

Abstract
RAS proteins represent critical drivers of tumor development and thus are the focus of intense efforts to pharmacologically inhibit these proteins in human cancer. Although recent success has been attained in developing clinically efficacious inhibitors to KRASG12C, there remains a critical need for developing approaches to inhibit additional mutant RAS proteins. A number of anti-RAS biologics have been developed which reveal novel and potentially therapeutically targetable vulnerabilities in oncogenic RAS. This review will discuss the growing field of anti-RAS biologics and potential development of these reagents into new anti-RAS therapies.
1. Introduction
The RAS family of proto-oncogenes (HRAS, KRAS, and NRAS) represent the most frequently mutated oncogenes in cancer, with KRAS being mutated in nearly 100% of pancreatic cancers (Moore, Rosenberg, McCormick, & Malek, 2020; Ryan & Corcoran, 2018; Waters & Der, 2018). The discovery of RAS as the human homolog of viral-Rat sarcoma (v-Ras) in the 1980s led to an explosion of efforts to understand its biology and role in tumor development and progression (Karnoub & Weinberg, 2008). Three decades of dedicated efforts to therapeutically target RAS passed without clinical success until the NCI RAS Initiative was launched in 2013 (Ryan & Corcoran, 2018). Now the fruits of those efforts are coming to bear. Several RAS-specific pharmacological inhibitors have entered the clinic with sotorasib (LUMAKRAS) gaining FDA accelerated approval for treatment of KRASG12C mutant non-small cell lung cancers in May 2021 (Hong et al., 2020). Unfortunately, these anti-RAS drugs inhibit only a single mutant RAS protein which is present in a small fraction of RAS-driven cancers (Zuberi, Khan, & O’Bryan, 2020). Thus, continued efforts to drug the “undruggable” RAS remain paramount.
RAS proteins are small molecular weight GTPases that function as regulated molecular switches (Fig. 1). RAS cycles between an inactive GDP-loaded state and the active GTP-loaded state. Although RAS possesses intrinsic GTPase activity, it is a rather poor enzyme and cooperates with GTPase activating/accelerating proteins (GAPs) to enhance the cleavage of GTP to GDP. Activation of RAS occurs through the action of guanine nucleotide exchange factors (GEFs) which bind to GDP-loaded RAS and destabilize the nucleotide binding pocket resulting in release of GDP. Given the high cellular concentration of GTP vs GDP, coupled with the high affinity of RAS for nucleotides, RAS reloads with GTP (Traut, 1994).
Fig. 1.
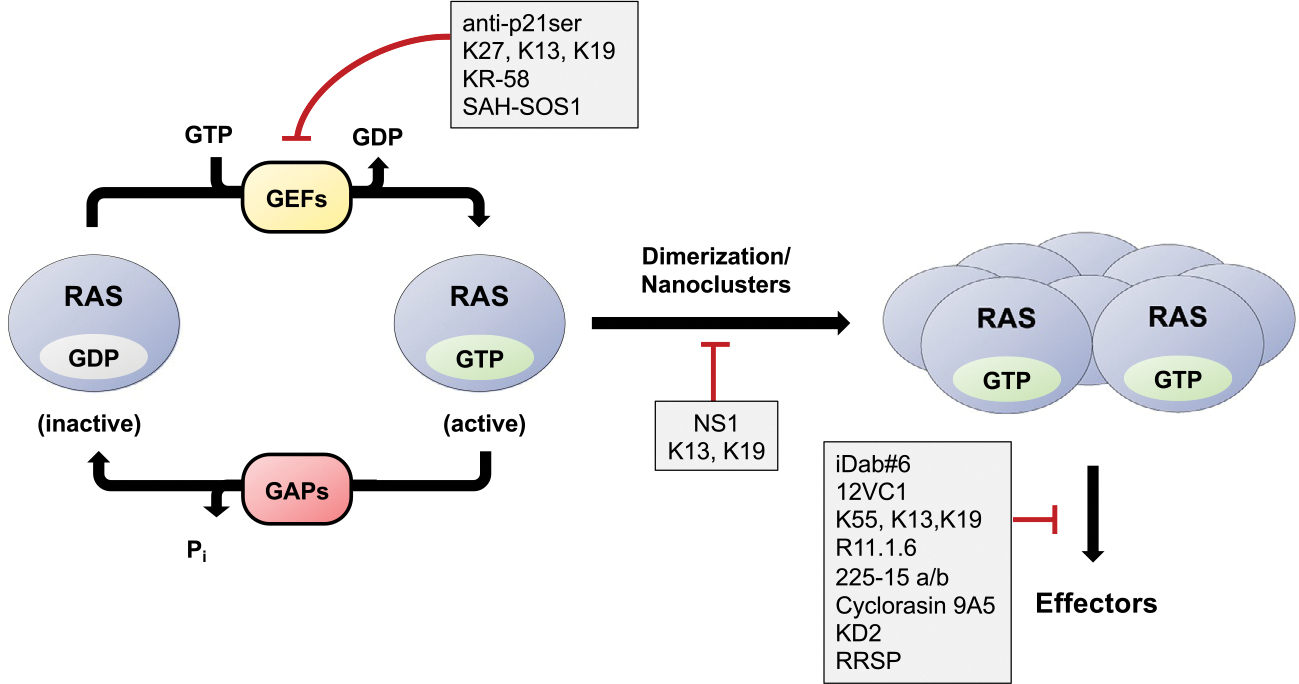
Targeting RAS with biologics. The cycling of RAS between the inactive GDP-bound state and active GTP-bound state is regulated by the action of GAPs and GEFs, respectively. Once activated, RAS assembles into nanoclusters resulting in recruitment and activation of effectors. Various anti-RAS biologics target different aspects of this cycle.
GTP-loaded RAS couples to targets containing RAS binding domains (RBDs) or RAS association domains (RA). Although these domains share limited sequence homology, they all adopt a similar ubiquitin-like topology. This interaction activates pathways such as the RAF–MEK–ERK mitogen-activated protein kinase (MAPK) and PI3K–AKT–mTOR pathways, resulting in enhanced cell proliferation and survival (Karnoub & Weinberg, 2008). In normal cells, RAS is activated upon stimulation of membrane bound receptors such as receptor tyrosine kinases (RTKs). However, mutations in RAS, predominantly at codons 12, 13, and 61, stabilize the GTP-bound state resulting in chronic activation of its downstream target pathways leading to uncontrolled cell proliferation and ultimately cancer.
Despite great efforts over the past three decades, pharmacological inhibition of RAS has proven difficult. Although pharmacological inhibition of kinases has been achieved through design of ATP analogs, pharmacological targeting of the nucleotide pocket of RAS has not seen success due to the picomolar affinity of RAS for GTP/GDP and the high cellular concentration of guanine nucleotides. Furthermore, structural analysis of RAS revealed a lack of deep hydrophobic pockets for binding of small molecules suggesting that RAS may be “undruggable”. The discovery of compounds that covalently bind and inhibit RASG12C by Kevan Shokat and colleagues has dispelled this premise and ushered in a newfound interest in pharmacologically targeting RAS (Ostrem, Peters, Sos, Wells, & Shokat, 2013). The success of these inhibitors is due to the unique chemical reactivity of the Cys12 mutation that allowed for development of chemically reactive warheads that irreversibly bind and lock KRASG12C in an inactive GDP-bound state. Based on these results, both Amgen and Mirati have developed KRASG12C-specific inhibitors, sotarasib (Fig. 2A) and adagrasib, respectively, with both demonstrating promising clinical results (Canon et al., 2019; Fell et al., 2020; Hallin et al., 2020; Hong et al., 2020; Skoulidis et al., 2021). Unfortunately, these drugs are limited to treating only KRASG12C mutant cancers. Thus, development of inhibitors targeting additional oncogenic mutants that lack similar chemical vulnerabilities remains a continuing challenge and unmet need.
Fig. 2.
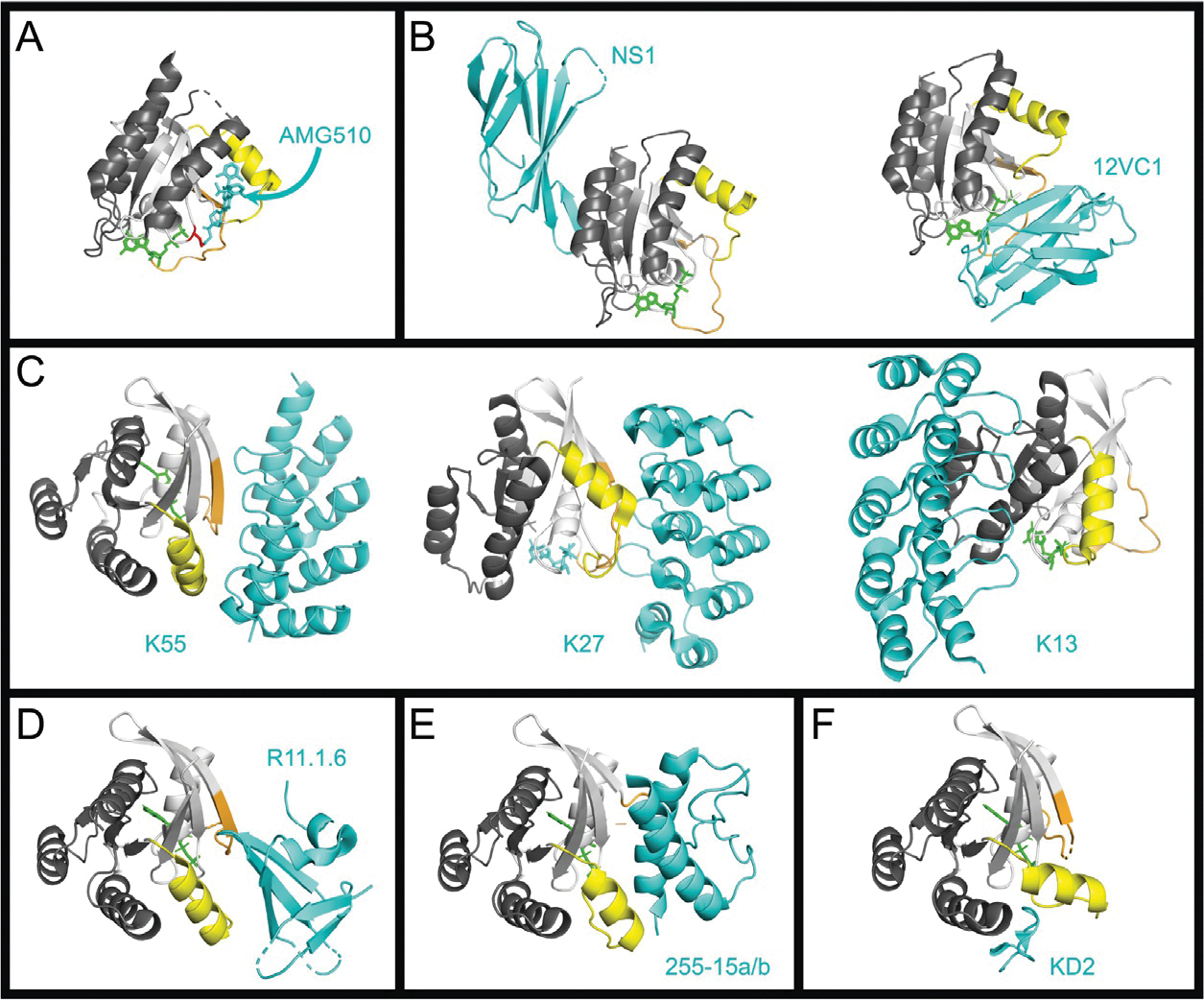
Binding interfaces of various RAS inhibitors. The crystal structures of RAS in complex with various inhibitors. For all RAS structures the effector lobe is highlighted light gray and the allosteric lobe, dark gray, Switch 1 is highlighted orange and Switch 2 highlighted yellow. All inhibitors are shown in cyan. Nucleotide is colored green. Metal ions and water molecules have been removed from structures. (A) Structure of KRASG12C bound to Sotorasib (AMG-510) (PDB 6OIM). The Cys12 is highlighted in red. (B) RAS Monobodies. HRAS:NS1, left (PDB 5E95), and HRAS(G12C):12VC1 (7L0G). (C) RAS DARPins. KRASG12V:K55, left (PDB 5MLA); KRASG12V:K27, center (PDB 5O2S); KRASG12V:K13, right (PDB 6H46). (D) Sso7d protein R11.1.6. KRASG12D:R11.1.6 (PDB RUFQ). (E) Miniprotein 225–15a/b. RASG12V:225–15a/b (PDB 5WLB). (F) KD2 cyclic peptide. KRASG12D:KD2 (PDB 6WGN).
As an alternative to small molecule pharmacologics, a number of groups have utilized biologics as an approach to identify vulnerabilities in RAS that can potentially be exploited to inhibit oncogenic RAS. Such biologics often inhibit their target by providing steric interference with critical protein–protein interactions (PPIs) (Hurd, Mott, & Owen, 2020; Pei, Chen, & Liao, 2018). In this review, we will describe the growing number of anti-RAS biologics that have been isolated, their mechanisms of action, and their potential for development into anti-RAS therapeutics.
2. Anti-RAS biologics
2.1. Immunoglobulin-based biologics
2.1.1. Antibodies
Antibodies have been harnessed in the therapeutic arena since 1986, when muromonab-CD3 became the first monoclonal antibody (mAb) to gain FDA approval for treatment of acute transplant rejection (Lu et al., 2020). Since then, there have been approximately 80 mAbs to receive FDA-approval, with an increasing number being evaluated in clinical trials (Lu et al., 2020). Therapeutic mAbs have several potential modes of action, including disruption of protein–protein or protein–ligand interactions essential to the function of the antigen (Weiner, 2007), allosteric inhibition, alteration of the subcellular localization of target antigens (Koschubs et al., 2012; Marschall & Dubel, 2016), and initiation of cell signaling cascades via interacting with receptor antigens (Weiner, 2007). These characteristics highlight the rationale, and versatility, of utilizing mAbs therapeutically.
The first mAbs generated against RAS were discovered in the lab of Edward Scolnick (Furth, Davis, Fleurdelys, & Scolnick, 1982). These mAbs were produced in rats bearing HRAS-induced tumors and further isolated from eight rat lymphocyte–myeloma hybrid cell lines (Furth et al., 1982). Three of these mAbs also recognized KRAS as well other mammalian RAS homologs (Furth et al., 1982). This work set the stage for the development of therapeutic mAbs against RAS.
Antibody-driven inhibition of KRAS-induced cellular transformation was first observed by Mulcahy et al. by microinjecting a KRAS-specific mAb, Y13–259 (Table 1), into KRAS-transformed NIH 3T3 cells (Mulcahy et al., 1985). Y13–259 decreased cell division whereas a control mAb had no effect (Mulcahy et al., 1985). Y13–259 inhibited RAS function without altering nucleotide binding suggesting that it recognized a region distinct from the nucleotide binding pocket of RAS (Lacal & Aaronson, 1986). Seminal work from the lab of Frank McCormick led to the generation of anti-p21ser (Table 1), an inhibitory antibody specific for amino acid 12 of the oncogenic mutant KRASG12S (Clark et al., 1985; Feramisco et al., 1985). Surprisingly, anti-p21ser competed with GDP or GTP for binding to KRASG12S (Clark et al., 1985). These studies provided critical evidence as to the location of the nucleotide binding pocket of RAS and supported the notion that GTP-binding was required for the oncogenic activity of RAS (Clark et al., 1985; Feramisco et al., 1985). In addition, these results suggested that targeting the nucleotide-free state of RAS might provide a feasible approach to inhibit oncogenic RAS.
Table 1.
Anti-RAS biologics.
| Biologic | Name | Mechanism of action | Binding specificity | Affinity (KD) | References |
|---|---|---|---|---|---|
| Antibodies | Y13–259 | RAS sequestration, delocalization from membrane | a.a. 70–81 of HRAS | N/A | Mulcahy, Smith, and Stacey (1985) |
| anti-p21ser | Inhibition of nucleotide binding | Ser12 of KRAS (G12S) | N/A | Clark, Wong, Arnheim, Nitecki, and McCormick (1985) | |
| Intrabodies | scFv Y13–259 | RAS sequestration, delocalization from membrane | a.a. 70–81 of HRAS | N/A | Cardinale, Lener, Messina, Cattaneo, and Biocca (1998) |
| iDab#6 | Blocks effector binding | Switch regions of GTP-loaded HRAS | 6.2 nM | Tanaka, Lobato, and Rabbitts (2003) | |
| KGHV500 | Cytokine-induced killer (CIK) cell-mediated, tumor-specific delivery | pan-RAS | N/A | Wang et al. (2018) | |
| inRas37 | Tumor-specific, cell-penetrating, full-length antibody | GTP-loaded RAS | N/A | Shin et al. (2020) | |
| Monobodies | NS1 | Allosteric disruption of dimerization/nanoclustering; inhibits GTP loading on WT | α4-α5 region of H/KRAS | 13–16nM (HRAS); 60–67 nM (kRAS) | Spencer-Smith et al. (2017) |
| 12VC1 | Blocks effector binding | Switch regions and mutant residues of KRAS (G12V/C) | 100nM | Teng et al. (2021) | |
| RasIn2 | Blocks effector binding | Switch I of GTP-loaded RAS | 120nM | Cetin et al. (2017) | |
| DARPins | K27 | Blocks nucleotide exchange | GDP-loaded RAS | 4nM | Guillard et al. (2017) |
| K55 | Blocks effector binding | GTP-loaded RAS | 167nM | Guillard et al. (2017) | |
| K13 | Blocks effector binding, nucleotide exchange, and dimerization/nanoclustering | α3-α4 (His95) region of KRAS | 30 nM | Bery et al. (2019) | |
| K19 | Blocks effector binding, nucleotide exchange, and dimerization/nanoclustering | α3-α4 (His95) region of KRAS | 10 nM | Bery et al. (2019) | |
| Sso7d | R11.1.6 | Blocks effector binding | Switch regions; specific RAS (G12) mutants | 2–6nM | Kauke et al. (2017) |
| Miniproteins | 225–15 a/b | Blocks effector binding | Binds as a dimer to RAS | 60 ± 20 pM for KRAS-GppNHp | McGee et al. (2018) |
| Toxin Peptidases | RRSP | Cleaves Switch I region of RAS; blocks effector binding | Cleaves between Y32 and D33 of Switch I | N/A | Antic, Biancucci, Zhu, Gius, and Satchell (2015) |
| Cyclic Peptides | Compound 12 | Blocks effector binding | Not specified | 0.83 μM for KRAS | Wu, Upadhyaya, Villalona-Calero, Briesewitz, and Pei (2013) |
| Cyclorasin 9A5 | Blocks effector binding | Switch I of GTP-loaded RAS | 0.44 μM for GTP-bound KRAS; 2.5 μM for GDP-bound KRAS | Upadhyaya et al. (2015) | |
| KS-58 | Blocks nucleotide exchange/binding; competes with SOS1 GEF binding | KRAS (G12D)-selective | N/A | Sakamoto, Masutani, and Hirokawa (2020) | |
| KD2 | Blocks effector binding | Switch II groove of GTP-loaded KRAS (G12D) | N/A | Zhang et al. (2020) | |
| Stapled peptides | SAH-SOS1 | Blocks nucleotide exchange/binding | Switch regions | 100–175 nM for KRAS | Leshchiner et al. (2015) |
The high target specificity and ease of production make mAbs an enticing therapeutic modality; however, certain properties limit their clinical application. First, mAbs are cell impermeable and as such are almost exclusively limited to extracellular targets (Lin, Chen, Hu, Chen, & Zhang, 2020; Lu et al., 2020; Marschall & Dubel, 2016; Stocks, 2004). Second, mAbs rely on disulfide bonds for stability and thus are generally unstable in the reducing environment of the cytosol (Cardinale et al., 1998; Lin et al., 2020; Lu et al., 2020; Marschall & Dubel, 2016; Stocks, 2004). Therefore, simply employing cell-penetrating peptide (CPP) sequences or other methods of cellular entry cannot ensure their efficacy in targeting intracellular antigens, such as RAS. However, methods to generate cell-penetrating IgG antibodies have been described (Choi et al., 2014) and used to target RAS (Shin et al., 2017). Shin et al. isolated a heavy chain variable fragment specific to activated KRAS and then used this to replace the VH region of a cell penetrating antibody, TMab4, to generate RT11, a cell-penetrating and PPI-interfering mAb against the GTP-bound state of all three RAS isoforms. RT11 bound active RAS isoforms with Kd values ranging from 4 to 17 nM (Shin et al., 2017). RT11 was further modified to confer tumor specificity by the addition of RGD10—a cyclic peptide that recognizes tumor-associated antigens—to the N-terminus of the light chain (Shin et al., 2017). This modified RT11 (RT11-i) preferentially localized to tumors in vivo after tail vein injection into athymic nude mice bearing SW480 xenografts (Shin et al., 2017). RT11-i was modified further (to inRas37; Table 1) to improve the biophysical and biochemical properties of the anti-RAS VH, increase cell penetration, and enhance endosomal escape (Shin et al., 2020). inRas37 exhibited 1.6–3.5-fold enhanced anti-tumor activity compared to RT11-i and demonstrated favorable pharmacokinetic and pharmacodynamic profiles (Shin et al., 2020). These studies provide a potential new therapeutic approach to inhibit oncogenic RAS in vivo using a pan-RAS inhibitor mAb. However, some caution remains as this antibody targeted both WT and mutant forms of GTP-loaded RAS, suggesting the potential for systemic toxicity, although no systemic toxicity was noted (Shin et al., 2020).
2.1.2. Intrabodies
Intrabodies are essentially any antibody (e.g., inRas37) or antibody fragment capable of acting intracellularly (Stocks, 2004) such as single-chain variable fragments (scFvs) consisting of both the heavy (VH) and light (VL) chain variable fragments. Huston et al. were the first to describe the generation of scFvs as modified versions of mAbs (Huston et al., 1988). By using a peptide linker to connect the VH and VL chains, scFvs eliminated the need for disulfide bonds while maintaining specificity and allowing for genetic encoding (Huston et al., 1988). Using this methodology, Biocca et al. developed the first anti-RAS scFvs using the mAb Y13–259 (Biocca, Pierandrei-Amaldi, Campioni, & Cattaneo, 1994; Biocca, Pierandrei-Amaldi, & Cattaneo, 1993). scFv Y13–259 (Table 1) inhibited insulin-stimulated meiosis of Xenopus laevis, colocalized with RAS at the cytosolic face of the plasma membrane (Biocca et al., 1993, 1994), and demonstrated anti-RAS activity in both mammalian cells (Cardinale et al., 1998; Montano & Jimenez, 1995; Werge, Baldari, & Telford, 1994) and tumor xenografts (Cochet et al., 1998). Subsequent studies suggested that scFv Y13–259 functioned by sequestering RAS into intracellular cytoplasmic aggregates (Cardinale et al., 1998).
Work from the laboratory of Terrence Rabbitts led to the discovery of intracellular single variable domains termed iDabs that maintain solubility, stability, and specificity in cells (Tanaka et al., 2003). Using antibody capture technology (Tse et al., 2002; Visintin et al., 2002), anti-RAS iDabs were identified that inhibited RAS-dependent transformation of NIH/3T3 cells (Tanaka & Rabbitts, 2003; Tanaka et al., 2003). Subsequent studies identified the iDab#6 VH intrabody which bound and inhibited HRASG12V (Table 1) (Tanaka, Williams, & Rabbitts, 2007). X-ray crystal structure analysis revealed that iDab#6 interacts with the Switch I (SW1) and Switch 2 (SW2) regions of HRASG12V (Tanaka et al., 2007). This was further corroborated using surface plasmon resonance (SPR), where iDab#6 bound GST-HRAS(GTPγS) in a concentration-dependent manner, but not with GDP-loaded HRAS (Tanaka et al., 2007). Collectively, iDab#6 inhibited RAS function by disrupting effector binding to the SW1 and SW2 regions (Tanaka & Rabbitts, 2010; Tanaka et al., 2007).
More recently, Pan et al. developed a dual-promotor-regulated recombinant oncolytic adenovirus, KGHV300, expressing a pan-RAS scFv (Yang et al., 2016; Pan et al., 2017). In this construct, expression of the E1a and E1b viral replication genes were regulated by the telomerase promotor and the hypoxia response element, respectively, both of which are upregulated in cancer cells, with the pan-RAS scFv gene controlled by the CMV promotor. Although KGHV300 only replicated in cancer cells, it infected all cell types and therefore required intratumoral administration. To increase cancer cell-specificity of KGHV300, its cilia gene was modified to bind cytokine-induced killer (CIK) cells to enhance tumor cell homing ability (Wang et al., 2018). Thus, the newly modified recombinant adenovirus (now referred to as KGHV500; Table 1) was hypothesized to essentially “hitch a ride” from the CIK cells to the tumor microenvironment (Wang et al., 2018). Injection of CIK cells carrying KGHV500 into mice bearing SGC7901 (RASWT, human gastric cancer) xenografts reduced tumor growth compared to injection of KGHV500 without CIK cells (Wang et al., 2018). Furthermore, both the pan-RAS scFv and the adenovirus hexon protein were restricted to tumor and spleen in the KGHC500 + CIK injected samples whereas KGHV500 injection alone resulted in wide expression of both proteins, demonstrating tissue-specificity of the treatment (Wang et al., 2018). Thus, this approach may lead to new therapeutic approaches to treat RAS-mutant cancers.
2.2. Non-immunoglobulin-based biologics
2.2.1. Monobodies
Monobodies (Mbs) are high affinity engineered proteins derived from the fibronectin type III (FN3) domain (Sha, Salzman, Gupta, & Koide, 2017). Mbs bind critical sites of their target and often inhibit the function of that target. The isolation of Mbs involves selection by phage display and yeast display formats from large combinatorial libraries, and this art was pioneered and refined by Shohei Koide and colleagues (Biancucci et al., 2018; Koide, Bailey, Huang, & Koide, 1998; Koide, Wojcik, Gilbreth, Hoey, & Koide, 2012). The smaller size and lack of disulfide bonds provides an advantage for the use of Mbs as genetically encoded reagents and facilitates their production in E. coli.
Our laboratory, in collaboration with the Koide laboratory, has applied this technology to identify anti-RAS biologics (Spencer-Smith et al., 2017; Teng et al., 2021). Our first success came with the isolation of the NS1 Mb which bound the α4-α5 interface on the allosteric lobe of RAS, a region implicated in RAS self-association and nanoclustering (Table 1; Fig. 2B) (Khan, Spencer-Smith, & O’Bryan, 2019). Indeed, ectopic expression of NS1 inhibited RAS dimerization and nanoclustering and inhibited activation of ERK and AKT by both growth factors and oncogenic RAS. NS1 bound HRAS and KRAS with low nanomolar affinity but did not interact with NRAS. This selectivity was due to a single amino acid difference in α4 helix of NRAS (K135) vs H- and KRAS (R135) resulting in disruption of multiple Mb-RAS contacts. In addition to inhibiting RAS signaling, NS1 inhibited the oncogenic activity of H/KRAS mutants but not NRAS or downstream oncogenic kinases such as BRAF(V600E) and MEK(DD) (Spencer-Smith et al., 2017).
Recently, we found that RAS inhibition with NS1 enhanced the anti-tumor immune response (Khan et al., 2021). Using PDAC tumor cells derived from the KPC mouse model (KrasG12D/+; TP53R172H/+) (Hingorani et al., 2005), we established a subclone (KPCNS1) that expressed NS1 in a chemically regulated manner. Injection of KPCNS1 cells into the pancreas resulted in rapid tumor development which was dramatically reduced upon induction of NS1 expression. Furthermore, we observed an increase in tumor infiltrating CD4+ T-cells upon NS1 induction. Surprisingly, there was no change in the number of CD8+ cytotoxic T-cells in the NS1-expressing cells versus controls. Numerous pre-clinical and clinical studies have demonstrated effective anti-tumor immunity of CD4+ cells without engaging CD8+ T-cells (Galaine et al., 2019; Kreiter et al., 2015; Linnemann et al., 2015; Ott et al., 2017; Sahin et al., 2017; Tay, Richardson, & Toh, 2021; Zhang et al., 2018). Interestingly, in a DOX-inducible KRASG12D colorectal cancer mouse model, extinguishing KRAS signaling upon DOX withdrawal led to increased CD4+ T cell infiltration (Liao et al., 2019). It is possible that different KRAS mutations mount different immunological responses in terms of T-cell signatures. This may account for the difference in T-cell response to NS1 vs KRASG12C inhibitors (Canon et al., 2019). Indeed, recent studies illustrate that different KRAS mutations provoke distinct biochemical, cellular, and pathological responses (Hobbs et al., 2020; Poulin et al., 2019).
There are more than 130 different reported missense mutations in the RAS oncogene in various cancers (Hobbs, Der, & Rossman, 2016). Thus, a pan-RAS inhibitory agent like NS1, rather than a mutant selective inhibitor, may represent a more efficacious approach to treat diverse RAS mutant cancer types. The broad-spectrum nature of such an inhibitor may be therapeutically important in light of reports describing the presence of multiple RAS mutations in a single patient and heterogenous mutations between primary and metastatic tumor (Kordiak et al., 2019; Lamy et al., 2011; Oltedal et al., 2011; Richman et al., 2011). As with other RAS biologics, specificity to differentiate oncogenic mutants from WT RAS, which is essential for cells, raises the question of possible toxicity with NS1. Although recent results suggest that NS1 lacks non-specific “off-target” toxicity, it remains possible that residual NRAS expression may be sufficient to avoid such toxicity (Khan et al., 2021).
Although the main mode of action of NS1 appears to be inhibition of RAS self-association/nanoclustering, additional studies reveal added complexity in the inhibitory action of NS1 (Spencer-Smith et al., 2019). For example, NS1 reduced interaction of oncogenic KRAS, but not HRAS, with RAF and reduced KRAS localization to the plasma membrane. The isoform specific effects of NS1 on RAS–RAF association were mediated by the distinct hypervariable regions of RAS isoforms. Further, NS1 reduced GTP loading on WT but not oncogenic RAS in cells suggesting that NS1 binding may alter the orientation of the G-domain with the plasma membrane, hence, perturbing RAS interaction with SOS (Spencer-Smith et al., 2019).
Following on the success with NS1, Teng et al. recently reported the isolation of a mutation selective Mb to RAS (Teng et al., 2021). This Mb, termed 12VC1, selectively bound and inhibited KRASG12C and KRASG12V (Table 1; Fig. 2B). Structural analysis revealed that 12VC1 directly recognized the mutant residues along with the Switch regions to inhibit KRAS–effector interaction. Regulated expression of 12VC1 also reduced the growth of PATU8902 PDAC cells (KRASG12V mutant) tumor cells in athymic nude mice. However, they further enhanced the activity of this Mb through fusion to the von Hippel Lindau E3 ligase (VHL) to generate a mutant selective degrading biologic (see Section 3 for further discussion). Indeed, this modified Mb resulted in proteosome-mediated degradation of KRASG12C and KRASG12V but not KRASG12D or WT RAS. Further, this RAS degrader resulted in sustained pERK inhibition compared to the isolate 12VC1 Mb, further establishing the efficacy of this RAS degrader.
Another genre of RAS-specific Mbs, RasIns, were identified by Cetin et al. (Cetin et al., 2017) using a Mb mRNA display selection against GTPγS-bound RAS, where the BC loop of the Mb was based on mutagenized sequences of the complementary determining region of iDab#6 (Tanaka et al., 2007). RasIn1 was GTP-state-selective for HRAS and KRAS and bound to the Switch I region of RAS (Cetin et al., 2017). Affinity maturation of RasIn1 produced RasIn2 (Table 1). While RasIn1 bound GTP-loaded HRASG12V with a KD of 2.1 μM, RasIn2 bound with a KD of 120 nM (Cetin et al., 2017). RasIn1 and RasIn2 also co-localized with RAS in COS-7 cells; however, their ability to inhibit RAS-mediated signaling was not assessed (Cetin et al., 2017). Together, these studies highlight the power of Mbs in both understanding RAS and inhibiting its oncogenic activity in vivo.
2.2.2. DARPins
Designed ankyrin repeat proteins (DARPins) represent an additional engineered affinity reagent that has been used to inhibit RAS. DARPins are single domain proteins (14 kDa) that are derived from ankyrin repeat binding proteins, which are naturally occurring in eukaryotic cells (Stumpp, Binz, & Amstutz, 2008). Like Mbs, they lack cysteine residues and hence are suitable as genetically encodable intracellular reagents (Kummer et al., 2012; Parizek et al., 2012). The first RAS-specific DARPin, K55, is a pan-RAS binder that preferentially binds to the active GTP-loaded state of RAS and inhibits RAS effector interactions (Table 1; Fig. 2C) (Guillard et al., 2017). K27 DARPin is also a pan-RAS binder but in contrast to K55 prefers the GDP-loaded state and inhibits RAS mainly by preventing nucleotide exchange (Table 1; Fig. 2C) (Guillard et al., 2017). Building on these results, Bery et al. isolated two KRAS specific DARPins, K13 (Table 1; Fig. 2C) and K19 (Table 1), that bind the ⍺3-⍺4 allosteric interface encompassing the region around His95, an amino acid unique to KRAS (Bery et al., 2019). K13 and K19 inhibited KRAS by impairing effector interactions, preventing nucleotide exchange, and inhibiting KRAS dimerization at the plasma membrane (Bery et al., 2019). Though K13 and K19 did not discriminate between mutant and WT KRAS, they did not bind to NRAS and HRAS owing to the selective interaction with His95. Thus, these DARPins provide a potential approach to selectively inhibit KRAS while sparing the function of both N- and HRAS thereby reducing the potential for toxicity due to inhibition of all RAS isoforms.
2.2.3. Affibodies
Affibodies are biologically active engineered protein reagents of approximately 6 kDa based on a scaffold of the three-α-helix bundle Z-domain (Friedman & Stahl, 2009; Nilsson & Tolmachev, 2007; Nygren, 2008). Affibodies typically lack cysteine residues and thus are immune to the redox environment of the cell (Nygren, 2008). HRAS-specific affibodies have been isolated that bound with high nanomolar affinity and inhibited HRAS–RAF interaction in vitro (Grimm et al., 2010). These affibodies inhibited cell proliferation and production of inflammatory mediators in synovial cells by impairing RAS activation of the MAPK signaling cascade (Shibasaki et al., 2014). Unlike Mbs and DARPins, the efficacy of affibodies in RAS addicted oncogenic lines or tumor models has yet to be demonstrated. Thus, further work is needed to demonstrate their utility as RAS targeting biologics in vivo.
2.2.4. Sso7d scaffold proteins
Antibodies and intrabodies, as highlighted above, are excellent candidates as therapeutic agents. However, the aforementioned hurdles thwart the development of effective antibody-based therapeutics against many sought-after intracellular targets such as RAS. Ideal binding peptides would exhibit smaller molecular masses, cell-penetrability, and lack disulfide bonds and glycosylation sites while maintaining stability, solubility, and specificity for their intended target (Traxlmayr et al., 2016). An example of a peptide binding scaffold displaying many of these qualities is Sso7d from the hyperthermophilic archaea Sulfolobus solfataricus (Kauke et al., 2017; Traxlmayr et al., 2016). In contrast to Mbs where the binding sites are typically located within flexible loops, the target binding site of Sso7d lies on the surface of a ridged β-sheet (Traxlmayr et al., 2016).
Kauke et al. isolated a RAS-specific Sso7d, R11.1 using GppNHp-loaded KRASG12D as bait (Traxlmayr et al., 2016). Affinity maturation of R11.1 yielded R11.1.6 (Table 1; Fig. 2D) which was eight-fold more selective for KRASG12D than WT KRAS with single-digit nanomolar affinity. However, this selectivity—but not affinity—was lost in the GDP-loaded state suggesting that R11.1.6 did not directly contact the mutant residue, but rather recognized a specific conformation of KRASG12D in the GppNHp-loaded state (Traxlmayr et al., 2016). Accordingly, R11.1.6 bound other GppNHp-loaded KRAS oncogenic mutants (G12V and G12C), as well as HRAS and NRAS, with comparable affinity to GppNHp-loaded KRASG12D. Structural analysis indicated that R11.1.6 bound distal to the nucleotide pocket, further supporting the hypothesis R11.1.6 binding is due to the conformation of mutant KRAS in the GppNHp-loaded state. The binding interface of R11.1.6/GppNHp–KRASG12D consisted of Switch II in a conformation that exposes hydrophobic residues that interact with aromatic residues of R11.1.6. This interaction at Switch II alters the orientation of Tyr32 and Glu61 at the active site of KRASG12D, rendering it catalytically incompetent. This was supported by the observation that R11.1.6 reduced the intrinsic hydrolysis of KRASG12D. Although the GTP-loaded state of KRASG12D would presumably be stabilized by its interaction with R11.1.6, this is compensated for by the ability of R11.1.6 to disrupt RAS effector interactions. Additionally, in the R11.1.6/GppNHp–KRASG12D complex, the conformation of Switch I of KRASG12D places Glu37 and Glu63 in a position that favors interaction with Lys32 and Lys40 of R11.1.6. Switch I of wild-type KRAS, in contrast, appears to orient Glu63 away from Lys32, thus making the R11.1.6/wild-type KRAS complex less favorable.
2.2.5. Miniproteins
McGee et al. employed a randomized yeast surface display screening approach, utilizing a select number of small, conformationally stable “miniproteins” based on the avian pancreatic polypeptide (aPP) miniprotein scaffold to identify peptides that bound specifically to KRAS (McGee et al., 2018). Three hits were obtained and subjected to directed evolution to optimize the affinity for KRAS resulting in two improvements: addition of PRR to the N-terminus and incorporation of a Y7C mutation. The former was due to primer slipping during PCR; the latter was discarded to avoid unwanted disulfide interactions. Following a third round of directed evolution, miniprotein 225–3 was chosen for further optimization. 225–3 bound GppNHp-loaded H, K, and NRASG12V with mid-nanomolar affinity, competed with RAF-RBD binding, and impaired nucleotide release.
Due to solubility issues, 225–3 was replaced with 225–1. HSQC NMR spectroscopy revealed that 225–1 interacted with the effector binding domain of RAS. Alanine scanning mutagenesis revealed that nearly every effector domain residue contributed to 225–1 interaction suggesting a complex binding mechanism. Furthermore, subsequent NMR analyses suggested that the miniprotein possibly bound KRAS as a dimer. Re-incorporation of Y7C to generate 255–11 resulted in a dimeric miniprotein that bound KRAS more strongly in the absence vs presence of reducing agent. However, DTT had no effect on 255-11-KRAS interaction when the C7 residue was replaced with selenocysteine. Crystal structure analysis revealed that 225–11 formed a disulfide-stabilized dimer, of which one protomer bound KRAS in an extended, open conformation that distorted the Switch I loop and β-strand 2 of the KRAS effector domain. Further optimization of 225–11 by A30R mutation led to a twofold selectivity of GppNHp-loaded over GDP-loaded KRAS.
The authors hypothesized that breaking the symmetry between the 255–11 dimer might improve RAS binding and therefore, allowed each protomer to evolve independently (McGee et al., 2018). The resulting miniprotein 225–15 a/b heterodimer (Table 1; Fig. 2E) bound GppNHp-loaded KRAS >10-fold better than 225–11 (Kd of ~60 nM) and interacted with all 3 RAS isoforms (H, K, and NRAS) as well as various oncogenic KRAS mutants (G12V, G13D, and Q61H) with subnanomolar affinity. However, the ability of these RAS-targeting miniproteins to interfere with RAS function in cells was not examined. Thus, further work will be needed to determine the usefulness as anti-RAS biologics.
2.2.6. Toxin peptidases
The multifunctional-autoprocessing repeats-in-toxin (MARTX) is a bacterial toxin capable of penetrating eukaryotic cell membranes and inducing cytotoxicity (Antic et al., 2015; Kwak, Jeong, & Satchell, 2011). Kwak et al. showed that the most virulent strains of MARTX produced by Vibrio vulnificus contained an effector domain, DUF5Vv, consisting of two subdomains: the membrane anchoring C1 subdomain and the cytotoxic C2 domain (Antic et al., 2015). To identify potential targets of DUF5Vv, a genome-wide, non-essential gene deletion library screen was performed on yeast strains expressing the cytotoxic C2 subdomain. Many of the hits from the yeast strains that survived were connected to the MAPK pathway. Indeed, DUF5Vv decreased ERK1/2 phosphorylation in HeLa cells. It was therefore postulated that a likely target of DUF5Vv was RAS, given the membrane localization of DUF5Vv via its C1 subdomain. It was further shown that DUF5Vv cleaves RAS (and RAP1) between Y32 and D33 in the Switch I region and was therefore dubbed a RAS/RAP1-specific protease (RRSP) (Table 1). Surprisingly, RAS nucleotide binding was not affected by RRSP cleavage; however, effector and GEF interactions were disrupted (Biancucci et al., 2018). Lastly, Vidimar et al. engineered a cell-penetrating RRSP by linking it to a catalytically inactive diphtheria toxin (RRSP-DTB), which binds the heparin-binding epidermal growth factor-like growth factor (HB-EGF) cell surface receptor and promotes cell uptake via endocytosis (Vidimar et al., 2020). When injected into athymic nude mice harboring human HB-EGF positive, triple negative breast cancer xenografts (WT RAS), RRSP-DTB effectively decreased tumor growth (Vidimar et al., 2020). Further studies will be necessary to confer mutant or isoform specificity as RRSP cleaves all RAS isoforms as well as RAP1.
2.3. Synthetic peptides
2.3.1. Cyclic peptides
Cyclic peptides, as opposed to traditional biologics, offer greater therapeutic potential as they are more amenable to modifications that increase bioavailability (Philippe, Craik, & Henriques, 2021). By combining the biophysical properties of small molecules and the high affinity of biologics, peptide cyclization may yield greater chemical stability and protease resistance, potentially increasing systemic half-life, as well as increased affinity for target ligand (Philippe et al., 2021). Importantly, cyclic peptides still retain the ability to bind and disrupt PPI interfaces (Santini and Zacharias, 2020). Properties like these increase the likelihood of developing desirable therapeutics such as Cyclosporin A, an orally available cyclic peptide used for treatment of immunoregulatory disorders (Nielsen et al., 2017; Philippe et al., 2021).
Wu et al. discovered the first cyclic peptide targeting RAS, Compound 12 (Table 1) (Wu et al., 2013). Although this molecule blocked RAS interaction with RBDs of various effectors, Compound 12 was unable to decrease MEK or ERK phosphorylation, likely due to poor membrane permeability (Wu et al., 2013). Upadhyaya et al. utilized a general methodology for synthesizing and screening bicyclic peptides to isolated KRASG12V binding compounds, termed Cyclorasins (Upadhyaya, Qian, Habir, & Pei, 2014). Although two such compounds disrupted the RAS–RAF interaction and competed with Compound 12 for RAS binding suggesting similar target sites on RAS, they failed to confer any effects in cells, again due to poor membrane permeability (Upadhyaya et al., 2014). Further refinements to this approach led to the isolation of Cyclorasin 9A5 (Table 1) with improved cell permeability and affinity for KRAS (Upadhyaya et al., 2015). Indeed, fluorescein isothiocyanate (FITC)-labeled Cyclorasin 9A5 displayed diffuse fluorescence throughout the cytoplasm along with weaker, punctate fluorescence indicative of endosomal entrapment (Upadhyaya et al., 2015). FITC-labeled Cyclorasin 9A5 bound GTP-, GppNHp-, and GDP-loaded KRASG12V with Kd values of 0.44, 0.64, and 2.5 μM, respectively, suggesting that Cyclorasin 9A5 preferentially binds KRAS-GTP (Upadhyaya et al., 2015). 1H—15N heteronuclear single quantum correlation (HSQC) NMR spectroscopy revealed that Cyclorasin 9A5 interacted with the Switch I loop of KRAS, consistent with the dose-dependent inhibition of RAS–RAF interaction in H358 lung cancer cells (KRASG12C) as well as MEK, ERK and AKT phosphorylation in H1299 cells (NRASQ61K) and H1650 cells (wild-type RAS; mutant EGFR) (Upadhyaya et al., 2015). The enhanced membrane permeability of Cyclorasin 9A5 may be due to its more compact, amphipathic structure in DMSO, with converged aromatic amino acids surrounded by arginine residues thereby allowing for more effective membrane penetration (Takeuchi et al., 2021).
KRASG12D represents the most frequent KRAS mutant in human tumors (Vatansever, Erman, & Gumus, 2019). Although there are no targeted therapeutics against KRASG12D in the clinic, recent research has provided hope for selective inhibition of KRASG12D. KRpep-2, a cyclic peptide, was the first reported KRASG12D-selective inhibitor (Sakamoto et al., 2017). KRpep-2 was identified by phage display screening against KRASG12D and found to compete with SOS1 binding. Affinity optimization of KRpep-2 was achieved by the addition of arginine residues to both the N- and C-termini, resulting in KRpep-2d (Sakamoto et al., 2017). Both KRpep-2 and KRpep-2d inhibited KRASG12D nucleotide exchange in a concentration-dependent manner in vitro and KRpep-2d decreased ERK activation in A427 cells (KRASG12D) but not in A549 cells (KRASG12C) (Sakamoto et al., 2017). However, due to poor membrane permeability and decreased inhibitory activity, KRpep-2d was not effective in vivo (Sakamoto et al., 2017). To address this concern, KRpep-2d was modified using three approaches: (1) replacing the disulfide bond with an amide bond to increase resistance to reductive cleavage, (2) introducing unnatural amino acids to the binding region to increase its affinity for KRASG12D, and (3) connecting the N- and C-termini via amide bond to introduce protease-resistant, membrane permeable cyclization (Sakamoto et al., 2020). This optimization led to KS-58 (Table 1), the first reported KRASG12D inhibitory peptide displaying in vivo anti-cancer activity (Sakamoto et al., 2020). Further development of this lead compound will be needed prior to moving into clinical trials.
Utilizing a different screening approach, Zhang et al. recently reported another KRASG12D-specific cyclic peptide, KD2 (Table 1). Random non-standard Peptides Integrated Discovery (RaPID) (Ong et al., 2017) was used to screen for cyclic peptides that bound GppNHp-loaded KRASG12D/T35S to minimize the probability of identifying peptides that bound RAS but did not disrupt effector binding or peptides that stabilized the effector binding-competent state (Zhang et al., 2020). Five total rounds of screening were performed against GppNHp-bound KRASG12D/T35S and compared to binding of GDP-bound KRASG12D/T35S and empty streptavidin beads for quantification (Zhang et al., 2020). KD2 inhibited the RAS–RAF interaction and, to a weaker extent, SOS1-mediated nucleotide exchange; however, unlike KRpep-2d, KD2 bound KRASG12D-GTP (Zhang et al., 2020). Structural analysis revealed that KD2 bound and expanded the Switch II groove by shifting the α2 helix and Switch II loop and appeared to make direct contact with Asp12 of KRASG12D via Thr10 (Fig. 2F) (Zhang et al., 2020). Structure-guided optimization led to a >10-fold increase in potency (IC50 of 0.80 μM) at inhibiting the RAS–RAF interaction in vitro (Zhang et al., 2020). While these results remain encouraging, KD2 was cell impermeable, thereby requiring further optimization to translate into cell-based and in vivo assays (Zhang et al., 2020).
2.3.2. Stapled peptides
Stapled peptides represent yet another group of macromolecules to disrupt RAS function. Stapled peptides usually consist of α-helical segments linked or “stapled” by side chains that can be covalently bonded (Ali, Atmaj, Van Oosterwijk, Groves, & Domling, 2019). This linkage renders the alpha helical structure more rigid, increasing stability and resistance to proteolysis in the cell. Furthermore, the incorporation of non-natural amino acids can further enhance both of these properties (Ali et al., 2019). In addition, the peptide nature of these compounds allows easier optimization to confer affinity and selectivity to the target of interest (Leshchiner et al., 2015).
Patgiri et al. created a SOS1 αH helix mimic, HBS3, using a hydrogen bond surrogate approach. HBS3 bound to nucleotide-free (Kd of ~28 μM) and GDP-loaded RAS (Kd of ~158 μM) near the Switch regions and nucleotide pocket where the αH helix of SOS1 normally binds (Patgiri, Yadav, Arora, & Bar-Sagi, 2011). HBS treatment of cells decreased RAS activation following EGF stimulation of HeLa cells without altering the phosphorylation status of EGFR (Patgiri et al., 2011). Leshchiner et al. used the primary sequence of the RAS-interacting α-helix of SOS1 to generate stabilized alpha helices of SOS1 (SAH-SOS1). Of the stapled peptides screened, one was further optimized by the addition of two arginine residues to the N-terminus to decrease its overall charge and increase its cell permeability (Leshchiner et al., 2015). The resulting stapled peptide, SAH-SOS1A (Table 1), bound all RAS mutants (both GDP- and GTP-bound) with nanomolar affinity (Kd of 100–175 nM) and competed with RAS–SOS1 binding (Leshchiner et al., 2015). SAH-SOS1A additionally blocked the association of nucleotide with both WT and KRASG12D in vitro. In cell-based assays, SAH-SOS1A dose-dependently impaired the viability of various KRAS-mutant cancer cell lines and dose-dependently decreased MEK, ERK, and AKT phosphorylation in EGF-stimulated Panc 10.5 cells (Leshchiner et al., 2015).
3. Targeting RAS using peptide-PROTACs: A new frontier for RAS inhibition
An emerging strategy to selectively degrade proteins utilizes the ubiquitin–proteasome system (UPS). In contrast to technologies such as RNAi and CRISPR, which knock-down or eliminate expression of proteins at a genetic level, proteolysis-targeting chimeras (PROTACs) result in the specific post-translational degradation of a target protein (Chopra, Sadok, & Collins, 2019; Pettersson & Crews, 2019). Traditionally, PROTACs are bifunctional small molecules containing two moieties: a ligand for a specific target of interest coupled to a ligand for a specific E3 ubiquitin ligase complex (Chopra et al., 2019; Pettersson & Crews, 2019). The PROTAC acts to recruit the ubiquitin ligase machinery directly to a target protein for polyubiquitination and subsequent proteasomal degradation (Chopra et al., 2019; Pettersson & Crews, 2019). Thus, PROTACs “hijack” the ubiquitin–proteasome machinery to specifically degrade target proteins. This approach holds advantages over genetic knockdown methods, especially when considering the long-lived half-life of certain proteins (Pettersson & Crews, 2019). An additional benefit of PROTACs is their event-driven mechanism of action (MOA) as opposed to traditional pharmacological occupancy-driven MOA (Pettersson & Crews, 2019). This is particularly beneficial for small molecule PROTACs: as they are not consumed in the reaction, they can carry out multiple rounds proteasomal targeting (Pettersson & Crews, 2019).
Recently, the Crews laboratory developed the first oncogenic RAS-targeting PROTAC, termed LC2, by combining the KRASG12C-specific inhibitor, MRTX849, with a VHL E3 ligase-recruiting ligand (Bond, Chu, Nalawansha, Li, & Crews, 2020). Although this represents an encouraging development in anti-RAS pharmacologics, the current KRASG12C inhibitors are already covalent, irreversible inhibitors of KRAS. Furthermore, the problem still remains that KRASG12C is present in only ~10% of KRAS-mutant tumors (Khan, Rhett, & O’Bryan, 2020) with a ~2.2% overall prevalence in cancer (Zuberi et al., 2020). Furthermore, the lack of high affinity small molecule inhibitors of other KRAS alleles limits the translation of these findings to additional KRAS mutants.
Peptide-PROTACs (also referred to as peptide degraders, biodegraders, bioPROTACs, and Affinity-Directed Protein Missiles) provide an alternative approach to small molecule PROTACs (Fig. 3). Peptide-PROTACs contain three main components: an E3 ubiquitin ligase, a high affinity protein binder specific for the target of interest, and a linker joining the two protein domains. The most commonly used E3 ligases thus far are VHL and CRBN (Jin et al., 2020; Lim et al., 2021). Gly/Ser linkers of varying lengths are most often used to connect the E3 ligase and protein-targeting ligands ( Jin et al., 2020). Peptide-PROTACs are advantageous as they can be genetically encoded ( Jin et al., 2020), include an E3 ligase or an E3 ligase-recruiting motif (i.e., degrons) ( Jin et al., 2020; Qu et al., 2020), and can include high-affinity biologics such as Mbs (Lim et al., 2021; Roth et al., 2020) or DARPins (Bery, Miller, & Rabbitts, 2020) as protein-targeting ligands when small molecule ligands are not an option.
Fig. 3.

RAS-targeting peptide-PROTACs. Anti-RAS biologics are genetically fused to an E3 ubiquitin ligase resulting in recruitment of the complex to RAS, polyubiquitination and subsequent proteasomal degradation. The peptide-PROTAC, however, can be recycled to initiate multiple rounds of RAS degradation.
Ma et al. effectively applied the peptide-PROTAC approach to inhibit KRAS (Ma et al., 2013). They coupled the RBD and cysteine-rich domain (CRD) of c-RAF to the U-Box E3 ligase domain of the carboxyl terminus of Hsc70 interacting protein (CHIP) to generate the peptide-PROTAC, RC-U (Ma et al., 2013). RC-U co-immunoprecipitated with FLAG-tagged KRAS in HEK 293T cells and decreased the expression of endogenous KRAS in PANC-1 cells in a polyubiquitination-dependent manner (Ma et al., 2013). Treatment with the proteasome inhibitor, MG-132, rescued KRAS expression in RC-U-transfected PANC-1 cells, demonstrating that the decrease in KRAS expression in RC-U-transfected cells was proteasome-dependent (Ma et al., 2013). Lastly, expression of RC-U decreased tumor volume of PANC-1 xenografts (Ma et al., 2013). Collectively, these results demonstrated that proteasomal targeting of KRAS was indeed possible using a peptide-PROTAC approach.
Another KRAS-targeting peptide-PROTAC, VHL-aHRAS, was recently reported by Roth et al. (Roth et al., 2020). This approach linked the NS1 Mb discussed in Section 2.2.1 (termed aHRAS in the study) to VHL. VHL-aHRAS or VHL-aGFP16 (a control specific for GFP) were retrovirally transduced in A549 cells bearing a homozygous knock-in of GFP-tagged KRAS (Roth et al., 2020). VHL-aHRAS reduced both endogenous KRAS (and HRAS) and GFP-KRAS protein levels whereas the VHL-aGFP16 only reduced GFP-KRAS protein levels (Roth et al., 2020). Furthermore, MG-132 and bortezomib both induced accumulation of polyubiquitinated proteins and rescued RAS protein expression in A549 cells transduced with VHL-aHRAS (Roth et al., 2020). Proteomic analysis revealed that HRAS was decreased >2-fold by VHL-aHRAS compared to VHL alone; however, KRAS was not significantly reduced (Roth et al., 2020). This same trend was observed (via Western blot analysis) in HT-29 and SW620 cell lines (Roth et al., 2020). This selectivity for HRAS is likely due to the higher affinity of NS1 for HRAS vs KRAS (Spencer-Smith et al., 2017). Nonetheless, VHL-aHRAS was effective at targeting HRAS, and KRAS to a lesser extent, for proteasomal degradation.
Rabbitts and colleagues also developed anti-RAS peptide-PROTACs using their RAS targeting iDabs and DARPins as described in Section 2.1.2 and 2.2.2, respectively (Bery et al., 2020). These anti-RAS biologics were genetically fused to either VHL or the U-Box domain of CHIP and examined for their ability to degrade endogenous H, K, and NRAS in HCT116 cells. Fusion of the U-Box domain to the COOH-terminus of the iDab (pan-RAS degrader) was more efficient than N-terminal fusion and was more potent than VHL fusions (Bery et al., 2020). Conversely, fusion of VHL to the N-terminus of the anti-KRAS DARPin (KRAS degrader) was the most effective peptide-PROTAC (Bery et al., 2020). These findings emphasize the importance of optimizing the orientation of the specific ubiquitin ligase domain relative to the specific targeting domain in the design of a particular peptide-PROTAC.
The pan-RAS PROTAC (i.e., iDab) reduced expression of H, K, and NRAS whereas the KRAS-specific PROTAC (i.e., DARPin) selectively reduced KRAS levels in a time- and dose-dependent manner in H358 cells resulting in a commensurate reduction in AKT, MEK, and especially ERK phosphorylation (Bery et al., 2020). RAS expression levels were rescued by inhibition of the proteasome suggesting that the mechanism of targeted knockdown was indeed proteasome-dependent degradation (Bery et al., 2020). Furthermore, the specificities of the pan-RAS and KRAS degraders were consistent in a panel of RAS mutant cell lines and recapitulated in vivo using doxycycline-inducible pan-RAS degrader and KRAS degrader, both of which reduced KRAS-mutant tumor growth (Bery et al., 2020). However, only the pan-RAS degrader elicited effects in non-oncogenic KRAS mice xenografts (Bery et al., 2020).
Lim et al. have also explored the use of peptide-PROTACs in targeting RAS degradation. They examined the effectiveness of 10 different E3 ligases and identified the speckle type POZ protein (SPOP) as the most robust E3 ligase for this format. SPOP was then fused to various anti-RAS biologics including NS1 Mb, K27 and K55 DARPins, the R11.1.6 Sso7d, and the c-RAF RBD-CRD (Lim et al., 2021). All peptide-PROTACs with the exception of K55 effectively depleted KRAS levels, with the RBD-CRD-SPOP and K27-SPOP promoting the most robust RAS depletion (Lim et al., 2021). In addition, K27-reduced endogenous KRAS expression in AsPC-1 PDAC tumor cells (KRASG12D) resulting in decreased proliferation and increased apoptosis. Furthermore, these studies provided novel insights into the prevalence of the inactive state of KRAS through the use of GDP-state specific biologics such as K27 DARPin. Consistent with the relative population of the GDP-bound state by each KRAS (Moore et al., 2020), K27-SPOP degraded KRASWT >KRASG12C > KRASG12D >KRASG12V. To further confirm the specificity of K27-SPOP for the GDP-bound state, treatment of KRASG12C expressing cells with AMG510, which traps KRASG12C in the GDP-bound state, enhanced K27-SPOP mediated degradation of KRAS but blocked the effects of the RBD-CRD-SPOP degrader. Interestingly the KRASQ61H mutant was efficiently degraded by K27-SPOP despite its reported low catalytic activity (Hunter et al., 2015). Finally, as discussed in Section 2.2.1, Teng et al. recently developed a mutation specific degrader (Mb 12VC1) that selectively binds and degrades KRASG12V and KRASG12C (Teng et al., 2021). Together, these studies highlight the potential power of using peptide-PROTACs to target oncogenic RAS in cells. Further development of this technology will be needed to allow for effective and efficient delivery of such degraders to tumor cells in a patient.
4. Conclusion and future outlook
RAS remains at the forefront of cancer research given its prominent role in driving tumorigenesis. Despite the recent success in developing anti-RAS pharmacologics, continued efforts are required to expand on this success. RAS biologics have proven effective at inhibiting oncogenic RAS function through multiple mechanisms including disruption of PPI with effectors or activators of RAS as well as allosteric inhibition. However, these biologics require additional optimization to enhance their cellular uptake and tumor specificity in order to become useful clinical agents. With the utilization of delivery mechanisms such as cell-penetrating peptides, this shift appears ever more realistic. Additionally, with the entrance of liposomal-mediated cellular delivery of mRNA in the vaccine arena, many of the aforementioned genetically encodable biologics may yet reach the clinic setting. Nevertheless, biologics have proven powerful tools to further understand the biology of RAS, and these new insights are unveiling additional vulnerabilities of the formerly “undruggable” RAS.
Acknowledgments
The authors wish to apologize in advance for any omissions of references to our colleagues’ work. M.W was supported by an NIH T32 (GM132055). J.P.O. was supported in part by a Merit Review Award (1I01BX002095) from the United States (US) Department of Veterans Affairs Biomedical Laboratory Research and Development Service, NIH awards (CA212608, CA138313, and GM103542), and start-up funds from the Hollings Cancer Center at MUSC. The contents of this article do not represent the views of the US. Department of Veterans Affairs or the United States Government.
Footnotes
Disclosures
J.P.O is listed as an inventor on a patent application on Monobodies targeting RAS filed by the Medical University of South Carolina and New York University (No. 62/862,924).
References
- Ali AM, Atmaj J, Van Oosterwijk N, Groves MR, & Domling A (2019). Stapled peptides inhibitors: A new window for target drug discovery. Computational and Structural Biotechnology Journal, 17, 263–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antic I, Biancucci M, Zhu Y, Gius DR, & Satchell KJF (2015). Site-specific processing of Ras and Rap1 switch I by a MARTX toxin effector domain. Nature Communications, 6, 7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bery N, Legg S, Debreczeni J, Breed J, Embrey K, Stubbs C, et al. (2019). KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nature Communications, 10, 2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bery N, Miller A, & Rabbitts T (2020). A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nature Communications, 11, 3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancucci M, Minasov G, Banerjee A, Herrera A, Woida PJ, Kieffer MB, et al. (2018). The bacterial Ras/Rap1 site-specific endopeptidase RRSP cleaves Ras through an atypical mechanism to disrupt Ras-ERK signaling. Science Signaling, 11, eaat8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biocca S, Pierandrei-Amaldi P, Campioni N, & Cattaneo A (1994). Intracellular immunization with cytosolic recombinant antibodies. Biotechnology (N Y), 12, 396–399. [DOI] [PubMed] [Google Scholar]
- Biocca S, Pierandrei-Amaldi P, & Cattaneo A (1993). Intracellular expression of anti-p21ras single chain Fv fragments inhibits meiotic maturation of xenopus oocytes. Biochemical and Biophysical Research Communications, 197, 422–427. [DOI] [PubMed] [Google Scholar]
- Bond MJ, Chu L, Nalawansha DA, Li K, & Crews CM (2020). Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Central Science, 6, 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature, 575, 217–223. [DOI] [PubMed] [Google Scholar]
- Cardinale A, Lener M, Messina S, Cattaneo A, & Biocca S (1998). The mode of action of Y13–259 scFv fragment intracellularly expressed in mammalian cells. FEBS Letters, 439, 197–202. [DOI] [PubMed] [Google Scholar]
- Cetin M, Evenson WE, Gross GG, Jalali-Yazdi F, Krieger D, Arnold D, et al. (2017). RasIns: Genetically encoded intrabodies of activated Ras proteins. Journal of Molecular Biology, 429, 562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DK, Bae J, Shin SM, Shin JY, Kim S, & Kim YS (2014). A general strategy for generating intact, full-length IgG antibodies that penetrate into the cytosol of living cells. MAbs, 6, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra R, Sadok A, & Collins I (2019). A critical evaluation of the approaches to targeted protein degradation for drug discovery. Drug Discovery Today: Technologies, 31, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R, Wong G, Arnheim N, Nitecki D, & McCormick F (1985). Antibodies specific for amino acid 12 of the ras oncogene product inhibit GTP binding. Proceedings of the National Academy of Sciences of the United States of America, 82, 5280–5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochet O, Kenigsberg M, Delumeau I, Virone-Oddos A, Multon MC, Fridman WH, et al. (1998). Intracellular expression of an antibody fragment-neutralizing p21 ras promotes tumor regression. Cancer Research, 58, 1170–1176. [PubMed] [Google Scholar]
- Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. (2020). Identification of the clinical development candidate MRTX849, a covalent KRAS(G12C) inhibitor for the treatment of cancer. Journal of Medicinal Chemistry, 63, 6679–6693. [DOI] [PubMed] [Google Scholar]
- Feramisco JR, Clark R, Wong G, Arnheim N, Milley R, & McCormick F (1985). Transient reversion of ras oncogene-induced cell transformation by antibodies specific for amino acid 12 of ras protein. Nature, 314, 639–642. [DOI] [PubMed] [Google Scholar]
- Friedman M, & Stahl S (2009). Engineered affinity proteins for tumour-targeting applications. Biotechnology and Applied Biochemistry, 53, 1–29. [DOI] [PubMed] [Google Scholar]
- Furth ME, Davis LJ, Fleurdelys B, & Scolnick EM (1982). Monoclonal antibodies to the p21 products of the transforming gene of Harvey murine sarcoma virus and of the cellular ras gene family. Journal of Virology, 43, 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaine J, Turco C, Vauchy C, Royer B, Mercier-Letondal P, Queiroz L, et al. (2019). CD4 T cells target colorectal cancer antigens upregulated by oxaliplatin. International Journal of Cancer, 145, 3112–3125. [DOI] [PubMed] [Google Scholar]
- Grimm S, Lundberg E, Yu F, Shibasaki S, Vernet E, Skogs M, et al. (2010). Selection and characterisation of affibody molecules inhibiting the interaction between Ras and Raf in vitro. New Biotechnology, 27, 766–773. [DOI] [PubMed] [Google Scholar]
- Guillard S, Kolasinska-Zwierz P, Debreczeni J, Breed J, Zhang J, Bery N, et al. (2017). Structural and functional characterization of a DARPin which inhibits Ras nucleotide exchange. Nature Communications, 8, 16111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. (2020). The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discovery, 10, 54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell, 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, Tran TH, et al. (2020). Atypical KRAS(G12R) mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discovery, 10, 104–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs GA, Der CJ, & Rossman KL (2016). RAS isoforms and mutations in cancer at a glance. Journal of Cell Science, 129, 1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (2020). KRAS(G12C) inhibition with sotorasib in advanced solid tumors. The New England Journal of Medicine, 383, 1207–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, & Westover KD (2015). Biochemical and structural analysis of common cancer-associated KRAS mutations. Molecular Cancer Research, 13, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Hurd CA, Mott HR, & Owen D (2020). Therapeutic peptides targeting the Ras superfamily. Peptide Science, 112, e24165. [Google Scholar]
- Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, et al. (1988). Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 85, 5879–5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Wu Y, Chen J, Shen Y, Zhang L, Zhang H, et al. (2020). The peptide PROTAC modality: A novel strategy for targeted protein ubiquitination. Theranostics, 10, 10141–10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnoub AE, & Weinberg RA (2008). Ras oncogenes: Split personalities. Nature Reviews. Molecular Cell Biology, 9, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauke MJ, Traxlmayr MW, Parker JA, Kiefer JD, Knihtila R, McGee J, et al. (2017). An engineered protein antagonist of K-Ras/B-Raf interaction. Scientific Reports, 7, 5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I, MarElia-Bennet C, Lefler J, Zuberi M, Denbaum E, Koide A, et al. (2021). Targeting the KRAS alpha4-alpha5 allosteric interface inhibits pancreatic cancer tumorigenesis. Small GTPases, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I, Rhett JM, & O’Bryan JP (2020). Therapeutic targeting of RAS: New hope for drugging the “undruggable”. Biochimica et Biophysica Acta, Molecular Cell Research, 1867, 118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I, Spencer-Smith R, & O’Bryan JP (2019). Targeting the alpha4-alpha5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene, 38, 2984–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A, Bailey CW, Huang X, & Koide S (1998). The fibronectin type III domain as a scaffold for novel binding proteins. Journal of Molecular Biology, 284, 1141–1151. [DOI] [PubMed] [Google Scholar]
- Koide A, Wojcik J, Gilbreth RN, Hoey RJ, & Koide S (2012). Teaching an old scaffold new tricks: Monobodies constructed using alternative surfaces of the FN3 scaffold. Journal of Molecular Biology, 415, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordiak J, Szemraj J, Grabska-Kobylecka I, Bialasiewicz P, Braun M, Kordek R, et al. (2019). Intratumor heterogeneity and tissue distribution of KRAS mutation in non-small cell lung cancer: Implications for detection of mutated KRAS oncogene in exhaled breath condensate. Journal of Cancer Research and Clinical Oncology, 145, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschubs T, Dengl S, Durr H, Kaluza K, Georges G, Hartl C, et al. (2012). Allosteric antibody inhibition of human hepsin protease. The Biochemical Journal, 442, 483–494. [DOI] [PubMed] [Google Scholar]
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. (2015). Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature, 520, 692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer L, Parizek P, Rube P, Millgramm B, Prinz A, Mittl PR, et al. (2012). Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries. Proceedings of the National Academy of Sciences of the United States of America, 109, E2248–E2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JS, Jeong HG, & Satchell KJ (2011). Vibrio vulnificus rtxA1 gene recombination generates toxin variants with altered potency during intestinal infection. Proceedings of the National Academy of Sciences of the United States of America, 108, 1645–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacal JC, & Aaronson SA (1986). Monoclonal antibody Y13–259 recognizes an epitope of the p21 ras molecule not directly involved in the GTP-binding activity of the protein. Molecular and Cellular Biology, 6, 1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy A, Blanchard F, Le Pessot F, Sesboue R, Di Fiore F, Bossut J, et al. (2011). Metastatic colorectal cancer KRAS genotyping in routine practice: Results and pitfalls. Modern Pathology, 24, 1090–1100. [DOI] [PubMed] [Google Scholar]
- Leshchiner ES, Parkhitko A, Bird GH, Luccarelli J, Bellairs JA, Escudero S, et al. (2015). Direct inhibition of oncogenic KRAS by hydrocarbon-stapled SOS1 helices. Proceedings of the National Academy of Sciences of the United States of America, 112, 1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. (2019). KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell, 35, 559–572 e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Khoo R, Juang YC, Gopal P, Zhang H, Yeo C, et al. (2021). Exquisitely specific anti-KRAS biodegraders inform on the cellular prevalence of nucleotide-loaded states. ACS Central Science, 7, 274–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Chen Z, Hu C, Chen ZS, & Zhang L (2020). Recent progress in antitumor functions of the intracellular antibodies. Drug Discovery Today, 25, 1109–1120. [DOI] [PubMed] [Google Scholar]
- Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. (2015). High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nature Medicine, 21, 81–85. [DOI] [PubMed] [Google Scholar]
- Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, et al. (2020). Development of therapeutic antibodies for the treatment of diseases. Journal of Biomedical Science, 27, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Gu Y, Zhang Q, Han Y, Yu S, Lu Z, et al. (2013). Targeted degradation of KRAS by an engineered ubiquitin ligase suppresses pancreatic cancer cell growth in vitro and in vivo. Molecular Cancer Therapeutics, 12, 286–294. [DOI] [PubMed] [Google Scholar]
- Marschall AL, & Dubel S (2016). Antibodies inside of a cell can change its outside: Can intrabodies provide a new therapeutic paradigm? Computational and Structural Biotechnology Journal, 14, 304–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee JH, Shim SY, Lee SJ, Swanson PK, Jiang SY, Durney MA, et al. (2018). Exceptionally high-affinity Ras binders that remodel its effector domain. The Journal of Biological Chemistry, 293, 3265–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano X, & Jimenez A (1995). Intracellular expression of the monoclonal anti-ras antibody Y13–259 blocks the transforming activity of ras oncogenes. Cell Growth & Differentiation, 6, 597–605. [PubMed] [Google Scholar]
- Moore AR, Rosenberg SC, McCormick F, & Malek S (2020). RAS-targeted therapies: Is the undruggable drugged? Nature Reviews. Drug Discovery, 19, 533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy LS, Smith MR, & Stacey DW (1985). Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature, 313, 241–243. [DOI] [PubMed] [Google Scholar]
- Nielsen DS, Shepherd NE, Xu W, Lucke AJ, Stoermer MJ, & Fairlie DP (2017). Orally absorbed cyclic peptides. Chemical Reviews, 117, 8094–8128. [DOI] [PubMed] [Google Scholar]
- Nilsson FY, & Tolmachev V (2007). Affibody molecules: New protein domains for molecular imaging and targeted tumor therapy. Current Opinion in Drug Discovery & Development, 10, 167–175. [PubMed] [Google Scholar]
- Nygren PA (2008). Alternative binding proteins: Affibody binding proteins developed from a small three-helix bundle scaffold. The FEBS Journal, 275, 2668–2676. [DOI] [PubMed] [Google Scholar]
- Oltedal S, Aasprong OG, Moller JH, Korner H, Gilje B, Tjensvoll K, et al. (2011). Heterogeneous distribution of K-ras mutations in primary colon carcinomas: Implications for EGFR-directed therapy. International Journal of Colorectal Disease, 26, 1271–1277. [DOI] [PubMed] [Google Scholar]
- Ong YS, Gao L, Kalesh KA, Yu Z, Wang J, Liu C, et al. (2017). Recent advances in synthesis and identification of cyclic peptides for bioapplications. Current Topics in Medicinal Chemistry, 17, 2302–2318. [DOI] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, & Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature, 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature, 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan XY, Liu XJ, Li J, Zhen SJ, Liu DX, Feng Q, et al. (2017). The antitumor efficacy of anti-p21Ras scFv mediated by the dual-promoter-regulated recombinant adenovirus KGHV300. Gene Therapy, 24, 40–48. [DOI] [PubMed] [Google Scholar]
- Parizek P, Kummer L, Rube P, Prinz A, Herberg FW, & Pluckthun A (2012). Designed ankyrin repeat proteins (DARPins) as novel isoform-specific intracellular inhibitors of c-Jun N-terminal kinases. ACS Chemical Biology, 7, 1356–1366. [DOI] [PubMed] [Google Scholar]
- Patgiri A, Yadav KK, Arora PS, & Bar-Sagi D (2011). An orthosteric inhibitor of the Ras-Sos interaction. Nature Chemical Biology, 7, 585–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei D, Chen K, & Liao H (2018). Targeting Ras with macromolecules. Cold Spring Harbor Perspectives in Medicine, 8, a031476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson M, & Crews CM (2019). PROteolysis TArgeting chimeras (PROTACs)—Past, present and future. Drug Discovery Today: Technologies, 31, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe GJB, Craik DJ, & Henriques ST (2021). Converting peptides into drugs targeting intracellular protein-protein interactions. Drug Discovery Today, 26, 1521–1531. [DOI] [PubMed] [Google Scholar]
- Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, et al. (2019). Tissue-specific oncogenic activity of KRAS(A146T). Cancer Discovery, 9, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Ren X, Xue F, He Y, Zhang R, Zheng Y, et al. (2020). Specific knockdown of alpha-synuclein by peptide-directed proteasome degradation rescued its associated neurotoxicity. Cell Chemical Biology, 27, 751–762 e754. [DOI] [PubMed] [Google Scholar]
- Richman SD, Chambers P, Seymour MT, Daly C, Grant S, Hemmings G, et al. (2011). Intra-tumoral heterogeneity of KRAS and BRAF mutation status in patients with advanced colorectal cancer (aCRC) and cost-effectiveness of multiple sample testing. Analytical Cellular Pathology (Amsterdam), 34, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, Macartney TJ, Konopacka A, Chan KH, Zhou H, Queisser MA, et al. (2020). Targeting endogenous K-RAS for degradation through the affinity-directed protein missile system. Cell Chemical Biology, 27, 1151–1163 e1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MB, & Corcoran RB (2018). Therapeutic strategies to target RAS-mutant cancers. Nature Reviews. Clinical Oncology, 15, 709–720. [DOI] [PubMed] [Google Scholar]
- Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. (2017). Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature, 547, 222–226. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Kamada Y, Sameshima T, Yaguchi M, Niida A, Sasaki S, et al. (2017). K-Ras(G12D)-selective inhibitory peptides generated by random peptide T7 phage display technology. Biochemical and Biophysical Research Communications, 484, 605–611. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Masutani T, & Hirokawa T (2020). Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Scientific Reports, 10, 21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini BL, & Zacharias M (2020). Rapid in silico design of potential cyclic peptide binders targeting protein-protein interfaces. Frontiers in Chemistry, 8, 573259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha F, Salzman G, Gupta A, & Koide S (2017). Monobodies and other synthetic binding proteins for expanding protein science. Protein Science, 26, 910–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki S, Karasaki M, Graslund T, Nygren PA, Sano H, & Iwasaki T (2014). Inhibitory effects of H-Ras/Raf-1-binding affibody molecules on synovial cell function. AMB Express, 4, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SM, Choi DK, Jung K, Bae J, Kim JS, Park SW, et al. (2017). Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nature Communications, 8, 15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SM, Kim JS, Park SW, Jun SY, Kweon HJ, Choi DK, et al. (2020). Direct targeting of oncogenic RAS mutants with a tumor-specific cytosol-penetrating antibody inhibits RAS mutant-driven tumor growth. Science Advances, 6, eaay2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. (2021). Sotorasib for lung cancers with KRAS p.G12C mutation. The New England Journal of Medicine, 384, 2371–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, et al. (2017). Inhibition of RAS function through targeting an allosteric regulatory site. Nature Chemical Biology, 13, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Smith R, Li L, Prasad S, Koide A, Koide S, & O’Bryan JP (2019). Targeting the alpha4-alpha5 interface of RAS results in multiple levels of inhibition. Small GTPases, 10, 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocks MR (2004). Intrabodies: Production and promise. Drug Discovery Today, 9, 960–966. [DOI] [PubMed] [Google Scholar]
- Stumpp MT, Binz HK, & Amstutz P (2008). DARPins: A new generation of protein therapeutics. Drug Discovery Today, 13, 695–701. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Misaki I, Tokunaga Y, Fujisaki M, Kamoshida H, Takizawa T, et al. (2021). Conformational plasticity of cyclic ras-inhibitor peptides defines cell permeabilization activity. Angewandte Chemie (International Ed. in English), 60, 6567–6572. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Lobato MN, & Rabbitts TH (2003). Single domain intracellular antibodies: A minimal fragment for direct in vivo selection of antigen-specific intrabodies. Journal of Molecular Biology, 331, 1109–1120. [DOI] [PubMed] [Google Scholar]
- Tanaka T, & Rabbitts TH (2003). Intrabodies based on intracellular capture frameworks that bind the RAS protein with high affinity and impair oncogenic transformation. The EMBO Journal, 22, 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, & Rabbitts TH (2010). Interfering with RAS-effector protein interactions prevent RAS-dependent tumour initiation and causes stop-start control of cancer growth. Oncogene, 29, 6064–6070. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Williams RL, & Rabbitts TH (2007). Tumour prevention by a single antibody domain targeting the interaction of signal transduction proteins with RAS. The EMBO Journal, 26, 3250–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay RE, Richardson EK, & Toh HC (2021). Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Therapy, 28, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng KW, Tsai ST, Hattori T, Fedele C, Koide A, Yang C, et al. (2021). Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nature Communications, 12, 2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW (1994). Physiological concentrations of purines and pyrimidines. Molecular and Cellular Biochemistry, 140, 1–22. [DOI] [PubMed] [Google Scholar]
- Traxlmayr MW, Kiefer JD, Srinivas RR, Lobner E, Tisdale AW, Mehta NK, et al. (2016). Strong enrichment of aromatic residues in binding sites from a charge-neutralized hyperthermostable Sso7d scaffold library. The Journal of Biological Chemistry, 291, 22496–22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse E, Lobato MN, Forster A, Tanaka T, Chung GT, & Rabbitts TH (2002). Intracellular antibody capture technology: Application to selection of intracellular antibodies recognising the BCR-ABL oncogenic protein. Journal of Molecular Biology, 317, 85–94. [DOI] [PubMed] [Google Scholar]
- Upadhyaya P, Qian Z, Habir NA, & Pei D (2014). Direct Ras inhibitors identified from a structurally rigidified bicyclic peptide library. Tetrahedron, 70, 7714–7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya P, Qian Z, Selner NG, Clippinger SR, Wu Z, Briesewitz R, et al. (2015). Inhibition of Ras signaling by blocking Ras-effector interactions with cyclic peptides. Angewandte Chemie (International Ed. in English), 54, 7602–7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatansever S, Erman B, & Gumus ZH (2019). Oncogenic G12D mutation alters local conformations and dynamics of K-Ras. Scientific Reports, 9, 11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidimar V, Beilhartz GL, Park M, Biancucci M, Kieffer MB, Gius DR, et al. (2020). An engineered chimeric toxin that cleaves activated mutant and wild-type RAS inhibits tumor growth. Proceedings of the National Academy of Sciences of the United States of America, 117, 16938–16948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin M, Settanni G, Maritan A, Graziosi S, Marks JD, & Cattaneo A (2002). The intracellular antibody capture technology (IACT): Towards a consensus sequence for intracellular antibodies. Journal of Molecular Biology, 317, 73–83. [DOI] [PubMed] [Google Scholar]
- Wang M, Hong Y, Feng Q, Pan X, Song S, Cui J, et al. (2018). Recombinant adenovirus KGHV500 and CIK cells codeliver anti-p21-Ras scFv for the treatment of gastric Cancer with wild-type Ras overexpression. Molecular Therapy Oncolytics, 11, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM, & Der CJ (2018). KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harbor Perspectives in Medicine, 8, a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner GJ (2007). Monoclonal antibody mechanisms of action in cancer. Immunologic Research, 39, 271–278. [DOI] [PubMed] [Google Scholar]
- Werge TM, Baldari CT, & Telford JL (1994). Intracellular single chain Fv antibody inhibits Ras activity in T-cell antigen receptor stimulated Jurkat cells. FEBS Letters, 351, 393–396. [DOI] [PubMed] [Google Scholar]
- Wu X, Upadhyaya P, Villalona-Calero MA, Briesewitz R, & Pei D (2013). Inhibition of Ras-effector interaction by cyclic peptides. MedChemComm, 4, 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JL, Liu DX, Zhen SJ, Zhou YG, Zhang DJ, Yang LY, et al. (2016). A novel anti-p21Ras scFv antibody reacting specifically with human tumour cell lines and primary tumour tissues. BMC Cancer, 16, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gao R, Hu Q, Peacock H, Peacock DM, Dai S, et al. (2020). GTP-state-selective cyclic peptide ligands of K-Ras(G12D) block its interaction with Raf. ACS Central Science, 6, 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. (2018). Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature, 564, 268–272. [DOI] [PubMed] [Google Scholar]
- Zuberi M, Khan I, & O’Bryan JP (2020). Inhibition of RAS: Proven and potential vulnerabilities. Biochemical Society Transactions, 48, 1831–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]