
Fig. 1.
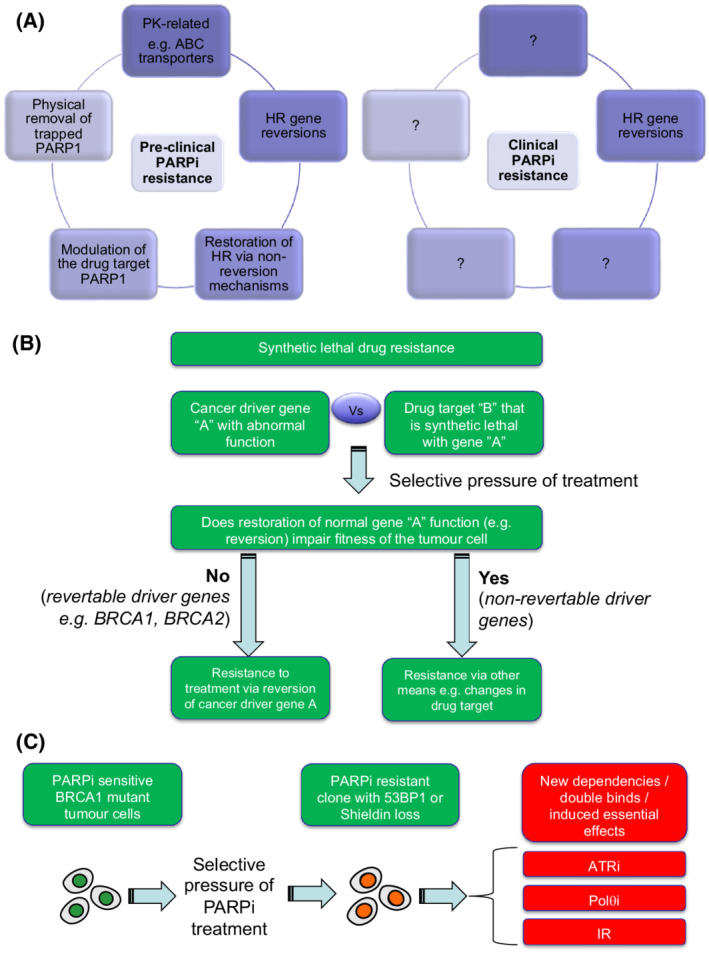
DDR inhibitor resistance. (A) In pre‐clinical models (e.g. tumour cell lines and genetically engineered mice, mechanisms of PARPi resistance have been identified that can be classified into four broad groups as shown on left. How many of these resistance mechanisms operate in the clinic is currently unclear. HR gene reversion mutations have been seen in multiple patients (right hand image), but do not explain all cases of PARPi resistance (see main text). Anecdotal numbers of PARPi resistant patients with either ABC transporter gene fusions, 53BP1 mutation or PARP1 mutation have been identified, but the true frequency of these mechanisms of resistance remain to be established. (B) Revertable and non‐revertable genes in synthetic lethal resistance. Synthetic lethal resistance describes the situation where drug resistance to a synthetic lethal treatment is caused by modulation of the synthetic lethal partner (e.g. reversion of BRCA2), as opposed to being caused by changes in the drug target. The potential for reversion emerging as a cause of synthetic lethal resistance must be determined by whether the reversing the dysfunction of the synthetic lethal partner (gene ‘a’) impairs the fitness of the tumour cell. The reversion of BRCA2 has no deleterious effects on tumour cell fitness and indeed reversion gives a tumour cell a fitness advantage in the face of PARPi treatment. Other driver genes, however, may not be revertable, as their continued dysfunction is essential for the tumour cell to survive (e.g. addicted oncogenes). For synthetic lethal interactions involving these non‐revertable genes, other forms of drug resistance might predominate, including alterations in the drug target itself. (C) New therapeutic vulnerabilities caused by drug resistance mechanisms. In some cases, the mechanism of cancer drug resistance that emerges upon treatment creates new therapeutic vulnerabilities, not previously present or as profound in the pre‐treated state. For example, in pre‐clinical models, PARPi resistance in BRCA1 mutant tumour cells can be caused by loss of 53BP1 or Shieldin complex function (see main text). These mechanisms of PARPi resistance, whilst giving the tumour cell a fitness advantage in the face of PARPi treatment, also impart a fitness disadvantage in the face of either ATRi, Polθi or ionising radiation (IR) exposure, an evolutionary double bind or induced essentiality effect.