
Figure 4.
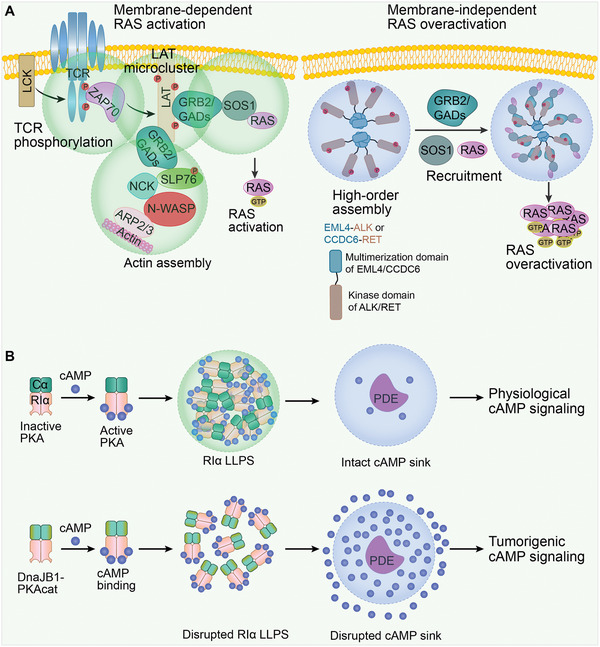
Misregulated signaling and transduction by LLPS in cancer. A) Left, membrane‐dependent RAS activation. A series of LLPS events results in RAS activation. Phosphorylated T cell receptor (TCR) activates the membrane kinase ZAP70, which then phosphorylates the tyrosine residues on linker for the activation of T cells (LAT). In turn, LAT microclusters form through enriching growth factor receptor‐bound protein 2 (GRB2) and GRB2‐related adaptor downstream of Shc (GADs). Subsequently, SOS1 is recruited for RAS activation. In addition, the actin effectors, non‐catalytic region of tyrosine kinase (NCK), neural Wiskott‐Aldrich syndrome protein (N‑WASP), and actin related protein (ARP) 2/3 complex can also be recruited for actin filament assembly. Right. membrane‐independent RAS overactivation. The oncogenic fusion protein, EML4‐ALK or CCDC6‐RET, obtains multimerization domains of echinoderm microtubule‐associated protein‐like 4 (EML4) or coiled‐coil domain‐containing protein 6 (CCDC6). These proteins lose membrane‐targeting sequences of anaplastic lymphoma kinase (ALK) or rearranged during transfection (RET) to form de novo membraneless cytoplasmic condensates. The RAS‐activating complexes GRB2/SOS1 are then concentrated in condensates, which leads to RAS overactivation. B) Upper, physiological cAMP signaling. In normal cells, RIα, a regulatory subunit of 3’, 5’‐cyclic adenosine monophosphate (cAMP)‐dependent protein kinase A (PKA), is capable of forming condensates and acting as a dynamic cAMP buffer. PKA activity is retained in the PDE sink. Lower, tumorigenic cAMP signaling. In fibrolamellar carcinoma, the native N‐terminus of PKA‐Cα is replaced by the J‐domain of DnaJB1. The resulting DnaJB1‐PKAcat fusion oncoprotein interferes with RIα LLPS, thus disrupting cAMP compartmentation by PDE. As a result, cAMP signaling is highly active.