

Abstract
Fungal natural products comprise a wide range of bioactive compounds including important drugs and agrochemicals. Intriguingly, bioinformatic analyses of fungal genomes have revealed that fungi have the potential to produce significantly more natural products than what have been discovered so far. It has thus become widely accepted that most biosynthesis pathways of fungal natural products are silent or expressed at very low levels under laboratory cultivation conditions. To tap into this vast chemical reservoir, the reconstitution of entire biosynthetic pathways in genetically tractable fungal hosts (total heterologous biosynthesis) has become increasingly employed in recent years. This review summarizes total heterologous biosynthesis of fungal natural products accomplished before 2020 using Aspergillus nidulans as heterologous hosts. We review here Aspergillus transformation, A. nidulans hosts, shuttle vectors for episomal expression, and chromosomal integration expression. These tools, collectively, not only facilitate the discovery of cryptic natural products but can also be used to generate high-yield strains with clean metabolite backgrounds. In comparison with total synthesis, total heterologous biosynthesis offers a simplified strategy to construct complex molecules and holds potential for commercial application.
Graphical Abstract
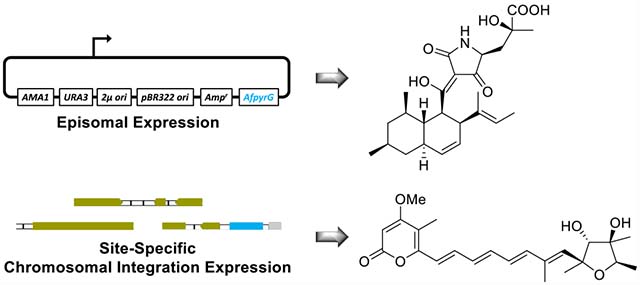
INTRODUCTION
Fungi live in complex ecosystems and are widely distributed in nature. They have evolved to produce bioactive natural products that either benefit their own growth or suppress the growth of their competitors such as bacteria or other fungi.1 Consequently, fungi have been invaluable sources of new leads for the pharmaceutical and agricultural industries. A number of important drugs and fungicides are produced or derived from fungal natural products.2
Typically, the carbon backbone of a fungal natural product is biosynthesized by one or two core enzyme(s) such as polyketide synthases (PKSs),3,4 nonribosomal peptide synthetases (NRPSs),5 terpene cyclases (TCs),6 or prenyltransferases.7 These core enzymes use primary metabolites such as acyl-CoAs (PKSs), amino acids (NRPSs), or isoprenoids (TCs and prenyltransferases) to generate carbon backbones which define the chemical class of the metabolite. The scaffolds synthesized by core enzymes are further modified by various tailoring enzymes, such as cytochromes P450 monooxygenases, flavin-dependent monooxygenases (FMOs), short-chain dehydrogenase/reductases (SDRs), and methyltransferases (MTs).8
Fungal Natural Product Discovery in the Postgenomic Era.
Early studies on the biosynthesis of fungal natural products showed that genes from the same biosynthetic pathway tend to be clustered in the genome.9 Clustering may facilitate co-regulation or horizontal gene transfer of all genes in a biosynthesis pathway. Later fungal genome projects revealed that biosynthetic gene clusters (BGCs) vastly outnumber known natural products.1,10 This observation indicated that most BGCs are “cryptic” (completely silent or expressed at a very low level) under laboratory cultivation conditions. Since the signals that trigger their expression are unknown, the chemical diversity of fungi remains largely untapped. Considering that there are millions of fungal species11 and that certain species contain up to 70 BGCs,12 there is tremendous potential for fungal natural product discovery.
In addition to the enzymes required for biosynthesis, some BGCs also encode major facilitator superfamily (MFS) transporters,13 cluster-specific regulators,14,15 and self-resistant enzymes.16,17 Most putative transporters have not been functionally characterized, but they are hypothesized to transport substrates, intermediates, or the final products of a biosynthesis pathway. Cluster-specific regulators modulate the expression of the whole cluster. Therefore, a common strategy for eliciting a silent BGC in a genetically tractable fungus is to induce the positive regulator.15 However, since not every BGC contains one, it is not a universal strategy to activate cryptic BGCs. Lastly, many BGCs produced toxic natural products that target metabolic enzymes. To prevent self-harm, some BGCs encode a mutated version of the original target that is resistant to the toxic natural product (a resistance enzyme). This feature has greatly facilitated functional assignment or target identification of the toxic natural product.17
The field of fungal natural product discovery has dedicated much effort in developing strategies to unlock cryptic natural products. While an in-depth review of the various strategies is out of the scope of this review, readers are referred to articles on this topic.15,18 Here, we focus on the reconstitution of biosynthetic pathways in genetically tractable hosts (total heterologous biosynthesis). This approach is particularly useful if the producer is uncultivable or has no genetic system for genome editing.19–23
Heterologous Expression Hosts.
E. coli and Saccharomyces cerevisiae (yeast) are widely used for the expression of fungal genes. They have several advantages such as well-established genetic systems, robust growth, and clean backgrounds due to lack of secondary metabolic pathways.
However, both species also have limitations. E. coli is a prokaryote and cannot perform RNA splicing and post-translational modification. As such, most studies using E. coli are limited to characterizing individual enzymes rather than full-length BGCs. Yeasts are unicellular eukaryotes and have the machinery for RNA splicing and post-translational modifications. The engineered yeast strain BJ5464-NpgA, with the npgA gene from A. nidulans integrated into its genome, is widely used for the expression of fungal PKS pathways.24 NpgA encodes for 4′-phosphopantetheine transferase that catalyzes the attachment of a 4′-phosphopantetheine, which converts an inactive apo PKS or NRPS to an active holo form. Although there are many advantages of using yeasts as heterologous hosts, yeasts lack specialized compartments required to produce certain metabolites.25 In addition, yeasts cannot splice the mRNA of filamentous fungi correctly, requiring accurate prediction of introns if cDNA cannot be obtained from the producer.24,26
In contrast, genetically tractable filamentous fungi have compatible transcription, translation, post-translational modification, and secretion machineries for the expression of foreign fungal genes and therefore are better suited for the heterologous expression of full-length BGCs. Intact BGCs of penicillin,27 citrinin,28 fusatins,29 W493,29 bikaverin,30 and flavoglaucin31 have been transferred and successfully expressed in fungal hosts. Moreover, Bok and Clevenger et al. developed fungal artificial chromosomes to introduce entire BGCs from three Aspergillus species into A. nidulans, and about 27% of the transferred BGCs produced detectable products.32,33
Despite these successful cases, however, most heterologous BGCs do not produce detectable products in filamentous fungal hosts, and even if they do, the titers are often low. The silence could be attributed to the lack of triggering signals, and the poor yield to suboptimal expression of the foreign genes.30 While it is possible to increase the titer by overexpression of the positive regulator, as previously discussed, not every fungal BGC harbors one.28,34,35 Against this background, BGC refactoring with strong promoters offers an alternative route to biosynthesizing compounds in high titers.
EXPRESSION OF HETEROLOGOUS GENES IN ASPERGILLUS
Aspergillus Transformation.
Aspergillus spp. can grow in a wide range of conditions and are widely used as heterologous hosts.36 Several engineered Aspergillus strains are commonly used to express BGCs.19,20 Since the fungal cell wall is an impenetrable barrier for exogenous DNA, the first step of gene delivery (transformation) is oftentimes enzymatic digestion of the fungal cell wall to generate protoplasts (Figure 1) (for details on the transformation of filamentous fungi, see refs 37 and 38). Once the cell wall has been sufficiently digested, the transforming DNA can be taken up by the cell via polyethylene glycol (PEG)-mediated delivery.
Figure 1.
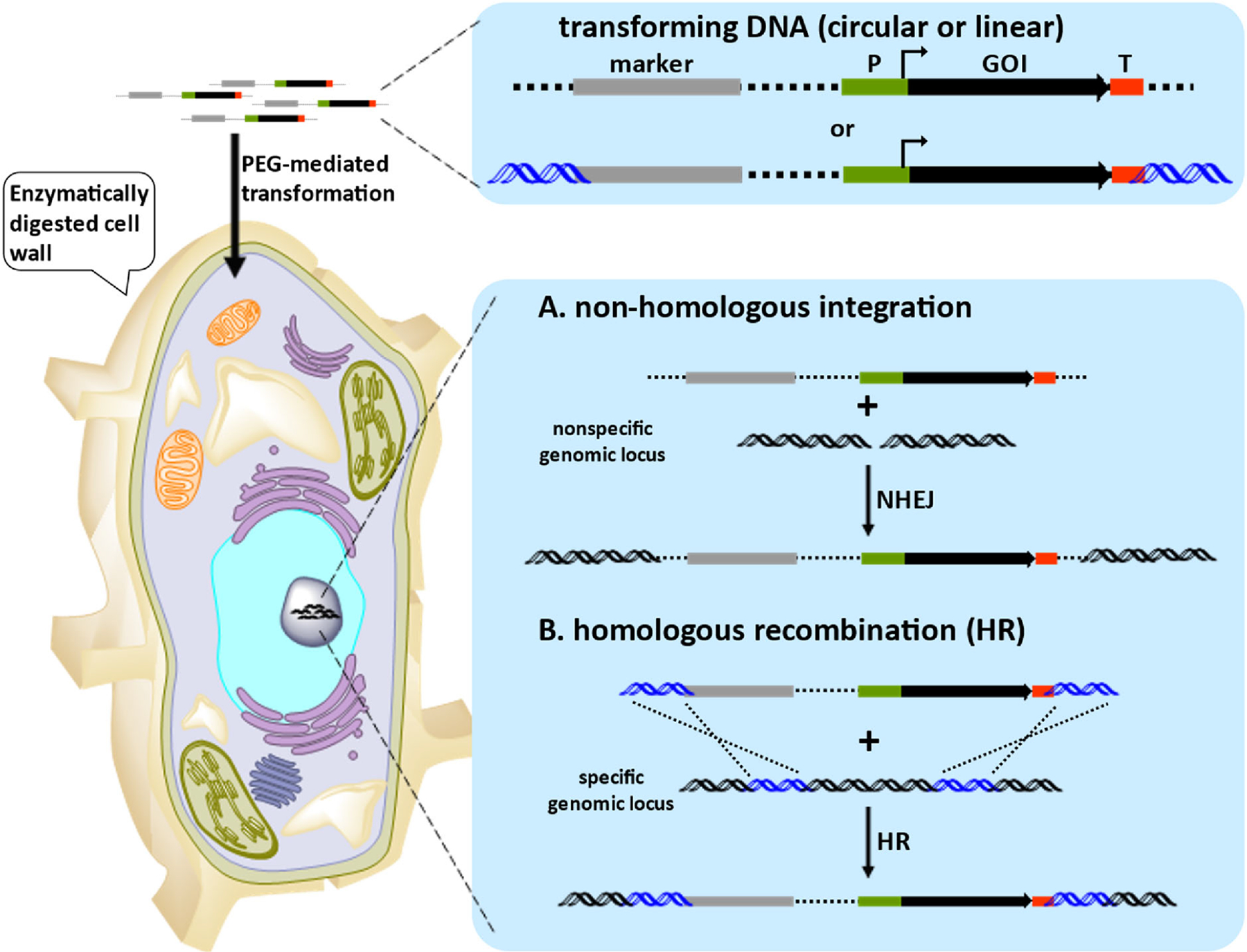
Overview of chromosomal integration expression. Enzymatic digestion of the fungal cell results in a protoplast, which allows transforming DNA to be delivered into the nucleus via PEG-mediated transformation. Transforming DNA contains a selectable marker and at least one expression cassette. Expression of a gene of interest (GOI) is driven by a constitutive or inducible promoter (P). The 3′ untranslated region (3′-UTR) of a GOI functions as a terminator (T), which ensures the releasing of the transcribed mRNA, so that the “GOI + T” fragment can be amplified from the gnomic DNA (gDNA) of the native producer. Once the transforming DNA reaches the nucleus, it can integrate into the host’s chromosome via (A) non-homologous end joining (NHEJ) or (B) homologous recombination (HR). NHEJ results in the integration of the transforming DNA into a nonspecific genomic locus, while recombination of homologous sequences (blue) results in the integration at a specific genomic locus.
After the transforming DNA reaches the nucleus, it can self-replicate episomally or integrate into the genome. For episomal replication, the autonomous fungal replication element, AMA1 (autonomous maintenance in Aspergillus), has been identified for extrachromosomal replication in A. nidulans (see section AMA1-Based Episomal Expression System).39 For chromosomal integration, either non-homologous integration or homologous recombination (HR) may occur (Figure 1). In the event of non-homologous integration, transforming DNA ligates into spontaneous double-strand breaks (DSBs) via non-homologous end joining (NHEJ), which is the major DNA repair mechanism of DSBs in many eukaryotes. It should be noted that it is not uncommon for multiple copies of the transforming DNA to integrate into different genomic loci with NHEJ. In the event of HR, recombination of the homologous sequences results in the integration of transforming DNA at a specific locus.
Elements in Transforming DNA.
Selection of successful transformants is achieved with the selectable marker in the transforming DNA.37 Either a dominant (resistance) or auxotrophic (nutritional) selectable marker can be used. Dominant markers (such as bar and hph) are genes encoding enzymes that confer resistance against toxic chemicals. For example, bar encodes phosphinothricin acetyltransferase able to detoxify glufosinate ammonium, and hph encodes hygromycin B phosphotransferase able to detoxify the antibiotic hygromycin B. In media containing a specific toxic chemical, only the fungal cells harboring the dominant marker can grow. Auxotrophic markers (such as pyrG, riboB, and pyroA) are based on nutritional genes. Auxotrophy is the inability of an organism to synthesize a specific metabolite required for growth. Growth can be restored with an auxotrophic marker that complements that defect. Therefore, nutritional selection requires an auxotrophic mutant with a specific deficiency in primary metabolism. For example, pyrG encodes orotidine 5′-monophosphate decarboxylase, and a pyrG mutation with dysfunctional PyrG leads to uridine/uracil auxotrophy, which requires uridine/uracil for the organism to grow (Figure S1A). Transforming DNA with functional pyrG can restore growth in media lacking uridine/uracil. Additionally, pyrG mutants are resistant to 5-fluoroorotic acid (5-FOA), which can be used for counter-selection. Strains with functional pyrG metabolize 5-FOA to the toxic metabolite 5-fluorouracil. pyrG mutants cannot metabolize 5-FOA to 5-fluorouracil and, therefore, can grow in a medium containing uridine/uracil and 5-FOA. Counter-selection enables pyrG to be recycled and used for subsequent rounds of transformation. Besides pyrG, riboB and pyroA are two other auxotrophic markers frequently used in A. nidulans transformation. RiboB is required for riboflavin biosynthesis, and a riboB mutant requires riboflavin to grow (Figure S1B). PyroA is required for pyridoxine biosynthesis, and a pyroA mutant requires pyridoxine to grow (Figure S1C).
To express a gene of interest, the transforming DNA also needs to carry an expression cassette. An expression cassette contains the coding region of a gene of interest placed downstream of a promoter and upstream of a terminator to drive expression and ensure the releasing of the synthesized RNA from the transcription machinery, respectively. Either a strong constitutive promoter (such as PgpdA) or an inducible promoter (such as PalcA, PamyB, and PglaA) can be used.40 gpdA encodes glyceraldehyde-3-phosphate dehydrogenase, and its expression is independent of transcriptional regulation. alcA encodes alcohol dehydrogenase I and can be induced by alcohols, aldehydes, or ketones. amyB and glaA encode α-amylase and glucoamylase, respectively, and both can be induced by maltose or starch. One advantage of using an inducible expression system is that biomass can be accumulated before induction of the genes of interest, which is especially useful if a reconstituted pathway produces a metabolite toxic to the host.
Aspergillus Hosts.
Today, A. oryzae and A. nidulans are the most popular fungal hosts for the reconstitution of fungal biosynthesis pathways. A. oryzae is identified as a GRAS (generally recognized as safe) organism by the U.S. Food and Drug Administration and has been used by the fermentation industry to produce sake, miso, and soy sauce for centuries.41 The heterologous expression system was developed based on the fact that amyB in A. oryzae is highly expressed in media containing maltose.42 As such, the promoter and terminator from amyB can be used to drive the expression of genes of interest. Genetically tractable strains A. oryzae M-2–3 (argB−)43 and NSAR1 (niaD−, sC−, argB−, adeA−)44 are widely used for ectopic integration to constitute biosynthesis pathways. A. oryzae M-2–3 was used by Heneghan et al. to achieve the first total biosynthesis of the fungal natural product tenellin in 2010.45 NSAR1 is a quadruple auxotrophic mutant that allows the transformation of at least four expression vectors. Furthermore, the development of multiple expression cassettes in a single vector enabled the expression of multiple genes of interest in one transformation using only one selectable marker.21,46 Reconstitution of meroterpenoid biosynthesis pathways in A. oryzae has recently been reviewed.47
In this review, we will focus on total biosynthesis in A. nidulans. The most popular A. nidulans host strains used are TN02A7 and its derivatives.48,49 TN02A7 contains the deletion of nkuA, a homologue of the human KU70 gene, which is essential for NHEJ. Deletion of nkuA greatly impairs NHEJ and, therefore, increases the probability of HR events. TN02A7 also carries three auxotrophic mutations (pyrG89, pyroA4, and riboB2) which can be complemented by homologue genes from A. fumigatus or A. terreus. Using homologue genes from other Aspergillus species prevents potential HR between the transformation DNA and the mutated gene. TN02A7 has been deposited at Fungal Genetics Stock Center (www.fgsc.net) with FGSC strain number A1145 (Table 1).
Table 1.
A. nidulans Hosts and Their Genotypes Used for Total Heterologous Biosynthesis
| host strain | genotypes | refs |
|---|---|---|
| A1145 (TN02A7) | argB::nkuAΔ; pyrG89; pyroA4; riboB2 | 48 |
| A1145ΔST | argB::nkuAΔ; stcAΔ; pyrG89; pyroA4; riboB2 | 53 |
| A1145ΔEM | argB::nkuAΔ; easAΔ; pyrG89; pyro0A4; riboB2 | 53 |
| A1145ΔSTΔEM | argB::nkuAΔ; stcAΔ; easAΔ; pyrG89; pyroA4; riboB2 | 53 |
| LO4389 | argB::nkuAΔ; stcΔ; pyrG89; pyroA4; riboB2 | 54 |
| LO7890 | argB::nkuAΔ; stcΔ; easΔ; afoΔ; mdpΔ; tdiΔ; ausΔ; orsΔ; pyrG89; pyroA4; riboB2 | 55 |
| LO8030 | argB::nkuAΔ; stcΔ; easΔ; afoΔ; mdpΔ; tdiΔ; ausΔ; orsΔ; aptΔ; pyrG89; pyroA4; riboB2 | 55 |
TN02A7 (A1145) produces sterigmatocystin and emericellamides in a variety of different culturing conditions. StcA50 and EasA51 are the nonreducing (NR)-PKS and NRPS responsible for the biosynthesis of sterigmatocystin and emericellamides, respectively. Using HR facilitated by CRISPR-Cas952 to delete stcA, easA, or both, the Tang group generated A1145ΔST, A1145ΔEM, and A1145ΔSTΔEM strains.53 Deletion of endogenous BGCs frees up substrates such as malonyl-CoA for the heterologous pathway and simplifies the metabolite background to facilitate detection and purification of new compounds. The Oakley group took the approach one step further and deleted the entire ST BGC stc (>50 kb) in A1145 to generate the strain LO4389.54 While deleting only the core gene stcA eliminates ST production, other genes in the stc pathway could potentially still interfere with the heterologous pathway. Deletion of the entire BGC eliminates any potential crosstalk. Lastly, the Oakley group created the genetically dereplicated strains LO7890 and LO803055 with seven and eight native A. nidulans BGCs removed, respectively (Table 1). They were originally created to unearth hidden metabolites buried by the background, but have also been used as hosts in total heterologous biosynthesis.
TOTAL HETEROLOGOUS BIOSYNTHESIS IN A. NIDULANS
This review highlights total heterologous biosynthesis work in A. nidulans accomplished before 2020 on BGCs with at least four enzymatic steps. A timeline of the highlighted work is depicted in Figure 2. As these works employed either the episomal or site-specific chromosomal integration expression (Table 2), we will examine each system in two separate sections. We recognize that there are numerous manuscripts that describe similar approaches for producing intermediates in biosynthesis pathways that are not covered here. We apologize to the authors whose work we missed in this review. We intended to target audiences with synthetic or natural product chemistry backgrounds and thus have focused this review on biosynthesis. For an overview of fungal heterologous expression systems, readers are directed to relevant literature in refs 19–23.
Figure 2.
Timeline of total heterologous biosynthesis in A. nidulans by publication date. aReiterative recombination. bCluster refactoring in the afo regulon.
Table 2.
Comparison between Episomal Expression and Site-Specific Chromosomal Integration Expression in A. nidulans
| expression system | transforming DNA | advantages | disadvantages |
|---|---|---|---|
| Episomal |
|
|
|
| Site-specific chromosomal integration |
|
|
|
I. AMA1-BASED EPISOMAL EXPRESSION SYSTEM
As mentioned, the AMA1 replicator supports extrachromosomal vector maintenance in Aspergillus spp.,39 and its utility has been demonstrated in DNA constructs >100 kb in A. nidulans.32 Based on this finding, the Watanabe, Tang, and Chooi groups constructed E. coli-yeast-fungal shuttle vectors for episomal expression in A. nidulans.
E. coli-Yeast-Fungal Shuttle Vectors pKW20088, pYTP, pYTR, and pYTU.
The Watanabe group created the E. coli-yeast-fungal shuttle vector pKW2008856 from pPTRII57 (Figure S2A). Besides the AMA1 replicator, pPTRII also contains pBR322 ori and Ampr for plasmid maintenance and selection in E. coli, respectively. Insertion of 2μ ori and URA3, which allow plasmid maintenance and selection in S. cerevisiae, respectively, into pPTRII resulted in pKW19030. S. cerevisiae is a powerful molecular tool for the assembly of BGCs because it can assemble DNA fragments with ~30 bp of overlap in vivo via HR with high efficiency and fidelity.58 The selectable marker from A. fumigatus, AfpyrG, was then inserted into pKW19030, resulting in pKW20088. The expression cassettes can be inserted between AMA1 and AfpyrG. Since pKW20088 contains elements for maintenance and selection in E. coli, yeast, and Aspergillus, it can assemble BGCs in yeast, propagate in E. coli, and episomally express genes of interest in Aspergillus.
Sch 210972 (1).
Based on the shuttle vector pKW20088, the Watanabe group generated pKW10027, pKW10029, and pKW10035 expression vectors (Figure S2B) to reconstitute the biosynthetic pathway of Sch 210972 (1) in the A1145 strain.59 1 is a tetramic acid-containing metabolite isolated from Chaetomium globosum (Figure 3A).60 It exhibits potent inhibitory activity against chemokine receptor CCR-5, a cell surface receptor that plays an important role in the attachment and entry of HIV-1 into cells.
Figure 3.
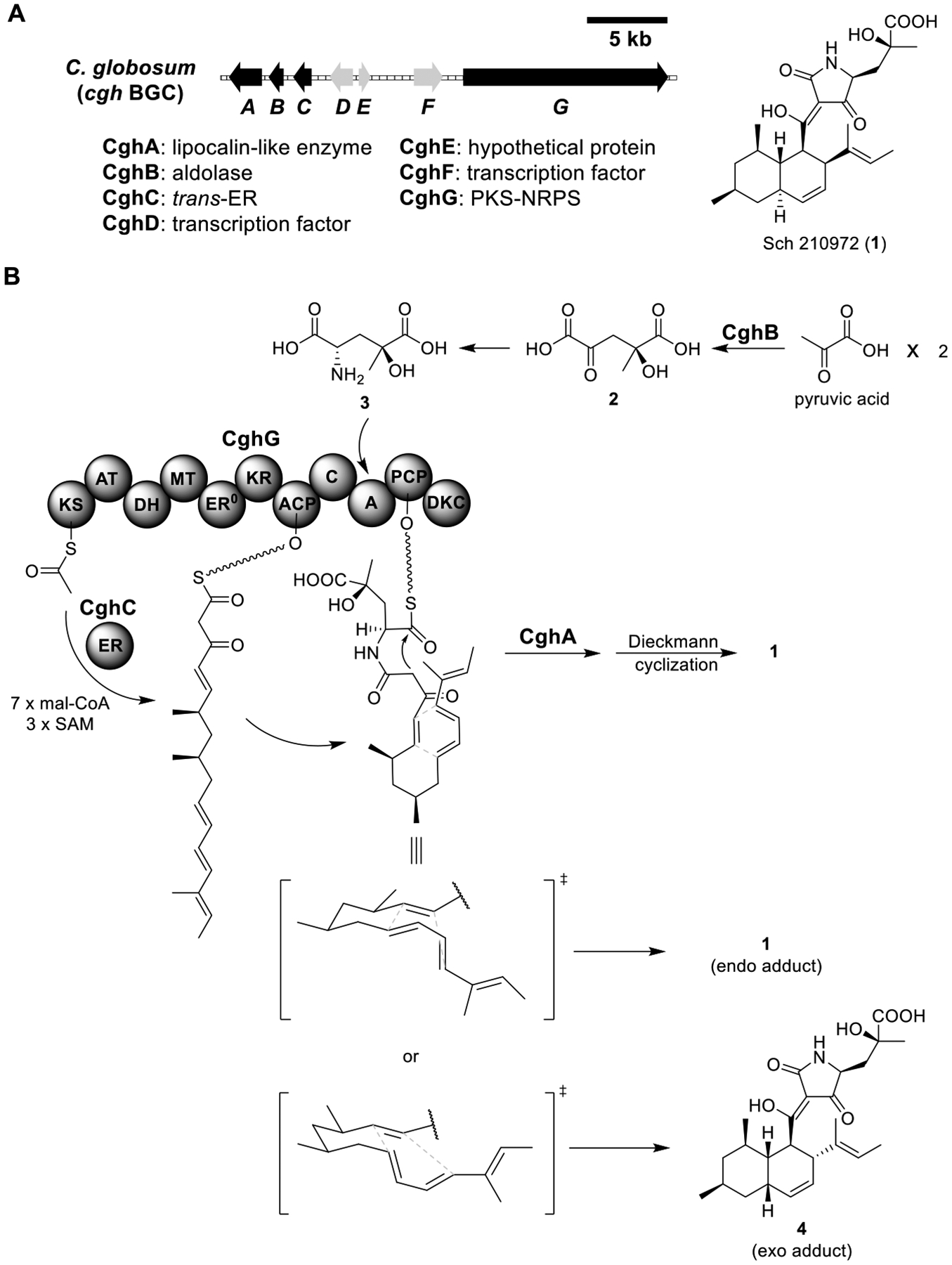
Total biosynthesis of Sch 210972 (1). (A) Organization and predicted gene functions of the cgh BGC. Black ORFs encode for enzymes involved in the biosynthesis of 1. (B) Proposed biosynthesis of 1.
Four genes, cghA (encoding a lipocalin-like enzyme), cghB (aldolase), cghC (trans-acting enoyl reductase (trans-ER)), and chgG (PKS-NRPS) in the Sch 210972 BGC were identified to be involved in the biosynthesis of 1 (Figure 3A).59 CghG and the partnering ER (trans-ER) CghC were proposed to assemble the backbone of 1. CghB was proposed to dimerize pyruvic acid via aldol reaction to generate 2, which then transaminated to 3 (Figure 3B). Interestingly, expression of all four genes resulted in the exclusive production of the endo adduct 1, while omission of CghA led to a mixture of endo and exo adducts 1 and 4 (Table S1). This suggested that CghA stereoselectively promoted the Diels–Alder reaction, leading to its assignment as a Diels–Alderase.
Using similar methodology to the construction of pKW20088, the Tang group utilized pYTU, pYTP, and pYTR expression vectors carrying AfpyrG, AfpyroA, and AfriboB auxotrophic markers, respectively, to reconstitute fungal biosynthesis pathways. Their total biosynthesis works using A. nidulans are summarized and organized according to the class of fungal natural products expressed, i.e., PKS (Figures 4 and 5), NRPS (Figure 6–8), PKS-NRPS hybrid (Figures 9–12), PKS and NRPS (Figures 14–16), and others (Figures 17 and 18).
Figure 4.
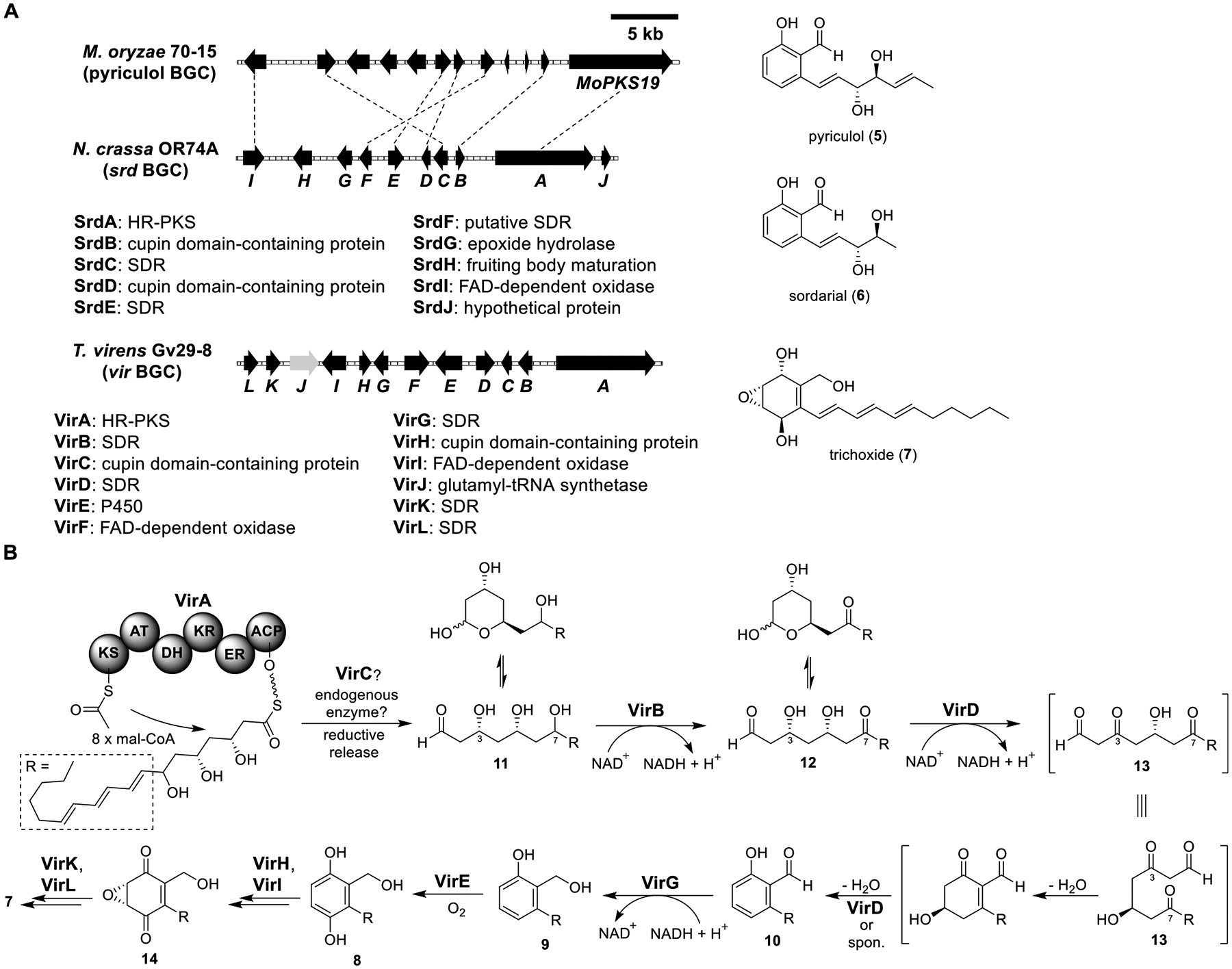
Total biosynthesis of trichoxide (7). (A) Organization and predicted gene functions of the pyriculol, srd, and vir BGCs. Black ORFs encode for enzymes involved in the biosynthesis of pyriculol (5), sordarial (6), and 7. (B) Proposed biosynthesis of 7.
Figure 5.
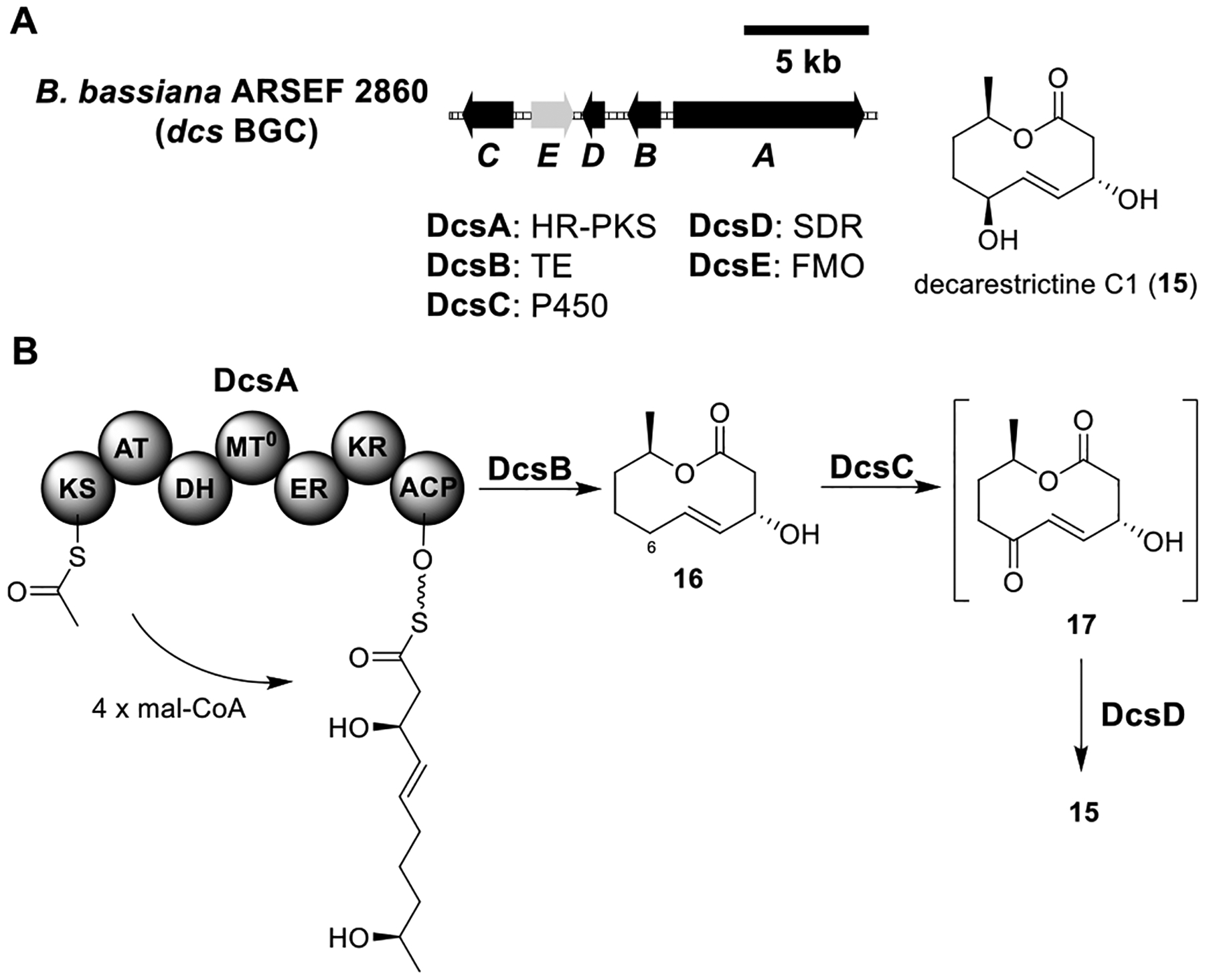
Total biosynthesis of decarestrictine C1 (15). (A) Organization and predicted gene functions of the dcs BGC. Black ORFs encode for enzymes involved in the biosynthesis of 15. (B) Proposed biosynthesis of 15.
Figure 6.
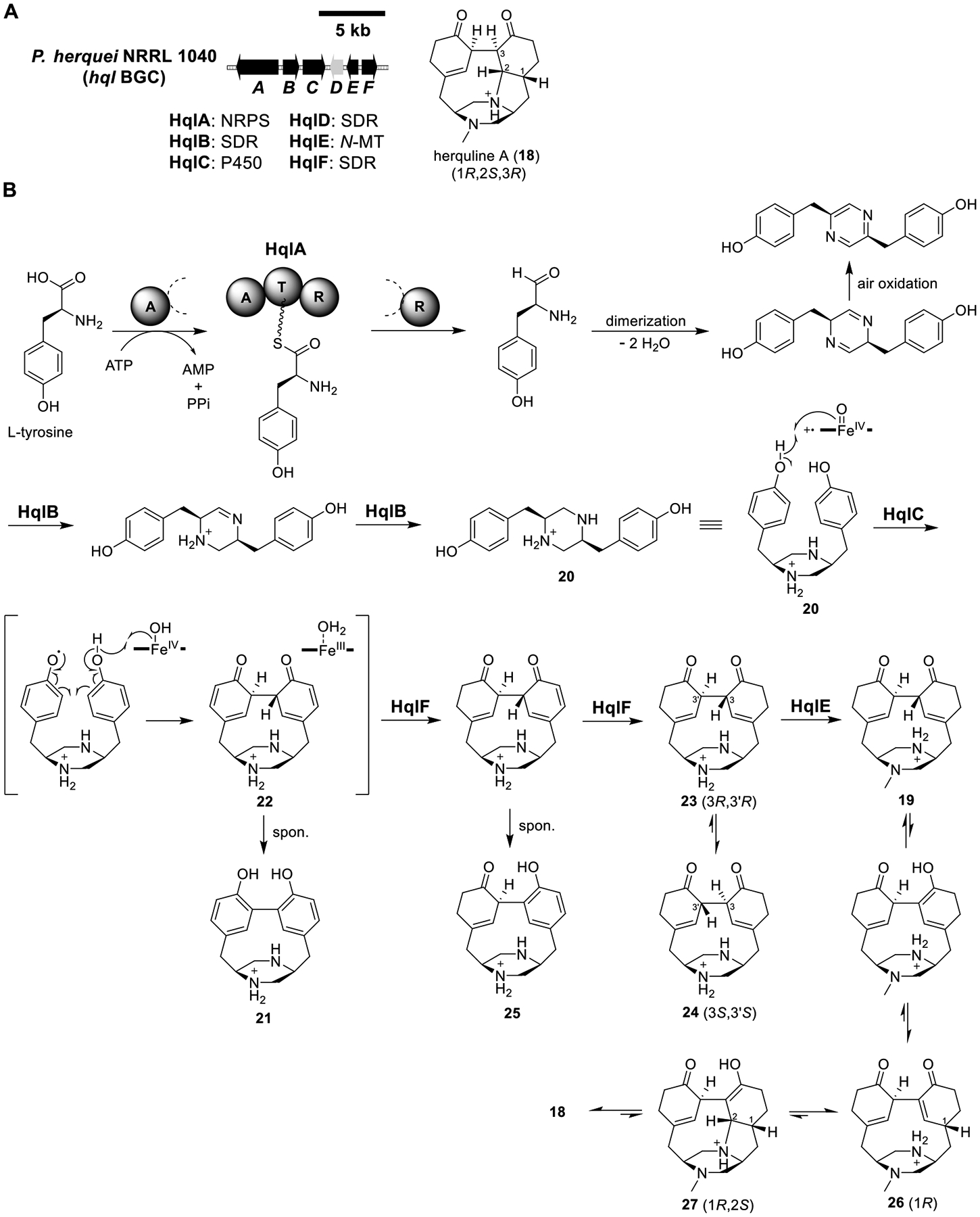
Total biosynthesis of herquline A (18). (A) Organization and predicted gene functions of the hql BGC. Black ORFs encode for enzymes involved in the biosynthesis of 18. (B) Proposed biosynthesis of 18.
Figure 8.
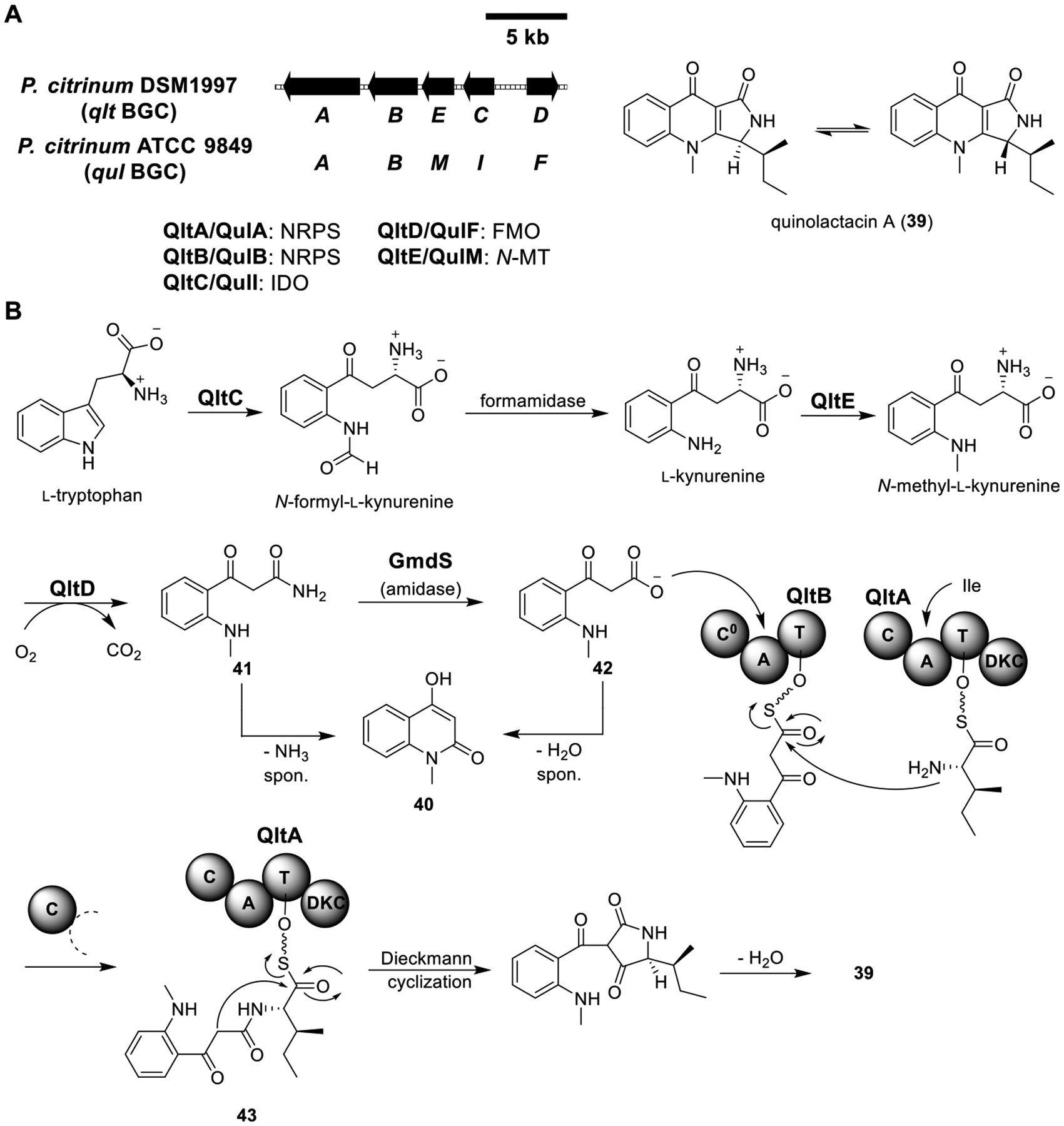
Total biosynthesis of quinolactacin A (39). (A) Organization and predicted gene functions of the qlt and qul BGCs. Black ORFs encode for enzymes involved in the biosynthesis of 39. (B) Proposed biosynthesis of 39.
Figure 9.
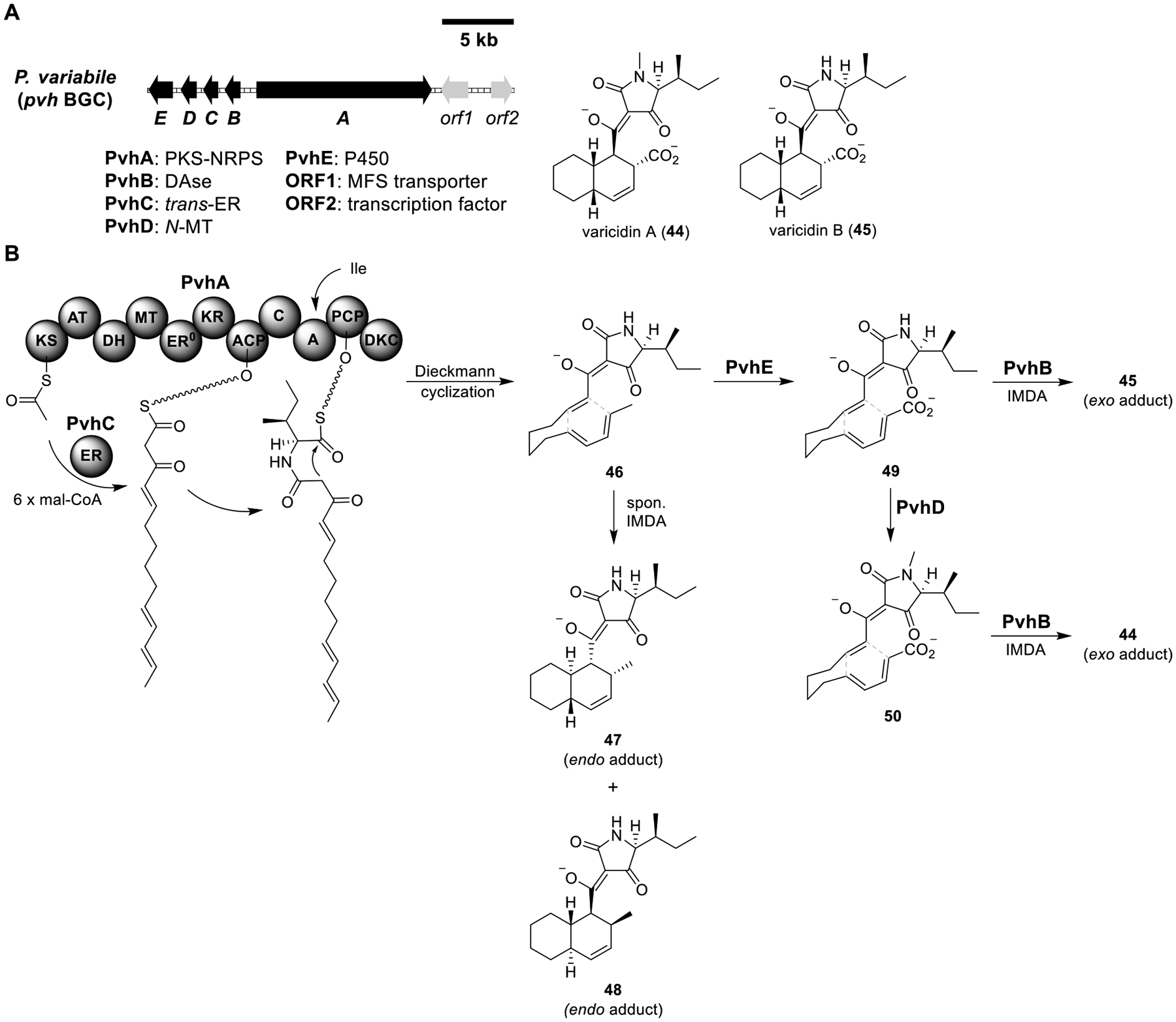
Total biosynthesis of varicidins A (44) and B (45). (A) Organization and predicted gene functions of the pvh BGC. Black ORFs encode for enzymes involved in the biosynthesis of 44 and 45. (B) Proposed biosynthesis of 44 and 45.
Figure 13.
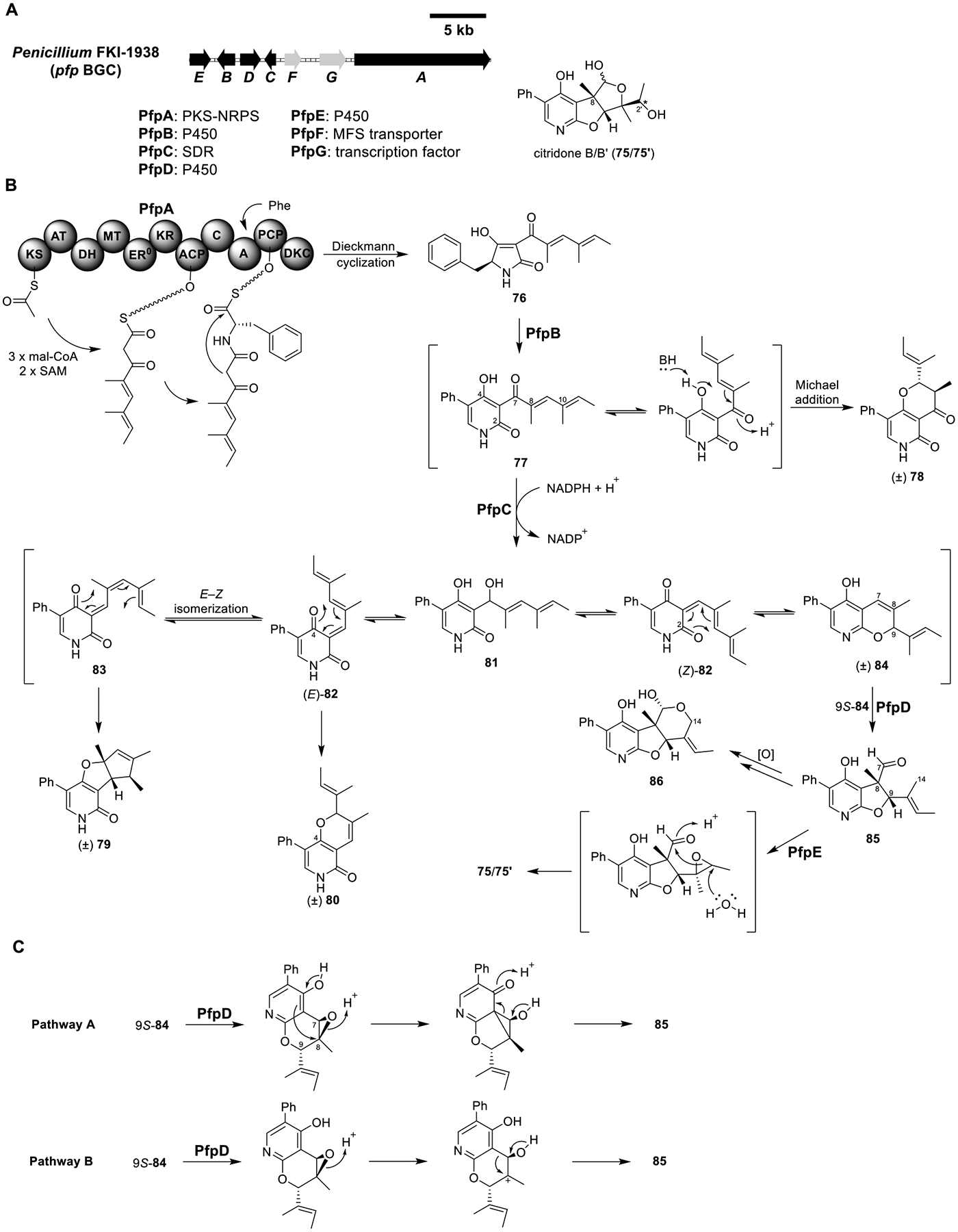
Total biosynthesis of citridone B/B (75/75′). (A) Organization and predicted gene functions of the pfp BGCs. Black ORFs encode for enzymes involved in the biosynthesis of 75/75′. (B) Proposed biosynthesis of 75/75′. (C) Proposed mechanisms for the formation of 85 via 9S-84. *The relative configuration of C-2′ in 75/75′ was not determined.
Figure 14.
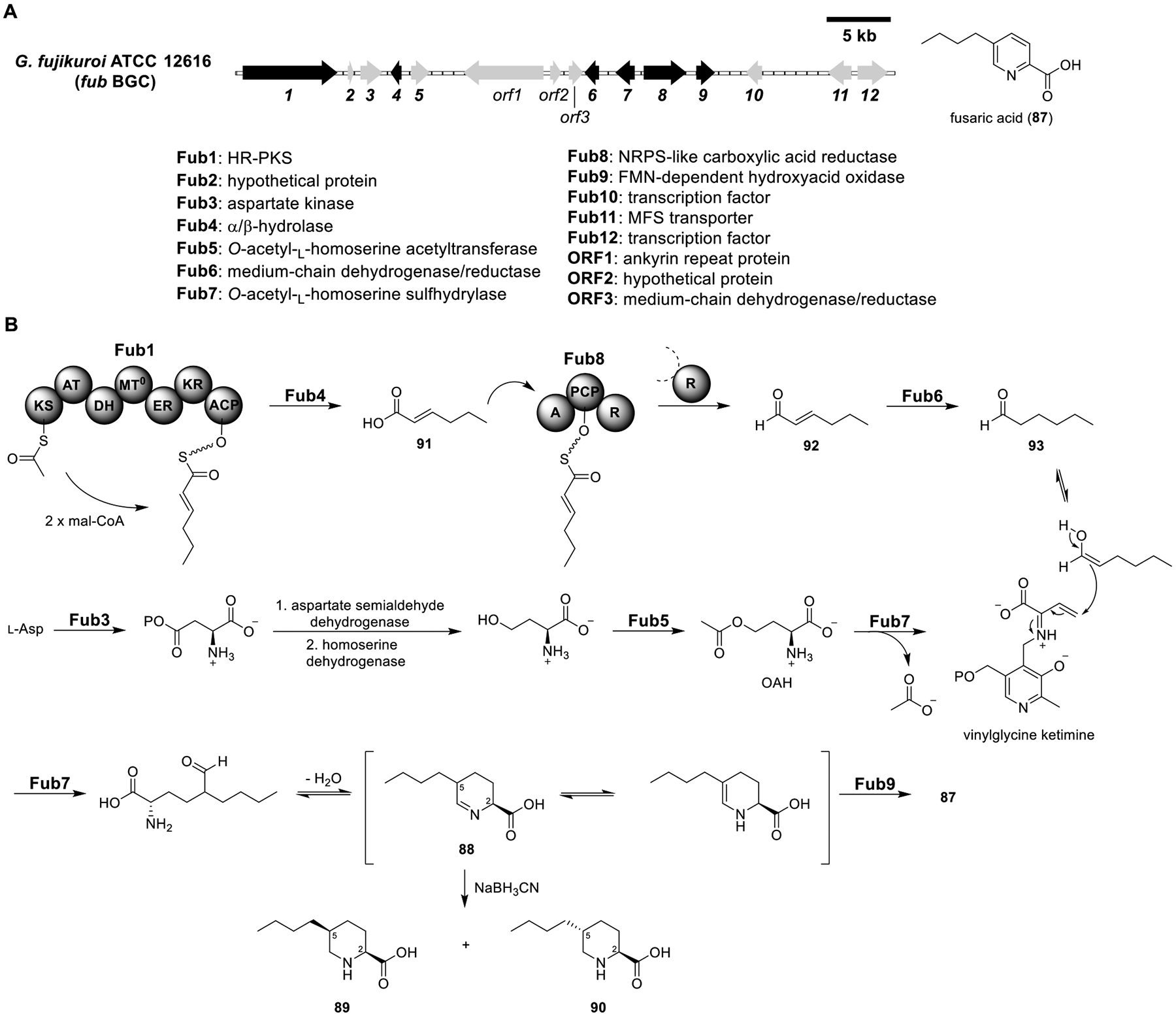
Total biosynthesis of fusaric acid (87). (A) Organization and predicted gene functions of the fub BGC. Black ORFs encode for enzymes involved in the biosynthesis of 87. (B) Proposed biosynthesis of 87.
Figure 16.
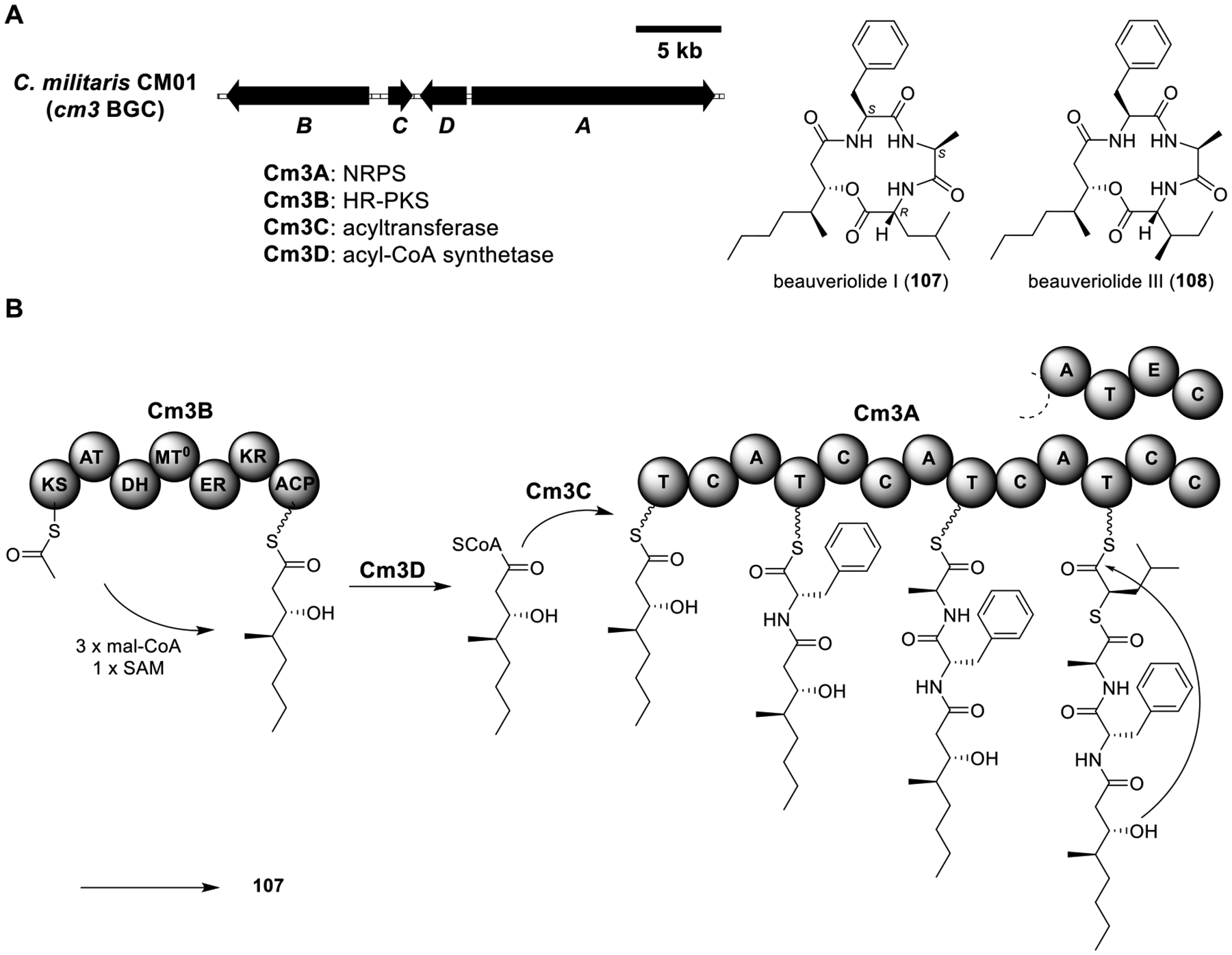
Total biosynthesis of beauveriolides I (107) and III (108). (A) Organization and predicted gene functions of the cm3 BGC. Black ORFs encode for enzymes involved in the biosynthesis of 107 and 108. (B) Proposed biosynthesis of 107.
Figure 17.
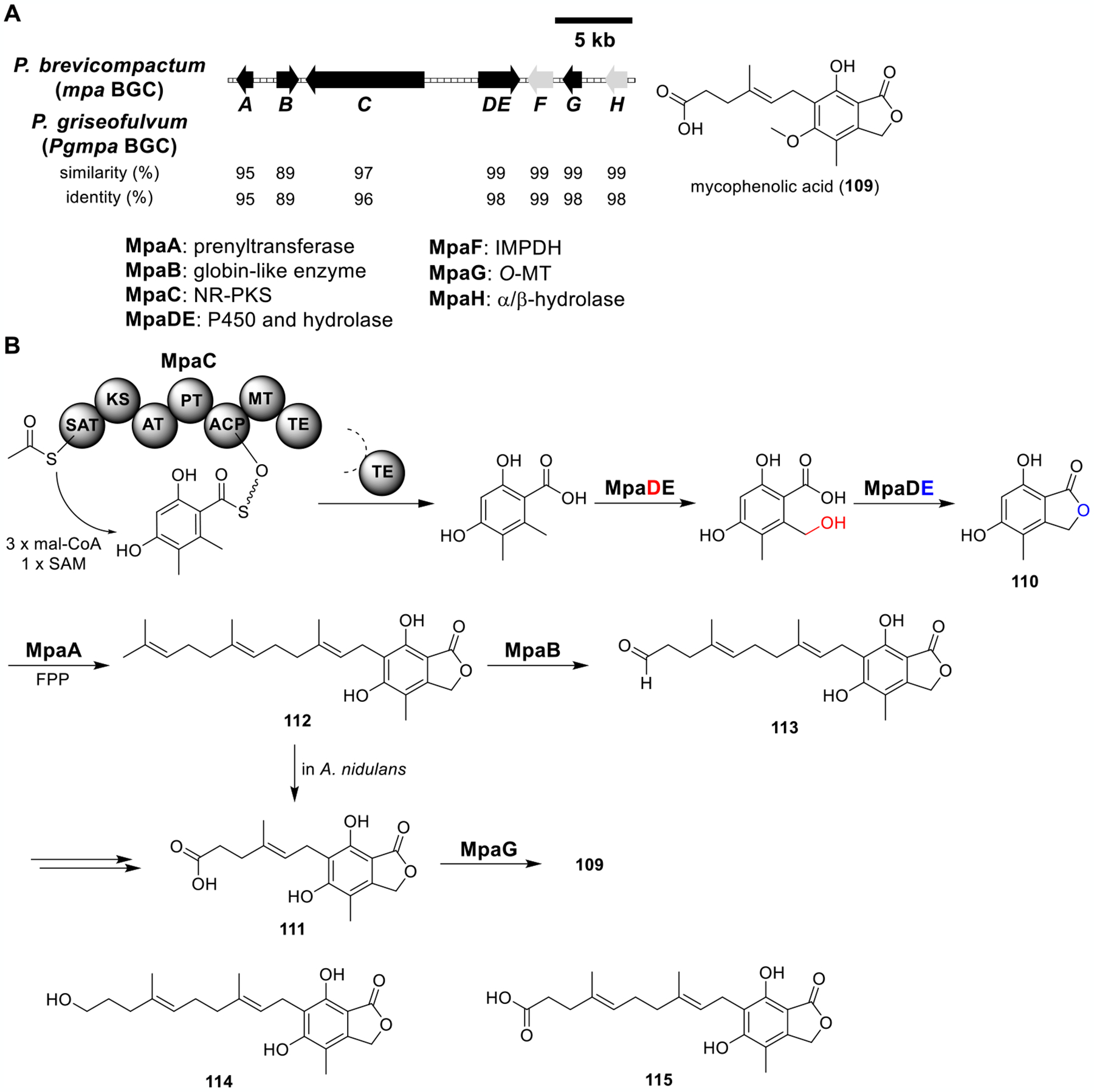
Total biosynthesis of mycophenolic acid (109). (A) Organization and predicted gene functions of the mpa and Pgmpa BGCs. Black ORFs encode for enzymes involved in the biosynthesis of 109. (B) Proposed biosynthesis of 109.
Figure 18.
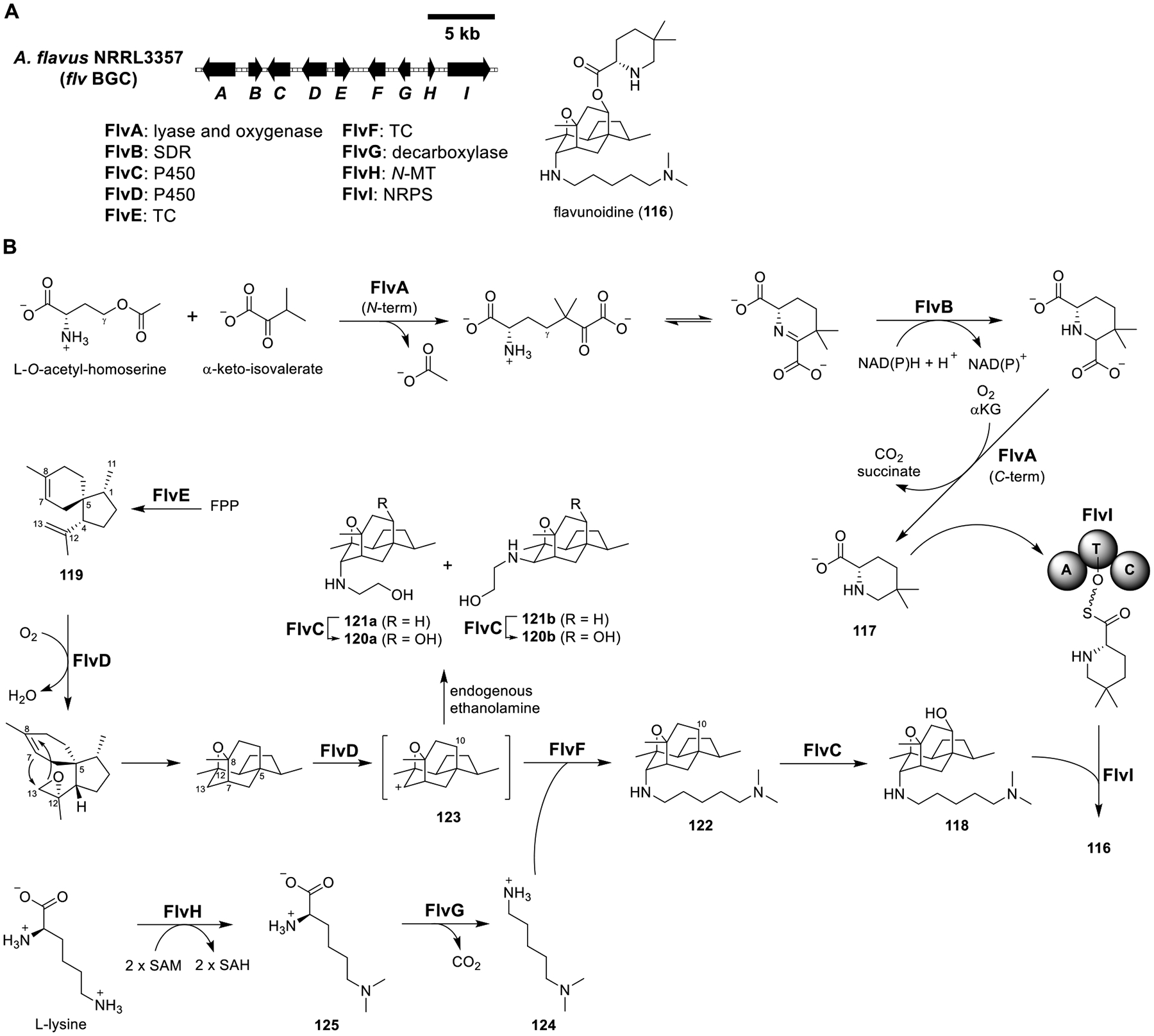
Total biosynthesis of flavunoidine (116). (A) Organization and predicted gene functions of the flv BGC. Black ORFs encode for enzymes involved in the biosynthesis of 116. (B) Proposed biosynthesis of 116.
Pyriculol (5), Sordarial (6), and Trichoxide (7).
Pyriculol (5) is a salicylaldehyde produced by Magnaporthe oryzae. It was found to induce lesion formation on rice leaves. A highly reducing (HR)-PKS (MoPKS19) was genetically verified to be involved in the biosynthesis of 5 (Figure 4A).61 It was surprising that an HR-PKS-containing BGC was responsible for the biosynthesis of 5 since nonreducing (NR)-PKSs or partially reducing (PR)-PKSs are well-recognized to synthesize aromatic polyketides.3 Using a comparative genomic approach, a BGC (srd) similar to the pyriculol BGC was identified in Neurospora crassa (Figure 4A).62 Heterologous expression of SrdA–J in the A1145ΔSTΔEM strain produced sordarial (6) (Figure 4A). While the detailed biosynthesis of 6 was not elucidated, this study confirmed that fungal salicylaldehydes indeed originated from HR-PKSs.
Using a similar approach, another BGC (vir) was identified from Trichoderma virens (Figure 4A).63 Heterologous expression of VirA–L in the A1145 strain led to a new compound, trichoxide (7), together with pathway intermediates virensol A (8), virensol B (9), and 5-deoxyaurocitrin (10) (Figure 4B and Table S2). To investigate how the HR-PKS (VirA) and its associated tailoring enzymes worked together to synthesize salicylaldehyde 10, different combinations of vir genes in A1145 were heterologously expressed.
Expression of VirA produced virensol C (11), which existed mostly as a pair of hemiacetals in solution. The production of 11 by VirA alone was surprising since VirA did not contain a reductive releasing (R) domain. When the cupin-domain enzyme VirC was coexpressed with VirA, the yield of 11 increased, suggesting that VirC may be involved in enhancing product turnover. After testing different binary combinations, only coexpression of the SDR VirB with VirA resulted in the disappearance of 11 and emergence of 12, which also existed as a pair of hemiacetals in solution. Coexpression of another SDR VirD with VirA/B led to the disappearance of 12 and the emergence of 10, with a trace amount of 9. Thus, VirB was proposed to dehydrogenate the C-7 alcohol and VirD the C-3 alcohol to generate 13, which then underwent intramolecular aldol condensation, dehydration, and aromatization to form the salicylaldehyde moiety in 10. These results demonstrated that two of the three hydroxy groups (C-3 and C-7) in 11 were oxidized back to ketones, which set up the formation of the salicylaldehyde moiety in 10. The trace amounts of 9 in the VirA/B/D or VirA–D strains likely came from alcohol dehydrogenation of 10 catalyzed by an endogenous enzyme in the host, since when VirG was added to the VirA–D strain exclusive formation of 9 was observed.
When the P450 VirE was introduced to the VirA–D/G strain, the hydroquinone 8 was made. Finally, the VirA–I strain afforded 14, suggesting the involvement of VirH (cupin domain-containing protein) and VirI (flavin-dependent oxidoreductase) in the oxidation of 8 to quinone and epoxidation to form 14. Since the VirA–L strain produced 7, the two remaining SDR enzymes (VirK and VirL) were proposed to be responsible for reducing the two ketones in 14.
Decarestrictine C1 (15).
Decarestrictine C1 (15), originally isolated from a Penicillium species, contains a 10-membered lactone.64 A BGC (dcs) responsible for the biosynthesis of 15 was identified from the genome of Beauveria bassiana ARSEF 2860 (Figure 5A).65 The dcs cluster encoded an HR-PKS (DcsA), a thioesterase (TE, DcsB), a P450 (DcsC), an SDR (DcsD), and an FMO (DcsE). Heterologous expression of the HR-PKS DcsA and TE DcsB in A1145 produced diplodialide B (16) (Figure 5B and Table S3), indicating that DcsA and DcsB were sufficient to synthesize the nonanolide. Coexpression of the P450 DcsC with DcsA/B led to the disappearance of 16 and no accumulation of other metabolites. Additional coexpression of the SDR DcsD led to the formation of 15. It was proposed that DcsC oxidized the allylic C6 position of 16 to form 17, which was metabolized by the host. DcsE was not found to be required for the biosynthesis of 15, so its function remained undetermined.
Remarkably, DcsB was shown to have broad substrate promiscuity to form medium-ring lactones. The yields of lactonization for linear substrates of different sizes (7–13 membered) range from 49% to 82%. Compared to a five-membered ring, lactonization of medium-sized rings is synthetically challenging. The characterization of enzymes such as DcsB that perform medium-ring lactonization could facilitate chemoenzymatic preparation of desired lactones.
Herquline A (18).
Herquline A (18) is a strained pentacyclic alkaloid isolated from Penicillium herquei with weak antiplatelet activity.66 Forging strained molecules is of great interest to synthetic chemists, and understanding nature’s strategy may lead to their biomimetic total syntheses. A six-gene BGC (hql) encoding an NRPS (HqlA), a P450 (HqlC), an N-MT (HqlE), and three SDRs (HqlB, -D, and -F) was identified in the genome of P. herquei (Figure 6A).67 Activation of all six genes (hqlA–F) in A1145 produced 18 and herquline B (19) (Figure 6B and Table S4A).
Expression of HqlA and HqlB afforded 20, which, when fed to an HqlC–F-expressing strain, yielded back 18 and 19 (Table S4A and S4B). Expression of HqlA–C produced 21, implicating HqlC in the formation of the strained biaryl linkage. A mechanism was proposed involving the aromatization of the dicyclohexadienone intermediate 22 to 21. However, feeding 21 to a HqlC–F-expressing strain did not give the final product 18, suggesting that 21 was a shunt product. Indeed, expression of HqlA–C/F generated 20, 23, 24, and 25, but not 21. Feeding 23 (3R,3′R) to a HqlC–F-expressing strain produced its diastereomer 24 (3S,3′S) along with 18 and 19, while feeding 24 to the same strain produced no new compounds. This result suggested that 23 was on-pathway, whereas its stereoisomer 24, which was calculated to be thermodynamically more stable, was not. Conversion of 23 to 19 was catalyzed by the N-MT HqlE as validated by recombinant HqlE.
Interestingly, the transformation of 19 to 18 was observed spontaneously, and the exclusion of HqlD, the only unassigned enzyme in the cluster, from the HqlA–F strain had no effect on the production of 18 and 19. These data suggested that the stereoselective cyclization of 19 to 18 involving the creation of three chiral centers was nonenzymatic. Notably, the 3R,3′R configuration in 19 appeared to have been locked by methylation, setting the stage for the subsequent steps to 18. Tautomerization would convert 19 to 26 (1R), which was shown to be more stable than its 1S stereoisomer by free energy calculations. Michael addition from the α phase of the cyclohexanone would produce 27 (1R,2S), which then tautomerized to the more stable (1R,2S,3R) configuration in 18. The nonenzymatic cascade after methylation of the piperazine core in 23 was fascinating and could serve as biomimetic inspiration for total synthesis.
FR901483 (28).
FR901483 (28) is a potent immunosuppressant isolated from Cladobotryum sp. No. 11231.68 Its mode of action, which was proposed to be the inhibition of purine nucleotide biosynthesis, is distinct from that of cyclosporine A and FK-506.
Structurally, 28 has a rigid 2-azabicyclo[3,3,1]nonane core, a spiro-fused pyrrolidine, and a phosphate ester at C4′ (Figure 7A). Since 28 contains a phosphate moiety, the phosphotransferase PsiK from the psilocybin biosynthesis pathway was used as a query to search for candidate BGCs of 28 in the genome of Cladobotryum sp. No. 11231.69 The hits were then refined by scanning for a nearby NRPS-encoding gene since 29 and (S,S)-dityrosyl-piperazine (30) (Figure 7B) were proposed to be key intermediates of 28 (see Figure 6 as an example).
Figure 7.
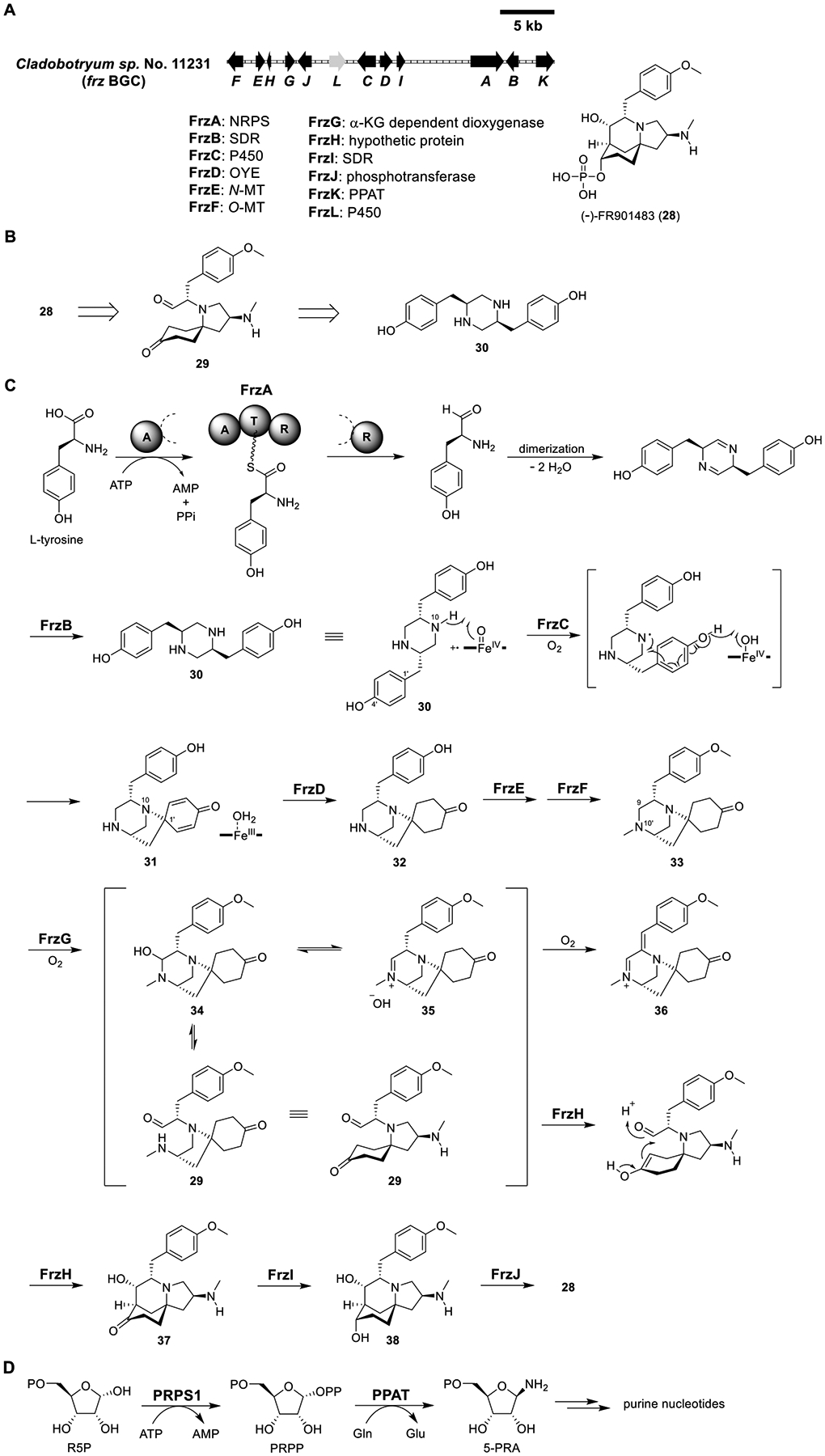
Total biosynthesis of FR901483 (28). (A) Organization and predicted gene functions of the frz BGC. Black ORFs encode for enzymes involved in the biosynthesis of 28. (B) Retrosynthetic analysis of 28. (C) Proposed biosynthesis of 28. (D) De novo purine biosynthesis.
The frz cluster was identified (Figure 7A), which contained 12 genes including a single-module NRPS (FrzA), two SDRs (FrzB and FrzI), two P450 (FrzC and FrzL), two MT (FrzE and FrzF), an old-yellow enzyme (OYE) like ene-reductase (FrzD), a nonheme, iron- and α-ketoglutarate (αKG)-dependent oxygenase (FrzG), a phosphotransferase (FrzJ), a hypothetic protein (FrzH), and a phosphoribosylpyrophosphate amidotransferase (PPAT, FrzK). The predicted functions of the encoded protein were consistent with the structure of 28.
As expected, heterologous expression of FrzA (58% sequence identity to HqlA, Figure 6) and FrzB (64% sequence identity to HqlB, Figure 6) in A1145ΔSTΔEM produced 30 (Figure 7C and Table S5). Coexpression with the P450 FrzC (45% sequence identity to HqlC, Figure 6) delivered 31, which contained a new N10–C1′ bond. The proposed reaction mechanism is shown in Figure 7C. Based on the structure of the proposed intermediate 29, the dienone portion of 31 must be reduced to cyclohexanone. The most likely enzyme for this transformation was FrzD, the OYE-like ene-reductase, and coexpression of FrzA–D indeed led to 32. Additional coexpression with FrzE and FrzF, which were predicted to be N-MT and O-MT, respectively, produced 33. The order of the two methylation steps appeared to be interchangeable, as adding either MT alone resulted in monomethylated products. To obtain the aldehyde 29, the C9–N10′ bond in 33 must be cleaved, likely via the hemiaminal intermediate 34. There were two enzyme candidates, the P450 FrzL and the αKG FrzG, for the hydroxylation of C9 in 33. Only coexpression of FrzG with FrzA–F produced a new product, 36, which was a possible shunt product from the oxidation of the dehydrate imine 35 that existed in equilibrium with 34 and 29.
For the next aldol reaction, there was no Frz candidate with sequence similarity to known aldolases. Interestingly, the hypothetical protein FrzH showed low sequence identity to ketosteroid isomerase (KSI), which catalyzes olefin isomerization of 3-oxo-Δ5 ketosteroids to the Δ4-enone isomers in steroid biosynthesis. The mechanism involves the formation of a dienolate stabilized by the KSI. Coexpression of FrzH with FrzA–G afforded 37, establishing FrzH to be an aldolase that likely acts through enolate intermediate formation. Additional coexpression of the SDR FrzI in a FrzA–H expressing strain led to the production of 3 mg/L of dephospho-FR901483 (38).
Surprisingly, when the phosphotransferase FrzJ was coexpresed with FrzA–I, neither 28 nor 38 was detected. Since 28 was proposed to inhibit purine nucleotide biosynthesis, it is possible that 28 was toxic to the host, which led to either its degradation or transcriptional repression of all or some of frzA–J. Interestingly, frzK encoded the only copy of PPAT in Cladobotryum sp. No 11231. PPAT catalyzes the first step of de novo purine biosynthesis in which 5-phosphoribosyl-1-pyrophosphate (PRPP) is converted to 5-phosphoribosyl-1-amine (5-PRA) (Figure 7D). FrzK could be, as aforementioned in the Introduction, a self-resistance enzyme encoded in a BGC to protect the natural producer.17 If so, PPAT could be the protein target of 28. If 28 was made by the FrzA–J strain, coexpression with FrzK could protect the host from its toxicity. Indeed, coexpression of FrzK with FrzA–J restored the production of 38 along with the accumulation of 28. In vitro assay using purified FrzJ, ATP, and MgCl2 in the presence of 38 also led to the formation of 28, confirming the phosphotransferase activity of FrzJ. Coexpression of the second P450 FrzL with FrzA–K did not deliver a new product, so the function of FrzL remained unknown. Although the function of FrzK as a resistance enzyme awaited biochemical validation, total biosynthesis of 28 was successfully achieved.
Quinolactacin A (39).
Quinolactacin A (39) is a quinolone-γ-lactam hybrid fungal natural product isolated from a Penicillium sp. It exhibits a broad spectrum of bioactivities, including the inhibition of TNF (tumor necrosis factor) production from stimulated macrophages.70–72 By analyzing the distribution patterns of 13C atoms in 39 from feeding experiments with 13C-labeled substrates, l-isoleucine and l-kynurenine were proposed to be precursors of 39.70 In addition, the N-methyl group was found to be derived from methionine, an installation likely catalyzed by a SAM-dependent MT. Guided by this information, Zhao et al. and Liu et al. independently identified the BGC of 39 from P. citrinum ATCC 9849 (qul) and DSM1997 (qlt), respectively (Figure 8A).71,72 Both BGCs have similar gene organization, including genes encoding two single-module NRPSs (QltA/QulA and QltB/QulB), an FMO (QltD/QulF), an MT(QltE/QulM), and an indoleamine-2,3-dioxygenase (IDO, QltC/QulI).
Heterologous expression of QltA–E in A1145ΔEM produced 39 (Figure 8B and Table S6).72 While excluding QltA, B, D, or E abolished the production of 39, excluding QltC, a putative IDO, did not alter the titer. These data suggest that the housekeeping IDO in A. nidulans could be complementing the function of QltC in converting tryptophan to kynurenine. Similarly, except for qulI, which putatively encodes for an IDO, deletion of each of the individual genes in the qul cluster abolished the production of 39.71
The biosynthesis of 39 was established by biochemical studies71 as well as reconstitution of the pathway in vitro, i.e., total enzymatic synthesis72 (Figure 8B). Incubation of l-kynurenine with all five enzymes (QltA–E) and necessary cofactors did not yield 39; instead, shunt product 40 was observed. It was then realized that an endogenous amidase (GmdS) was essential to completing the biosynthesis. GmdS hydrolyzed the terminal amide in 41 to an β-keto acid 42, allowing 42 to be loaded onto the tridomain NRPS QltB. The tetra-domain NRPS QltA activated and loaded l-isoleucine and catalyzed condensation with 42-S-QltB to yield 43. Finally, intramolecular Dieckmann cyclization and subsequent dehydration resulted in 39.
Varicidins A (44) and B (45).
An emerging class of fungal Diels–Alderase has sequence homology to lipocalin-like proteins which bind to steroids and other hydrophobic molecules (see the biosynthesis of Sch 210972 in Figure 3).59,73 Using lipocalin-like fungal Diels–Alderase as a search query, a BGC (pvh) from Penicillium variabile was identified (Figure 9A).74 Heterologous expression of PvhA–E in the A1145 strain produced varicidin A (44) (Figure 9B and Table S7). Excluding the N-MT PvhD resulted in varicidin B (45). The cis-decalin moieties of 44 and 45 were presumably formed by exo-IMDA (intramolecular Diels–Alder) reactions, which were most likely catalyzed by the lipocalin-like Diels–Alderase PvhB. Expression of the PKS-NRPS PvhA and its partnering trans-ER PvhC produced 46, 47, and 48. Based on the biosynthesis logic of decalin-containing natural products, the polyketide acyl chain was likely synthesized by the PKS portion of PvhA and the partnering trans-ER PvhC. The polyketide chain was then condensed with l-isoleucine and underwent Dieckmann cyclization to yield a released 46. When 46 was left in a pH = 7.0 buffer, the trans-decalin 47 and 48 along with other minor compounds were detected, indicating nonenzymatic endo-IMDA formation of 47 and 48 from 46.
Coexpression of PvhB with PvhA/C did not change the metabolite profile, indicating that 46 was not a substrate of PvhB. Coexpression of the P450 PvhE with PvhA/C produced 49, confirming the function of PvhE in catalyzing oxidation of the terminal methyl group. Coexpression of PvhD with PvhA/C/E yielded 50. These results elucidated the biosynthesis of 44 and 45. Notably, installing the electron-withdrawing carboxylate in 49 (and 50) significantly suppressed nonenzymatic IMDA reactions. This phenomenon could be explained by the electrostatic repulsion between the two anions in neutral pH solution. In addition, the electron-deficient diene and electron-deficient dienophile increased the reaction barrier, which was overcome by the Diels–Alderase PvhB.
Leporin B (51).
Leporins are 2-pyridone metabolites that exhibit antiinsectan and antifeedant activities from Aspergillus species (Figure 10A).75,76 Based on the structure of leporin B (51), Ohashi et al. hypothesized that the leporin BGC lep (previously identified and verified in A. flavus76) encodes for an enzyme that can catalyze the hetero-Diels–Alder (HDA) reaction to construct the heterocycle dihydropyran core of 51.77 To identify the HDA-catalyzing enzyme, they heterologously expressed the six lep genes in A1145. Similar to the biosynthesis of tenellin,45 three enzymesa PKS-NRPS LepA, a partnering trans-ER LepG, and a ring-expansion P450 LepHwere shown to be sufficient to construct the 2-pyridone core, as evidenced by the emergence of compound 52 (Figure 10B and Table S8). Additional coexpression of the SDR LepF, which was hypothesized to reduce 52 to 53, led to a mixture of HDA and IMDA products (54–58). Among these products, 54 and 55 were proposed to derive from the quinone methide (E)-59, while 56–58 were from (Z)-59. There results highlighted the need for enzymatic stereoselectivity for the dehydration of 53 to (E)-59 and the subsequent HDA reaction to the desired compound 54.
Figure 10.
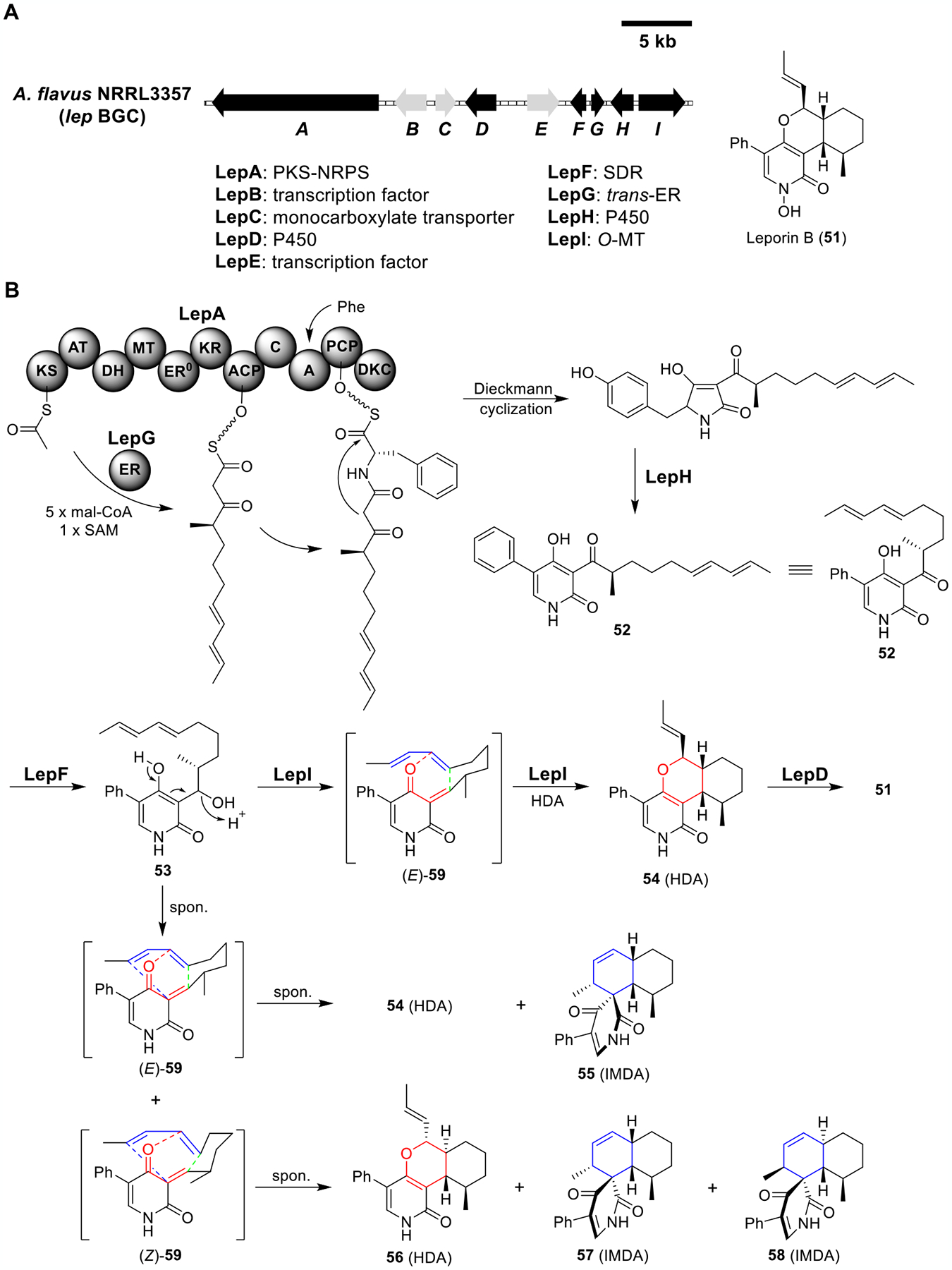
Total biosynthesis of leporin B (51). (A) Organization and predicted gene functions of the lep BGC. Black ORFs encode for enzymes involved in the biosynthesis of 51. (B) Proposed biosynthesis of 51.
Surprisingly, coexpression of LepI, a predicted O-MT with a conserved SAM binding site, in the LepA/F/G/H strain led to the production of 54 exclusively. These data, along with biochemical assays and computational calculations, provided evidence that LepI was responsible for the stereoselective pericyclic transformation of 53 to 54. Interestingly, SAM was essential for the activity of LepI in vitro, indicating the versatility and importance of SAM in metabolism. Lastly, addition of the P450 LepD yielded the final product leporin B (51).
Ilicicolin H (60).
Ilicicolin H (60) was first isolated from Cylindrocladium ilicicola strain MFC-870.78 It exhibits potent and broad antifungal activities by inhibiting the cytochrome bc1 complex.79 Based on the 4-hydroxy-2-pyridone moiety in the structure of 60, a five-gene BGC (ncc) in the genome of a producing fungus Nectria sp. B-13 was identified (Figure 11A).80 Searching for the homologues of this five-gene BGC in the NCBI database resulted in 41 different fungal strains minimally containing these five genes. Among them, the cDNA library of Penicillium variabile was available to the Tang group; therefore, the BCG icc from P. variabile was used for the study (Figure 11B and Table S9).
Figure 11.
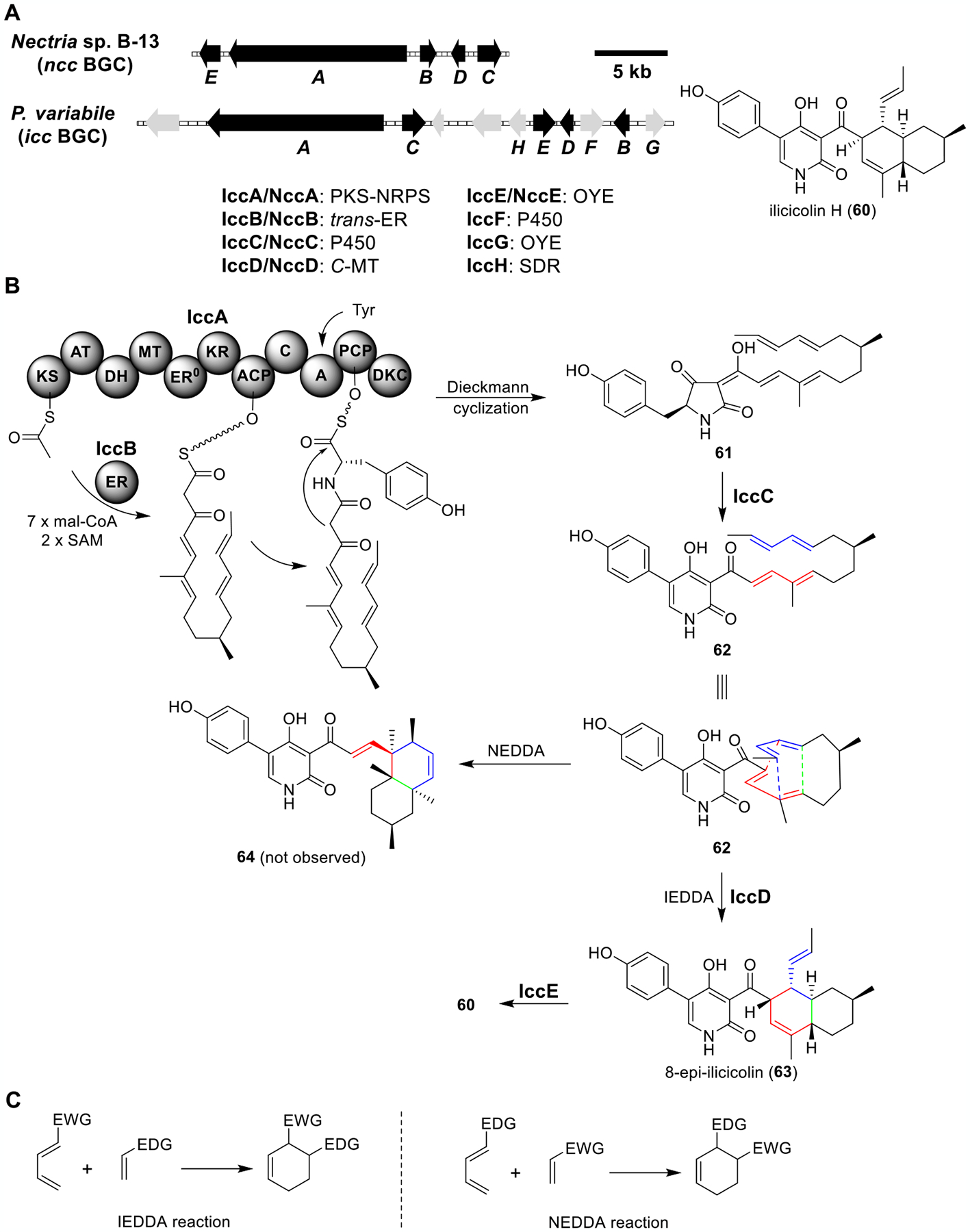
Total biosynthesis of ilicicolin H (60). (A) Organization and predicted gene functions of the ncc and icc BGCs. Black ORFs encode for enzymes involved in the biosynthesis of 60. (B) Proposed biosynthesis of 60. (D) Inverse-electron-demand Diels–Alder (IEDDA) and normal-electron-demand Diels–Alder (NEDDA) reactions. EWG: electron-withdrawing group. EDG: electron-donating group.
As expected, expression of the PKS-NRPS IccA and partnering trans-ER IccB produced 61. Further coexpression of the ring-expansion P450 IccC afforded 61, 62, and trace amounts of 8-epi-ilicicolin H (63). Adding IccD to the IccA–C strain produced 61 and 63, confirming that IccD was a Diels–Alderase. The IccA–E expression strain produced 60, suggesting that IccE was an epimerase. These results indicated that 62 was relatively unreactive under culturing conditions, requiring the Diels–Alderase IccD to produce the trans-decalin 63. IccD selectively catalyzed the inverse-electron-demand Diels–Alder (IEDDA) but not the normal-electron-demand Diels–Alder (NEDDA) reaction to afford 64 (Figure 11B and C). Moreover, unlike LepI in the leporin B biosynthesis pathway (Figure 10), biochemical analysis indicated that IccD-catalyzed IEDDA was SAM-independent. This work expanded the catalytic repertoire of naturally occurring pericyclases.
Harzianopyridone (65) and Atpenin B (66).
Harzianopyridone (65) and atpenin A5 (67) structurally resemble the electron carrier ubiquinone (coenzyme Q, Figure 12A) and are nanomolar inhibitors of mammalian succinate ubiquinone oxidoreductase (mitochondrial complex II).81 Isotope labeling studies suggested that 65 was derived from a polyketide-amino acid-containing tetramic acid.82 This knowledge, together with the fact that dephenylated 2-pyridone is a shunt product of the aspyridone A biosynthesis pathway,83 allowed researchers to identify a BGC (har) encoding several enzymes homologous to those encoded in the aspyridone A BGC from the genome of Trichoderma harzianum, a producer of 65 (Figure 12A).84 This BGC also encoded proteins homologous to those in the lep BGC (Figure 10), such as the PKS-NRPS HarA, trans-ER HarE, ring expansion P450 HarG, and N-hydroxylase HarD. In addition, two FMOs (HarC and HarF) and an O-MT (HarB) were also encoded in har. The predicted functions of genes in har were consistent with the structure of 65, except for the putative N-hydroxylase (HarD).
Figure 12.
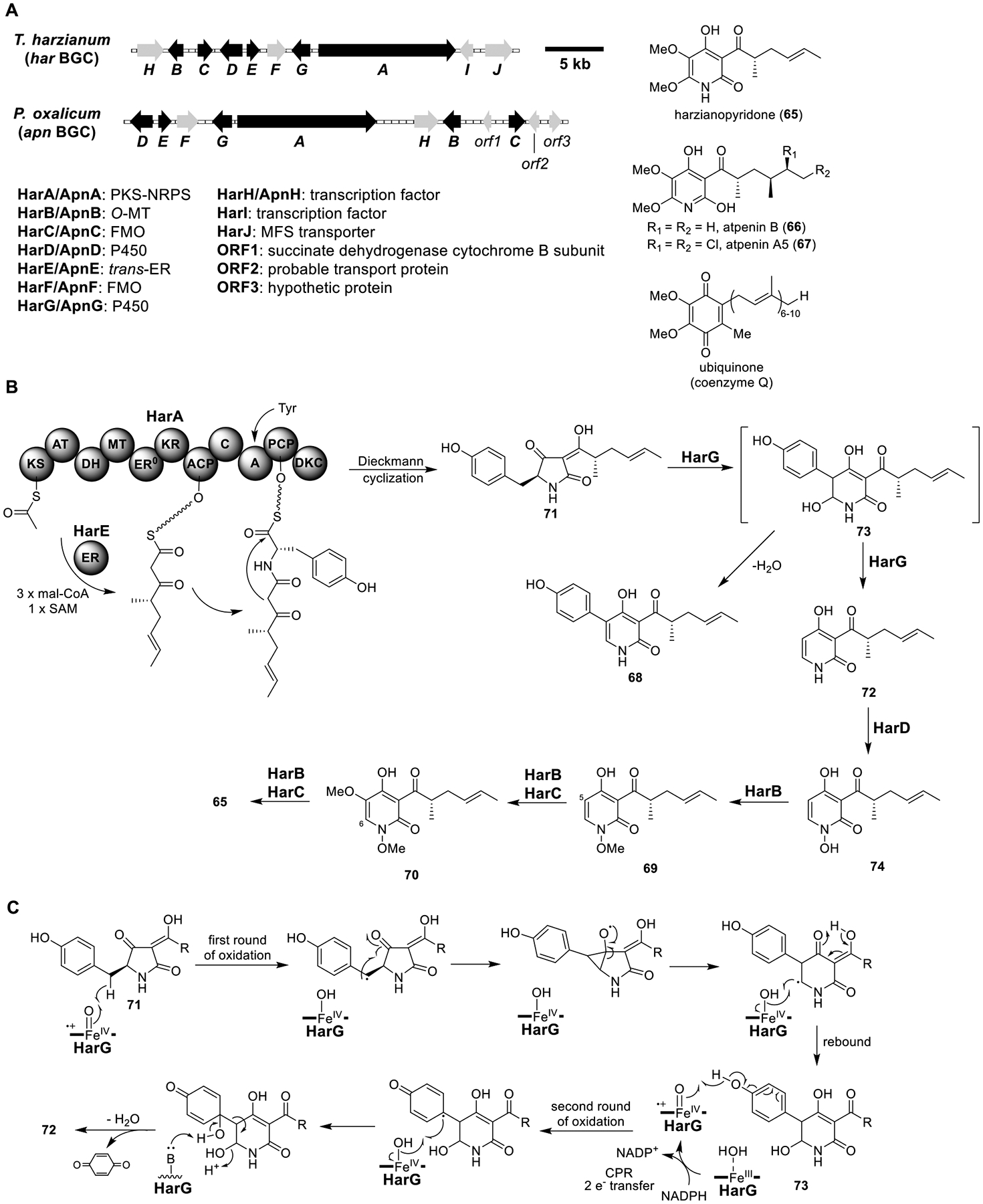
Total biosynthesis of harzianopyridone (65) and atpenin B (66). (A) Organization and predicted gene functions of the har and apn BGCs. Black ORFs encode for enzymes involved in the biosynthesis of 65 and 66 in the har and apn BGCs, respectively. (B) Proposed biosynthesis of 65. (C) Proposed mechanism of ring expansion and phenyl cleavage for the formation of 72 via radical intermediates.
Heterologous expression of the seven genes harA–G in the A1145ΔEM strain yielded 65 along with 68 and two N-methoxylated compounds, 69 and 70 (Figure 12B and Table S10). The presence of an N-methoxy in 69 and 70 was surprising, as 65 did not contain this moiety, although this finding agreed with the function of HarD. To elucidate the biosynthesis of 65, different combinations of har genes were expressed. The HarA/E expression strain produced 71, consistent with the assigned function of the two enzymes. The HarA/E/G strain produced 68, 71, and 72, indicating HarG was responsible for both ring expansion and phenyl cleavage. Feeding 68 to the HarG expression strain did not lead to the cleavage of the phenyl moiety, suggesting that 68 was a shunt product. A possible reaction mechanism from 71 to 72 via radical intermediates was proposed (Figure 12C). After ring expansion, dehydration of the 6-hydroxy-dihydro-pyridone (73) led to 68. If the P450 heme-iron could be reduced prior to dehydration, HarG could further catalyze oxidation of the phenyl, which could lead to the loss of the quinone and give 72.
To determine the functions of the remaining enzymes, HarB, -C, and -D were individually coexpressed with HarA/E/G. Only coexpression of HarD with HarA/E/G led to the production of a new product, N-hydroxy pyridone 74. Further coexpression of HarB led to the emergence of 69 and 70. While methylation of 74 to 69 fitted with the predicted function of HarB, methoxylation of 69 to 70 was not expected. 65 was produced when HarC was coexpressed with HarA/B/D/E/G strain, demonstrating HarF was not essential for the biosynthesis of 65.
To further clarify the har biosynthetic pathway, the authors conducted feeding experiments. Feeding 69, 70, or 74 to yeast expressing HarB/C led to the formation of 65, indicating 69, 70, and 74 were bona fide biosynthetic intermediates. However, 72 was not consumed by the HarB/C expression yeast strain. Therefore, N-methoxylation was required for the formation of 65. Moreover, feeding 74 to yeast expressing HarB only produced 69, indicating that emergence of 70 in A. nidulans expressing HarA/B/D/E/G was likely due to crosstalk with endogenous monooxygenases. These results also demonstrated that HarB was responsible for all methylation reactions and HarC was required for C-5 and C-6 hydroxylation.
Guided by the har BGC, a homologous cluster (apn) in Penicillium oxalicum, an atpenin A5 (67) producer, was identified (Figure 12A). Heterologous expression of ApnA–E/G in A. nidulans led to the production of atpenin B (66) at 0.3 mg/L. The apn BGC contained additional genes possibly involved in the chlorination steps.
Citridone B/B′ (75/75′).
Citridones are a family of pyridone-containing fungal natural products that can potentiate the activity of miconazole against Candida albicans.85 The pyridone moiety in citridones suggested a PKS-NRPS origin. By using PKS-NRPS and ring expansion P450 sequences as queries, the citridone BGC (pfp) was identified from the genome of the natural producer Penicillium FKI-1938 (Figure 13A).86 Besides the PKS-NRPS PfpA and ring expansion P450 PfpB, pfp also encoded a SDR PfpC and the two additional P450s PfpD and PfpE. The absence of a trans-ER enzyme indicated a possible polyene product from PfpA.
Heterologous expression of PfpA in A1145 produced 76 (Figure 13B and Table S11). Coexpression of PfpA with PfpB was expected to form the ring expansion product 77, but instead the major metabolite detected was (±)-citridone E (78), a Michael addition product of 77. The formation of 78 was attributed to the two methyl substitutions at C-8 and C-10, which caused a nonplanar conformation of the diene due to steric repulsion. Disruption of the conjugation facilitated the Michael addition. Coexpression of PfpA–C produced (±)-citridone A (79) and (±)-tersone D (80). PfpC belonged to the SDR superfamily, and its homologues in other 2-pyridone pathways reduce the C-7 ketone to alcohol, which can dehydrate spontaneously to form E or Z ortho-quinone methide (see LepF in Figure 10 as an example). Therefore, PfpC was proposed to catalyze the reduction of 77 to 81, which then dehydrated to (E)-82 and (Z)-82. (E)-82 and (Z)-82 could undergo electrocyclization to produce C4-O-pyranopyridone 80 or C2-O-pyranopyridone 84, respectively. E–Z isomerization of (E)-82 to 83 set up a nonenzymatic cycloisomerization to give 79. The production of 79 and 80 but not 84 in the PfpA–C strain suggested that 79 and 80 are the thermodynamically favored products.
To produce the C2-O-furopyridine moiety in 75/75′, enzymatic reaction on the thermodynamically unfavored intermediate 84 was necessary to push the pathway forward. Considering the appearance of a quaternary carbon (C-8) connected to the pyridine ring in 75/75′, a rearrangement involving the formation of an epoxide at the C-7–C-8 olefin was proposed (Figure 13C). Coexpression of PfpA–D produced CJ-15696 (85) and CJ-16173 (86), along with decreased levels of previously observed metabolites. Although the absolute configurations at C-8 and C-9 were not determined, both 85 and 86 were enantiomerically pure. Since the putative substrate 84 was a racemate, PfpD must stereospecifically epoxidize the 9S-84 isomer so that after carbon–carbon rearrangement 85 could be formed (Figure 13C). Hydroxylation of C-14 in 85 by PfpD or an endogenous oxidase, followed by hemiacetal formation, yielded 86. Lastly, coexpression of all five enzymes PfpA–E produced 75/75′, isolated as a mixture. Feeding 85 to the PfpE expression strain also produced 75/75′. Therefore, PfpE was proposed to stereospecifically epoxidize 85. After water-mediated epoxide opening, 75/75′ were formed.
Fusaric Acid (87).
Fusaric acid (87) is a mycotoxin isolated from Fusarium species that exhibits strong phytotoxicity and causes wilt symptoms.87 The BGC of 87 has been identified in several Fusarium species, and gene knockout studies indicated that five genes (fub1, fub4, and fub6–8) were essential for its biosynthesis (Figure 14A).88 To characterize the biosynthesis of 87, all five essential genes were reconstituted in the A1145ΔEM strain.89 However, instead of 87, an unstable compound with m/z = 184 ([M + H]+, proposed structure 88) was detected (Figure 14B and Table S12). To support the proposed structure of 88, the crude 88 was reduced by sodium cyanoborohydride, which led to the identification of (2S,5S)-and (2S,5R)-5-butyl-l-pipeoclic acid (89 and 90). This result implied that 88 was epimerized at C-5, presumably through tautomerization. The production of 88 from heterologous expression of the five essential genes indicated that the enzyme responsible for the last oxidation step was missing. An FMN-dependent oxidase encoded in the fub BGC, Fub9, seemed like a plausible candidate, and, indeed, its coexpression with Fub1/4/6/7/8 led to the accumulation of 87. This discrepancy with the gene inactivation studies suggested that an endogenous pathway in Fusarium compensated for the activity of Fub9.
The functions of Fub6-9 were biochemically characterized. Recombinant Fub8, a predicted NRPS-like carboxylic acid reductase, reduced trans-2-hexenoic acid (91) to trans-2hexenal (92) in the presence of ATP, NADPH, and MgCl2. Incubation of Fub6, a putative medium-chain dehydrogenase/reductase, with 92 and NADPH afforded n-hexanal (93), indicating that it was an ene-reductase. Incubation of Fub7, a predicted PLP-dependent enzyme, with 93 and O-acetyl-l-homoserine (OAH) afforded 88, showing that Fub7 catalyzed C–C bond formation. As expected, its activity was abolished when Fub7 was pretreated with hydroxylamine, an inhibitor for PLP-dependent enzymes. Lastly, including Fub9 in the Fub7 reaction mixture using 93 and OAH as substrates produced 87. Biochemical characterization of Fub6-9 allowed total enzymatic synthesis of 87 in one pot from 91 and OAH in the presence of Fub6-9 and the necessary cofactors.
Thermolides A–E (94–98).
Thermolides A–E (94–98) (Figure 15A) are 13-membered macrolactones discovered from the extreme thermophilic fungi Talaromyces thermophilus YM 3–4.90 Thermolides A (94) and B (95) exhibit potent toxicity toward nematodes with LC50 value of 0.5–1 μg/mL. Genome sequenced strain T. thermophilus NRRL 2155 was found to produce 94–98, providing the opportunity to identify the BGC of Thermolides.91 Based on the structures of 94–98, a seven-gene BGC (thm) encoding an HR-PKS (ThmA), a single-module NRPS (ThmB), an SDR (ThmC), a flavin-dependent oxidase (ThmD), an acetyltransferase (ThmE), a glycosylhydrolase (ThmF), and a glycotransferase (ThmG) was identified (Figure 15A).92
Figure 15.
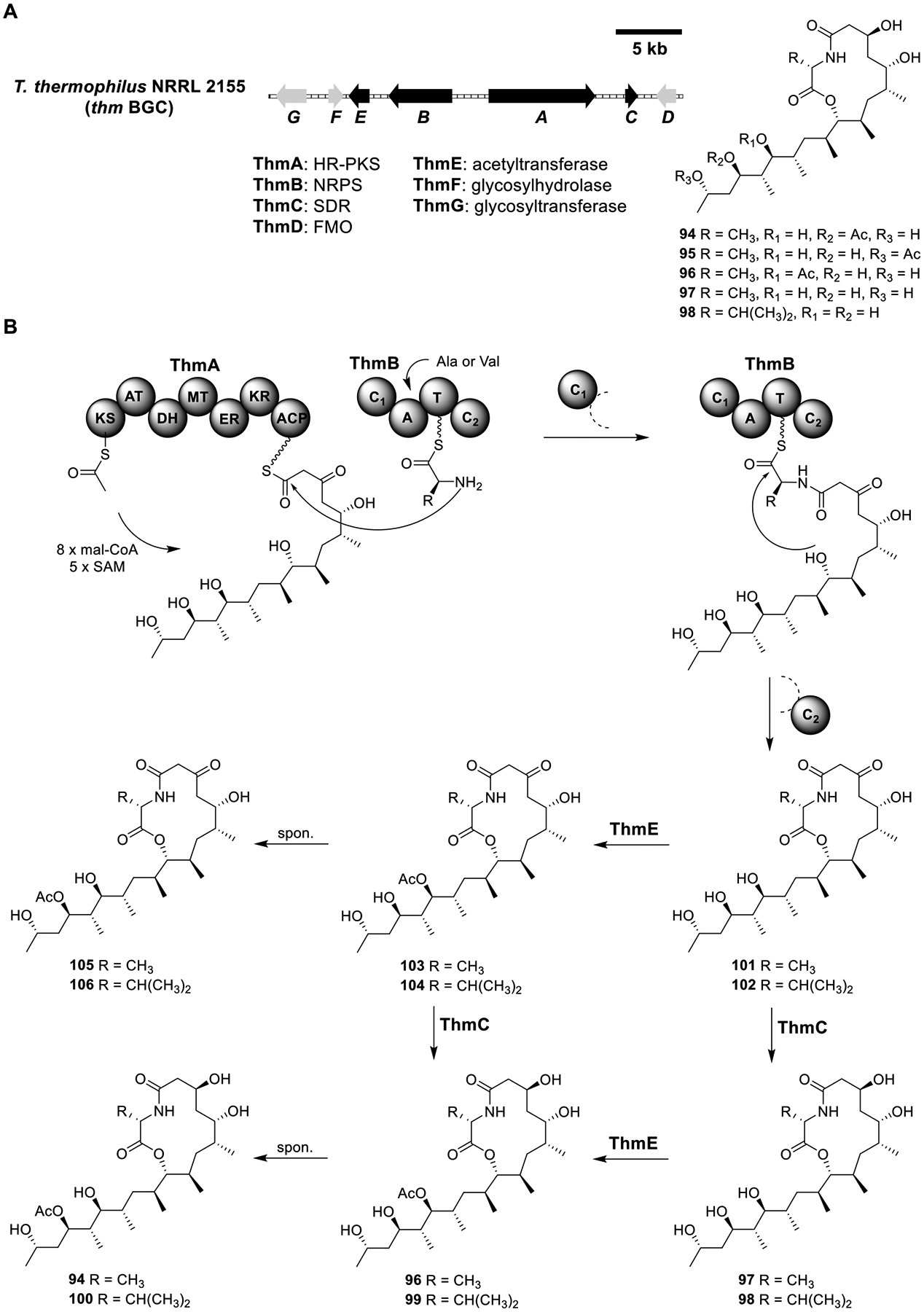
Total biosynthesis of thermolides A–E (94–98). (A) Organization and predicted gene functions of the thm BGC. Black ORFs encode for enzymes involved in the biosynthesis of thermolides. (B) Proposed biosynthesis of thermolides.
Heterologous expression of ThmA–G in A1145 produced 94, 96, 99, and 100 (Figure 15B and Table S13). To investigate the functions of the Thm enzymes, different combinations were expressed. Heterologous expression of the HR-PKS ThmA and NRPS ThmB produced 101 and 102, indicating that ThmA and -B were sufficient to synthesize the backbone. Coexpression of ThmC with ThmA/B produced 97 and 98, while coexpression of ThmE with ThmA/B produced 103–106, indicating that both 101 and 102 can be the substrate of the SDR ThmC and the acetyltransferase ThmE. Conversion of 103 to 105 and 104 to 106 were likely spontaneous since 106 could be formed within 12 h in a pH = 7.5 Tris buffer containing 104. The ThmA–C/E strain produced 94, 96, 99, and 100, demonstrating that only four genes were required to complete the total biosynthesis.
Beauveriolides I (107) and III (108).
Cordyceps militaris is an entomopathogenic fungus that has been widely used in traditional Chinese medicine for centuries.93 Genome mining of C. militaris CM01 led to the identification of the cm3 BGC encoding an NRPS (Cm3A), an HR-PKS (Cm3B), an acyltransferase (Cm3C), and an acyl-CoA synthetase (Cm3D)94 (Figure 16A). Heterologous expression of Am3A–D in the A1145 strain produced beauveriolides I (107) and III (108) (Table S14). Beauveriolides have been identified from Beauveria species and were found to inhibit acyl-CoA:cholesterol acyltransferases.95 The biosynthesis pathway of 107 was proposed as shown in Figure 16B. The HR-PKS Cm3B iteratively catalyzes the formation of the linear polyketide, which is then transferred to the first T domain of the NRPS Cm3A facilitated by Cm3D and Cm3C. Cm3A then activates and incorporates l-phenylalanine, l-alanine, and l-leucine into the growing peptide chain followed by intramolecular cyclization to produce 107. The intramolecular cyclization is catalyzed by the ester-bond-forming C domain in the C terminal of Cm3A.96 However, considering that the leucine and allo-leucine residues in 107 and 108, respectively, are both d form and the fact that epimerization (E) domains show sequence and structure homology to C domains,96 the domain architecture of the last module of Cm3A is likely A-T-E-C rather than A-T-C-C as originally proposed.
Mycophenolic Acid (109).
Mycophenolic acid (109) isolated from Penicillium brevicompactum is an active component of immunosuppressants used in organ transplantations. It is a potent inhibitor of inosine-5′-monophosphate dehydrogenase (IMPDH), a rate-limiting enzyme in guanosine monophosphate (GMP) biosynthesis.97 The mpa BGC responsible for producing 109 had been identified from P. brevicompactum (Figure 17A).98 The mpa BGC encoded an NR-PKS (MpaC), a prenyltransferase (MpaA), a globin-like enzyme (MpaB) with unknown function, a P450 (MpaD), a hydrolase (MpaE), an IMPDH (MpaF, a putative self-resistance enzyme), an O-MT (MpaG), and a putative protein (MpaH) containing an α/β-hydrolase fold. mpaD and mpaE were later demonstrated to be transcribed as a single gene that encoded a single polypeptide MpaDE, and coexpression of MpaC and MpaDE in A. nidulans produced 5,7-dihydroxy-4-methylphthalide (110) (Figure 17B).99
The mpa BGC was also identified from P. griseofulvum, a high producer of mycophenolic acid.100 The mpa clusters from the two strains showed high similarity/identity (Figure 17A). Reconstitution of PgMpaA/C/DE in A1145 produced 110 and demethylmycophenolic acid (111) (Figure 17B and Table S15). Feeding 112 to A1145 produced 111, indicating that the oxidative cleavage step from 112 to 111 in A1145 was an mpa cluster-independent process. PgMpaA was a membrane-bound prenyltransferase. Incubation of 110, FPP, Mg2+, and PgMpaA prepared from the microsome fraction of the PgMpaA-expressing yeast strain produced 112, which confirmed that MpaA catalyzed the regioselective farnesylation. When 112 was incubated with the cell-free lysate containing PgMpaB, 113–115 were detected. 114 and 115 were likely derived from 113 in the presence of cell-free lysate. Therefore, 113 was proposed to be the real product from 112 catalyzed by PgMpaB. Feeding 113 to the P. griseofulvum mpaCΔ mutant restored the biosynthesis of 109, confirming 113 to be an intermediate in the mpa pathway. Lastly, coexpression of PgMpaA–C/DE/G produced 109, 110, and a trace amount of 112, which completed the total biosynthesis of 109.
Flavunoidine (116).
Among the known fungal NPs, the hybrid TC/NRPS core is underrepresented as suggested by the prevalence of TC/NRPS BGCs in sequenced fungal genomes.101 Yee et al. investigated one such uncharacterized nine-gene BGC (flv) from Aspergillus flavus (Figure 18A). This BGC was conserved in several fungal species and encoded two TCs (FlvE and FlvF) and one NRPS (FlvI). Heterologous expression of FlvA–I in A1145 led to the identification of the novel compound flavunoidine (116) (Figure 18B and Table S16A). The omission of FlvI resulted in the disappearance of 116 but the emergence of its component precursors 5,5-dimethyl-l-pipecolic acid (117) and 118. Feeding 117 and 118 to A1145-expressing FlvI yielded back 116 (Figure 18B and Table S16B), revealing that FlvI catalyzed their esterification.
The biosynthesis of pipecolic acid has been shown to involve a PLP-dependent enzyme and a reductase.102 FlvA was a didomain enzyme: the N-terminal domain was predicted to be a PLP-dependent lyase, while the C-terminal domain was predicted to be an α-ketoglutarate-dependent oxygenase. Thus, FlvA and FlvB (a predicted SDR) were proposed to be involved in the biosynthesis of 117. This hypothesis was validated when the FlvA/B expression strain produced only 117, the FlvC–H strain produced only 118, and the FlvA/B/I strain produced 116 from the feeding of 118 (Table S16A and S16B). The PLP-dependent lyase and oxygenase domains of FlvA were proposed to catalyze the γ-replacement and decarboxylation reactions, respectively (Figure 18B).
To examine the functions of the two TCs (FlvE and FlvF), each gene was removed from the flvC–H strain separately. The removal of flvE abolished the related metabolites, suggesting that FlvE was involved in the formation of the terpene core. The function of FlvE was confirmed by heterologous expression of the enzyme in S. cerevisiae, which afforded (1R,4R,5S)-(+)-acoradiene (119). On the other hand, the removal of flvF led to accumulation of 120a and 120b, which indicated that FlvF was involved in making the C–N bond in 118. To determine if oxidative modifications of 119 could generate the terpene core in 118, FlvC and FlvD were coexpressed with FlvE. While the FlvC/D/E strain produced 120a and 120b, the FlvD/E strain produced 121a and 121b. Moreover, the FlvD–H strain produced 122. Collectively, these results indicated that FlvD was responsible for the oxidative conversion of 119 into the tetracyclic terpene core and FlvC was responsible for the hydroxylation of the C-10.
Feeding 119 to the FlvD/F/G/H strain generated 122, while feeding 121a/121b to the same strain did not (Table S16B), confirming that 119 was a precursor and 121a/121b were shunt products. Endogenous ethanolamine might have entered the active site of FlvD and quenched the carbocation intermediate 123 to form 121a/121b. When FlvF was expressed, the quenching was suppressed and dimethyl cadaverine (124) could stereoselectively quench 123 to afford 122. To test the function of FlvF, 124 was fed to the FlvD/E and FlvD–F strains, and only the latter produced 122. The two remaining enzymes in the pathway, FlvH and FlvG, were verified to synthesize 124 from l-lysine. Removing FlvH from the FlvC–H strain, trace amounts of 118 were formed. The titer of 118 was restored upon feeding of Nε,Nε-dimethyl-l-lysine (125). When 125 was fed to the FlvC–F strain, 118 was not detected and only 120a/120b were observed. Feeding of 124 restored 118. These results indicated that FlvH methylated l-lysine to give 125, which was then decarboxylated by FlvG to afford 124. Therefore, in the flv pathway, the TC FlvE and the P450 FlvD constructed the tetracyclic core, while the second TC FlvF attached the C-13 axial dimethyl cadaverine (124). The NRPS FlvI acylated the terpenoid core with dimethylpipecolate (117).
E. coli–Yeast–Fungal Shuttle Vectors pYFAC-CH2, pYFAC-CH3, and pYFAC-CH4.
The Chooi group renamed pKW20088 to pYFAC-pyrG (Figure S2A; YFAC stands for yeast fungal artificial chromosome).103 Insertion of different alcohol-inducible promoters into pYFAC-pyrG yielded pYFAC-CH2 (Figure S2C).34 The pYFAC-CH2 vector contains an alcA promoter, a bidirectional alcS/M promoter,104 and an aldA promoter105 along with terminators in between. Replacing AfpyrG with AfriboB and AfpyroA generated pYFAC-CH3 and pYFAC-CH4, respectively. Since each vector contains four expression cassettes, this system is capable of expressing up to 12 genes in a pyrG89, pyroA4, and riboB2 mutant strain.
Elsinochrome A (126).
Perylenequinones are a group of reactive oxygen species (ROS)-generating photosensitizers that function as phytotoxins and have potential application in photodynamic cancer therapy.106 Using the YFAC-CH system, the biosynthesis of the elc pathway in the wheat pathogen Parastagonospora nodorum was elucidated (Figure 19).34 Heterologous expression of the NR-PKS ElcA in the genetically dereplicated strain LO789055 (Table 1) yielded nor-toralactone (127) (Figure 19B and Table S17).107 The product template (PT) domain of ElcA was proposed to catalyze C4–C9 and C2–C11 cyclization, while the thioesterase (TE) domain to catalyze pyrone formation.108 ElcB was a didomain enzyme containing an N-terminal O-MT and a C-terminal FMO domain.34,109 Coexpression of ElcA with the truncated ElcB containing only the O-MT domain produced toralactone (128) (Figure 19C). Coexpression of ElcA with the full-length ElcB yielded 129, 130, and unstable intermediate 131. 129 and 130 were likely derived from 132 via the glutathione S-transferase detoxification pathway in the host. Coexpression of the O-MT ElcD with ElcA/B produced the major compound 133 with a mass almost three times that of the putative product 134. The chemical structure of 133 was proposed as shown in Figure 19B but has not been confirmed.
Figure 19.
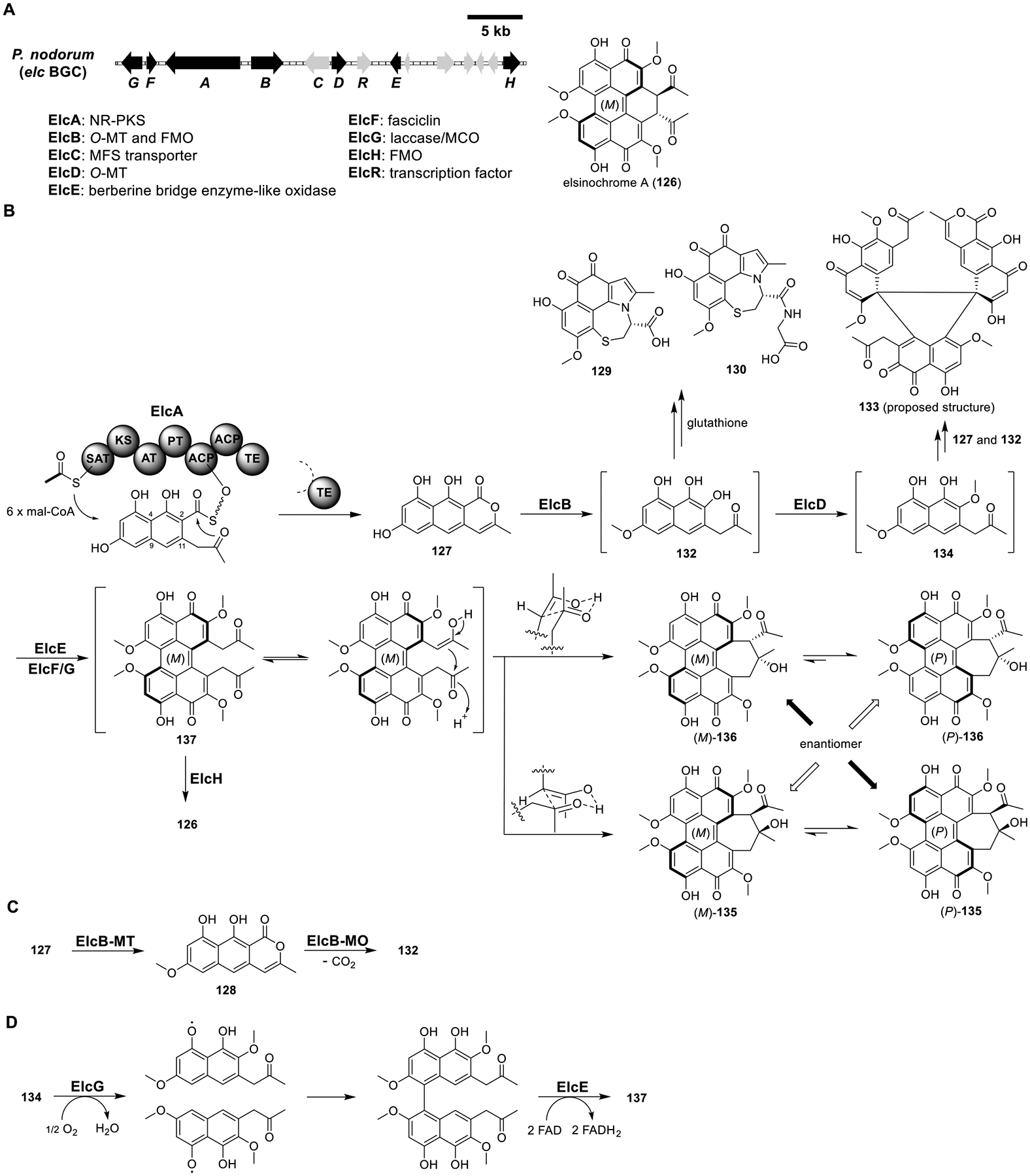
Total biosynthesis of elsinochrome A (126). (A) Organization and predicted gene functions of the elc BGC. Black ORFs encode for enzymes involved in the biosynthesis of 126. (B) Proposed biosynthesis of 126. (C) Function of the didomain enzyme ElcB. (D) Proposed biosynthesis of 137 from 134.
Coexpression of ElcA/B/D/E had little change to the metabolic profile compared to ElcA/B/D. Only the ElcA/B/D/E/F/G strain made (P)-135 (hypocrellin A) and (P)-136. The (P) helicity of 135 and 136 was confirmed by electronic circular dichroism (ECD) spectra. Intriguingly, some ElcA/B/D/E/G transformants produced less amounts of (P)-135 and (P)-136, while others produced no (P)-135 and (P)-136 at all.
Therefore, ElcE and ElcG were proposed to be involved in the dimerization of 134, leading to the formation of the pentacyclic perylenequinone core in 137 (Figure 19D). In addition, a putative FMO gene, elcH, located ~8 kb upstream of the elc cluster was found to be co-upregulated with the elc genes. Expression of ElcH in a strain producing (P)-135 and (P)-136 resulted in the formation of elsinochrome A (126), confirming the involvement of ElcH in the biosynthesis of 126. Interestingly, 126 had (M) helicity, while the isolated 135 and 136 had (P) helicity. These data suggested that 137 was first converted to (M)-135 and (M)-136 via transannular syn aldol reactions followed by spontaneous atropisomerization to afford (P)-135 and (P)-136.
(M)-Viriditoxin ((M)-138).
(M)-Viriditoxin ((M)-138) is a biaryl mycotoxin first isolated from A. viridinutans.110 (M)-138 and its derivatives possess antibacterial and cytotoxic activities.111 A BLASTp search using the NR-PKS ElcA from the elsinochrome pathway (see Figure 19) as a query led to the identification of a candidate BGC of (M)-138, vdt, from the producer Paecilomyces variotii CBS101075 (Figure 20A).112 Heterologous expression of the NR-PKS VdtA in LO7890 produced the naphtho-α-pyrone 139, the predicted core of (M)-138 (Figure 20B and Table S18). Coexpression of the O-MT VdtC with VdtA yielded 140. Coexpression of the SDR VdtF with VdtA/C resulted in 141. Next, coexpression of the Baeyer–Villiger monooxygenase (BVMO) VdtE with VdtA/C produced 142 and 143, while coexpression of VdtE with VdtA/C/F produced 144 and 145. These results confirmed that VdtE functioned as a BVMO. However, BVMOs have never been reported to possess hydrolyzing activity.
Figure 20.
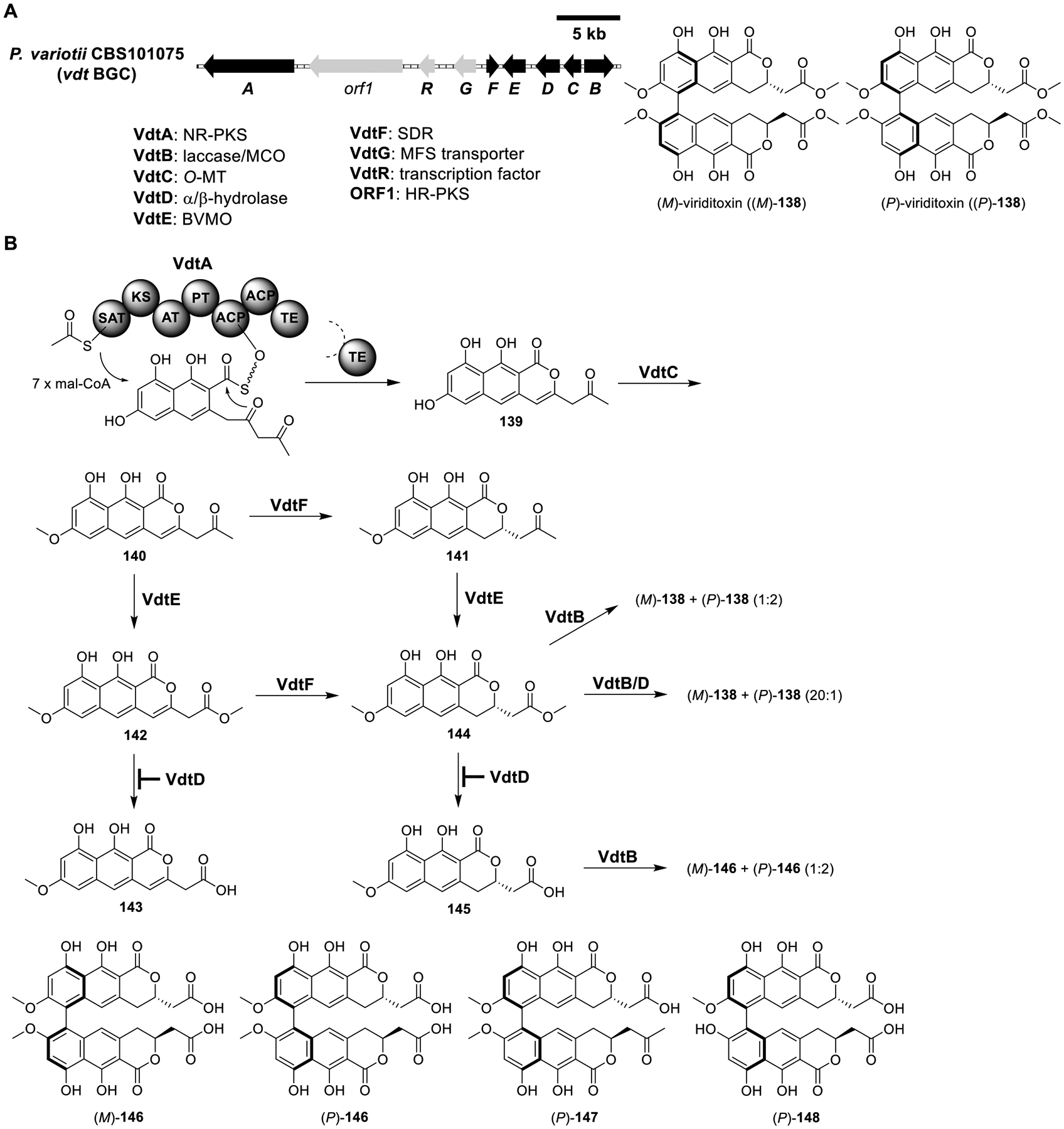
Total biosynthesis of (M)-viriditoxin ((M)-138). (A) Organization and predicted gene functions of the vdt BGC. Black ORFs encode for enzymes involved in the biosynthesis of (M)-138. (B) The proposed biosynthesis of (M)-138.
To characterize the function of VdtE, recombinant VdtE was assayed, and it was found to fully convert 140 to 142 and 141 to 144 in the presence of flavin adenine dinucleotide (FAD) and NADPH. The result indicated that VdtE was a methyl ester-forming BVMO, which had not been reported before. The formation of 143 and 145 was likely due to the activity of an endogenous hydrolase in the host. The Baeyer–Villiger reaction catalyzed by VdtE was intriguing, as the formation of a methyl ester required the migration of the less electron-rich methyl group, against migratory aptitude.
Since VdtD was a predicted hydrolase, the authors initially proposed that coexpression of VdtD with VdtA/C/E would produce more carboxylic acid derivative 143. Interestingly, the VdtA/C/D/E strain produced more 142 and the VdtA/C/D/E/F strain produced exclusively 144. These results suggested that instead of functioning as a hydrolase, VdtD possessed the opposite role. Analysis of the VdtD protein sequence identified a mutation in the catalytic Ser-(Glu/Asp)-His triad, in which the Ser was mutated to Asp. Therefore, VdtD might be able to bind to the methyl esters 142 and 144 and protect them from hydrolyzation by endogenous hydrolases. Addition of the multicopper oxidase (MCO) VdtB to the VdtA–F strain produced (M)-138 and its atropisomer (P)-viriditoxin ((P)-138) at a 20:1 ratio. Interestingly, without VdtD, the VdtA/B/C/E/F strain generated a mixture of bisnaphthopyrones along with trace (M)-138 and (P)-138. Among them, the major metabolites isolated were (M)-146, (P)-146, 147, and 148 (Figure 20B). The ratio of (M)-138:(P)-138 and (M)-146: (P)-146 produced by the VdtA/B/C/E/F strain was 1:2 (Table S18). It appeared that in the absence of VdtD, VdtB lost the stereoselectivity and substrate fidelity for the oxidative coupling reaction.
To corroborate the functions of VdtB and VdtD, cell-free lysates containing VdtB and VdtD were prepared separately from A. nidulans. When 144 was used as a substrate, cell-free extracts containing VdtB alone converted 144 to (M)-138 and (P)-138 at a 1:2 ratio, while VdtB and VdtD converted 144 to (M)-138 predominantly. The in vitro data demonstrated VdtB as a regioselective coupling MCO and that VdtD controlled the stereoselectivity in the formation of (M)-138. Therefore, VdtD was a noncatalytic hydrolase with a function analogous to the dirigent protein, which could orient phenoxy radicals to direct stereospecific formation of biaryl products.113 Lastly, coexpression of the HR-PKS ORF1 with VdtA–F did not change the metabolic profile, indicating ORF1 was not involved in the biosynthesis of (M)-138.
Stemphyloxin II (149).
The sth BGC from the wheat pathogen Parastagonospora nodorum was found to be highly expressed during plant infection (Figure 21A).114 It had high homology to the previously identified bet cluster responsible for betaenones B (150) and C (151) biosynthesis,115 hinting that sth might produce betaenone-like molecules (Figure 21B). Indeed, overexpression of the putative regulator sthR in P. nodorum led to the accumulation of the phytotoxin stemphyloxin II (149), which was originally isolated from Stemphylium botryosum along with stemphyloxin I (152).116 In order to elucidate the biosynthesis of the unique tricyclo[6.2.2.02,7] system in 149, sth was reconstituted in LO7890 using the YFAC-CH vectors (Table S19).
Figure 21.
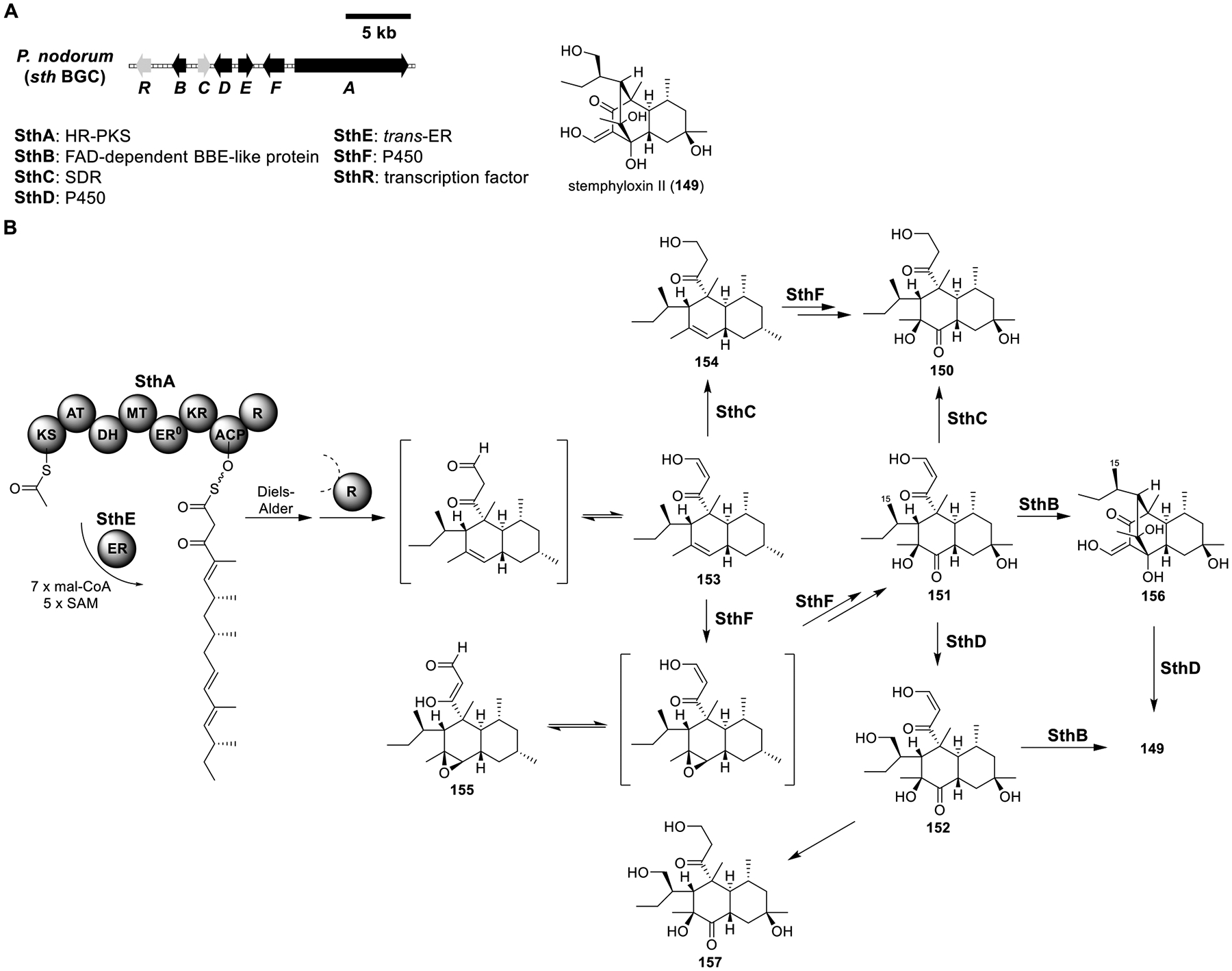
Total biosynthesis of stemphyloxin II (149). (A) Organization and predicted gene functions of the sth BGC. Black ORFs encode for enzymes involved in the biosynthesis of 149. (B) Proposed biosynthesis of 149.
Coexpression of the HR-PKS SthA and its partner trans-ER SthE produced dehydroprobetaenone I (153). Coexpression of the SDR SthC with SthA/E yielded probetaenone I (154), indicating the aldehyde reductase activity of SthC. Coexpression of the P450 SthF with SthA/E produced 153, small amounts of 150 and 151, and epoxybetaenone (155). The production of 155 led to the hypothesis that SthF catalyzed the transformation of 153 to 151 by successive epoxidation, oxidative epoxide opening, and hydroxylation. The small amount of 150 likely came from the reduction of 151 by an endogenous reductase. Coexpression of SthA/C/E/F resulted in the exclusive accumulation of 150.
Sequence analysis of SthB revealed that SthB comprised a FAD-binding domain and a berberine bridge enzyme (BBE) domain. Coexpression of SthB with SthA/E/F gave a new compound, 156, which contained the tricyclo[6.2.2.02,7] system in 149. Incubation of 151 with the cell-free lysate of A. nidulans containing SthB also yielded 156, indicating that SthB was responsible for the intramolecular aldol reaction that generated the bridged tricyclic system. Lastly, coexpression of the second P450 SthD with SthA/B/E/F generated 149, confirming that SthD hydroxylated the C-15 methyl group. Surprisingly, when SthA/E/F/D were coexpressed, instead of stemphyloxin I (152), dihydrostemphyloxin I (157) was obtained. Formation of 157 from 152 was likely due to an endogenous reductase.
The discovery of SthB as an aldolase in the sth pathway represented the first example of a dedicated aldolase in the biosynthesis of a fungal secondary metabolite. However, the function of the FAD-binding domain in SthB remained unknown, awaiting further biochemical characterization.
Nanangelenin A (158).
Nanangelenin A (158) was isolated from A. nanangensis, a rare Aspergillus species from Australia.117 It exhibited moderate cytotoxicity and no significant antifungal, antibacterial, or antiparasitic activity. Notably, it contained a rare 1-benzazepine scaffold. Retrobiosynthesis analysis suggested that 158 was derived from anthranilic acid and l-kynurenine. l-Kynurenine is an intermediate in the metabolism of l-tryptophan. Conversion of l-tryptophan to l-kynurenine is first catalyzed by tryptophan-2,3-dioxygenase (TDO) or IDO, as aforementioned in the quinolactacin A (39) biosynthesis pathway (Figure 8), to generate N-formyl-l-kynurenine, which is then hydrolyzed spontaneously118 or by kynurenine formamidase to form l-kynurenine.
Genomic and retrobiosynthetic analyses led to the identification of a BGC (nan) encoding an IDO (NanC), an NRPS (NanA), a prenyltransferase (NanD), an N-MT (NanE), an FAD oxidoreductase (NanF), and an acetyltransferase (NanB) in the genome of Aspergillus nanangensis (Figure 22A).117 Heterologous expression of the NRPS NanA in LO7890 did not produce any new metabolite, which was likely due to the absence of the substrate l-kynurenine. Gratifyingly, coexpression of the IDO NanC with NanA yielded nanangelenin B (159) (Figure 22B and Table S20). These data, together with the fact that feeding l-kynurenine to the NanA expression strain yielded 159, indicated that NanC converted l-tryptophan to N-formyl-l-kynurenine, which then hydrolyzed spontaneously to l-kynurenine. NanA then activated and condensed anthranilic acid and l-kynurenine to generate 159. Coexpression of the prenyltransferase NanD with NanA/C yielded nanangelenin C (160), while coexpression of the N-MT NanE with NanA/C led to partial conversion of 159 to nanangelenin D (161). When NanA/C/D/E were expressed together, a new product, nanangelenin E (162), was formed.
Figure 22.
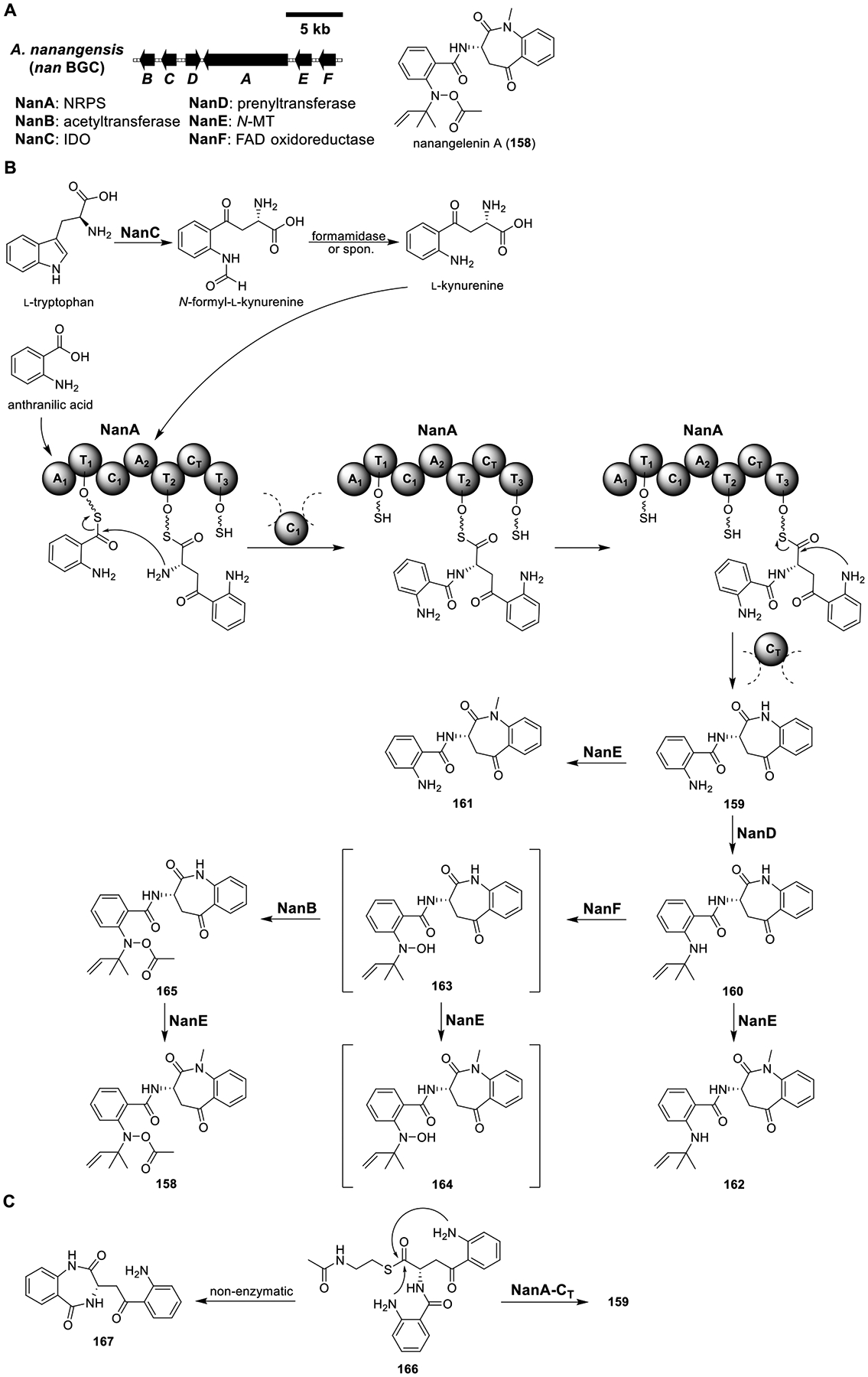
Total biosynthesis of nanangelenin A (158). (A) Organization and predicted gene functions of the nan BGC. Black ORFs encode for enzymes involved in the biosynthesis of 158. (B) Proposed biosynthesis of 158. (C) Enzymatic and nonenzymatic cyclization of 166.
To assess the function of the FAD oxidoreductase NanF (proposed to be an N-hydroxylase) and the acetyltransferase NanB, NanA/C/F- and NanA/B/C/F-expressing strains were constructed. However, both strains still produced 159, suggesting that N-hydroxylation and acetylation could not occur before prenylation. Indeed, the NanA/C/D/F and NanA/C/D/E/F strains produced putative MS peaks corresponding to the N-hydroxylated intermediates 163 and 164, respectively. However, due to low yield and instability, purification of both compounds for structural characterization was unsuccessful. Lastly, the NanA–D/F- and NanA–F-expressing strains delivered nanangelenin F (165) and 158, respectively.
To characterize the C-terminal CT-T3 didomain of NanA, point mutations were introduced. Coexpression of NanC with NanA-H2106A (containing a point mutation of the catalytic histidine in the CT domain) lost the ability to synthesize 159, and coexpression of NanC with NanA-S2401A (containing a point mutation of the active site serine in the T3 domain) greatly reduced the yield of 159. These results indicated that the CT domain was required for cyclization release to give 159 and that the T3 domain enhanced the catalytic efficiency of CT. In addition, incubation of the N-acetylcysteamine thioester 166 with recombinant Nan-CT produced 159, while incubation of 166 with boiled Nan-CT produced 167, which was thought to be formed spontaneously (Figure 22C). These results indicated that Nan-CT catalyzed the regioselective cyclization of 166 to afford 159.
Burnettramic Acid A (168).
Burnettramic acids A (168) and B (169), together with their aglycones (170 and 171), were isolated from A. burnettii (Figure 23A).119 168 and 169 are unusual bolaamphiphilic pyrrolizidinediones with antibiotic activities. Genome sequencing and comparative genomic analysis led to the identification of a BGC (bua) that encoded a proline hydroxylase (BuaE), a PKS-NRPS (BuaA), a trans-ER (BuaC), two P450s (BuaD and BuaG), and a glycosyltransferase (BuaB). To verify that bua encoded for the biosynthesis of 168, strains expressing BuaA/C, BuaA/C/E, BuaA–C/E/F/G, and BuaA–G were constructed from LO7890 (Table S21). While the BuaA/C and BuaA/C/E strains did not produce new metabolites, the BuaA–C/E/F/G and BuaA–G strains yielded 168, 170, and 171. 168 converted to 169, 170, and 171 in acidic condition, raising the prospect that the minor epimers 169 and 171 were artifacts from the isolation process. Based on these results, a tentative biosynthesis pathway of 168 was proposed (Figure 23B). First, BuaE hydroxylated proline to form 4-hydroxyproline, which was then activated and transferred to the PKS-NRPS/trans-ER BuaA/BuaC to produce the intermediate 172. Multiple hydroxylations of 172 by the P450 BuaG yielded the aglycone 170, which was then mannosylated by the glycosyltransferase BuaB to produce 168. The reason that the BuaA/C/E strain did not accumulate a detectable amount of 172 remained unclear.
Figure 23.
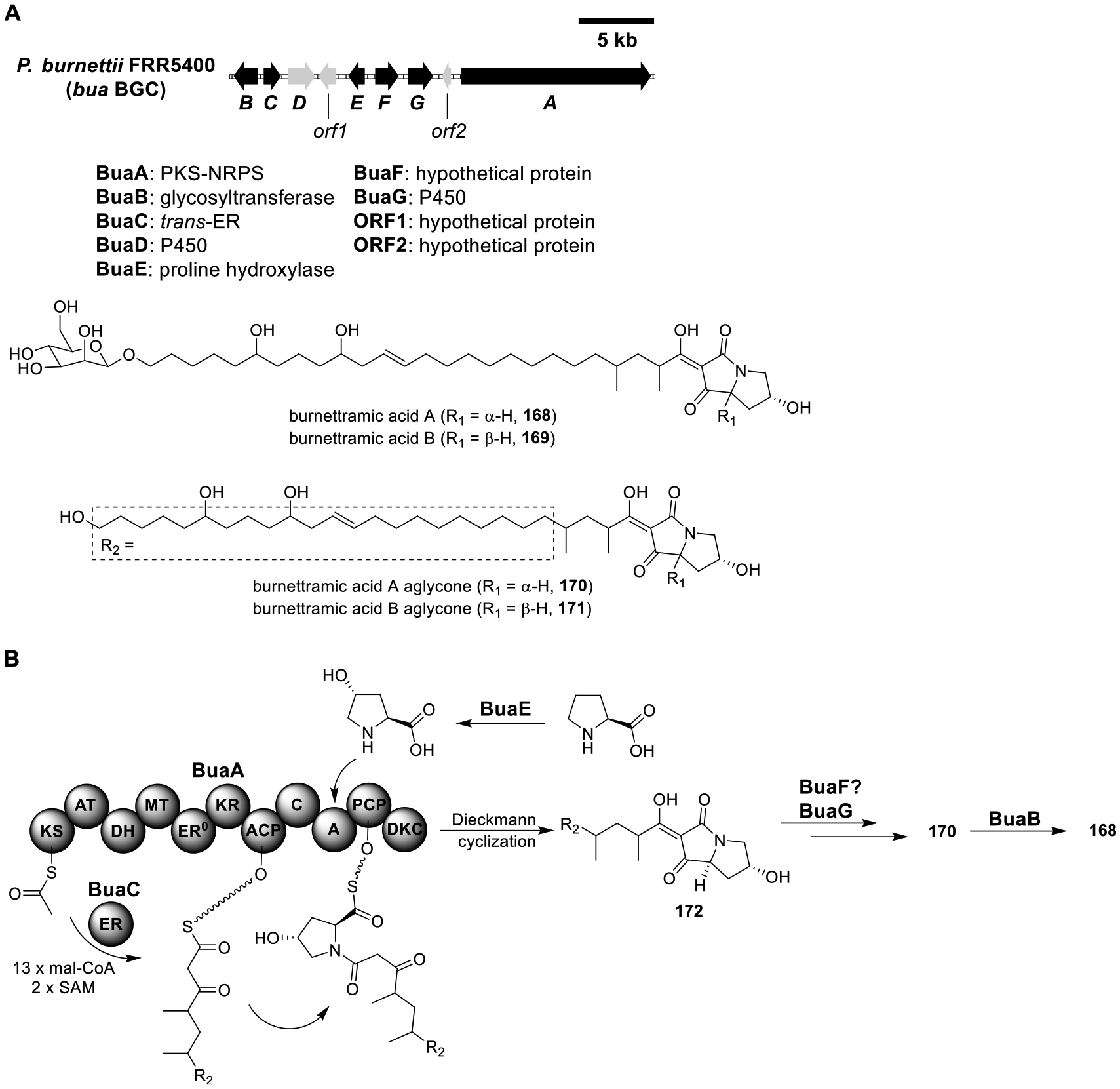
Total biosynthesis of burnettramic acid A (168). (A) Organization and predicted gene functions in the bua BGC. Black ORFs encode for enzymes involved in the biosynthesis of 168. (B) Proposed biosynthesis of 168.
II. SITE-SPECIFIC CHROMOSOMAL INTEGRATION EXPRESSION SYSTEM
In contrast to the heterologous expression systems that use ectopic integration and extrachromosomal replication described above, the Oakley group and our group used homologous recombination to integrate genes of interest into specific sites in the chromosome. The high-efficiency gene targeting system in A. nidulans nkuAΔ strains48,49 allows heterologous genes to be inserted scarlessly (i.e., without restriction site) into a defined locus. There are at least two advantages to this approach. First, a defined integration site circumvents positional effects and allows further strain engineering to be designed more rationally. Second, and most importantly, chromosomal integration provides intrinsic genetic stability, which is crucial for large-scale and long-term bioproduction.120 The wA and yA loci were chosen as the sites of integration because both genes are involved in the biosynthesis of green conidial pigment. wA, an NR-PKS, synthesizes a yellow-colored polyketide, which is converted to green pigment by yA, a conidial laccase. Therefore, wA and yA mutants produce white and yellow spores, respectively, allowing correct transformants to be quickly identified visually.
Reiterative Recombination.
In our workflow, transforming fragments are generated by PCR, bypassing cloning and yeast assembly. Because PKS and NRPS genes are large and sometimes difficult to amplify, we often generate two smaller transforming fragments that are fused by HR in vivo to yield the full-length coding sequence (gene A) under the control of PalcA (Figure 24A).54 Each fragment carries a selection marker, and transformants carrying both markers (markers 1 and 2) are selected and verified by diagnostic PCR. To integrate another gene (gene B), we apply an iterative recombination strategy (Figure 24B).121 Insertion of gene B simultaneously removes marker 2; thus transformants carrying markers 1 and 3 are selected. Since a selection marker is recycled with each transformation, in principle, an unlimited number of heterologous genes can be sequentially integrated.
Figure 24.
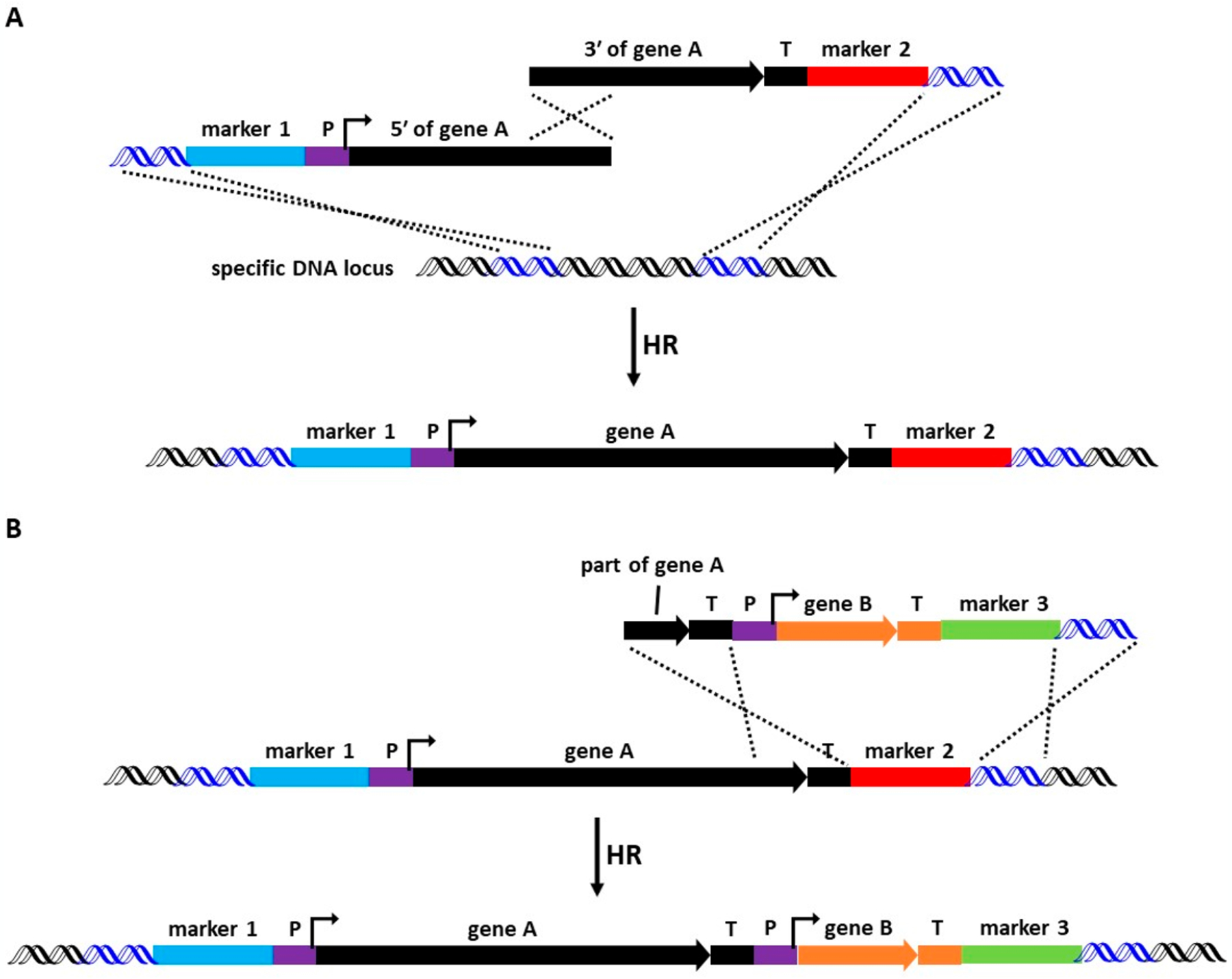
Site-specific chromosomal integration with iterative recombination and maker recycling. (A) Two transforming DNA fragments can be obtained by fusion PCR. The first DNA fragment contains a ~1 kb sequence (blue) homologous to the integration site of the host genome, a selectable marker (maker 1), an inducible PalcA (P), and the 5′ portion of gene A. The second DNA fragment contains the 3′ portion of gene A overlapping by ~1 kb with the 5′ portion and extending ~100 bp downstream of the stop codon (T), marker 2, and another ~1 kb sequence (blue) homologous to the host genome. HR of the two transforming DNA fragments (cotransfomation) results in the integration of the complete expression cassette into the host genome. (B) HR of transforming DNA containing ~900 bp of the 3′ portion of gene A plus ~100 bp of T, a P, a smaller gene (gene B) plus ~100 bp of T, marker 3, and a ~1 kb homologous sequence (blue) results in the integration of the expression cassette of gene B and recycling of marker 2.
Asperfuranone (173).
The utility of this system was demonstrated in the reconstitution of the Ateafo biosynthesis pathway from A. terreus in LO4389 (Figure 25A).54 A homologous BGC (afo) existed in A. nidulans, which we have previously activated by placing the positive regulator afoA under the control of PalcA and identified the encoded metabolite, asperfuranone (173).122 We first deleted afo from the host strain to eliminate possible crosstalk between the homologous pathways. The Ateafo genes were then introduced sequentially into the wA locus using the reiterative recombination strategy.
Figure 25.
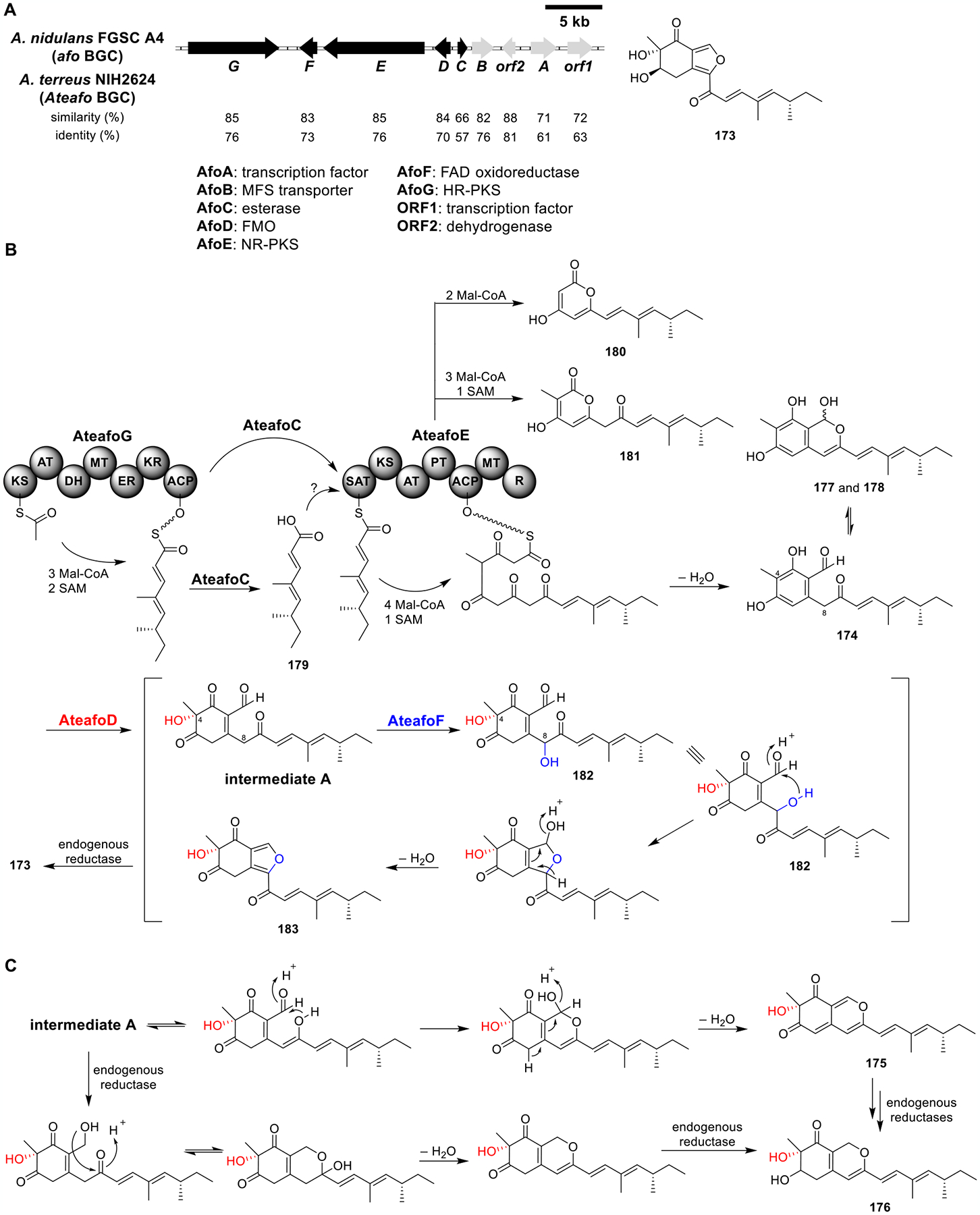
Total biosynthesis of asperfuranone (173). (A) Organization and predicted gene functions of the afo and Ateafo BGCs. Black ORFs encode for enzymes involved in the biosynthesis of 173. (B) Proposed biosynthesis of 173.
While expression of the NR-PKS AteAfoE alone yielded no product, coexpression with the HR-PKS AteAfoG produced asperbenaldehyde (174) (Figure 25B and Table S22). This data confirmed our previous hypothesis that the HR-PKS synthesized a highly reduced polyketide side chain, which was transferred to the SAT domain of the NR-PKS for extension, aldol condensation, aromatization, and reductive release to generate 174.122 Coexpression of the FAD-dependent oxygenase AteAfoF with AteAfoE/G did not result in a new product, indicating that AteafoF did not modify 174. On the other hand, coexpression of the monooxygenase AteAfoD with AteAfoE/G resulted in preasperpyranone (175) and asperpyranone (176), both of which contained a hydroxy group at C-4, suggesting that AteAfoD dearomatized 174 to generate intermediate A. This reaction has also been demonstrated by Davison et al.123 and Zabala et al.124
Interestingly, coexpression of AteAfoC greatly increased the yield of 174 and its two hemiacetal derivatives 177 and 178. AteAfoC and its homologue CtnB were initially misassigned as oxidoreductases,28,122 but Zabala et al. revised their function to be esterases/lipases.124 To investigate whether AteAfoC could release the polyketide side chain 179 from AteafoG, we created an AteAfoG/C coexpression strain. Even though 179 was produced, it was at a very low level (~1 mg/L). Alternatively, AteAfoC could be responsible for transferring 179 from AteAfoG to AteAfoE, considering the high yield of 174 (~300 mg/L) in the AteAfoG/E/C strain. Coexpression of AteAfoC/D/E/G resulted in increased yields of 175 and 176 compared to AteAfoD/E/G. Adding another copy of AteafoC further increased the yield about 1.5-fold, and we were able to isolate minor shunt products proasperfuranone A (180) and B (181) from this strain. Lastly, coexpression of the FAD-dependent oxygenase AteAfoF with AteAfoC/D/E/G produced 173, completing the total biosynthesis of asperfuranone (173). Based on these data, we proposed that AteAfoF oxidized C-8 to generate 182, which abolished enol formation and six-membered ring cyclization (Figure 25B and C). Fivemembered ring cyclization and dehydration of 182 yielded 183, which was then reduced to 173. The fact that no 183 had been identified indicated that the conversion of 183 to 173 was catalyzed by an endogenous reductase. The afo and Ateafo clusters did contain a dehydrogenase (orf 2), but its deletion did not alter the yield of 173.122 If orf 2 was involved in the transformation of 183 to 173, its function was likely compensated by an unknown endogenous reductase.
We created three strains with different orders of AteafoC, AteafoD, and AteafoF (Table S22). All three strains had nearly identical metabolite profiles, suggesting that positional effects were minimal or nonexistent. Coexpression of the putative efflux pump AteAfoB with AteAfoC–G did not change the metabolite profile, so its function remained unknown.
Citreoviridin (184).
Citreoviridin (184) is a mycotoxin that binds the β-subunit of F1-ATPase.125 It is an uncompetitive inhibitor of ATP hydrolysis and a noncompetitive inhibitor of ATP synthesis catalyzed by membrane-bound F1-ATPase.126 The A. terreus var. aureus CBS503.65 strain was a known citreoviridin producer, but its genome was not publicly available at the time of the study. Searching instead the published genome of A. terreus NIH2624, we identified a candidate BGC of 184.127 However, 184 was undetectable in A. terreus NIH2624 in various culturing conditions, and heterologous expression of the putative HR-PKS gene did not lead to the production of the polyene precursor. Thus, we cloned the BGC of 184 from the CBS503.65 strain (ctv, Figure 26A) using primers designed from the sequence of the NIH2624 strain.
Figure 26.
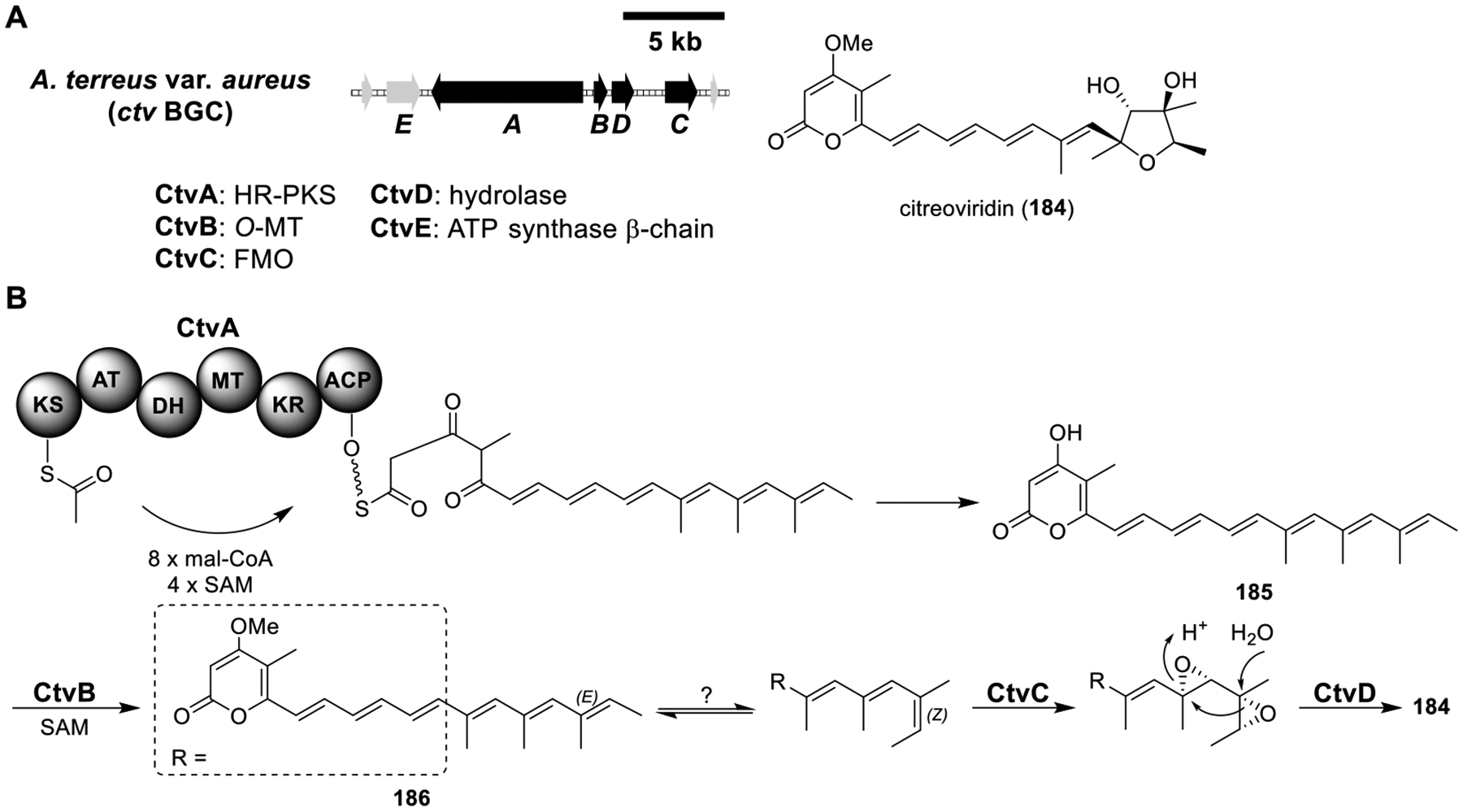
Total biosynthesis of citreoviridin (184). (A) Organization and predicted gene functions of the ctv BGC. Black ORFs encode for enzymes involved in the biosynthesis of 184. (B) Proposed biosynthesis of 184.
Five genes were successfully amplified, and they were found to share >85% DNA sequence identities and >84% amino acid identities with their homologues in the NIH2624 strain. Heterologous expression of the PKS CtvA in the yA locus in LO8030 produced 185 (Figure 26B and Table S23). While coexpression of the FMO CtvC with CtvA did not produce a new product, coexpression of the O-MT CtvB with CtvA yielded citreomontanin (186). This indicated that methylation of 185 preceded epoxidation. Coexpression of CtvA–C led to the emergence of many peaks on the HPLC including 184. Since epoxide is a reactive functional group, spontaneous hydrolysis might be responsible for these peaks. When the hydrolase CtvD was coexpressed with CtvA–C, only 184 was produced, indicating that CtvD regioselectively hydrolyzed the epoxide of 186. Interestingly, ctvE encoded an ATP synthase β-chain, which was likely a resistance enzyme; however, its function awaits validation.
Cluster Refactoring with the afo Regulon.
While the heterologous expression systems described so far employ promoters from housekeeping or primary metabolic genes, several groups have instead used promoters of high-titer secondary metabolites.128 the rationale is that placing genes of a target BGC under the control of promoters of a high-yield secondary metabolite would similarly result in abundant production of the target molecule. This idea was proposed in a pioneer review paper, where Lubertozzi and Keasling propounded heterologous expression by swapping the coding regions of a native BGC with the coding regions from a target BGC.36
In line with this concept, we developed a cluster refactoring strategy to assemble heterologous biosynthesis pathways using the afo transcriptional regulatory elements (regulon).129 As mentioned above, induction of the positive regulator afoA activated the afo genes and resulted in good yield of asperfuranone (173) (Figure 25). By swapping the afo genes with genes of interest from a target BGC, we aimed to obtain a good yield of the encoded molecule by induction of afoA. Accordingly, we constructed the chassis strain YM87 (derived from LO4389) (Figure 27A), in which the promoter of afoA was replaced by PalcA and the sequences from afoC to afoG by AfriboB.
Figure 27.
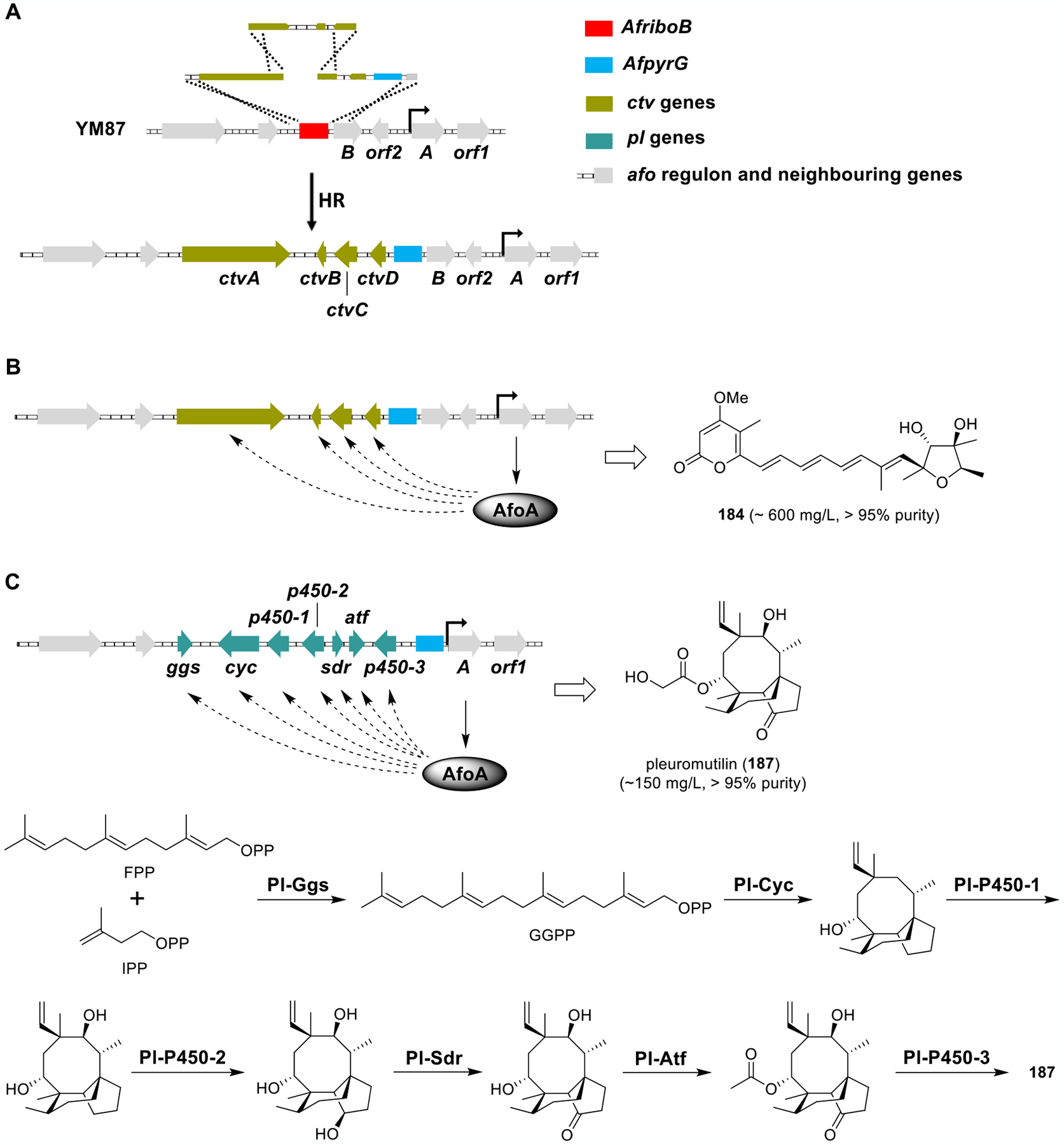
Cluster refactoring with the afo regulon. (A) Reconstitution of ctv biosynthesis in the afo regulon. HR of the three transforming fragments with the afo locus of the recipient strain (YM87) replaced the coding sequences of afo with ctv genes (yellow). Transforming fragments were obtained by PCR amplification of the Gibson assembly products (see text for details). (B) Induction of the positive regulator afoA resulted in the activation of ctvA–D and the production of citreoviridin (184). (C) Using a similar approach, seven afo coding sequences (afoB–G and orf 2, see Figure 25A) were replaced by the seven pl genes (teal). Induction of afoA resulted in the production of pleuromutilin (187).
The transforming DNA fragments were generated by (1) amplifying the intergenic regions of afo (the afo regulon) and the genes of interest by PCR, (2) purifying the amplified fragments by gel extraction, (3) assembling those purified fragments by Gibson assembly, and (4) amplifying large transforming fragments by PCR using templates from step 3. Gibson assembly can isothermally assemble multiple DNA fragments in 1 h, and therefore, transforming fragments can be obtained within 1 day. The transforming fragments were then assembled in vivo via HR with high efficiency in the nKuAΔ strains, allowing the simultaneous introduction of multiple genes of interest in one transformation.
We first refactored the ctv genes with the afo regulon and successfully obtained citreoviridin (184) with high yield (~600 mg/L) and high purity (>95%) after a liquid–liquid partition of the culture broth (Figure 27A and B). Notably, we were able to introduce all four ctv genes (18.7 kb) in just one transformation, in contrast to the reiterative recombination strategy that would have required four transformations. Using the same strategy, we also refactored the seven-gene pleuromutilin biosynthesis pathway (pl) from the Basidiomycete Clitopilus passeckerianus130 (Figure 27C). Pleuromutilin derivatives are a new class of antibiotics used against topical infections, and their total biosynthesis has been achieved from A. oryzae.131 Gratifyingly, pleuromutilin (187) was produced with good yield (~150 mg/L) and obtained with high purity (>95%) after liquid–liquid partition.
The afo refactoring system thus is a robust and efficient fungal platform for total heterologous biosynthesis. The genes of interest are refactored into a defined genomic locus, minimizing positional effects caused by differential integration sites and facilitating further rational design of strain engineering. Moreover, the ability to introduce multiple genes of interest in a single step using transforming fragments generated by Gibson assembly and PCR allows us to quickly generate producing strains. With our platform, we aim to engineer “microbial factory” strains that produce high-value fungal natural products with high yield and purity. This “one-strain one-compound” approach would greatly simplify the downstream purification processes and lower the cost of production. It remains elusive why the refactored strains only produced the final metabolite without the accumulation of intermediates or shunt products. One possibility is that the downstream enzymes were catalytically more efficient and readily converted intermediates to the final metabolite. Another possibility is that the pathway enzymes formed a complex through protein–protein interactions to keep the intermediates protected as they were passed between the catalytic pockets until ejection in the final step.
CONCLUDING REMARKS
Total synthesis of structurally complex fungal natural products faces challenges in steps that require stereo- or chemoselective transformations, such as the construction of stereocenters and site-specific functionalization.132 Biosynthesis, on the other hand, employs pathway-specific enzymes with remarkable efficiency and selectivity to generate enantiomerically pure molecules. Yet the low yields and complex metabolite backgrounds of natural producers often render the costs for obtaining sufficient material for downstream development prohibitively high.
In recent years, reconstitution of biosynthetic pathways in genetically tractable hosts has emerged as a feasible alternative to accessing complex natural products apart from total synthesis or natural producers.20–22,133 Total heterologous biosynthesis leverages the selectivity of pathway enzymes while benefiting from a dereplicated host and from high-yielding regulatory elements. Moreover, it holds the promise of unlocking cryptic corners of the vast fungal BGC universe. The platforms and tools developed in A. nidulans described in this review represent significant advancements toward the efficient exploration of that space; the challenge is now prioritizing the most valuable biosynthesis dark matter for investigation.134 With continued advancements in high-throughput genome sequencing, bioinformatics, genetics, molecular biology, and strain engineering, the potential for total heterologous biosynthesis to transform pharmaceutical and agrochemical lead discovery and production awaits to be realized.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of Allergy and Infectious Diseases (Grant R21AI127640).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.2c00487.
Vectors used and additional information (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jnatprod.2c00487
The authors declare no competing financial interest.
DEDICATION
This review is dedicated to Professor Yueh-Hsiung Kuo on the occasion of his 80th birthday.
Contributor Information
Yi-Ming Chiang, Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, Los Angeles, California 90089, United States; Department of Pharmacy, Chia Nan University of Pharmacy and Science, Tainan 71710, Taiwan.
Tzu-Shyang Lin, Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, Los Angeles, California 90089, United States.
Clay C. C. Wang, Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy and Department of Chemistry, Dornsife College of Letters, Arts, and Sciences, University of Southern California, Los Angeles, California 90089, United States.
REFERENCES
- (1).Brakhage AA Nat. Rev. Microbiol 2013, 11 (1), 21–32. [DOI] [PubMed] [Google Scholar]; Keller NP Nat. Rev. Microbiol 2019, 17 (3), 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bills GF; Gloer JB Microbiol Spectr 2016, 4 (6). DOI: 10.1128/microbiolspec.FUNK-0009-2016 [DOI] [PubMed] [Google Scholar]; Newman DJ; Cragg GM J. Nat. Prod 2020, 83 (3), 770–803. [DOI] [PubMed] [Google Scholar]; Sparks TC; Hahn DR; Garizi NV Pest Manag Sci 2017, 73 (4), 700–715. [DOI] [PubMed] [Google Scholar]
- (3).Chooi YH; Tang YJ Org. Chem 2012, 77 (22), 9933–9953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Herbst DA; Townsend CA; Maier T Nat. Prod Rep 2018, 35 (10), 1046–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hur GH; Vickery CR; Burkart MD Nat. Prod Rep 2012, 29 (10), 1074–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Quin MB; Flynn CM; Schmidt-Dannert C Nat. Prod Rep 2014, 31 (10), 1449–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]; Christianson DW Chem. Rev 2017, 117 (17), 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chang HY; Cheng TH; Wang AH IUBMB Life 2021, 73 (1), 40–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hoffmeister D; Keller NP Nat. Prod Rep 2007, 24 (2), 393–416. [DOI] [PubMed] [Google Scholar]; Wang X; Jarmusch SA; Frisvad JC; Larsen TO Nat. Prod. Rep 2022, DOI: 10.1039/D1NP00074H. [DOI] [PubMed] [Google Scholar]
- (9).Keller NP; Hohn TM Fungal Genet Biol 1997, 21 (1), 17–29. [PubMed] [Google Scholar]
- (10).Keller NP; Turner G; Bennett JW Nat. Rev. Microbiol 2005, 3 (12), 937–947. [DOI] [PubMed] [Google Scholar]
- (11).Blackwell M Am. J. Bot 2011, 98 (3), 426–438. [DOI] [PubMed] [Google Scholar]
- (12).Inglis DO; Binkley J; Skrzypek MS; Arnaud MB; Cerqueira GC; Shah P; Wymore F; Wortman JR; Sherlock G BMC Microbiol 2013, 13, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]; Robey MT; Caesar LK; Drott MT; Keller NP; Kelleher NL Proc. Natl. Acad. Sci. U. S. A 2021, 118 (19). DOI: 10.1073/pnas.2020230118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mousa JJ; Bruner SD Nat. Prod Rep 2016, 33 (11), 1255–1267. [DOI] [PubMed] [Google Scholar]
- (14).Bergmann S; Schü J; Scherlach K; Lange C; Brakhage AA; Hertweck C Nat. Chem. Biol 2007, 3 (4), 213–217. [DOI] [PubMed] [Google Scholar]
- (15).Lyu HN; Liu HW; Keller NP; Yin WB Nat. Prod Rep 2020, 37 (1), 6–16. [DOI] [PubMed] [Google Scholar]
- (16).Yeh HH; Ahuja M; Chiang YM; Oakley CE; Moore S; Yoon O; Hajovsky H; Bok JW; Keller NP; Wang CC; et al. ACS Chem. Biol 2016, 11 (8), 2275–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yan Y; Liu N; Tang Y Nat. Prod Rep 2020, 37 (7), 879–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wiemann P; Keller NPJ Ind. Microbiol Biotechnol 2014, 41 (2), 301–313. [DOI] [PubMed] [Google Scholar]; Scherlach K; Hertweck C Nat. Commun 2021, 12 (1), 3864. [DOI] [PMC free article] [PubMed] [Google Scholar]; Brakhage AA; Schroeckh V Fungal Genet Biol 2011, 48 (1), 15–22. [DOI] [PubMed] [Google Scholar]
- (19).Anyaogu DC; Mortensen UH Front Microbiol 2015, 6, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]; Meng X; Fang Y; Ding M; Zhang Y; Jia K; Li Z; Collemare J; Liu W Biotechnology Advances 2022, 54, 107866. [DOI] [PubMed] [Google Scholar]
- (20).He Y; Wang B; Chen W; Cox RJ; He J; Chen F Biotechnol Adv 2018, 36 (3), 739–783. [DOI] [PubMed] [Google Scholar]
- (21).Lazarus CM; Williams K; Bailey AM Nat. Prod Rep 2014, 31 (10), 1339–1347. [DOI] [PubMed] [Google Scholar]
- (22).Qiao Y-M; Yu R-L; Zhu P RSC Adv 2019, 9 (60), 35124–35134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Greco C; Keller NP; Rokas A Curr. Opin Microbiol 2019, 51, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bond C; Tang Y; Li L Fungal Genet Biol 2016, 89, 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kistler HC; Broz K Front Microbiol 2015, 6, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]; Roze LV; Chanda A; Linz JE Fungal Genet Biol 2011, 48 (1), 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tsunematsu Y; Ishiuchi K; Hotta K; Watanabe K Nat. Prod Rep 2013, 30 (8), 1139–1149. [DOI] [PubMed] [Google Scholar]
- (27).Smith DJ; Burnham MK; Edwards J; Earl AJ; Turner G Biotechnology (N Y) 1990, 8 (1), 39–41. [DOI] [PubMed] [Google Scholar]
- (28).Sakai K; Kinoshita H; Shimizu T; Nihira TJ Biosci Bioeng 2008, 106 (5), 466–472. [DOI] [PubMed] [Google Scholar]
- (29).Nielsen MR; Wollenberg RD; Westphal KR; Sondergaard TE; Wimmer R; Gardiner DM; rensen JL Fungal Genet Biol 2019, 132, 103248. [DOI] [PubMed] [Google Scholar]
- (30).de Reus E; Nielsen MR; Frandsen RJ N. Mol. Microbiol 2019, 112 (6), 1684–1700. [DOI] [PubMed] [Google Scholar]
- (31).Nies J; Ran H; Wohlgemuth V; Yin WB; Li SM Org. Lett 2020, 22 (6), 2256–2260. [DOI] [PubMed] [Google Scholar]
- (32).Bok JW; Ye R; Clevenger KD; Mead D; Wagner M; Krerowicz A; Albright JC; Goering AW; Thomas PM; Kelleher NL; et al. BMC Genomics 2015, 16, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Clevenger KD; Bok JW; Ye R; Miley GP; Verdan MH; Velk T; Chen C; Yang K; Robey MT; Gao P; et al. Nat. Chem. Biol 2017, 13 (8), 895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hu J; Sarrami F; Li H; Zhang G; Stubbs KA; Lacey E; Stewart SG; Karton A; Piggott AM; Chooi YH Chem. Sci 2019, 10 (5), 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Nielsen MT; Nielsen JB; Anyaogu DC; Holm DK; Nielsen KF; Larsen TO; Mortensen UH PLoS One 2013, 8 (8), e72871. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yin WB; Chooi YH; Smith AR; Cacho RA; Hu Y; White TC; Tang Y ACS Synth. Biol 2013, 2 (11), 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang P; Wang X; Fan A; Zheng Y; Liu X; Wang S; Zou H; Oakley BR; Keller NP; Yin WB Mol. Microbiol 2017, 105 (3), 469–483. [DOI] [PubMed] [Google Scholar]; Ma Z; Li W; Zhang P; Lyu H; Hu Y; Yin WB Appl. Microbiol. Biotechnol 2018, 102 (1), 297–304. [DOI] [PubMed] [Google Scholar]
- (36).Lubertozzi D; Keasling JD Biotechnol Adv 2009, 27 (1), 53–75. [DOI] [PubMed] [Google Scholar]
- (37).Lichius A; Ruiz DM; Zeilinger S Genetic transformation of filamentous fungi: achievements and challenges. In Grand Challenges in Fungal Biotechnology; Nevalainen H, Ed.; Springer International Publishing, 2020; pp 123–164. [Google Scholar]
- (38).Li D; Tang Y; Lin J; Cai W Microb Cell Fact 2017, 16 (1), 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Aleksenko A; Clutterbuck AJ Fungal Genet Biol 1997, 21 (3), 373–387. [DOI] [PubMed] [Google Scholar]
- (40).Fleissner A; Dersch P Appl. Microbiol. Biotechnol 2010, 87 (4), 1255–1270. [DOI] [PubMed] [Google Scholar]
- (41).Barbesgaard P; Heldt-Hansen HP; Diderichsen B Appl. Microbiol. Biotechnol 1992, 36 (5), 569–572. [DOI] [PubMed] [Google Scholar]
- (42).Tada S; Gomi K; Kitamoto K; Takahashi K; Tamura G; Hara S Mol. Gen Genet 1991, 229 (2), 301–306. [DOI] [PubMed] [Google Scholar]; Tada S; Gomi K; Kitamoto K; Kumagai C; Tamura G; Hara S Agric. Biol. Chem 1991, 55 (7), 1939–1941. [PubMed] [Google Scholar]
- (43).Gomi K; Iimura Y; Hara S Agric. Biol. Chem 1987, 51 (9), 2549–2555. [Google Scholar]
- (44).Jin FJ; Maruyama J; Juvvadi PR; Arioka M; Kitamoto K FEMS Microbiol Lett 2004, 239 (1), 79–85. [DOI] [PubMed] [Google Scholar]
- (45).Heneghan MN; Yakasai AA; Halo LM; Song Z; Bailey AM; Simpson TJ; Cox RJ; Lazarus CM Chembiochem 2010, 11 (11), 1508–1512. [DOI] [PubMed] [Google Scholar]
- (46).Pahirulzaman KA; Williams K; Lazarus CM Methods Enzymol 2012, 517, 241–260. [DOI] [PubMed] [Google Scholar]
- (47).Awakawa T; Abe I; et al. J. Fungi (Basel) 2021, 7 (6), 468–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Nayak T; Szewczyk E; Oakley CE; Osmani A; Ukil L; Murray SL; Hynes MJ; Osmani SA; Oakley BR Genetics 2006, 172 (3), 1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Szewczyk E; Nayak T; Oakley CE; Edgerton H; Xiong Y; Taheri-Talesh N; Osmani SA; Oakley BR; Oakley B Nat. Protoc 2006, 1 (6), 3111–3120. [DOI] [PubMed] [Google Scholar]
- (50).Brown DW; Yu JH; Kelkar HS; Fernandes M; Nesbitt TC; Keller NP; Adams TH; Leonard TJ Proc. Natl. Acad. Sci. U. S. A 1996, 93 (4), 1418–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Chiang YM; Szewczyk E; Nayak T; Davidson AD; Sanchez JF; Lo HC; Ho WY; Simityan H; Kuo E; Praseuth A; et al. Chem. Biol 2008, 15 (6), 527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Nø CS; Nielsen JB; Kogle ME; Mortensen UH PLoS One 2015, 10 (7), No. e0133085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Liu N; Hung YS; Gao SS; Hang L; Zou Y; Chooi YH; Tang Y Org. Lett 2017, 19 (13), 3560–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Chiang YM; Oakley CE; Ahuja M; Entwistle R; Schultz A; Chang SL; Sung CT; Wang CC; Oakley BR J. Am. Chem. Soc 2013, 135 (20), 7720–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Chiang YM; Ahuja M; Oakley CE; Entwistle R; Asokan A; Zutz C; Wang CC; Oakley BR Angew. Chem., Int. Ed. Engl 2016, 55 (5), 1662–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Tsunematsu Y; Ishikawa N; Wakana D; Goda Y; Noguchi H; Moriya H; Hotta K; Watanabe K Nat. Chem. Biol 2013, 9 (12), 818–825. [DOI] [PubMed] [Google Scholar]
- (57).Kubodera T; Yamashita N; Nishimura A Biosci Biotechnol Biochem 2002, 66 (2), 404–406. [DOI] [PubMed] [Google Scholar]
- (58).Hua SB; Qiu M; Chan E; Zhu L; Luo Y Plasmid 1997, 38 (2), 91–96. [DOI] [PubMed] [Google Scholar]; Oldenburg KR; Vo KT; Michaelis S; Paddon C Nucleic Acids Res 1997, 25 (2), 451–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Sato M; Yagishita F; Mino T; Uchiyama N; Patel A; Chooi YH; Goda Y; Xu W; Noguchi H; Yamamoto T; et al. Chembiochem 2015, 16 (16), 2294–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Yang SW; Mierzwa R; Terracciano J; Patel M; Gullo V; Wagner N; Baroudy B; Puar M; Chan TM; McPhail AT; et al. J. Nat. Prod 2006, 69 (7), 1025–1028. [DOI] [PubMed] [Google Scholar]
- (61).Jacob S; Grötsch T; Foster AJ; Schü A; Rieger PH; Sandjo LP; Liermann JC; Opatz T; Thines E Microbiology 2017, 163 (4), 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhao Z; Ying Y; Hung YS; Tang YJ Nat. Prod 2019, 82 (4), 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Liu L; Tang MC; Tang YJ Am. Chem. Soc 2019, 141 (50), 19538–19541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Grabley S; Granzer E; Hütter K; Ludwig D; Mayer M; Thiericke R; Till G; Wink J; Philipps S; Zeeck AJ Antibiot (Tokyo) 1992, 45 (1), 56–65. [DOI] [PubMed] [Google Scholar]; Göhrt A; Zeeck A; Hütter K; Kirsch R; Kluge H; Thiericke R J. Antibiot (Tokyo) 1992, 45 (1), 66–73. [DOI] [PubMed] [Google Scholar]
- (65).Gao DW; Jamieson CS; Wang G; Yan Y; Zhou J; Houk KN; Tang YJ Am. Chem. Soc 2021, 143 (1), 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Omura S; Hirano A; Iwai Y; Masuma RJ Antibiot (Tokyo) 1979, 32 (8), 786–790. [DOI] [PubMed] [Google Scholar]
- (67).Yu X; Liu F; Zou Y; Tang MC; Hang L; Houk KN; Tang YJ Am. Chem. Soc 2016, 138 (41), 13529–13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Sakamoto K; Tsujii E; Abe F; Nakanishi T; Yamashita M; Shigematsu N; Izumi S; Okuhara MJ Antibiot (Tokyo) 1996, 49 (1), 37–44. [DOI] [PubMed] [Google Scholar]
- (69).Zhang Z; Tamura Y; Tang M; Qiao T; Sato M; Otsu Y; Sasamura S; Taniguchi M; Watanabe K; Tang YJ Am. Chem. Soc 2021, 143 (1), 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Sasaki T; Takahashi S; Uchida K; Funayama S; Kainosho M; Nakagawa AJ Antibiot (Tokyo) 2006, 59 (7), 418–427. [DOI] [PubMed] [Google Scholar]
- (71).Zhao F; Liu Z; Yang S; Ding N; Gao X Angew. Chem., Int. Ed. Engl 2020, 59 (43), 19108–19114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Liu M; Ohashi M; Tang Y Org. Lett 2020, 22 (16), 6637–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Flower DR; North AC; Sansom CE Biochim. Biophys. Acta 2000, 1482 (1–2), 9–24. [DOI] [PubMed] [Google Scholar]
- (74).Tan D; Jamieson CS; Ohashi M; Tang MC; Houk KN; Tang YJ Am. Chem. Soc 2019, 141 (2), 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Tepaske M; Gloer J; Wicklow D; Dowd P Tetrahedron Lett 1991, 32 (41), 5687–5690. [Google Scholar]
- (76).Cary JW; Uka V; Han Z; Buyst D; Harris-Coward PY; Ehrlich KC; Wei Q; Bhatnagar D; Dowd PF; Martens SL; et al. Fungal Genet Biol 2015, 81, 88–97. [DOI] [PubMed] [Google Scholar]
- (77).Ohashi M; Liu F; Hai Y; Chen M; Tang MC; Yang Z; Sato M; Watanabe K; Houk KN; Tang Y Nature 2017, 549 (7673), 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Hayakawa S; Minato H; Katagiri KJ Antibiot (Tokyo) 1971, 24 (9), 653–654. [DOI] [PubMed] [Google Scholar]
- (79).Gutierrez-Cirlos EB; Merbitz-Zahradnik T; Trumpower BL J. Biol. Chem 2004, 279 (10), 8708–8714. [DOI] [PubMed] [Google Scholar]
- (80).Zhang Z; Jamieson CS; Zhao YL; Li D; Ohashi M; Houk KN; Tang YJ Am. Chem. Soc 2019, 141 (14), 5659–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Miyadera H; Shiomi K; Ui H; Yamaguchi Y; Masuma R; Tomoda H; Miyoshi H; Osanai A; Kita K; Omura S Proc. Natl. Acad. Sci. U. S. A 2003, 100 (2), 473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Dickinson J; Hanson J; Hitchcock P; Claydon N Journal of the Chemical Society-Perkin Transactions 1 1989, No. 11, 1885–1887. [Google Scholar]
- (83).Wasil Z; Pahirulzaman K; Butts C; Simpson T; Lazarus C; Cox R Chemical Science 2013, 4 (10), 3845–3856. [Google Scholar]
- (84).Bat-Erdene U; Kanayama D; Tan D; Turner WC; Houk KN; Ohashi M; Tang YJ Am. Chem. Soc 2020, 142 (19), 8550–8554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Fukuda T; Yamaguchi Y; Masuma R; Tomoda H; Omura SJ Antibiot (Tokyo) 2005, 58 (5), 309–314. [DOI] [PubMed] [Google Scholar]; Fukuda T; Tomoda H; Omura SJ Antibiot (Tokyo) 2005, 58 (5), 315–321. [DOI] [PubMed] [Google Scholar]
- (86).Zhang Z; Qiao T; Watanabe K; Tang Y Angew. Chem., Int. Ed. Engl 2020, 59 (45), 19889–19893. [DOI] [PubMed] [Google Scholar]
- (87).Bacon CW; Porter JK; Norred WP; Leslie JF Appl. Environ. Microbiol 1996, 62 (11), 4039–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bani M; Rispail N; Evidente A; Rubiales D; Cimmino AJ Agric. Food Chem 2014, 62 (12), 2574–2580. [DOI] [PubMed] [Google Scholar]; Singh VK; Singh HB; Upadhyay RS Plant Physiol Biochem 2017, 118, 320–332. [DOI] [PubMed] [Google Scholar]
- (88).Brown DW; Butchko RA; Busman M; Proctor RH Fungal Genet Biol 2012, 49 (7), 521–532. [DOI] [PubMed] [Google Scholar]; Niehaus EM; von Bargen KW; Espino JJ; Pfannmü A; Humpf HU; Tudzynski B Appl. Microbiol. Biotechnol 2014, 98 (4), 1749–1762. [DOI] [PubMed] [Google Scholar]; Brown DW; Lee SH; Kim LH; Ryu JG; Lee S; Seo Y; Kim YH; Busman M; Yun SH; Proctor RH; et al. Mol. Plant-Microbe Interact 2015, 28 (3), 319–332. [DOI] [PubMed] [Google Scholar]; Studt L; Janevska S; Niehaus EM; Burkhardt I; Arndt B; Sieber CM; Humpf HU; Dickschat JS; Tudzynski B Environ. Microbiol 2016, 18 (3), 936–956. [DOI] [PubMed] [Google Scholar]
- (89).Hai Y; Chen M; Huang A; Tang YJ Am. Chem. Soc 2020, 142 (46), 19668–19677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Guo JP; Zhu CY; Zhang CP; Chu YS; Wang YL; Zhang JX; Wu DK; Zhang KQ; Niu XM J. Am. Chem. Soc 2012, 134 (50), 20306–20309. [DOI] [PubMed] [Google Scholar]
- (91).Niu X; Chen L; Yue Q; Wang B; Zhang J; Zhu C; Zhang K; Bills GF; An Z Org. Lett 2014, 16 (14), 3744–3747. [DOI] [PubMed] [Google Scholar]
- (92).Zhang JM; Wang HH; Liu X; Hu CH; Zou YJ Am. Chem. Soc 2020, 142 (4), 1957–1965. [DOI] [PubMed] [Google Scholar]
- (93).Yue K; Ye M; Zhou Z; Sun W; Lin XJ Pharm. Pharmacol 2013, 65 (4), 474–493. [DOI] [PubMed] [Google Scholar]
- (94).Wang X; Gao YL; Zhang ML; Zhang HD; Huang JZ; Li LJ Biotechnol 2020, 309, 85–91. [DOI] [PubMed] [Google Scholar]
- (95).Namatame I; Tomoda H; Ishibashi S; Omura S Proc. Natl. Acad. Sci. U. S. A 2004, 101 (3), 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Bloudoff K; Schmeing TM Biochim Biophys Acta Proteins Proteom 2017, 1865 (11), 1587–1604. [DOI] [PubMed] [Google Scholar]
- (97).Bentley R Chem. Rev 2000, 100 (10), 3801–3826. [DOI] [PubMed] [Google Scholar]
- (98).Regueira TB; Kildegaard KR; Hansen BG; Mortensen UH; Hertweck C; Nielsen J Appl. Environ. Microbiol 2011, 77 (9), 3035–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Hansen BG; Mnich E; Nielsen KF; Nielsen JB; Nielsen MT; Mortensen UH; Larsen TO; Patil KR Appl. Environ. Microbiol 2012, 78 (14), 4908–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Chen X; Wang L; Zhang J; Jiang T; Hu C; Li D; Zou Y Acta Pharm. Sin B 2019, 9 (6), 1253–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Yee DA; Kakule TB; Cheng W; Chen M; Chong CTY; Hai Y; Hang LF; Hung YS; Liu N; Ohashi M; et al. J. Am. Chem. Soc 2020, 142 (2), 710–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Hartmann M; Kim D; Bernsdorff F; Ajami-Rashidi Z; Scholten N; Schreiber S; Zeier T; Schuck S; Reichel-Deland V; Zeier J Plant Physiol 2017, 174 (1), 124–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Li H; Hu J; Wei H; Solomon PS; Vuong D; Lacey E; Stubbs KA; Piggott AM; Chooi YH Org. Lett 2018, 20 (19), 6148–6152. [DOI] [PubMed] [Google Scholar]
- (104).Fillinger S; Felenbok B Mol. Microbiol 1996, 20 (3), 475–488. [DOI] [PubMed] [Google Scholar]
- (105).Flipphi M; Mathieu M; Cirpus I; Panozzo C; Felenbok BJ Biol. Chem 2001, 276 (10), 6950–6958. [DOI] [PubMed] [Google Scholar]
- (106).Olivo M; Chin WJ Environ. Pathol Toxicol Oncol 2006, 25 (1–2), 223–237. [DOI] [PubMed] [Google Scholar]; Daub ME; Herrero S; Chung KR Antioxid Redox Signal 2013, 19 (9), 970–989. [DOI] [PubMed] [Google Scholar]
- (107).Chooi YH; Zhang G; Hu J; Muria-Gonzalez MJ; Tran PN; Pettitt A; Maier AG; Barrow RA; Solomon PS Environ. Microbiol 2017, 19 (5), 1975–1986. [DOI] [PubMed] [Google Scholar]
- (108).Newman AG; Vagstad AL; Belecki K; Scheerer JR; Townsend CA Chem. Commun. (Camb) 2012, 48 (96), 11772–11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Newman AG; Townsend CA J. Am. Chem. Soc 2016, 138 (12), 4219–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Weisleder D; Lillehoj E Tetrahedron Lett 1971, 12 (48), 4705–4706. [Google Scholar]; Suzuki K; Nozawa K; Nakajima S; Kawai K Chem. Pharm. Bull 1990, 38 (11), 3180–3181. [Google Scholar]
- (111).Wang J; Galgoci A; Kodali S; Herath KB; Jayasuriya H; Dorso K; Vicente F; González A; Cully D; Bramhill D; et al. J. Biol. Chem 2003, 278 (45), 44424–44428. [DOI] [PubMed] [Google Scholar]; Liu Y; Kurtán T; Yun Wang C; Han Lin W; Orfali R; ller WE; Daletos G; Proksch PJ Antibiot (Tokyo) 2016, 69 (9), 702–706. [DOI] [PubMed] [Google Scholar]; Kundu S; Kim TH; Yoon JH; Shin HS; Lee J; Jung JH; Kim HS Int. J. Oncol 2014, 45 (6), 2331–2340. [DOI] [PubMed] [Google Scholar]
- (112).Hu J; Li H; Chooi YH J. Am. Chem. Soc 2019, 141 (20), 8068–8072. [DOI] [PubMed] [Google Scholar]
- (113).Davin LB; Wang HB; Crowell AL; Bedgar DL; Martin DM; Sarkanen S; Lewis NG Science 1997, 275 (5298), 362–366. [DOI] [PubMed] [Google Scholar]
- (114).Li H; Hu J; Wei H; Solomon PS; Stubbs KA; Chooi YH Chemistry 2019, 25 (66), 15062–15066. [DOI] [PubMed] [Google Scholar]
- (115).Ugai T; Minami A; Fujii R; Tanaka M; Oguri H; Gomi K; Oikawa H Chem. Commun. (Camb) 2015, 51 (10), 1878–1881. [DOI] [PubMed] [Google Scholar]
- (116).Manulis S; Kashman Y; Netzer D; Barash I Phytochemistry 1984, 23 (10), 2193–2198. [Google Scholar]
- (117).Li H; Gilchrist CLM; Phan CS; Lacey HJ; Vuong D; Moggach SA; Lacey E; Piggott AM; Chooi YH J. Am. Chem. Soc 2020, 142 (15), 7145–7152. [DOI] [PubMed] [Google Scholar]
- (118).Kurnasov O; Jablonski L; Polanuyer B; Dorrestein P; Begley T; Osterman A FEMS Microbiol Lett 2003, 227 (2), 219–227. [DOI] [PubMed] [Google Scholar]
- (119).Li H; Gilchrist CLM; Lacey HJ; Crombie A; Vuong D; Pitt JI; Lacey E; Chooi YH; Piggott AM Org. Lett 2019, 21 (5), 1287–1291. [DOI] [PubMed] [Google Scholar]
- (120).Li L; Liu X; Wei K; Lu Y; Jiang W Biotechnol Adv 2019, 37 (5), 730–745. [DOI] [PubMed] [Google Scholar]
- (121).Wingler LM; Cornish VW Proc. Natl. Acad. Sci. U. S. A 2011, 108 (37), 15135–15140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Chiang YM; Szewczyk E; Davidson AD; Keller N; Oakley BR; Wang CC J. Am. Chem. Soc 2009, 131 (8), 2965–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Davison J; al Fahad A; Cai M; Song Z; Yehia SY; Lazarus CM; Bailey AM; Simpson TJ; Cox RJ Proc. Natl. Acad. Sci. U. S. A 2012, 109 (20), 7642–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Zabala AO; Xu W; Chooi YH; Tang Y Chem. Biol 2012, 19 (8), 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Gause EM; Buck MA; Douglas MG J. Biol. Chem 1981, 256 (2), 557–559. [PubMed] [Google Scholar]
- (126).Sayood SF; Suh H; Wilcox CS; Schuster SM Arch. Biochem. Biophys 1989, 270 (2), 714–721. [DOI] [PubMed] [Google Scholar]
- (127).Lin TS; Chiang YM; Wang CC Org. Lett 2016, 18 (6), 1366–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Kakule TB; Jadulco RC; Koch M; Janso JE; Barrows LR; Schmidt EW ACS Synth. Biol 2015, 4 (5), 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gressler M; Hortschansky P; Geib E; Brock M Front Microbiol 2015, 6, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wiemann P; Soukup AA; Folz JS; Wang PM; Noack A; Keller NP Fungal Biol. Biotechnol 2018, 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Chiang YM; Lin TS; Chang SL; Ahn G; Wang CC C. ACS Synth. Biol 2021, 10 (1), 173–182. [DOI] [PubMed] [Google Scholar]
- (130).Bailey AM; Alberti F; Kilaru S; Collins CM; de MattosShipley K; Hartley AJ; Hayes P; Griffin A; Lazarus CM; Cox RJ; et al. Sci. Rep 2016, 6, 25202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Alberti F; Khairudin K; Venegas ER; Davies JA; Hayes PM; Willis CL; Bailey AM; Foster GD Nat. Commun 2017, 8 (1), 1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Roberts AA; Ryan KS; Moore BS; Gulder T Top Curr. Chem 2010, 297, 149–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Alberti F; Foster GD; Bailey AM Appl. Microbiol. Biotechnol 2017, 101 (2), 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Gilchrist CLM; Li H; Chooi YH Org. Biomol Chem 2018, 16 (10), 1620–1626. [DOI] [PubMed] [Google Scholar]; Tran PN; Yen MR; Chiang CY; Lin HC; Chen PY Appl. Microbiol. Biotechnol 2019, 103 (8), 3277–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.