair them. Caspase-2 (Casp2) is an essential component of this pathological cascade. Although it is believed that Casp2 exerts its effects by hydrolyzing tau at aspartate-314, forming Δtau314, it is also possible that a non-catalytic mechanism is involved, since catalytically dead Casp2 is biologically active in at least one relevant cellular pathway, i.e., autophagy. To decipher whether the pathological effects of Casp2 on synaptic function are due to its catalytic or non-catalytic properties, we discovered and characterized a new Casp2 inhibitor, compound 1 (pKi (Casp2) = 8.12), which is 123-fold selective versus Casp3 and >2000-fold selective versus Casp1, Casp6, Casp7, and Casp9. In an in vitro assay based on Casp2-mediated cleavage of tau, compound 1 blocked the production of Δtau314. Importantly, compound 1 prevented tau from accumulating excessively in dendritic spines and rescued excitatory neurotransmission in cultured primary rat hippocampal neurons expressing the P301S tau variant linked to FTDP-17, a familial tauopathy. These results support the further development of small molecule Casp2 inhibitors to treat synaptic deficits in tauopathies.
Keywords: Caspase-2, Alzheimer’s Disease, neurodegenerative disorders, tauopathies, dementia
Graphical Abstract
INTRODUCTION
Dementia is the most common clinical manifestation of tauopathies, defined as neurodegenerative diseases with tau histopathology. Tauopathies are the most prevalent form of neurodegenerative diseases. Tau is a neuronal cytoskeletal and scaffold protein best known for its ability to bind tubulin and stabilize microtubules; it is also known for forming pathological, hyper-phosphorylated cellular inclusions in over two dozen neurodegenerative disorders, called tauopathies, and it accumulates excessively in dendritic spines in multiple models of tauopathies1–7. In models of tauopathies expressing mutant human tau, this accumulation depends on Casp24 and tau phosphorylation8.
Caspases comprise a family of cysteine proteases found only in multicellular organisms. Some caspases (initiators) are activated by intra- or extracellular signals and cleave other caspases (executioners) which degrade vital structural proteins resulting in cell death. Other caspases both sense the triggers and implement the responses of the functions they serve. Caspase-2 (Casp2) belongs in the latter category, being capable of transducing a variety of incoming signals into proteolytic cascades9. In the brain, only two physiological roles for Casp2 are known. During development, Casp2 likely mediates neuronal apoptosis10,11. In the mature brain, Casp2 facilitates long-term depression, a synapse-weakening form of long-lasting synaptic plasticity12.
Casp2 also performs pathological roles. Lowering Casp2 levels restores memory function in mouse models of frontotemporal dementia4, Alzheimer’s disease13, and Huntington’s disease14. While it is assumed that lowering Casp2 rescues synaptic and cognitive deficits by reducing its ability to hydrolyze its substrates, one of which is believed to be tau4, it is also possible that it exerts its effects via a non-catalytic mechanism. For example, dead Casp2 has been shown to negatively regulate autophagy15. Therefore, another mechanism by which Casp2 might mediate cognitive deficits is by inhibiting autophagy, thereby causing misfolded proteins and pathological aggregates to accumulate in the brain.
We sought to resolve whether the pathological effects of Casp2 on synaptic function are due to its catalytic or non-catalytic properties using a Casp2 inhibitor. Because there have been no reports of a neuroactive, selective Casp2 inhibitor, we designed a new, reversible covalent Casp2 inhibitor based on published structure-activity relationships. Previous studies have shown that the placement of sterically demanding groups in the S2 subsite of the canonical pentapeptide structure increases the Casp2-selectivity of reversible, covalent inhibitors16. In addition, specific groups in the S4 and S5 subsites were shown to increase the selectivity of irreversible, covalent inhibitors of Casp217.
RESULTS AND DISCUSSION
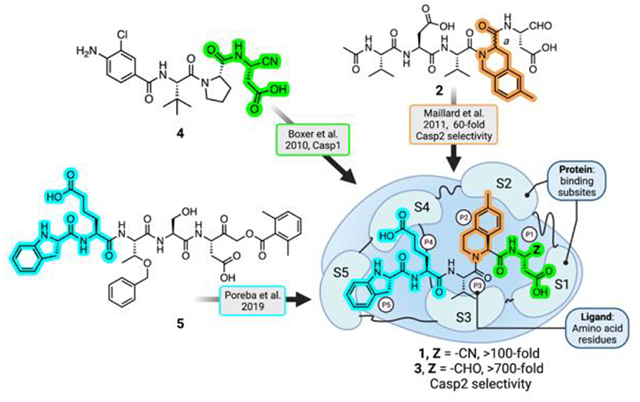
Design and characterization of Casp2 selective inhibitors
We prepared new, potent, and, reversible pentapeptide Casp2 inhibitors (Figure 1) by employing a rational-design strategy. That is, we combined the structural elements known to lead to selectivity and potency which had been identified in previous structure-activity-relationship (SAR) studies. In 2011, Maillard et al. reported a moderately selective Casp2 inhibitor 2 by introducing racemic 6-methyltetrahydroisoquinoline-1-carboxylic acid (6-Me-THIQ-COOH) at P2 position of the previously known non-selective Casp2 inhibitor AcVDVAD-CHO16. We prepared 2a and 2b (pure P2 epimers of compound 2) by employing enantiopure (R)-6-Me-1-THIQ-COOH and (S)-6-Me-1-THIQ-COOH, respectively (Figure 1). In a fluorescence-based in vitro biochemical assay18 compound 2a containing (R)-THIQ did not show any notable affinity for Casp2 and Casp3 (pKi (Casp2) = 4.98, pKi (Casp3) < 4; cf. Table 1). In contrast, 2b bearing (S)-THIQ showed significant affinity for Casp2 with 70-fold Casp2/Casp3 selectivity (pKi (Casp2) = 7.89, pKi (Casp3) = 6.04; cf. Table 1).
Figure 1.
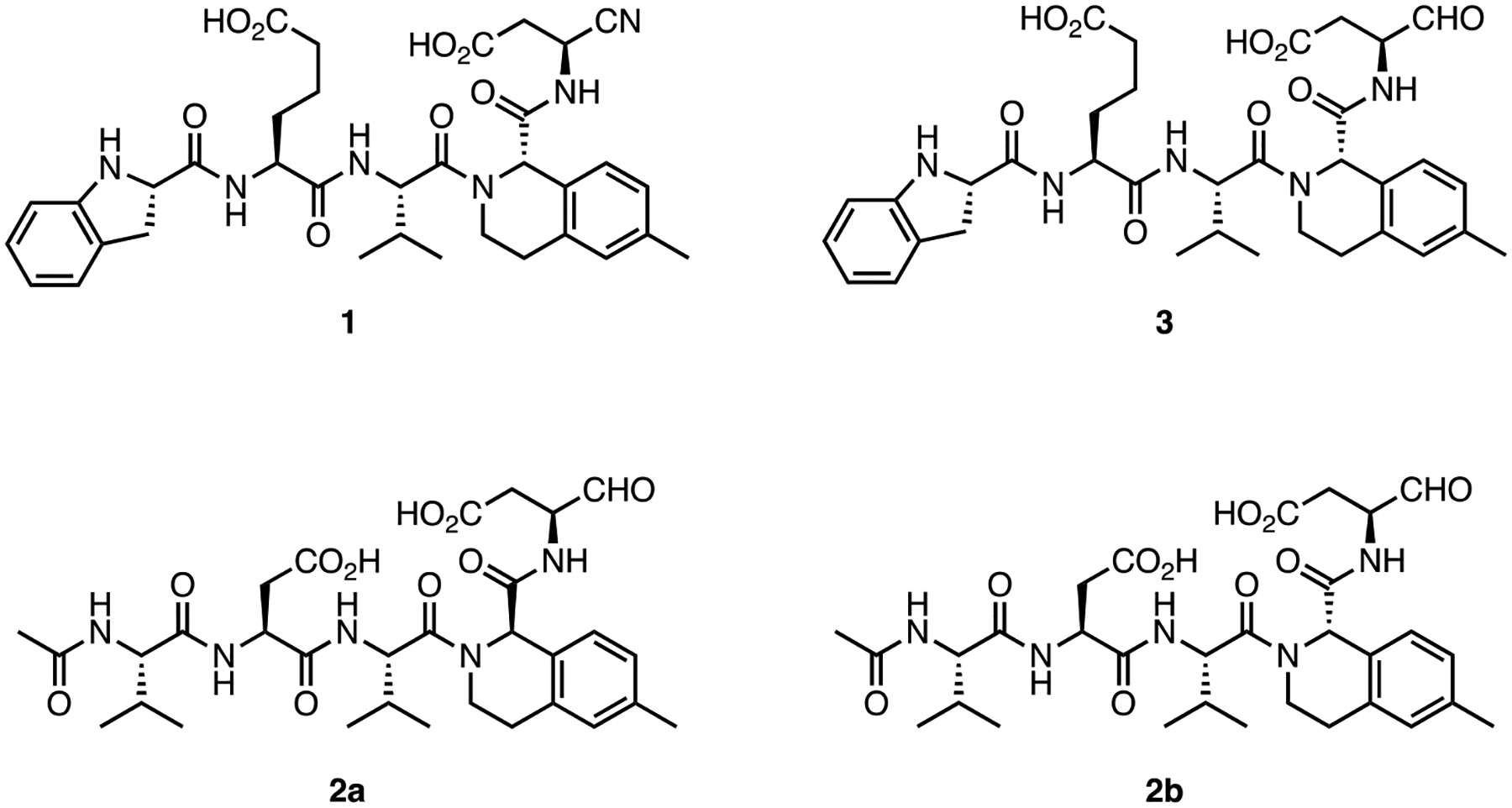
Structures of caspase-2 inhibitors 1, 2a, 2b, and 3.
Table 1.
Binding data (pKi values) of pentapeptides 1, 2a, 2b, and 3 at Casp2 and Casp3. Selectivity expressed as Ki (Casp3) / Ki (Casp2).
| Cmpd. | pKi ± SEM | Casp2 Selectivity |
|
|---|---|---|---|
| Casp 2 | Casp 3 | ||
| 1 | 8.12 ± 0.10 | 6.03 ± 0.06 | 123 |
| 2a | 4.98 ± 0.20 | < 4 | > 9 |
| 2b | 7.89 ± 0.02 | 6.04 ± 0.06 | 70 |
| 3 | 7.40 ± 0.11 | < 4.5 | 794 |
Data shown are mean values ± SEM of N ≥ 3 independent experiments, each performed in duplicate or triplicate. Data were analyzed by nonlinear regression and were best fitted to sigmoidal concentration−response curves using four parameter logistic (4PL) regression using GraphPad Prism.
Poreba et al. have shown that pentapeptides with an extended side chain carboxylic acid-based residue at P4 (h-L-glutamic acid) and a cyclic non-acylated amino acid at P5 (indoline-2(S)-carboxylic acid), show significant affinity and selectivity for Casp217. Taking (S)-THIQ-based 2b as our lead structure, we further replaced P4 L-aspartic acid moiety with h-L-glutamic acid and introduced (S)-indoline-2-carboxylic acid in place of N-acetyl-L-valine at P5. These modifications resulted in compound 3 with 794-fold Casp2 selectivity versus Casp3 (Figure 1; Table 1). Reversible peptide- and peptidomimetic protease inhibitors featuring aldehyde (-CHO) reactive groups are popular for early SAR studies since they are readily prepared by solid-phase synthesis and used in known drugs. Notably, the -CHO warhead is a prominent feature of the recently approved drug voxelotor19, a treatment for sickle cell anemia that is dosed at 1.5 g QD. Typically, however, the R-CHO reactive group is seen as undesirable due to its high electrophilicity and its susceptibility to oxidative metabolism. On the other hand, nitrile-based covalent inhibitors are also clinically validated and known for their safety and efficacy in humans (20, a covalent inhibitor of the SARS-CoV-2 main protease) and have been used successfully in inhibitors of other caspases21. Therefore, we prepared 1, the nitrile analog of 3, using solution-phase chemistry (Scheme 1, Figure 1). This compound was fully characterized and used in all subsequent experiments. Compound 1 showed high-affinity at Casp2 (pKi = 8.12; cf. Table 1), a promising selectivity profile in an in vitro caspase panel assay (123-fold selective versus Casp3 and >2000-fold selective versus Casp1, 6, 7, 9; cf. Table 2) and, evinced only weak activity at thrombin and cathepsin B proteolytic enzymes (See Table 2).
Scheme 1.
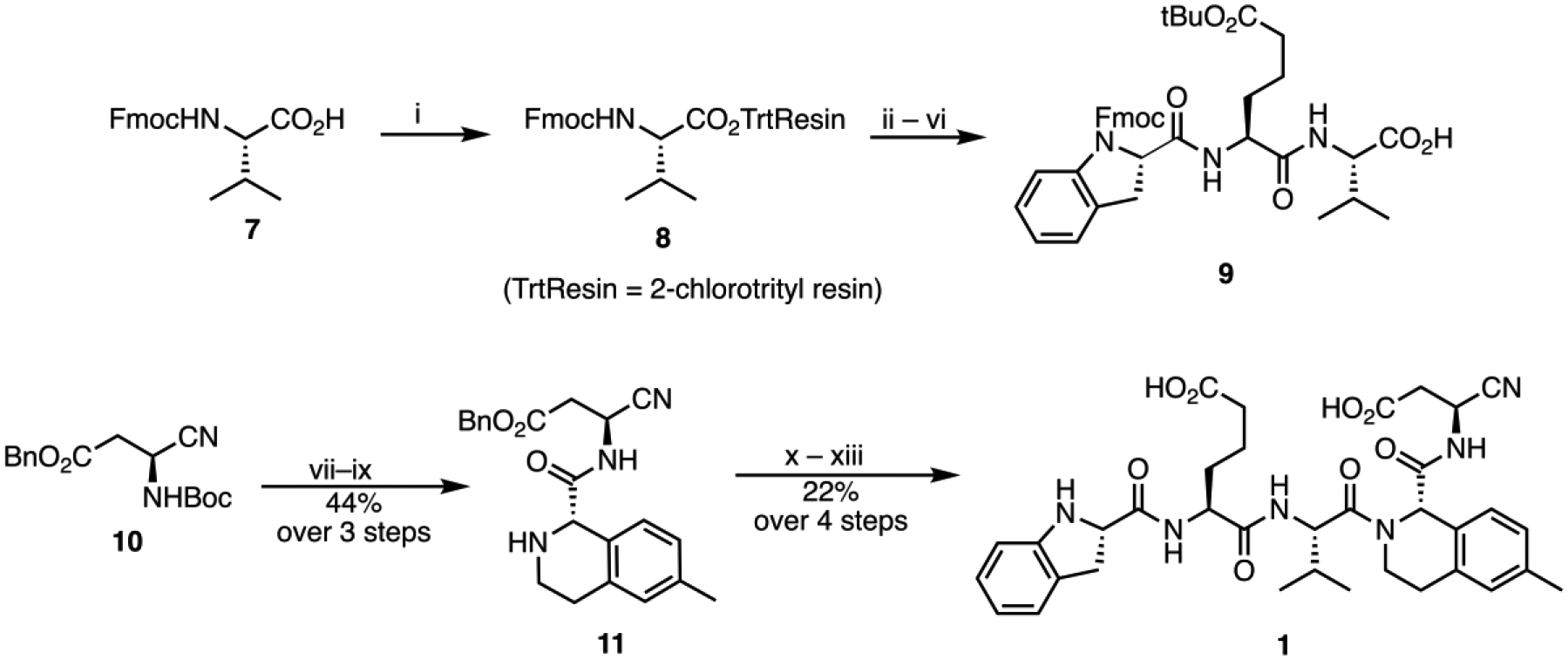
Synthesis of caspase-2 inhibitor 1. (i) 2-chlorotrityl chloride resin, collidine, DCM; (ii) 20% piperidine in DMF; (iii) Fmoc-hGlu(OtBu)-OH, HATU, DMF; (iv) 20% piperidine in DMF; (v) (S)-Fmoc-IdcOH, HATU, DMF; (vi) 20% HFIP in DCM; (vii) H3PO4; (viii) (S)-6, HATU, collidine, ACN; (ix) 30% Diethylamine in ACN; (x) 9, HATU, collidine; (xi) H3PO4; (xii) 30% Diethylamine in ACN; (xiii) 10% Pd/C.
Table 2.
Binding data of compounds 1, 2a, 2b, and 3 at Casp1, Casp2, Casp3, Casp6, Casp7, Casp9 (pKi values), Cathepsin B and Thrombin a (IC50 values in nM).
| Target | pKi ± SEM | |||
|---|---|---|---|---|
| 1 | 2a | 2b | 3 | |
| Casp1 | < 4 | < 4 | 5.56 ± 0.07 | 6.41 ± 0.12 |
| Casp2a | 8.12 ± 0.10 | 4.98 ± 0.20 | 7.89 ± 0.02 | 7.40 ± 0.11 |
| Casp3a | 6.03 ± 0.06 | < 4 | 6.04 ± 0.06 | < 4.5 |
| Casp6 | < 4 | < 4 | < 4 | < 4 |
| Casp7 | 4.80 ± 0.06 | < 4 | < 4.5 | < 4 |
| Casp9 | < 4.5 | < 4 | < 4.5 | 5.12 ± 0.04 |
| Cathepsin B | 18,620b | n.d. | 38,000b | 14,300b |
| Thrombin a | > 100,000b | n.d. | n.d. | n.d. |
Data shown are mean values ± SEM of N ≥ 2 independent experiments, each performed in duplicate or triplicate. Data were analyzed by nonlinear regression and were best fitted to sigmoidal concentration−response curves using four parameter logistic (4PL) regression using GraphPad Prism.
Data at Casp2 and Casp3 are also shown in Table 1.
Data shown are IC50 values (nM) of N = 1 independent experiments.
Casp2-selective inhibitors block enzyme-catalyzed tau cleavage at D314
To understand the effects of Casp2-selective inhibitors on enzyme-catalyzed site-specific cleavage of tau, a naturally occurring substrate in the brain, we performed in vitro Casp2 cleavage assays using purified recombinant proteins. The presence of Δtau314, the soluble truncated tau fragment ending C-terminally at D3144, was verified by mass spectrometry (MS) (Figure 2A). Both Casp2-selective compounds, 1 and 2b, inhibited enzyme-catalyzed Δtau314 production at 10 μM but with different efficacies (96% for 1 versus 68% for 2b). In contrast, 2a, the epimer of 2b, did not inhibit Δtau314 production at 10 μM (Figure 2B). 1 inhibited tau-cleavage at D314 with an IC50 of 2.02 μM (Figure 2C).
Figure 2.
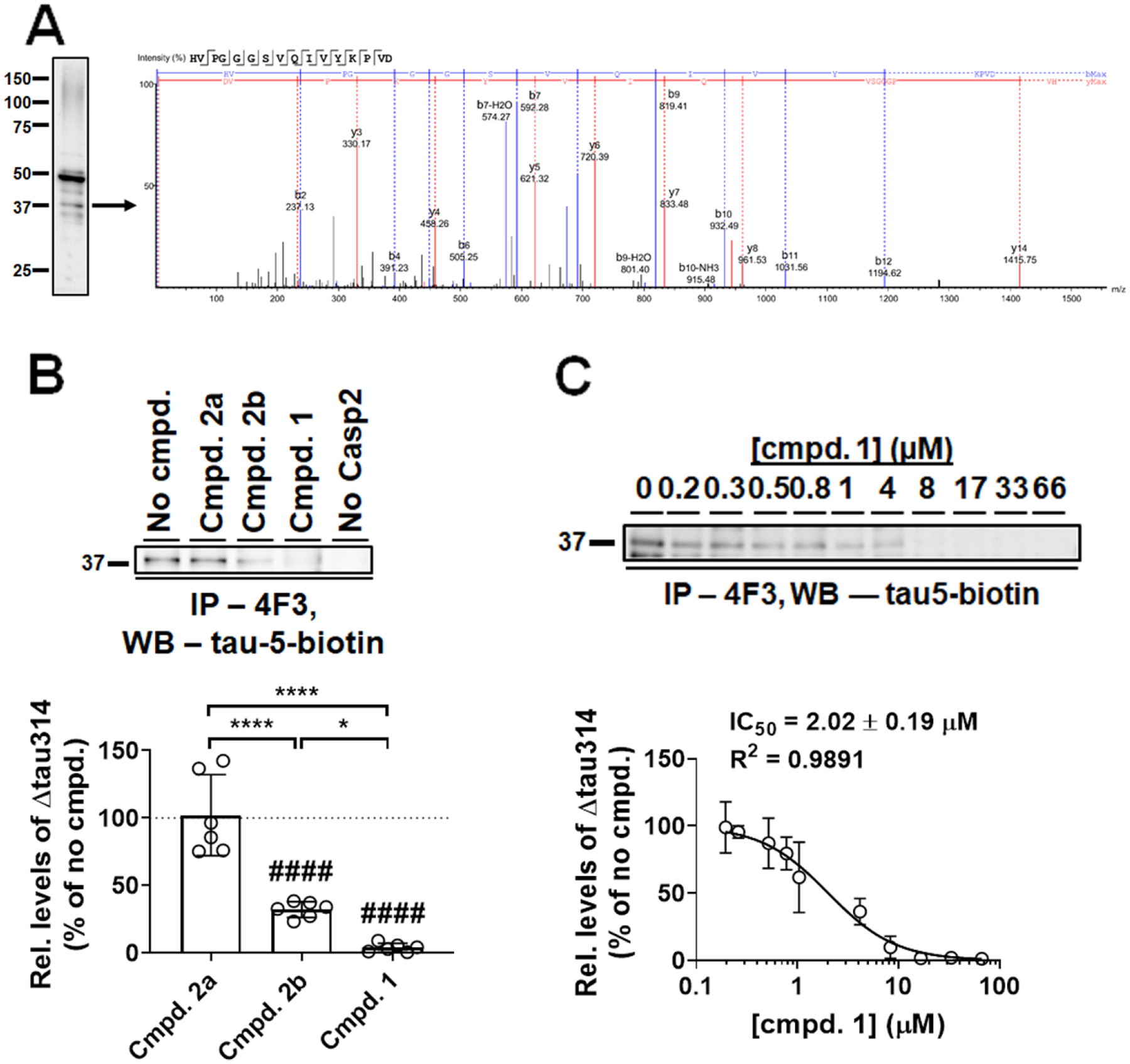
Casp2–selective inhibitors block enzyme-catalyzed Δtau314 production in vitro A) Mass spectrometry (MS) analysis confirmed the identity of a ~37-kDa tau cleavage product as Δtau314. A representative Western blot (WB) of Casp2-cleaved tau proteins immunoprecipitated by the Δtau314-specific 4F3 antibody was probed using biotin-conjugated pan-tau antibody tau-5 (tau-5-biotin). Liquid chromatography-MS/MS analysis identified the presence of Δtau314 in the ~37 kDa product as indicated by a representative MS spectrum of its trypsinized signature peptide at the C-terminus. B) Hybrid compounds inhibited Casp2-mediated Δtau314 production with different efficacies. Upper panel, a representative immunoprecipitation (IP, 4F3)/Western blotting (WB, tau-5-biotin) showing Δtau314 in the presence of various hybrid compounds at 10 μM. Δtau314 produced in the absence of any compound (No cmpd.) and a reaction in the absence of enzyme (No Casp2) served as positive and negative controls, respectively. Lower panel, quantification. The level of Δtau314 produced in the absence of any tested compound was defined as 100% (dotted line). At 10 μM, compounds 1 and 2b inhibited tau cleavage at D314 by 96% and 68%, respectively, whereas compound 2a showed no inhibition; compound 1 inhibited Δtau314 production more efficiently than compound 2b. Experiments were repeated six times. Individual values (open circles), means (histograms), and standard deviations (SDs, error bars) are shown. A single-sample t-test was performed to compare the effect of each compound to that of no compound (####, p < 0.0001). One-way ANOVA was performed to compare effects of tested compounds (F(2, 15) = 48.50, P < 0.0001), followed by Tukey’s post hoc test (*, p < 0.05; ****, p < 0.0001). C) Determining the IC50 of compound 1. Upper panel, a representative IP (4F3)/WB (tau-5-biotin) showing Δtau314 in the presence of various concentrations of compound 1. Lower panel, levels of Δtau314 (normalized to the no-compound control) and compound concentrations were fitted to a dose-response curve; the IC50 of compound 1 was determined to be 2.02 ± 0.19 μM. Experiments were performed in duplicate. Means (open circles) and SDs (error bars) are shown.
Compound 1 prevents the disproportionate accumulation of P301S tau in dendritic spines of cultured rat hippocampal neurons
The disproportionate accumulation in dendritic spines of mutant tau linked to FTDP-17 depends upon tau-truncation by Casp24. As shown in Figure 2, of the compounds tested the one that blocks tau-truncation most effectively is 1. Here, we examined the P301S tau mutation linked to FTDP-176,22. To determine whether 1 can prevent the accumulation of tau in dendritic spines, we cultured dissociated hippocampal neurons from postnatal day 0–1 rat pups. After 5–7 days in vitro (DIV), we co-transfected the neurons with plasmids encoding DsRed (to label neuronal morphology) and GFP-tagged wild-type or P301S mutant tau proteins (Figure 3A), as previously described1. Neurons at 17–18 DIV were incubated with 10 μM compound 1 for 3 days and live neurons were imaged at 20–21 DIV.
Figure 3.
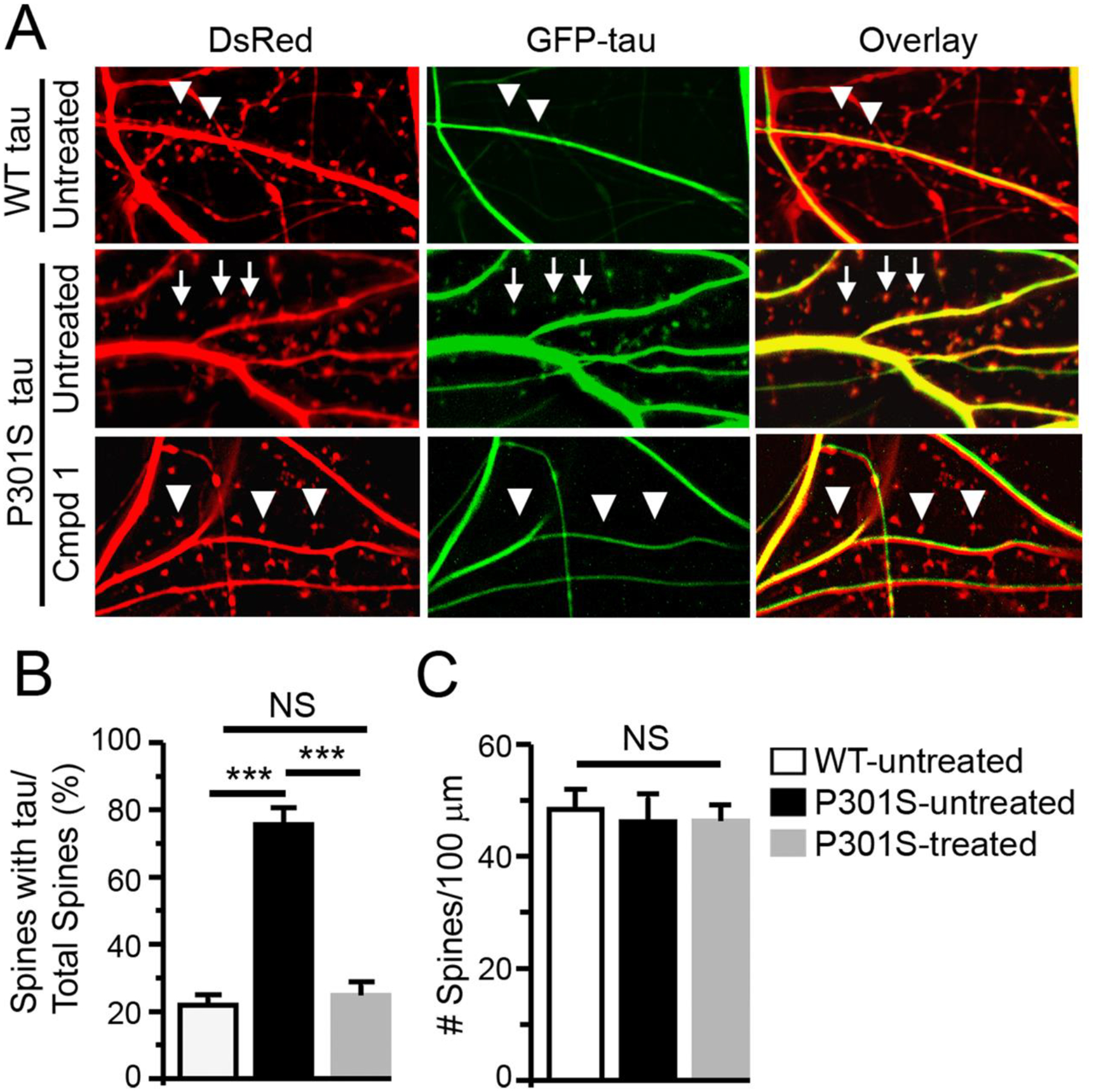
Compound 1 blocks the hyper-accumulation of tau caused by the P301S mutation in dendritic spines. A) Untreated neurons co-expressing DsRed and GFP-tagged wild-type tau (top row) were compared with neurons co-expressing DsRed and GFP-tagged P301S tau in the absence (middle row) or presence of compound 1 (bottom row). Arrows denote dendritic spines containing tau. Triangles denote spines devoid of tau. B) Comparison of the average percentage of dendritic spines labeled by DsRed containing GFP-tagged tau between the three groups. n = 8 neurons in each group; ANOVA, ***, p < 0.001. C) Comparison of the density of dendritic spines (the number of DsRed-labelled dendritic spines/100 μm length of dendrites) between the three groups. n = 8 neurons in each group; ANOVA, NS (not significant), P > 0.05.
As shown in Figure 3A, in neurons co-expressing DsRed and wild-type tau, relatively few dendritic spines contain GFP-tagged tau proteins (top row). In contrast, in neurons co-expressing DsRed and GFP-tagged P301S tau, most of the dendritic spines contain GFP-tagged tau (middle row). When 1 was applied to neurons expressing GFP-tagged P301S tau, a minority of dendritic spines contain GFP-tagged tau (bottom row). The percentage of dendritic spines containing GFP-tagged tau proteins in neurons significantly increased from 21.9% in neurons expressing wild-type tau to 75.5% in neurons expressing P301S tau; 1 reduced the percentage to 24.8% (Figure 3B; n=8 neurons in each group; ANOVA, F = 82.1; Bonferroni post-hoc test: P < 0.01 comparing untreated wild-type and P301S tau, and then comparing untreated and treated P301S tau). There were no significant differences in the density of dendritic spines between the three groups (Figure 3C; ANOVA, F = 0.13, P>0.05), indicating that neither the P301S mutation nor 1 caused loss of dendritic spines. Overall, these results indicate that 1 blocks the excessive accumulation of tau induced by the P301S mutation in dendritic spines, supporting its efficacy in reversing the molecular pathology of dendritic spines in a cellular model of tauopathy.
Compound 1 rescues P301S tau-induced functional deficits in dendritic spines
Our previous studies in models of several different tauopathies demonstrated that the excessive accumulation of tau in dendritic spines is associated with a reduction in postsynaptic excitatory neurotransmission due to the internalization of AMPA receptors1–5,7,8. Therefore, we compared the ability of 1, 2a, and 2b to rescue functional synaptic deficits in a cellular model of tauopathy.
For these studies, we used PS19 transgenic mice expressing P301S mutant tau, one of the most used models of FTDP-17. To detect functional deficits, we cultured primary hippocampal neurons from heterozygous transgenic mice overexpressing P301S tau23,24 and littermate controls, (denoted Tau −/+ and Tau −/− respectively in Figure 4), and recorded miniature excitatory synaptic currents (mEPSCs). 1, 2a (inactive analog of 2b), or 2b (final concentrations = 10 μM) were directly applied to the culture media (Figure 4A) and incubated for 3 days. In the two groups treated with 2a, the average amplitude of mEPSC responses in Tau −/+ neurons is significantly smaller than that in Tau −/− neurons (Figure 4B, ANOVA, F = 3.3; Bonferroni post hoc test: P < 0.05), indicating that the expression of P301S tau impaired postsynaptic function of the excitatory synapses by reducing the number of functional AMPA receptors on post-synaptic membranes. In contrast, the average amplitudes of mEPSC responses in Tau −/+ and Tau −/− neurons treated with 1 do not differ significantly, suggesting that 1 rescues impaired mEPSC amplitudes (ANOVA, F = 3.3, P>0.05). The average amplitudes of mEPSC responses of Tau −/+ neurons treated with 1 are significantly larger than those treated with 2a (Figure 4B, F = 3.3; Bonferroni After Test: P < 0.01) or 2b (Figure 4B, F = 3.3; Bonferroni post-hoc test: P < 0.05), indicating that 1 rescues post-synaptic neurotransmission more effectively than 2a or 2b. There were no significant differences in the average frequency of mEPSCs between the six groups (Figure 4C, ANOVA, F = 0.6, P > 0.05), indicating no changes in presynaptic function.
Figure 4.
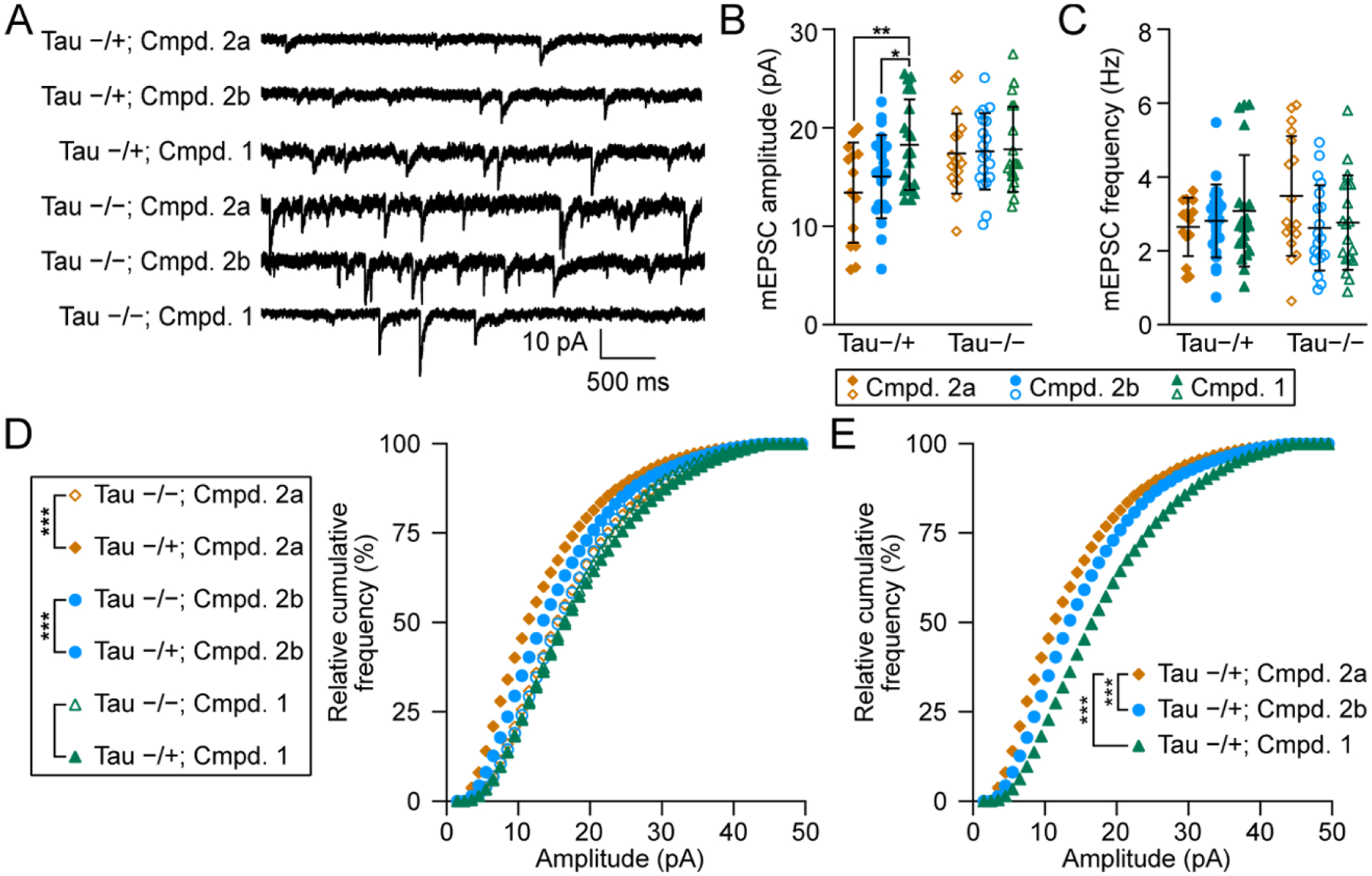
Treatment with 1 rescue synaptic dysfunction caused by the P301S mutation. A. Representative traces of mEPSCs recorded in Tau−/+ and Tau−/− neurons that have been treated with 2a, 2b, and 1. B and C. Comparisons between the means of mEPSC amplitude (B) or frequency (C) between the above six groups. n = 31 neurons in Compound 1 treated groups (−/− and −/+); n = 41 in Compound 2b treated groups; n = 39 in Compound 2a treated groups; ANOVA, *, p < 0.05; **, p < 0.01. D. Comparisons of cumulative curves of mEPSC amplitudes recorded in the above six groups. E. A comparison between the rescuing efficacy between 1 and 2b in Tau −/+ neurons. For D and E: Mann-Whitney U Test, *, p < 0.05; ***, p, 0.001. For B-E, orange circles, blue circles, and blue triangles denote treatment with 2a, 2b, and 1, respectively. Closed shapes denote Tau−/+ mice and open shapes denote Tau−/− mice.
To confirm and extend the results in Figure 4B, we analyzed the cumulative curves of mEPSC amplitudes. The cumulative curve of mEPSC amplitudes of 2a-treated neurons from Tau −/+ compared with Tau −/− mice is shifted to the left (Figure 4D, Mann-Whitney U test: P < 0.001), indicating that the P301S mutation decreases the amplitudes of mEPSCs. The cumulative curve of mEPSC amplitudes of 2b-treated neurons from Tau −/+ compared with Tau −/− mice is shifted to the left but less markedly (Figure 4D, Mann-Whitney U test: P < 0.001), indicating that 2b partially blocks the synaptic impairment caused by the P301S mutation. In contrast, the cumulative curve of mEPSC amplitudes of 1-treated neurons from Tau −/+ compared with Tau −/− mice is not significantly shifted (Figure 4D, Mann-Whitney U test: P > 0.05), which indicates that 1 completely blocks the synaptic impairment caused by the P301S mutation. Comparison of the cumulative curves of Tau −/+ neurons treated with each of the three compounds indicates that 1 shifted the cumulative curve of mEPSC amplitudes more to the right than 2b, further supporting that 1 is the more effective compound (Figure 4E, Mann-Whitney U test: P < 0.001).
Discussion and Summary
We designed and prepared 1, a new, potent, and selective Casp2 inhibitor, and found that it is significantly more effective than 2b, the P2 pure epimer of the previously reported Casp2 inhibitor 2, in restoring excitatory postsynaptic neurotransmission in a cellular model of tauopathy. We designed 1 with the following features in mind: potency at Casp2, selectivity at Casp2 versus Casp3 (discussed in Maillard, et al.16), and reversibility of the covalent reactive group (CRG), anticipating that we could fine-tune the warhead in later optimization work. Additionally, we sought physicochemical properties that were consistent with cell entry and an overall structure that contained moieties that would allow more careful optimization in future structure-activity studies. Thus, we incorporated several known selectivity elements in the design of 1 and were delighted to discover that our initial selection of features for the pentapeptide inhibitor25 yielded a potent, selective compound. While ultimately the Casp2/Casp3 selectivity of 3 (CRG = -CHO) was several-fold greater than that of 1 (CRG = -CN), the 10-fold increase in potency offered by 1 outweighed its slightly diminished selectivity.
The first studies of Casp2 in the nervous system focused on its apoptotic roles in development10 and neurodegeneration caused by the withdrawal of trophic factors and the application of amyloid-β peptides26,27. The presence of Casp2 in post-mitotic neurons in the adult brain indicated that it presumably also performs non-apoptotic roles, but it was not until quite recently that its role in the endocytosis of AMPA receptors in response to the expression of tau variants was discovered4. Subsequently, Xu et al. demonstrated the requirement of Casp2 for the endocytosis of AMPA receptors in response to the induction of long-term depression12, echoing its pathological effects in tauopathy.
Although it was the search for genes mediating apoptosis that led to the discovery of caspases28, most of the functions that the family of caspases perform are non-apoptotic9. Casp2 is unusual in that it can mediate some non-apoptotic processes through a non-catalytic mechanism. For example, dead Casp2 mutated at its catalytic cysteine negatively regulates autophagy15. The precise mechanism by which this occurs is unknown. One possibility involves the role of catalytically dead Casp2 as a translational co-factor to produce cyclin-dependent kinase inhibitor p2129. p21, in turn, has been shown to negatively regulate autophagy in various cell types under different stress conditions30,31. Therefore, it is possible that lowering the levels of Casp2, which was how Zhao et al. demonstrated its role in tauopathy4, might have rescued neurotransmission by non-catalytically enhancing autophagy and thereby lowering the levels of misfolded and aggregated forms of tau. Distinguishing between a catalytic versus non-catalytic mechanism underlying the pathological effects of Casp2 had not been done until the present studies. It is important to eliminate the possibility that its effects were non-catalytic, to solidify the rationale for developing small-molecule Casp2 inhibitors. Our current results, in combination with previous studies4, clearly and definitively demonstrate that Casp2 exerts its pathological effects on synaptic function catalytically, by hydrolyzing tau at aspartate-314.
Compound 1 rescues tau-induced synaptic dysfunction by blocking the excessive accumulation of tau in dendritic spines (Figures 3 and 4). This mechanism has been shown to mediate synaptic dysfunction repeatedly in models of tauopathy ranging from Alzheimer’s disease, Lewy body dementia, and FTDP-17 to chronic traumatic encephalopathy1–5,7,8. The dysfunctional process begins when tau redistributes from axons to dendrites in response to various pathological triggers32,33. Our current working model stipulates that the phosphorylation of serine-396 or serine-404 in the C-terminal tail of tau by GSK3β and/or CDK5 concomitantly with the cleavage of tau by Casp2 at aspartate-314 in the third microtubule domain4,5,8 causes tau to accumulate in dendritic spines. A minority of dendritic spines normally contains tau34, but under pathological conditions tau is found in the vast majority of dendritic spines. This occurs presumably because non-physiological, pathological triggers activate or elevate the levels of Casp2 and relevant kinases. The presence of tau in dendritic spines stimulates calcineurin-dependent endocytosis of AMPARs, but only if one or more of five specific residues in the proline-rich domain of tau are phosphorylated1,2,5.
Elevated levels of Δtau314 and Casp2 found in brain specimens of individuals with Alzheimer’s disease4,35, Lewy body dementia36, and Huntington’s disease37 underscore the relevance of the Casp2-tau signaling pathway in tauopathies. The results reported here using 1 establish the feasibility of using a small-molecule Casp2 inhibitor to restore excitatory neurotransmission and justify efforts to develop brain-penetrant Casp2 inhibitors to treat patients with dementia due to tauopathies.
Based on structural attributes of the previously reported Casp2 inhibitors, we identified a reversible Casp2 inhibitor 1 with an excellent selectivity profile. Subsequently, we confirmed in an in vitro assay that 1 effectively inhibits Casp2-mediated cleavage of tau and blocks the production of Δtau314, the N-terminal truncation product formed by hydrolysis at aspartate-314. Using cultured rat and mouse hippocampal neurons expressing P301S tau, we showed that 1 prevents the disproportionate accumulation of P301S tau in dendritic spines. Finally, we showed that the expression of P301S tau impairs postsynaptic function of excitatory synapses, determined by recording miniature excitatory synaptic currents (mEPSCs), and that 1 completely normalizes the mEPSCs and rescues synaptic function.
METHODS AND MATERIALS
General Methods and Materials (Chemistry)
All solution phase experiments were carried out under nitrogen in oven-dried glassware. All starting materials, solvents, and reagents were purchased from commercial sources and used without further purification. Aspartic acid-derived semi-carbazide Merrifield resin was prepared by using the previously reported procedure38. The intermediates were purified by flash chromatography using 230–400 mesh silica gel. 1H NMR spectra were recorded on Bruker 400 MHz spectrometer in deuterated solvents. Final compounds for the biological assays were purified by preparative HPLC. Chemical shifts are reported in parts per million (ppm). Final compounds 1 and 3 were purified by preparative-HPLC, utilizing a Waters Atlantis Zoraba × SB C18 column, gradient 0–100% B (A = water, B = acetonitrile) over 18 min, injection volume 1 mL, flow = 20 ml/min. UV spectra were recorded at 215 nm using a Gilson 119 UV/vis detector system. LCMS purity was determined on Acquity UPLC (Waters Corporation) equipped with an Acquity BEH UPLC C18 column (2.1 × 50 mm, 1.7 μm) for separation. Compound absorbance was detected at 214 nm using a photo diode array detector. Mass data was acquired using a Micromass ZQ mass spectrometer. LC was completed using a gradient method with each of the following mobile phase systems: Mobile Phase A2: 95% Water, 5% Acetonitrile, 0.1% Formic Acid; Mobile Phase B2: 95% Acetonitrile, 5% Water, 0.1% Formic Acid; Mobile Phase A1: 95% Water, 5% Methanol, 0.1% Formic Acid; Mobile Phase B1: 95% Methanol, 5% Water, 0.1% Formic Acid. Analytical HPLC-MS was performed on Shimadzu LCMS-2010 EV systems using reverse phase Atlantis C18 columns, gradient 5–100% B (A = water/0.1% formic acid, B = acetonitrile/0.1% formic acid) over 6 min, flow = 0.6 mL/min. Mass spectra were obtained over the range m/z 150–850 at a sampling rate of 2 scans per second using Waters LCT or analytical HPLC-MS systems using reverse phase Water Atlantis C18 columns gradient 5–100% B (A = water/0.1% formic acid, B = acetonitrile/0.1% formic acid).
For the compounds 2a and 2b, preparative HPLC was performed on a Knauer system (consisting of two K-1800 pumps, a K-2001 detector) using a Phenomenex Gemini C18 column (250 × 21 mm, 5 μm) at a flow rate of 15 mL/min. As a mobile phase, mixtures of MeCN and 0.1% TFA in water were used. The UV detection was carried out at 220 nm. Purity analysis of the compounds 2a and 2b was performed on an Agilent 1100 HPLC system (equipped with an Instant Pilot controller, a G1312A binary pump, a G1329A ALS autosampler, a G1379A vacuum degasser, a G1316A column compartment, and a G1315B diode array detector) using a Phenomenex Kinetex XB-C18 column (250 × 4.6 mm, 5 μm) at 30 °C oven temperature using mixtures of MeCN and 0.1% TFA in water as mobile phase. Absorbance was detected at 220 nm. Purity control for 2a and 2b was performed as recently reported by Bresinsky et al.18.
Racemic 6-methyl-tetrahydroisoquinoline-1-carboxylic acid (6-Me-1-THIQCOOH) was obtained from a commercial source and was outsourced for chiral resolution by supercritical fluid chromatography (SFC) (Figure S1). The enantiopure amino acids P1 and P2 were subjected to N-Fmoc protection to obtain (S)-6 and (R)-6 respectively (the absolute configurations of (S)-6 and (R)-6 were determined by comparison of the sign of the reported specific rotation of the commercially available enantiopure N-Fmoc-tetrahydroisoquinoline-1-carboxylic acids. Compounds 2a, 2b, and 3 were prepared via solid-phase peptide synthesis using aspartic acid modified semi-carbazide resin38 as described by Maillard and Bresinsky16,18.
Experimental data of compound 1 and 2b
(S)-6-(((S)-1-((S)-1-(((S)-2-carboxy-1-cyanoethyl)carbamoyl)-6-methyl-3,4-dihydroisoquinolin-2(1H)-yl)-3-methyl-1-oxobutan-2-yl)amino)-5-((S)-indoline-2-carboxamido)-6-oxohexanoic acid (1).
To a solution of tripeptide carboxylic acid 9 (267 mg, 0.39 mmol) in 5 mL acetonitrile were added HATU (102 mg, 0.39), 2,4,6-collidine (1.56 mmol, 0.21 mL), and 11 (100 mg, 0.26 mmol). The reaction mixture was stirred at rt for 6 h and the solvent was evaporated. The crude residue was diluted with water (20 mL) and extracted with ethyl acetate (4 × 20 mL). The combined organic layer was sequentially washed with sat. aq. NaHCO3 (20 mL), ice-cold 10% aq. KHSO4 (20 mL), and brine (20 mL). The organic layer was collected over anhyd. Na2SO4, filtered, and concentrated under reduced pressure to obtain the crude peptide. The crude residue was dissolved in a minimum volume of dichloromethane (3 mL) and transferred to vigorously stirring phosphoric acid (85%, 3 mL). The completion of the reaction was monitored by UPLC-MS. The reaction mixture was diluted with water (5 mL) and extracted with ethyl acetate (4 × 10 mL). The organic layer was dried over anhyd. Na2SO4, filtered, and concentrated to obtain the crude carboxylic acid. The crude product was dissolved in acetonitrile (10 mL) and diethyl amine (3 mL) was added to it. The reaction mixture was stirred at rt for 3 h and was concentrated under reduced pressure. The crude residue was triturated sequentially with hexane, diethyl ether, and the solvent decanted. The residue was dried under reduced pressure and dissolved in DMF (3 mL). Palladium (10%, 10 mg) was added, and the reaction mixture was purged with hydrogen gas for 5 minutes and stirred under hydrogen (1 atm). The completion of the reaction was monitored by UPLC-MS. The reaction mixture was filtered over a thin layer of celite. The celite layer was washed with 2 mL of DMF. The solvent was evaporated and the residue was purified by preparative HPLC. Lyophilization of the pure fractions provided the product 1 as a colorless amorphous powder (38 mg, 22% yield). 1H NMR (400 MHz, MeOD) δ 9.02 − 8.72 (m, 0.5H), 8.43 (m, 0.7H), 8.13 (m, 1H), 7.64 − 7.29 (m, 1H), 7.25 − 6.43 (m, 7H), 5.58 (s, 0.6H), 5.40 (s, 0.4H), 4.63 − 4.15 (m, 3H), 4.07 − 3.83 (m, 1H), 3.81 − 3.61 (m, 1H), 3.60 − 3.3 (m, 1H), 3.13 − 2.45 (m, 5H), 2.38 − 1.96 (m, 5H), 1.95 − 1.08 (m, 5H), 0.99 − 0.67 (m, 5H), 0.55 (d, J = 6.8 Hz 1H). HRMS (ESI) m/z [M+H+] calculated for C35H43N6O8+: 675.3137, found 675.3. HPLC purity 99%.
(S)-3-((S)-2-acetamido-3-methylbutanamido)-4-(((S)-1-((S)-1-(((S)-1-carboxy-3-oxopropan-2-yl)carbamoyl)-6-methyl-3,4-dihydroisoquinolin-2(1H)-yl)-3-methyl-1-oxobutan-2-yl)amino)-4-oxobutanoic acid (2b)
The title compound was synthesized according to the general procedure described from Bresinsky et al.3 using 300 mg Asp-resin (0.188 mmol) and yielding a fluffy white solid (43 mg, 36% yield). 1H NMR (400 MHz, D6-DMSO) δ9.51 − 8.73 (m, 1H), 8.29 − 8.22 (m, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.73 − 7.60 (m, 1H), 7.47 − 7.16 (m, 1H), 7.09 − 6.92 (m, 2H), 5.97 − 5.16 (m, 2H), 4.80 − 3.33 (m, 6H), 3.06 − 2.43 (m, 5H), 2.75 (s, 3H), 2.10 − 1.876 (m, 2H), 1.85 (s, 3H), 0.97 − 0.58 (m, 12H). HRMS (ESI) m/z [M+H+] calculated for C31H44N5O10+: 646.3083, found 646.3096. HPLC purity >99%.
Animal Care and Usage
Rat and mouse pups in postnatal day 0 or 1 were harvested to make primary hippocampal cultures. Sprague-Dawley timed-pregnancy adult rats were housed in facilities of Research Animal Resources (RAR) at the University of Minnesota (UMN) after being delivered from Envigo (www.envigo.com). The rats were fed a diet of regular chow before giving birth. Rat pups were euthanized by decapitation to harvest brain tissues in postnatal day 0 or 1. The appropriate transgenic mice were also housed and bred in RAR facilities at UMN. All work was conducted in accordance with the American Association for the Accreditation of Laboratory Animal Care and Institutional Animal Care and Use Committee (IACUC) at the University of Minnesota (protocol #1211A23505). We performed all procedures of euthanasia strictly according to the guidelines of the IACUC at the UMN.
Materials (Biology)
All common chemical reagents and cell culture supplies were purchased from Sigma-Aldrich, Fisher Scientific, Promega, and Thermo-Fisher Scientific/Invitrogen/Life Technologies unless otherwise indicated.
In vitro caspase-2-catalyzed tau cleavage assay
Purified recombinant Casp2 (final concentration during pre-incubation: 67 μM) was pre-incubated with various concentrations of inhibitory compounds at 4 °C for 72 h. Pre-treated enzyme was then incubated with purified recombinant tau at a molar ratio of 1:1 in 37 °C water bath, 1x reaction buffer (25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 0.1% (w/v) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 10 mM dithiothreitol (DTT), pH 7.5) for 7 h. The final volume of each reaction was 100 μL. At the end of the 7-h incubation, 0.1% (v/v) protease inhibitor cocktail (MilliporeSigma) was added to stop the reactions.
Immunoprecipitation (IP) / Western blotting (WB)
Immediately after the reaction was stopped, the 100 μL enzyme-product mixture was diluted in 400 μL IP buffer (50 mM Tris-HCl (pH 7.4) and 150 mM NaCl, containing 0.1 mM phenylmethylsulfonyl fluoride, 0.2 mM 1,10-phenanthroline monohydrate, and protease inhibitor cocktail (MilliporeSigma)), and then incubated with 10 μg of Δtau314-specific monoclonal antibody 4F3 and 50 μL of Protein G Sepharose 4 Fast Flow resin (GE Healthcare) at 4 °C for 14–16 h. Subsequent resin wash and protein elution were performed as described previously39. WB was performed according to a previously published protocol37.
Mass spectrometry (MS)
In-gel trypsin digestion, liquid chromatography-MS/MS, mass spectral database search, and data interpretation were performed as previously described35, except that Peaks Studio Xpro (Bioinformatics Solutions, Inc, Waterloo, Ontario, Canada) was used for interpretation of mass spectra.
Electrophysiology
Miniature excitatory postsynaptic currents (mEPSCs) were recorded from cultured dissociated mouse hippocampal neurons at 17–21 DIV with a glass pipette (resistance ~ 5 MΩ) at holding potentials of −65 mV on an Axopatch 200B amplifier (output gain = 0.5; filtered at 1 kHz) as we previously described5. Input and series resistances were assessed and found to have no significant difference before and after recording time (5–20 mins). Recording sweeps lasted 200 ms and were sampled for every 1 s (pClamp, v10, RRID:SCR_011323). Neurons were bathed in bubble-oxygenated artificial cerebrospinal fluid (ACSF) at 23 °C with 100 μM APV (NMDA receptor antagonist), 1 μM TTX (sodium channel blocker), and 100 μM picrotoxin (GABAa receptor antagonist). Passive oxygen perfusion was established with medical-grade 95% O2–5% CO2. ACSF contained (in mM) 119 NaCl, 2.5 KCl, 5.0 CaCl2, 2.5 MgCl2, 26.2 NaHCO3, 1 NaH2PO4, and 11 D-glucose. The internal solution of the glass pipettes contained (in mM) 100 cesium gluconate, 0.2 EGTA, 0.5 MgCl2, 2 ATP, 0.3 GTP, and 40 HEPES. The pH of the internal solution was normalized to 7.2 with cesium hydroxide and diluted to a trace osmotic deficit in comparison to ACSF (~300 mOsm). All analysis of recordings was performed using an automated detection software suit (Clampfit, 11.0.3, Molecular Devices, San Jose, CA, USA). Minimum analysis parameters were set at greater than 1 min stable recording time and events with amplitudes greater than 3 pA and smaller than 40 pA were included. A mEPSC event was identified by using a template which included a distinct fast-rising depolarization and slow-decaying repolarization. Combined individual events were used to form relative cumulative frequency curves; whereas the means of all events from individual recordings were treated as single samples for further statistical analysis. Example traces were exported from Clampfit and live-traced, simplified, and united in vector editing software (Adobe Illustrator CS5 and Affinity Designer).
Supplementary Material
ACKNOWLEDGMENTS AND FUNDING SOURCES
This work was supported by the Tau Pipeline Enabling Program (T-PEP) of the Alzheimer’s Association and Rainwater Charitable Foundation, T-PEP-18-578206C (Ashe, PI), the Edward N. and Della L. Thome Memorial Foundation Awards Program in Alzheimer’s Disease Drug Discovery Research (Ashe, PI), NIH Grant R01AG0623199 (Ashe PI, Walters Co-PI), R01AG60766 (Ashe PI), the Lucas Brothers Foundation, gifts from Beverly Grossman, Karin Moe and Family, and the Tom and Pat Grossman Family Foundation. Steffen Pockes was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, Forschungsstipendium 436921318). TOC graphic was created with BioRender.com.
Footnotes
SUPPORTING INFORMATION
Fluorescent enzyme assay protocols, Chiral-SFC chromatograms of 6-methyl-tetrahydrosioquinoline-1-carboxylic acid (Figure S1), Fmoc protection of tetrahydrosioquinoline carboxylic acids, Synthesis of compound 9, 11, 2a, and 3, Copies of 1H NMR spectra (Figure S2–S7), HPLC chromatograms of the compounds 1–3 (Figure S8–S11), Competition binding experiments of 1, 2a, 2b, and 3 in the fluorometric enzyme assay (Figure S12–S35), Expression and purification of recombinant caspase-2 and tau for the in vitro caspase-2-catalyzed tau cleavage assay, Antibody immobilization on protein G magnetic beads, Preparation of samples for mass spectrometry, Plasmids and PCR mutagenesis, Primary hippocampal neuron cultures and Low-efficiency calcium-phosphate transfection are shown in the Supporting Information.
CONFLICTS OF INTEREST
K.H.A. consults for AC Immune.
DATA AVAILABILITY STATEMENT
All raw and quantified data will be made available by the authors upon reasonable request.
REFERENCES
- (1).Hoover BR; Reed MN; Su J; Penrod RD; Kotilinek LA; Grant MK; Pitstick R; Carlson GA; Lanier LM; Yuan L-L; Ashe KH; Liao D Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron 2010, 68 (6), 1067–1081. 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Teravskis PJ; Covelo A; Miller EC; Singh B; Martell-Martínez HA; Benneyworth MA; Gallardo C; Oxnard BR; Araque A; Lee MK A53T Mutant Alpha-Synuclein Induces Tau-Dependent Postsynaptic Impairment Independently of Neurodegenerative Changes. Journal of Neuroscience 2018, 38 (45), 9754–9767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Singh B; Covelo A; Martell-Martínez H; Nanclares C; Sherman MA; Okematti E; Meints J; Teravskis PJ; Gallardo C; Savonenko AV; Benneyworth MA; Lesné SE; Liao D; Araque A; Lee MK Tau Is Required for Progressive Synaptic and Memory Deficits in a Transgenic Mouse Model of α-Synucleinopathy. Acta Neuropathologica 2019, 138 (4), 551–574. 10.1007/s00401-019-02032-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhao X; Kotilinek LA; Smith B; Hlynialuk C; Zahs K; Ramsden M; Cleary J; Ashe KH Caspase-2 Cleavage of Tau Reversibly Impairs Memory. Nature medicine 2016, 22 (11), 1268. [DOI] [PubMed] [Google Scholar]
- (5).Miller EC; Teravskis PJ; Dummer BW; Zhao X; Huganir RL; Liao D Tau Phosphorylation and Tau Mislocalization Mediate Soluble Aβ Oligomer-induced AMPA Glutamate Receptor Signaling Deficits. European Journal of Neuroscience 2014, 39 (7), 1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bugiani O; Murrell JR; Giaccone G; Hasegawa M; Ghigo G; Tabaton M; Morbin M; Primavera A; Carella F; Solaro C Frontotemporal Dementia and Corticobasal Degeneration in a Family with a P301S Mutation in Tau. Journal of neuropathology and experimental neurology 1999, 58 (6), 667–677. [DOI] [PubMed] [Google Scholar]
- (7).Braun NJ; Yao KR; Alford PW; Liao D Mechanical Injuries of Neurons Induce Tau Mislocalization to Dendritic Spines and Tau-Dependent Synaptic Dysfunction. Proceedings of the National Academy of Sciences 2020, 117 (46), 29069–29079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Teravskis PJ; Oxnard BR; Miller EC; Kemper L; Ashe KH; Liao D Phosphorylation in Two Discrete Tau Domains Regulates a Stepwise Process Leading to Postsynaptic Dysfunction. The Journal of physiology 2021, 599 (9), 2483–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Vigneswara V; Ahmed Z The Role of Caspase-2 in Regulating Cell Fate. Cells 2020, 9 (5), 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kumar S; Kinoshita M; Noda M; Copeland NG; Jenkins NA Induction of Apoptosis by the Mouse Nedd2 Gene, Which Encodes a Protein Similar to the Product of the Caenorhabditis Elegans Cell Death Gene Ced-3 and the Mammalian IL-1 Beta-Converting Enzyme. Genes & development 1994, 8 (14), 1613–1626. [DOI] [PubMed] [Google Scholar]
- (11).Di Donato N; Jean YY; Maga AM; Krewson BD; Shupp AB; Avrutsky MI; Roy A; Collins S; Olds C; Willert RA Mutations in CRADD Result in Reduced Caspase-2-Mediated Neuronal Apoptosis and Cause Megalencephaly with a Rare Lissencephaly Variant. The American Journal of Human Genetics 2016, 99 (5), 1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xu Z-X; Tan J-W; Xu H; Hill CJ; Ostrovskaya O; Martemyanov KA; Xu B Caspase-2 Promotes AMPA Receptor Internalization and Cognitive Flexibility via MTORC2-AKT-GSK3β Signaling. Nature communications 2019, 10 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pozueta J; Lefort R; Ribe EM; Troy CM; Arancio O; Shelanski M Caspase-2 Is Required for Dendritic Spine and Behavioural Alterations in J20 APP Transgenic Mice. Nature Communications 2013, 4 (1). 10.1038/ncomms2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Carroll JB; Southwell AL; Graham RK; Lerch JP; Ehrnhoefer DE; Cao L-P; Zhang W-N; Deng Y; Bissada N; Henkelman RM Mice Lacking Caspase-2 Are Protected from Behavioral Changes, but Not Pathology, in the YAC128 Model of Huntington Disease. Molecular neurodegeneration 2011, 6 (1), 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tiwari M; Sharma LK; Vanegas D; Callaway DA; Bai Y; Lechleiter JD; Herman B A Nonapoptotic Role for CASP2/Caspase 2: Modulation of Autophagy. Autophagy 2014, 10 (6), 1054–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Maillard MC; Brookfield FA; Courtney SM; Eustache FM; Gemkow MJ; Handel RK; Johnson LC; Johnson PD; Kerry MA; Krieger F; Meniconi M; Muñoz-Sanjuán I; Palfrey JJ; Park H; Schaertl S; Taylor MG; Weddell D; Dominguez C Exploiting Differences in Caspase-2 and −3 S2 Subsites for Selectivity: Structure-Based Design, Solid-Phase Synthesis and in Vitro Activity of Novel Substrate-Based Caspase-2 Inhibitors. Bioorganic & Medicinal Chemistry 2011, 19 (19), 5833–5851. 10.1016/j.bmc.2011.08.020. [DOI] [PubMed] [Google Scholar]
- (17).Poreba M; Rut W; Groborz K; Snipas SJ; Salvesen GS; Drag M Potent and Selective Caspase-2 Inhibitor Prevents MDM-2 Cleavage in Reversine-Treated Colon Cancer Cells. Cell Death & Differentiation 2019. 10.1038/s41418-019-0329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bresinsky M; Strasser JM; Vallaster B; Liu P; McCue WM; Fuller J; Hubmann A; Singh G; Nelson KM; Cuellar ME; Wilmot CM; Finzel BC; Ashe KH; Walters MA; Pockes S Structure-Based Design and Biological Evaluation of Novel Caspase-2 Inhibitors Based on the Peptide AcVDVAD-CHO and the Caspase-2-Mediated Tau Cleavage Sequence YKPVD314. ACS Pharmacology & Translational Science 2022, 5 (1), 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Metcalf B; Chuang C; Dufu K; Patel MP; Silva-Garcia A; Johnson C; Lu Q; Partridge JR; Patskovska L; Patskovsky Y Discovery of GBT440, an Orally Bioavailable R-State Stabilizer of Sickle Cell Hemoglobin. ACS medicinal chemistry letters 2017, 8 (3), 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Owen DR; Allerton CM; Anderson AS; Aschenbrenner L; Avery M; Berritt S; Boras B; Cardin RD; Carlo A; Coffman KJ An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374 (6575), 1586–1593. [DOI] [PubMed] [Google Scholar]
- (21).Boxer MB; Quinn AM; Shen M; Jadhav A; Leister W; Simeonov A; Auld DS; Thomas CJ A Highly Potent and Selective Caspase 1 Inhibitor That Utilizes a Key 3-Cyanopropanoic Acid Moiety. ChemMedChem 2010, 5 (5), 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lossos A; Reches A; Gal A; Newman JP; Soffer D; Gomori JM; Boher M; Ekstein D; Biran I; Meiner Z Frontotemporal Dementia and Parkinsonism with the P301S Tau Gene Mutation in a Jewish Family. Journal of neurology 2003, 250 (6), 733–740. [DOI] [PubMed] [Google Scholar]
- (23).Feinstein S Opinions Recommendation of Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Faculty Opinions. Post-Publication Peer Review of the Biomedical Literature 2007. [Google Scholar]
- (24).Takeuchi H; Iba M; Inoue H; Higuchi M; Takao K; Tsukita K; Karatsu Y; Iwamoto Y; Miyakawa T; Suhara T P301S Mutant Human Tau Transgenic Mice Manifest Early Symptoms of Human Tauopathies with Dementia and Altered Sensorimotor Gating. PloS one 2011, 6 (6), e21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Talanian RV; Quinlan C; Trautz S; Hackett MC; Mankovich JA; Banach D; Ghayur T; Brady KD; Wong WW Substrate Specificities of Caspase Family Proteases. Journal of Biological Chemistry 1997, 272 (15), 9677–9682. [DOI] [PubMed] [Google Scholar]
- (26).Troy CM; Stefanis L; Greene LA; Shelanski ML Nedd2 Is Required for Apoptosis after Trophic Factor Withdrawal, but Not Superoxide Dismutase (SOD1) Downregulation, in Sympathetic Neurons and PC12 Cells. Journal of Neuroscience 1997, 17 (6), 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Troy CM; Rabacchi SA; Friedman WJ; Frappier TF; Brown K; Shelanski ML Caspase-2 Mediates Neuronal Cell Death Induced by β-Amyloid. Journal of Neuroscience 2000, 20 (4), 1386–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ellis HM; Horvitz HR Genetic Control of Programmed Cell Death in the Nematode C. Elegans. Cell 1986, 44 (6), 817–829. [DOI] [PubMed] [Google Scholar]
- (29).Sohn D; Budach W; Jänicke RU Caspase-2 Is Required for DNA Damage-Induced Expression of the CDK Inhibitor P21WAF1/CIP1. Cell Death & Differentiation 2011, 18 (10), 1664–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fujiwara K; Daido S; Yamamoto A; Kobayashi R; Yokoyama T; Aoki H; Iwado E; Shinojima N; Kondo Y; Kondo S Pivotal Role of the Cyclin-Dependent Kinase Inhibitor P21WAF1/CIP1 in Apoptosis and Autophagy. Journal of Biological Chemistry 2008, 283 (1), 388–397. [DOI] [PubMed] [Google Scholar]
- (31).Capparelli C; Chiavarina B; Whitaker-Menezes D; Pestell TG; Pestell RG; Hulit J; Andò S; Howell A; Martinez-Outschoorn UE; Sotgia F CDK Inhibitors (P16/P19/P21) Induce Senescence and Autophagy in Cancer-Associated Fibroblasts,“Fueling” Tumor Growth via Paracrine Interactions, without an Increase in Neo-Angiogenesis. Cell cycle 2012, 11 (19), 3599–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Teravskis PJ; Ashe KH; Liao D The Accumulation of Tau in Postsynaptic Structures: A Common Feature in Multiple Neurodegenerative Diseases? The Neuroscientist 2020, 26 (5–6), 503–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tracy TE; Gan L Tau-Mediated Synaptic and Neuronal Dysfunction in Neurodegenerative Disease. Current opinion in neurobiology 2018, 51, 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kimura T; Whitcomb DJ; Jo J; Regan P; Piers T; Heo S; Brown C; Hashikawa T; Murayama M; Seok H Microtubule-Associated Protein Tau Is Essential for Long-Term Depression in the Hippocampus. Philosophical Transactions of the Royal Society B: Biological Sciences 2014, 369 (1633), 20130144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Liu W; Lin H; He X; Chen L; Dai Y; Jia W; Xue X; Tao J; Chen L Neurogranin as a Cognitive Biomarker in Cerebrospinal Fluid and Blood Exosomes for Alzheimer’s Disease and Mild Cognitive Impairment. Translational psychiatry 2020, 10 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Smith BR; Nelson KM; Kemper LJ; Leinonen-Wright K; Petersen A; Keene CD; Ashe KH A Soluble Tau Fragment Generated by Caspase-2 Is Associated with Dementia in Lewy Body Disease. Acta Neuropathologica Communications 2019, 7 (1). 10.1186/s40478-019-0765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Liu P; Smith BR; Huang ES; Mahesh A; Vonsattel JPG; Petersen AJ; Gomez-Pastor R; Ashe KH A Soluble Truncated Tau Species Related to Cognitive Dysfunction and Caspase-2 Is Elevated in the Brain of Huntington’s Disease Patients. Acta neuropathologica communications 2019, 7 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Black C; Grimm EL; Isabel E; Renaud J Nicotinyl Aspartyl Ketones as Inhibitors of Caspase-3. WO01/27085A1, 2001. [DOI] [PubMed]
- (39).Liu P; Smith BR; Montonye ML; Kemper LJ; Leinonen-Wright K; Nelson KM; Higgins L; Guerrero CR; Markowski TW; Zhao X A Soluble Truncated Tau Species Related to Cognitive Dysfunction Is Elevated in the Brain of Cognitively Impaired Human Individuals. Scientific reports 2020, 10 (1), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and quantified data will be made available by the authors upon reasonable request.