

Abstract
Introduction
While the Dominantly Inherited Alzheimer Network Trials Unit (DIAN‐TU) was ongoing, external data suggested higher doses were needed to achieve targeted effects; therefore, doses of gantenerumab were increased 5‐fold, and solanezumab was increased 4‐fold. We evaluated to what extent mid‐trial dose increases produced a dose‐dependent treatment effect.
Methods
Using generalized linear mixed effects (LME) models, we estimated the annual low‐ and high‐dose treatment effects in clinical, cognitive, and biomarker outcomes.
Results
Both gantenerumab and solanezumab demonstrated dose‐dependent treatment effects (significant for gantenerumab, non‐significant for solanezumab) in their respective target amyloid biomarkers (Pittsburgh compound B positron emission tomography standardized uptake value ratio and cerebrospinal fluid amyloid beta 42), with gantenerumab demonstrating additional treatment effects in some downstream biomarkers. No dose‐dependent treatment effects were observed in clinical or cognitive outcomes.
Conclusions
Mid‐trial dose escalation can be implemented as a remedy for an insufficient initial dose and can be more cost effective and less burdensome to participants than starting a new trial with higher doses, especially in rare diseases.
Highlights
We evaluated the dose‐dependent treatment effect of two different amyloid‐specific immunotherapies.
Dose‐dependent treatment effects were observed in some biomarkers.
No dose‐dependent treatment effects were observed in clinical/cognitive outcomes, potentially due to the fact that the modified study may not have been powered to detect such treatment effects in symptomatic subjects at a mild stage of disease exposed to high (or maximal) doses of medication for prolonged durations.
Keywords: autosomal dominant Alzheimer's disease, Dominantly Inherited Alzheimer Network, dose escalation, gantenerumab, solanezumab
1. INTRODUCTION
The Dominantly Inherited Alzheimer Network Trials Unit (DIAN‐TU)‐001 is a phase II/III randomized, double‐blind, placebo‐controlled, cognitive endpoint, international study of potential disease‐modifying therapies (gantenerumab and solanezumab) in individuals with dominantly inherited Alzheimer's disease (DIAD) mutations. The design 1 of this trial incorporated several innovative strategies such as: a platform trial with a master protocol to concurrently and sequentially investigate multiple drugs, 1 , 2 a shared placebo group between active arms allowing for a 3:1 randomization in each arm, extended follow‐up with a common‐close design for all participants instead of a fixed study duration (4 years up to 7 years in the placebo‐controlled period), the use of a multivariate cognitive endpoint; an interim biomarker analysis, and dose escalation. The main findings of the primary and secondary analyses were a lack of cognitive benefit but significant amyloid beta (Aβ) target engagement of gantenerumab (fibrillar Aβ) and solanezumab (soluble Aβ). 3 In addition, gantenerumab had a significant effect on downstream biomarkers (e.g., decreased cerebrospinal fluid [CSF] measures of phosphorylated tau [p‐tau], total tau, and neurofilament light chain [NfL]). 3 Based on these findings, the trial continued in an exploratory open label extension for gantenerumab at the higher dose used in the double‐blind period. The goal of this report is to: (1) present details of the dose escalation procedure and (ii) evaluate and quantify the dose‐dependent effects of gantenerumab and solanezumab in clinical, cognitive, imaging, and CSF biomarker endpoints. We hypothesized dose‐dependent treatment benefits for gantenerumab and solanezumab in clinical, cognitive, imaging, and CSF biomarker outcomes.
2. METHOD
2.1. Study oversight
The study was conducted in accordance with the Declaration of Helsinki and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Good Clinical Practice guidelines and had ethics committee approval at each participating site. All participants provided written informed consent.
2.2. Study participants
The details about the trial participants have been reported previously. 3 Briefly, 194 participants enrolled in DIAN‐TU‐001, of whom 144 were DIAD mutation carriers (52 on gantenerumab, 52 on solanezumab, and 40 on placebo). These participants were either cognitively normal (Clinical Dementia Rating [CDR= 0]) or had early‐stage disease (CDR 0.5 or 1 representing very mild or mild dementia) at enrollment.
2.3. Dose escalation
The initial dose in the DIAN‐TU‐001 study was 225 mg of gantenerumab administered subcutaneously every 4 weeks, and 400 mg of solanezumab administered intravenously every 4 weeks. Midway through the DIAN‐TU‐001 study, results of concurrent phase II and III trials with the same drugs in sporadic Alzheimer's disease (AD) were made public, 4 , 5 and indicated the initial doses for both drugs in the DIAN‐TU‐001 trial were unlikely to yield sufficient reduction in amyloid deposition or demonstrate significant clinical/cognitive benefit at these doses. Therefore, dose escalation was proposed as a potential remedy. After obtaining regulatory approval, all active participants in the study at that time initiated dose escalation. Because amyloid‐related imaging abnormalities (ARIAs) were more common for gantenerumab compared to solanezumab in previous studies, the dose escalation and safety magnetic resonance imaging (MRI) schedules were drug specific: the gantenerumab dose was increased stepwise every 8 weeks to a maximum dose of 1200 mg (titrating up after two doses at each of the following dose levels: 225, 450, 675, and 900 mg), or the highest dose that an individual can tolerate, with safety MRIs scheduled at each step (Table 1). The solanezumab dose was increased stepwise every 4 weeks to a maximum dose of 1600 mg (titrating up from 400 mg to 800 mg every 4 weeks for two doses, to 1600 mg every 4 weeks), or the highest dose that an individual can tolerate, with a safety MRI scheduled after the second dose of 800 mg (Table 1).
TABLE 1.
DIAN‐TU‐001 gantenerumab/solanezumab dose escalation and safety MRI schedule
| Titration step 1 | Titration step 2 | Titration step 3 | Titration step 4 | Titration step 5 | |
|---|---|---|---|---|---|
| Gantenerumab |
|
|
|
|
|
| Solanezumab |
|
|
Abbreviations: DIAN‐TU, Dominantly Inherited Alzheimer Network Trials Unit; MRI, magnetic resonance imaging.
2.4. Clinical, cognitive, imaging, and CSF biomarker outcomes
The clinical outcomes analyzed included the CDR‐Sum of Boxes 6 (CDR‐SB) and Functional Assessment Scale (FAS) 7 ; the cognitive outcomes included the Mini‐Mental State Examination (MMSE), 8 the Wechsler Memory Scale‐Revised Logical Memory Delayed Recall Test (Logical Memory), 9 the Wechsler Adult Intelligence Scale Digit Symbol Substitution Test (Digit Symbol), 9 and the International Shopping List Test (ISLT) Delayed Recall score; 10 , 11 the imaging and CSF biomarker outcomes included Pittsburgh compound B positron emission tomography (PiB‐PET) composite standardized uptake value ratio (SUVR), fluorodeoxyglucose (FDG)‐PET SUVR, MRI‐derived volumetrics, CSF total Aβ42 (defined as free plus bound Aβ42) for solanezumab, CSF Aβ42 for gantenerumab, CSF total tau, CSF p‐tau181, and CSF NfL. Methods used for imaging biomarkers were described previously. 12 , 13 , 14 CSF biomarker methods are described in the supporting information.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources and meeting abstracts and presentations. Publications related to dose escalation for Alzheimer's disease clinical trials have been identified and properly cited.
Interpretation: The Dominantly Inherited Alzheimer Network Trials Unit (DIAN‐TU)‐001 platform trial successfully and safely implemented a dose escalation procedure for both study drug arms based on results from concurrent studies in sporadic AD while the trial was still ongoing. To optimize participant safety and characterize the safety profile of the higher doses, participants were carefully monitored during dose escalation. Compared to the initial dose, the high doses led to larger treatment effects in each drug's biomarker of target engagement of amyloid beta (Aβ; 90% more decrease in Pittsburgh compound B positron emission tomography standardized uptake value ratio with high‐dose gantenerumab, 41% more increase in total Aβ42 with high‐dose solanezumab).
Future directions: Future AD clinical trials with an insufficient initial dose may use the DIAN‐TU approach to conduct dose escalation during the trial as a remedy. Additional measures may be needed to facilitate the treatment effect evaluation after mid‐trial dose escalation such as “re‐start the clock” of the follow‐up duration to increase the high‐dose exposure and enroll additional participants to account for the early dropouts.
2.5. Statistical analysis plan
Because dose escalation was initiated while the study was ongoing and at different times for gantenerumab and solanezumab arms, each participant had a variable duration of exposure to the high doses (defined as any dose higher than the initial dose) depending on how long that individual had already been on the low dose (the initial dose). In some instances, participants withdrew from the study before dose escalation. To accommodate the individual‐specific low‐ and high‐dose treatment durations, generalized linear mixed effects (LME) models were used to estimate the annual rates of change in each outcome during the low‐ and high‐dose periods simultaneously in a single model including the gantenerumab, solanezumab, and placebo arms. 15 The placebo arm included the pooled placebos from both gantenerumab and solanezumab. Statistical inference on treatment efficacy in each outcome was made by comparing the annual rates of change of each treatment arm to that of the placebo arm, whereas the gantenerumab arm and the solanezumab arm were not to be compared per protocol. For clinical and cognitive measures, all data collected until the conclusion of this common‐close study were included in the model, whereas for biomarker measures, data were collected only up to year 4. The LME models included random intercepts and random slopes at the individual level to account for the correlation among repeated measures. The variances of these random effects and the residual variance were modeled separately for asymptomatic (CDR 0 at baseline) and symptomatic (CDR 0.5–1 at baseline) participants (Statistical Supplemental Materials for details). The normality assumption was examined using the residual plots from the LME models. When the assumption was determined to not be sufficiently met, a log transformation was applied.
All analyses were conducted with the use of SAS software, version 9.4. Nominal P‐values were presented from two‐sided t‐tests with type I error of 0.05. All confidence intervals (CIs) were 95% CIs.
3. RESULTS
3.1. Demographics
The demographics at baseline were reported previously 3 and were well balanced among the gantenerumab arm, the solanezumab arm, and the placebo arm. Gantenerumab started dose escalation an average of 1.8 (standard deviation [SD] 0.61) years after baseline, and 47 out of 52 (90%) had at least one high‐dose administration with an average duration of 2.4 (SD 0.55) years; solanezumab started an average of 3.2 (SD 0.67) years after baseline, and 37 out of 52 (71%) received at least one high‐dose administration with an average duration of 1.3 (SD 0.42) years. Of the 52 gantenerumab participants, 43 (83%) received an average of 26.3 (SD 9.9) administrations of the maximum dose (1200 mg) and 37 (71%) of the 52 solanezumab participants received an average of 18.6 (SD 4.8) administrations of the maximum dose (1600 mg). Table 2 shows the differences in clinical/cognitive outcomes at baseline and at visits closest (either before or after) to dose escalation. Table S1 in supporting information presents the clinical/cognitive/biomarker outcomes before the dose escalation (the last visit on low dose) and after the dose escalation (the last visit on high dose).
TABLE 2.
Clinical/cognitive/biomarker outcomes of participants at baseline and at visits closest to dose escalation
| Baseline a | Visit closest to dose escalation b | |||||
|---|---|---|---|---|---|---|
| Outcomes | GantenerumabN = 52 | SolanezumabN = 50 | SharedplaceboN = 40 | GantenerumabN = 47 | SolanezumabN = 37 | SharedplaceboN = 34 |
|
CDR c 0 CDR 0.5 CDR = 1 CDR > 1 N (%) |
31 (60) 15 (29) 6 (12) 0 (0) |
30 (60) 13 (26) 7 (14) 0 (0) |
22 (55) 15 (38) 3 (8) 0 (0) |
26 (55) 13 (28) 5 (11) 3 (6) |
22 (59) 8 (22) 2 (5) 5 (14) |
18 (55) 7 (21) 6 (18) 2 (6) |
| Digit Symbol d | 46.96 ± 20.56 | 46.06 ± 19.94 | 46.63 ± 19.12 | 48.13 ± 23.83 | 50.69 ± 23.34 | 54.74 ± 21.64 |
| MMSE e | 27.10 ± 3.45 | 26.72 ± 4.11 | 26.68 ± 3.97 | 25.91 ± 5.52 | 26.06 ± 6.45 | 25.70 ± 6.17 |
| Logical Memory f | 9.90 ± 6.33 | 9.86 ± 6.86 | 9.40 ± 6.45 | 11.55 ± 7.37 | 13.38 ± 7.96 | 12.12 ± 7.59 |
| ISLT g | 5.96 ± 4.04 | 6.56 ± 3.95 | 5.80 ± 4.42 | 6.13 ± 4.55 | 5.67 ± 4.26 | 6.24 ± 4.70 |
| CDR‐SB h | 1.33 ± 2.08 | 1.37 ± 2.01 | 1.43 ± 1.87 | 2.13 ± 3.13 | 2.61 ± 4.46 | 2.30 ± 3.39 |
| PiB‐PET composite SUVR | 2.64 ± 1.23 | 2.75 ± 1.32 | 2.62 ± 1.20 | 2.63 ± 1.25 | 2.76 ± 1.29 | 2.58 ± 1.14 |
Note: Plus‐minus values are means ± SD. PiB‐PET composite SUVR refers to brain amyloid burden measured by the average SUVR of cortical regions of interest (superior frontal, rostral middle frontal, superior temporal, middle temporal, lateral orbito‐frontal, medial orbito‐frontal and precuneus), assessed by PiB‐PET.
Fifty‐two participants were randomized to the solanezumab arm; two did not have post‐baseline data and were excluded from the modified intent‐to‐treat population.
Because the clinical and cognitive assessments were administered annually or every 6 months, the timing of these assessments did not match the starting time of dose escalation, for example, participants started dose escalation in between two visits.
CDR scores range from 0 to 3, with higher scores indicating worse cognition and daily function.
Digit Symbol Substitution Test (Digit Symbol) scores range from 0 to 93, with lower scores indicating poorer cognitive performance.
MMSE scores range from 0 to 30, with lower scores indicating poorer cognitive performance.
Logical Memory Delayed Recall Test (Logical Memory) scores range from 0 to 25, with lower scores indicating poorer cognitive performance.
ISLT scores range from 0 to 12, with lower scores indicating poorer cognitive performance.
CDR‐SB scores range from 0 to 18, with higher scores indicating worse cognition and daily function.
Abbreviations: CDR, Clinical Dementia Rating Scale; CDR‐SB, Clinical Dementia Rating Sum of Boxes; ISLT, International Shopping List Test‐Delayed Recall; MMSE, Mini‐Mental State Examination; PiB‐PET, positron emission tomography with Pittsburgh compound B; SD, standard deviation; SUVR, standardized uptake value ratio.
3.2. Low‐ and high‐dose treatment effects for clinical and cognitive outcomes
The estimated annual treatment effects for cognitive and clinical outcomes (defined as the difference in the annual change between the treatment group and the placebo group) before and after dose escalation are shown in Figure 1 and Table S2 in supporting information. Compared to the placebo, neither the low‐ nor the high dose of either study drug arm yielded significant improvement on any of the cognitive or clinical outcomes except that the gantenerumab low dose slightly improved the Digit Symbol Substitution Test by 1.89/year with 95% CI 0.11, 3.68 compared to the placebo arm (Figure 1 and Table S2). Analyses of participants who completed at least 4 years of treatment showed similar results (Figure S1 in supporting information).
FIGURE 1.
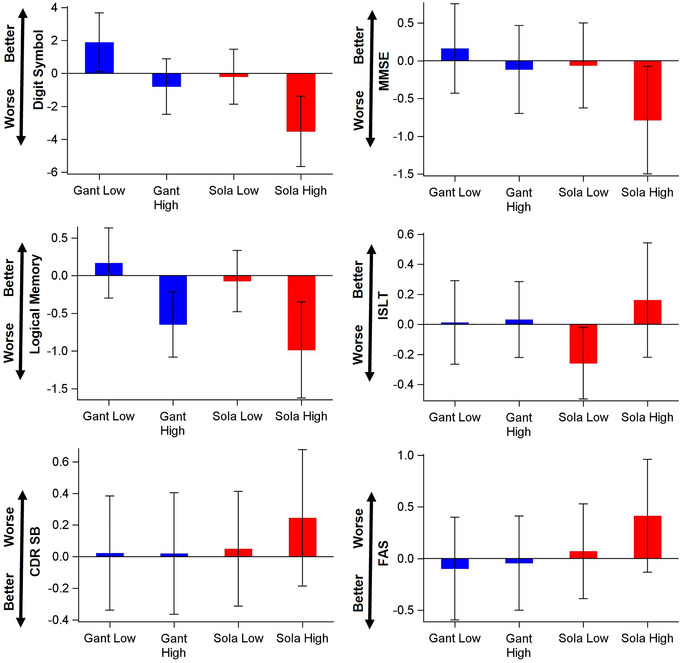
Demonstration of the estimated annual low‐ and high‐dose treatment effects and 95% confidence interval (CI) relative to the placebo. A bar with 95% CI covering 0 indicates a non‐significant treatment effect for a type I error of 0.05. Better, better than placebo; CDR SB, Clinical Dementia Rating Sum of Boxes; Digit Symbol, Digit Symbol Substitution Test; FAS, Functional Assessment Scale; Gant, gantenerumab; ISLT, International Shopping List Test‐Delayed Recall; Logical Memory, Logical Memory Delayed Recall Test; MMSE, Mini‐Mental State Examination; Sola, solanezumab; Worse, worse than placebo.
3.3. Low‐ and high‐dose treatment effects for imaging and CSF biomarker outcomes
Figure 2 demonstrates the estimated annual treatment effects before and after dose escalation (see Table S3 in supporting information for more details) for imaging and CSF biomarker outcomes relative to the placebo. Compared to the placebo, gantenerumab produced significant treatment effects in both low and high doses for PiB‐PET composite SUVR, CSF Aβ42, and CSF p‐tau181; and in high dose only for CSF total tau; and no significant treatment effects in CSF NfL, MRI hippocampus volumes, and MRI precuneus thickness (Figure 2 and more statistical details in Table S3). However, the gantenerumab treatment effect difference between the low dose and the high dose were only significant in PiB‐PET composite SUVR and CSF p‐tau181. The gantenerumab high dose reduced the PiB‐PET composite SUVR by 0.101 more per year than the lose dose (P‐value = 0.0095, 95% CI [−0.177, −0.025]), and reduced CSF p‐tau181 by 5.20 pg/ml more per year (P‐value = 0.010, 95% CI [−9.15, −1.25]).
FIGURE 2.
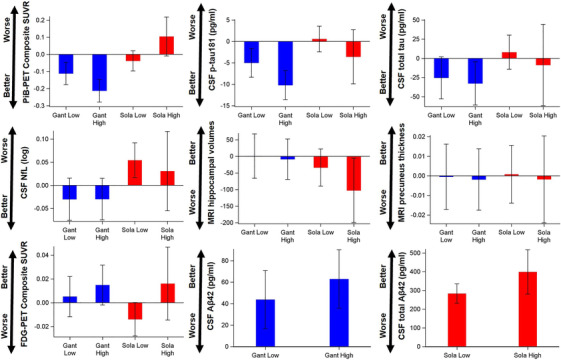
Demonstration of the estimated annual low‐ and high‐dose treatment effects and 95% confidence interval (CI) relative to the placebo. A bar with 95% CI covering 0 indicates a non‐significant treatment effect for a type I error of 0.05. Better, better than placebo; CSF total Aβ42, cerebrospinal fluid free plus bound amyloid beta 42; FDG‐PET, fluorodeoxyglucose positron emission tomography; Gant, gantenerumab; MRI, magnetic resonance imaging; NfL, neurofilament light; PiB‐PET SUVR, Pittsburgh compound B positron emission tomography standardized uptake value ratio; p‐tau, phosphorylated tau; Sola, solanezumab; Worse, worse than placebo.
Compared to the placebo, solanezumab produced significant treatment effects in both low and high doses for CSF total Aβ42 (free plus bound Aβ42); no significant treatment effects in PiB‐PET composite SUVR, CSF p‐tau181, CSF total tau, and MRI precuneus thickness; and significant worsening effects in low dose for CSF NfL and in high dose for MRI hippocampus volumes (Figure 2 and more statistical details in Table S3). The solanezumab high dose did not significantly improve the treatment effect in any imaging or CSF biomarker outcomes compared to the low dose although the increase in CSF total Aβ42 approached significance. The solanezumab high dose increased CSF total Aβ by 115.4 pg/ml more per year than the lose dose (P‐value = 0.0785. 95% CI [−13.3, 244.1]).
Tables S4 and S5 in supporting information demonstrate the treatment effects for the low dose and the high dose by baseline disease status (asymptomatic vs. symptomatic). Neither treatment showed significant beneficial treatment effect for asymptomatic cohort or symptomatic cohort in clinical and cognitive outcomes. Compared to the placebo, gantenerumab produced similar treatment effects in both low and high doses for PiB‐PET composite SUVR for both cohorts, larger treatment effect in high dose for asymptomatic cohort and similar, non‐significant treatment effect in both low and high doses for symptomatic cohort for CSF p‐tau181 and for CSF total tau; and no treatment effects in CSF NfL, MRI hippocampus volumes, MRI precuneus thickness, and FDG‐PET composite SUVR. Compared to the placebo, solanezumab produced significant treatment effect in both low and high doses for both asymptomatic and symptomatic cohorts in CSF total Aβ42 (free + bound Aβ42) and the high dose had larger treatment effect than the low dose in both cohorts; and produced no consistent treatment effects in either cohort for PiB‐PET composite SUVR, CSF p‐tau181, for CSF total tau, CSF NfL, MRI hippocampus volumes, MRI precuneus thickness, and FDG‐PET composite SUVR. Because of the small sample size for each categorical combination (ranging from 8 to 10 for symptomatic high dose to 16 to 24 for asymptomatic low dose) and the larger dropout in the symptomatic group, the estimated treatment effects should be interpreted with caution.
4. DISCUSSION
The DIAN‐TU‐001 platform trial successfully and safely implemented drug‐specific dose escalation procedures mid‐trial based on results from concurrent studies in sporadic AD. 4 , 5 Results from gantenerumab trials indicated that the 225 mg dose was not fully removing amyloid plaque, and that the dose used had acceptable safety profile to further increase the dose. For solanezumab, the completed phase 3 study indicated a lack of substantial cognitive benefit, while having a safety profile that would allow further increase. To optimize participant safety and characterize the safety profile of the higher doses, participants were carefully monitored during dose escalation, and the schedule of dose escalation and safety assessment was designed to ensure participants’ safety. The dose escalation achieved its intended goal for both drugs by demonstrating dose‐dependent treatment effects in target engagement (fibrillar Aβ for gantenerumab and soluble Aβ for solanezumab). Compared to the initial dose, the high doses led to larger treatment effects in each drug's Aβ target biomarker (90% more decrease in PiB PET SUVR with high‐dose gantenerumab, 41% more increase in CSF total Aβ42 [free + bound] with high‐dose solanezumab). The differential treatment effect between low‐ and high dose was statistically significant for gantenerumab, but not for solanezumab. The lack of significance for solanezumab can be potentially attributed to the shorter duration on high dose (e.g., an average of only 0.8 years before the year‐4 biomarker assessment), smaller sample size for high dose due to early dropout, and ceiling effect (e.g., CSF total Aβ42 [free + bound] had been restored to normal or close to normal level during the low‐dose treatment period and high dose yielded diminishing treatment effect). Additionally, compared to its low dose, the gantenerumab high dose also resulted in larger treatment effects in downstream biomarkers such as CSF total tau and CSF p‐tau181. Although gantenerumab did not achieve significant treatment effect in the reduction of the annual CSF NfL (log scale) change, the accumulated treatment at year 4 reached significance. 3 This dose‐dependent response indicates a possible causal relationship between amyloid reduction and prevention of downstream biomarker progression. However, the augmented high‐dose treatment effect demonstrated in biomarkers did not translate into cognitive or clinical outcomes in the double‐blind portion of the trial compared to placebo. The slight improvement over placebo in Digit Symbol Substitution Test observed during the gantenerumab low‐dose period needs to be interpreted with caution given the relatively small sample size as well as the magnitude of improvement. The faster decline over placebo observed during the solanezumab high‐dose period in CDR‐SB, MMSE, Digit Symbol Substitution Test, Logical Memory Delayed Recall Test and during the solanezumab low‐dose period in ISLT, as noted previously, 16 was contrary to the trend toward clinical and cognitive benefits seen in three large phase III solanezumab trials in sporadic AD, 5 , 17 and might be attributed to the small sample size and the more severe disease progression around dose escalation compared to placebo (14% participants with CDR > 1 in solanezumab vs. 6% in placebo, Table 2), the drug worsening progression in DIAD, or other unknown factors.
These findings suggest that dose escalation can be implemented during an ongoing phase IIb/III trial when concurrent studies indicated the initial dose may have been insufficient. Although the ideal is to maintain the same dose or to prespecify dose escalation for clinical trials, escalating the dose to achieve greater target engagement 18 , 19 based on new data is a better alternative than continuation of the trial without any change or restarting the trial due to futility given the logistical and financial burdens to launch a new trial. This is especially critical for clinical trials in rare disease such as the DIAN‐TU‐001 platform trial with limited enrollment capacity. A potential improvement for future trials with a similar situation is to “re‐start the clock” of the follow‐up duration at the initiation of dose escalation instead of maintaining the pre‐planned overall trial duration. Re‐starting the clock increases high‐dose drug exposure to amplify the potential treatment effect and ameliorates the exposure difference among participants, 19 which will likely increase the power of the study. However, to maintain similar power levels, additional enrollment would be needed for the increased dose period.
There are some noticeable limitations of the dose escalation procedure. First, variability in individual high‐dose durations and participant dropout preceding the administration of high doses will make the evaluation of both the low‐ and high‐dose treatment effects less interpretable. Second, the disease progression of symptomatic participants could diminish the observed treatment effect of the high dose at the group level if the investigational drug is most efficacious at earlier disease stages. Third, the variable low‐ and high‐dose durations limit the types of statistical models that can be used to analyze the treatment effects. For instance, the popular mixed model for repeated measures with the time variable being categorical is less appropriate than the LME model for estimating the treatment effect accurately.
The lack of clinical and cognitive treatment effects in the presence of larger Aβ biomarker target engagement at the higher dose can be attributed to several reasons as explained previously, including but not limited to the lack of clinical and cognitive decline in the asymptomatic cohort, the heterogeneous and short durations of high‐dose exposure, the small sample size, and the practice effects of some tests (e.g., Logical Memory Delayed Recall Test) in the asymptomatic group. These are also the overall limitations of the DIAN‐TU‐001 study. Furthermore, due to the small sample size, the results by disease status (asymptomatic vs. symptomatic) should be interpreted with caution.
5. COLLABORATORS
5.1. DIAN‐TU Study Team
The DIAN‐TU acknowledges the many individuals who have contributed to the DIAN‐TU including funding partners, leadership team, core leaders, project arm leaders, study sites, and institutional study partners listed in the following pages.
5.1.1. DIAN‐TU Leadership Team
Randall Bateman, MD, Director and Principal Investigator.
Eric McDade, DO, Associate Director.
David Clifford, MD, Associate Director and Medical Director.
Stephen Salloway, MD, Project Arm Leader, Gantenerumab.
Martin Farlow, MD, Project Arm Leader, Solanezumab.
Lon Schneider, MD, Project Arm Leader.
5.1.2. DIAN‐TU Core Leaders
Randall Bateman, MD, Administrative Core Leader, Washington University School of Medicine.
Anne Fagan, PhD, Biomarker Core Leader, Washington University School of Medicine.
Chengjie Xiong, PhD, Biostatistics Core Leader, Washington University School of Medicine.
Guoqiao Wang, PhD, Biostatistics Co‐Core Leader, Washington University School of Medicine.
Jason Hassenstab, PhD, Cognition Core Leader, Washington University School of Medicine.
Alison Goate, DPhil, Genetics Core Leader, Mt. Sinai School of Medicine.
Carlos Cruchaga, PhD, Genetics Core Leader, Washington University School of Medicine.
Tammie Benzinger, MD, Imaging Core Leader, Washington University School of Medicine.
Rick Perrin, MD, Neuropathology Core Leader, Washington University School of Medicine.
Jorge Llibre‐Guerra, MD, MSc, Post‐Doctoral Associate.
Cliff Jack, MD, MRI, Mayo Clinic.
Robert Koeppe, MD, PET Imaging, University of Michigan.
Nigel Cairns, MD (retired), Neuropathology Core Leader, Washington University School of Medicine.
Peter Snyder, PhD, (former Cognition Core Leader), Brown University.
5.1.3. DIAN Expanded Registry (DIAN‐EXR)
Eric McDade, Director.
Randall Bateman, Associate Director.
Jorge Llibre‐Guerra, Post‐Doctoral Associate.
Ellen Ziegemeier, Senior Clinical Research Coordinator.
Jennifer Petranek, Clinical Coordinator I.
Sarah Adams, Clinical Research Coordinator II.
Susan Brandon, Clinical Research Coordinator II.
5.1.4. DIAN‐TU faculty and staff
Amanda Fulbright, Grant Specialist, Administration Core.
Ron Hawley, IT Audiovisual and Interactive Web Designer, Administration Core.
Jacki Mallmann, Senior Grant Specialist, Administration Core.
Karen McCann, Financial Accounting Assistant, Administration Core.
Julie Murphy, Accounting/Purchasing Assistant, Administration Core.
Anna Santacruz, Administrative Director, Administration Core.
Jeanette Schillizzi, Research Administrator, Administration Core.
Wendy Simpson, Financial Accounting Assistant, Administration Core.
Shannon Sweeney, Finance Analyst, Administration Core.
Kelley Coalier, Lead Scientist, Biomarker Core.
Fatima Amtashar, QC Technician, Biomarker Core.
Sushila Sathyan, Archivist, Biomarker Core.
Jennifer Stauber, Research Specialist, Biomarker Core.
Susan Mills, Director of Clinical Operations.
Nicole Kelley, Associate Director of Clinical Operations.
Stephanie Belyew, Clinical Trial Manager, Site, Vendor and GCP Management, Clinical Operations.
Angela Fuqua, Clinical Trial Manager, Clinical Scale, Drug Supply and Vendor Management, Clinical Operations.
Inbal Meshulam, Clinical Trials Manager, Clinical Operations.
Annette Stiebel, Clinical Trial Manager, Regulatory, Clinical Operations.
Jeanine Portell, Clinical Trial Manager, MRI Sites, Clinical Operations.
Bettina Bell, Clinical Trial Manager, Brain Donation, Clinical Operations.
Caryll Bentley, Contract Project Manager, Clinical Operations.
Sharon Cirello, Senior Contract Data Manager, Clinical Operations.
Nithyanjali Devarapalli, Contract Specialist Clinical Data Manager, Clinical Operations.
Arthur Gipson, Contract Specialist Clinical Data Manager, Clinical Operations.
JaNeen Wisner, Contract Project Coordinator, Clinical Operations
Tayona Mayhew, Data Management Specialist, Clinical Operations.
Zenobia Bridgewater, Clinical Research Coordinator, Clinical Operations.
Dana Burgdorf, Research Nurse Coordinator II, Clinical Operations.
Molly Fitzgerald, Clinical Research Study Assistant I, Clinical Operations.
Erica Fowler, Clinical Research Coordinator I, Clinical Operations.
Dottie Heller, Research Nurse Coordinator II, Clinical Operations.
Miranda Jany, Clinical Research Coordinator I, Clinical Operations.
Latoya Jones, Clinical Research Coordinator II, Clinical Operations.
Michelle Jorke, Clinical Research Coordinator II, Clinical Operations.
Paulette MacDougall, Research Nurse Coordinator II, Clinical Operations.
Eugene Rubin, QC, Clinical Operations.
Jessi Smith, Senior Clinical Research Coordinator, Clinical Operations.
Mary Wolfsberger, Clinical Research Coordinator, Clinical Operations.
Andy Aschenbrenner, Assistant Professor, Cognition Core.
Jennifer Smith, Professional Rater III, Cognition Core.
Marisol Tahan, Clinical Research Coordinator, Cognition Core.
Theresa Butler, Research Lab Manager, Imaging Core.
Lisa Cash, Senior Clinical Research Coordinator, Imaging Core.
Jon Christensen, Staff Scientist, Imaging Core.
Aylin Dince, Research Assistant, Imaging Core.
Tony Durbin, Senior Clinical Research Coordinator, Imaging Core.
Shaney Flores, Research Assistant, Imaging Core.
Karl Friedrichsen, Research Assistant, Imaging Core.
Brian Gordon, Co‐Investigator, Imaging Core.
Russ Hornbeck, Project Manager, Imaging Core.
Nelly Joseph‐Mathurin, Post‐doc, Imaging Core.
Sarah Keefe, Research Assistant, Imaging Core.
Lakisha Lloyd, Research Coordinator, Imaging Core.
Laura Marple, Research Technician II, Imaging Core.
Austin McCullough, Graduate Research Assistant, Imaging Core.
Stephanie Schultz, Pre‐Doctoral Trainee, Imaging Core.
Sally Schwarz, Co‐Investigator, Imaging Core.
Yi Su, Co‐Investigator, Imaging Core.
Andrei Vlassenko, Co‐Investigator, Imaging Core.
Qing Wang, Post‐doc, Imaging Core.
Jinbin Xu, Co‐Investigator, Imaging Core.
Erin Franklin, Research Coordinator, Neuropathology Core.
5.1.5. Eli Lilly and Company and Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly and Company
Eli Lilly and Company
Isabella Velona, Clinical Trial Project Manager
Scott Andersen, Biostatistician
Michele Mancini, GPS
Brian Willis, PK/PD Project Leader
Sonia Nijampatnam, Pharmaceutical Project Management
Kathryn Broderick, Global Regulatory Affairs
Julie Bush, Product Delivery
Shamrock Garrett, Sr. CTMA
Traci Peddie, Data Sciences & Solutions
Natalie Vantwoud, Data Management
Barbara Lightfoot‐Owens, Medical Writer
John Brad‐Holmes, Central Lab
Avid Radiopharmaceuticals
Michael Devous
Erica Elephant
Laura Harper
Marybeth Howlett
Mark Mintun
Michael Pontecorvo
Former Team Members
Phyllis Ferrell Barkman, Russ Barton, Lauren Brunke, Robert Dean, Deanilee Deckard, Ann Catherine Downing, Ganapathy Goppalrathnam, David Henley, Janice Hitchcock, Tracie Peddie, Melissa Pugh, Tami Jo Rayle, Shiloh Scott, James Senetar, Gopalan Sethuraman, Eric Siemers, Brian Steuenwald, Connie Tong, Jim Vandergriff
5.1.6. F. Hoffman‐LaRoche Team
Monika Baudler, LifeCycle Leader.
Rachelle Doody, Global Head of Neurodegneration.
Paul Delmar, Principal Statistical Scientist.
Carsten Hofmann, Clinical Pharmacologist.
Michaela Jahn, Global Biometrics Team Leader.
Geoff Kerchner, Global Development Leader.
Gregory Klein, Biomarker Experimental Medicine Leader.
Smiljana Ristic, Associate Group Medical Director.
Alison Searle, Operations Program Leader.
Marco Sonderegger, Technical Development Leader.
Roz Sutton, EU Regulatory Partner.
Janette Turner, US Regulatory Partner.
Jaku Wojtowicz, Safety Science Director.
Susan Yule, Global Regulatory Leader.
5.1.7. Former team members
Elizabeth Ashford, Operations Program Leader; Bogdon Balas, Safety Science Leader; Estelle Vester‐ Blokland, LifeCycle Leader; Stephanie Capo‐Chichi, EU Regulatory Partner; David Agnew, Global Study Manager; Ernest Dorflinger, Translational Medicine Leader; Efe Egharevba, Global Study Manager; Christelle Laroche, Technical Development Leader; Isabelle Bauer Dauphin, Technical Development Leader; Rob Lasser, Global Development Leader; Ferenc Martenyi, Global Development Leader; Glenn Morrison, Global Development Leader; Tania Nikolcheva, Biomarker Experimental Medicine Leader; Michael Rabbia, Statistical Scientist; Juha Savola, Project Leader; Janice Smith, Clinical Science Leader; Dietmar Volz, Statistical Scientist.
5.1.8. DIAN‐TU DSMB members
Gary Cutter, PhD, DSMB Chairperson, University of Alabama at Birmingham.
Steve Greenberg, MD, PhD, Massachusetts General Hospital, Boston.
Scott Kim, MD, PhD, National Institutes of Health, Bethesda.
David Knopman, MD, Mayo Clinic, Rochester.
Willis Maddrey, MD (retired), UT Southwestern, Dallas.
Kristine Yaffe, MD, University of California, San Francisco.
Karl Kieburtz, MD, PhD, (DSMB Chairperson, retired), University of Rochester, NY.
Allan Levey, MD, PhD (retired), Emory University, Atlanta.
5.1.9. DIAN‐TU therapy evaluation committee
Randall Bateman, MD, Chair, Washington University School of Medicine, St. Louis.
Eric McDade, DO, Co‐Chair, Washington University School of Medicine, St. Louis.
Paul Aisen, MD, Alzheimer's Therapeutic Research Institute, USC.
Jasmeer Chhatwal, MD, PhD, MMSc, Massachusetts General Hospital, Harvard Medical School.
David Clifford, MD, Washington University School of Medicine, St. Louis.
David Cribbs, MD, UC Irvine, CA.
Nick Fox, MD, FRCP, FMedSci, Dementia Research Centre.
Serge Gauthier, Serge Gauthier, CM, MD, FRCPC, Director, AD & Related Disorders Unit,
McGill Centre for Studies in Aging.
David Holtzman, MD, Washington University School of Medicine.
Matthias Jucker, PhD, Hertie Institute for Clinical Brain Research, DZNE, Germany.
Jeff Kelly, MD, Scripps University, California.
Virginia Lee, PhD, University of Pennsylvania, Perelman School of Medicine.
Simon Mead, FRCP, PhD, Institute of Prion Diseases, London.
Cath Mummery, PhD, FRCP, Dementia Research Centre, London.
Erik Musiek, MD, PhD, Washington University School of Medicine.
Erik Roberson, MD, PhD, University of Alabama.
Mathias Staufenbiel, PhD, Hertie Institute for Clinical Brain Research, DZNE, Tubingen, Germany.
Robert Vassar, PhD, Northwestern University, IL.
5.1.10. Former TEC members
Bart DeStrooper, PhD; William Klunk, MD, PhD; Cynthia Lemere, MD; John C. Morris, MD.
5.1.11. DIAN Clinical Trials Committee (CTC) members
Randall Bateman, Washington University in St. Louis School of Medicine.
John Morris, Washington University in St. Louis School of Medicine.
Chengjie Xiong, Washington University in St. Louis School of Medicine.
Denise Heinrichs, DIAN Family Representative.
John Ringman University of California, Los Angeles.
Laurie Ryan, Division of Science, National Institute on Aging.
Neil Buckholtz, National Institute on Aging.
Reisa Sperling, Director, Center for Alzheimer Research and Treatment.
Stephen Salloway, Butler Hospital.
Paul Aisen, University of California, San Diego.
Anna Santacruz, Washington University in St. Louis School of Medicine.
Gabrielle Strobel, Alzheimer Research Forum.
Bill Klunk, University of Pittsburgh.
William Thies, Alzheimer's Association.
Anne Fagan, Washington University in St. Louis School of Medicine.
Mark Mintun, Washington University in St. Louis School of Medicine.
Natalie Ryan, University College London.
Virginia Buckles, Washington University in St. Louis School of Medicine.
David Hawver, Food and Drug Administration.
Martin Farlow, Indiana University.
Maritza Ciliberto, DIAN Family Representative.
Ralph Martins, Edith Cowan University.
Jennifer Williamson, Columbia University.
5.1.12. Study sites
Australia: Neuroscience Research Australia—W. Brooks, M.J. Fulham, J. Bechara, D. Foxe; Australian Alzheimer's Research Foundation—R. Clarnette, N. Reynders, P. Mather; University of Melbourne—C. Masters, C. Rowe, B. Clinch, D. Baxendale.
Canada: McGill University—S. Gauthier, P. Rosa‐Neto, C. Mayhew, L. Robb; University of British Columbia—R. Hsiung, D. Worsley, M. Assaly, E. Nicklin; Sunnybrook Research Institute—M. Masellis, K. Sharp, S. Hetherington.
France: Hopital Charles Nicolle—D. Wallon, D. Hannequin, A. Morin, A. Zarea, E. Gerardin, P. Bohn, M. Chastan, P. Vera, M. Colnot, N. Donnadieu, M. Quillard‐Muraine, C. Bergot, S. Jourdain; Groupe Hospitalier Pitie‐Salpetriere—B. Dubois, M. Habert, N. Younsi; Hopital Pierre Wertheimer—M. Formaglio, D. Lebars, N. El Kfif, A. Jullien; Hopital Purpan—J. Pariente, P. Payoux, C. Thalamas, A. Driff, E. Pomies, P. Gauteul; Hôpital Roger Salengro—F. Pasquier, A. Rollin‐Sillaire, F. Semah, L. Breuilh, M. Laforce; Orsay Imaging—M. Bottlaender.
Spain: Hospital Clinic i Provincial de Barcelona—R. Sanchez‐Valle, M. Balasa, A. Lladó, B. Bosch, N. Bargalló, I. Banzo, A. Perisinotti.
United Kingdom: University College London Hospital—C. Mummery, I. Kayani, J. Douglas, M. Grilo.
United States: Washington University School of Medicine—B.J. Snider, T. Benzinger, W. Sigurdson, T. Donahue, P. Kelly; Emory University—J. Lah, C. Meltzer, G. Schwartz, P. Vaughn, L. Piendel; University of Pittsburgh—S. Berman, J. Mountz, L. Macedonia, S. Ikonomovic, S. Goldberg, E. Weamer, J. Ruskiewiecz, S. Hegedus, L. Tarr, T. Potter, G. Valetti; University of Alabama at Birmingham—E. Roberson, D. Geldmacher, M. Love, A. Watkins, L. Ashley; Indiana University—J. Brosch, A. Kohn, N. McClaskey, J. Buck, J. Fletcher; Butler Hospital—G. Surti, R. Noto, C. Bodge, W. Menard; University of Puerto Rico—I. Jimenez Valazquez, J. Diaz, K. Aleman; Yale University—C. van Dyck, M. Chen, N. Diepenbrock, A. Mecca, S. Good; University of California San Diego—D. Galasko, C. Hoh, D. Szpak, S. Peackock; University of Washington—S. Jayadev, D. Lewis, Y. Tutterow.
5.1.13. DIAN‐TU collaborators and advisers
John Morris, MD, Senior Advisor, Washington University School of Medicine, St. Louis, MO.
David Holtzman, MD, Senior Advisor, Washington University School of Medicine, St. Louis, MO.
Laura Swisher, MS, Deputy Director, Washington University School of Medicine, St. Louis, MO.
Alisha Daniels, MD, MHA, Executive Director, DIAN, Washington University School of Medicine, St. Louis, MO.
Janice Hitchcock, PhD, Hitchcock Regulatory Consulting Inc.
Thomas Bird, MD, University of Washington, Seattle.
Dennis Dickinson, MD, Mayo Clinic, Jacksonville, FL.
M. Marsel Mesulam, MD, Cognitive Neurology and Alzheimer's Disease Center, Northwestern University, Chicago, IL.
Cornelia Kamp, MBA, University of Rochester, New York.
Ron Thomas, PhD, ADCS, University of California, San Diego.
Paul Aisen, MD, ADCS, University of Southern California, Los Angeles.
5.1.14. DIAN observational study site investigators
James Noble, MD, Columbia University, New York.
Martin Farlow, MD, Indiana University, Indianapolis, IN.
Jasmeer Chhatwal, MD, PhD, Brigham and Women's Hospital–Massachusetts GH, Charlestown, MA.
Stephen Salloway, MD, Butler Hospital, Warren Alpert School of Medicine, Brown University.
Sarah Berman, MD, PhD, University of Pittsburgh, PA.
Gregg Day, MD, Mayo Clinic Jacksonville, FL.
Hiroyuki Shimada, MD, PhD, Osaka City University, Japan.
Takeshi Ikeuchi, MD, PhD, Brain Research Institute, Nigata, Japan.
Kazushi Suzuki, MD, PhD, The University of Tokyo, Japan.
Peter Schofield, PhD, DSc, Neuroscience Research Australia, Sydney.
Ralph Martins, BSc, PhD, Edith Cowan University, Nedlands, Western Australia.
Nick Fox, MD, FRCP, FMedSci, Dementia Research Centre, University College London, United Kingdom.
Johannes Levin, MD, PhD, German Center for Neurodegenerative Diseases (DZNE), Munich, Germany.
Mathias Jucker, PhD, German Center for Neurodegenerative Diseases (DZNE), Tubingen, Germany.
Raquel Sanchez Valle, MD, Hospital Clinic i Provincial de Barcelona, Spain.
Patricio Chrem, MD, Fundación para la Lucha contra las Enfermedades Neurológicas de la Infancia (FLENI), Buenos Aires, Argentina.
5.1.15. DIAN EXR referring clinicians, researchers, and partner sites
Neelum T. Aggarwal, MD, Rush University Medical Center, Chicago IL.
Tom Ala, Center for Alzheimer's Disease and Related Disorders, Southern Illinois University School of Medicine Thomas Bird, University of Washington, Seattle.
Sandra E. Black, Sunnybrook Health Sciences Centre, University of Toronto, Canada.
William J. Burke, MD, Banner Alzheimer's Institute.
Cynthia M. Carlsson, MD, MS, University of Wisconsin School of Medicine and Public Health.
Andrew Frank MD B.Sc.H. F.R.C.P.(C), Bruyere Continuing Care, Ottawa, Ontario, Canada.
James E. Galvin, MD, MPH, Charles E. Schmidt College of Medicine, Florida Atlantic University.
Alvin C Holm, MD, Bethesda Hospital, St. Paul, MN.
John S.K. Kauwe, Brigham Young University.
David Knopman MD, Mayo Clinic, Rochester MN.
Sarah Kremen, MD, University of California, Los Angeles.
Alan J. Lerner, University Hospitals Cleveland Medical Center.
Barry S. Oken, MD, PhD, Oregon Health & Science University.
Hamid R. Okhravi, Eastern Virginia Medical School.
Ronald C. Petersen, Mayo Clinic, Rochester, MN.
Aimee L. Pierce, MD, University of California Irvine.
Marsha J. Polk, MED, University of Texas Health Science Center at San Antonio.
John M. Ringman, MD, MS, University Southern California.
Peter St. George Hyslop, MD, FRS, FRSC, FRCPC, University of Toronto.
Sanjeev N. Vaishnavi, MD, PhD, University of Pennsylvania.
Sandra Weintraub, Northwestern University Feinberg School of Medicine, IL.
CONFLICTS OF INTEREST
Guoqiao Wang, PhD, is the biostatistics core co‐leader for the DIAN‐TU. He reports serving on a Data Safety Committee for Eli Lilly and Company and as a statistical consultant for Alector. Eric McDade, DO, is the Associate Director of the DIAN‐TU. He reports serving on a Data Safety Committee for Eli Lilly and Company and Alector; as a scientific consultant for Eisai and Eli Lilly and Company; receiving institutional grant support from Eli Lilly and Company, F. Hoffmann‐La Roche, Ltd., and Janssen. Anne M. Fagan, PhD, is the Biomarker Core Leader of the DIAN‐TU. She is a member of the scientific advisory boards for Roche Diagnostics, Genentech, and DiademRes and also consults for DiamiR and Siemens Healthcare Diagnostics, Inc. Tammie L.S. Benzinger, MD, PhD, has investigator‐initiated research funding from the NIH, the Alzheimer's Association, the Barnes‐Jewish Hospital Foundation, and Avid Radiopharmaceuticals. Dr. Benzinger participates as a site investigator in clinical trials sponsored by Avid Radiopharmaceuticals, Eli Lilly and Company, Biogen, Eisai, Jaansen, and F. Hoffmann‐La Roche, Ltd. She serves as an unpaid consultant to Eisai and Siemens. She is on the Speaker's Bureau for Biogen. David B. Clifford, MD, is Medical Director of the DIAN‐TU and serves as scientific consultant to Biogen, Takeda, Millennium, Genzyme, Amgen, F. Hoffmann‐La Roche, Ltd./Genentech, Glaxo Smith Kline, Serono, Inhibikase, Dr Reddy's Lab, Bristol Myers Squibb, Atara, Mitsubishi Tanabe, Excision BioTherapeutics, Up to Date, and Wolters Kluwer; on DSMB/Data Monitoring Committees for Genentech/ F. Hoffmann‐La Roche, Ltd., Wave, EMD Serono, Shire, Pfizer, Sanofi; does legal consulting: Cook County, State Farm, Wilke & Wilke PC, Shevlin Smith, Sal Indomenico PC. He receives research support from NIH NINDS, NIMH, NIAID, NCATS, and NIA. Andrew J. Aschenbrenner, PhD, has served as a consultant for Biogen Inc, and H. Lundbeck HS. Jason Hassenstab, PhD, is a paid consultant for F. Hoffmann‐La Roche, Ltd., Takeda, and Lundbeck, and is on the Data Safety and Monitoring Board for Eisai. Catherine Mummery, MD, is a consultant for Biogen. Mario Masellis, MD, is a consultant to Arkuda Therapeutics, Ionis, and Alector and receives research funding from F. Hoffmann‐La Roche, Ltd., Novartis, and Alector. Serge Gauthier, MD, FRCPC, is a member of the Scientific Advisory Board for Alzheon, Biogen, Eli Lilly and Company, and TauRx and a member of the Data Safety Monitoring Board for ADCS, ATRI, and Banner Health. Scott Andersen, MS; Karen C. Holdridge, MPH; Saptarshi Chatterjee, PhD; John R. Sims, MD; and Roy Yaari, MD are employees and shareholders of Eli Lilly and Company. Randall J. Bateman, MD, is the Director of the DIAN‐TU and Principal Investigator of the DIAN‐TU‐001. He receives research support from the National Institute on Aging of the National Institutes of Health, DIAN‐TU Trial Pharmaceutical Partners (Eli Lilly and Company, F. Hoffman‐La Roche, Ltd., and Avid Radiopharmaceuticals), Alzheimer's Association, GHR Foundation, Anonymous Organization, DIAN‐TU Pharma Consortium (Active: Biogen, Eisai, Eli Lilly and Company, Janssen, F. Hoffmann‐La Roche, Ltd./Genentech. Previous: AbbVie, Amgen, AstraZeneca, Forum, Mithridion, Novartis, Pfizer, Sanofi, United Neuroscience). He has been an invited speaker for Novartis and serves on the Advisory Board for F. Hoffman La Roche, Ltd. Barbara A. Wendelberger, PhD; Susan L. Mills BS; Anna M. Santacruz BS; Kelley A. Coalier, MS; Brian A. Gordon, PhD; Jorge J. Libre‐Guerra, MD; Austin McCullough; Nelly Joseph‐Mathurin, PhD; Charlie Chen; Yan Li, PhD; Chengji Xiong, PhD, have no conflicts of interest to disclose. Paul Delmar, Geoffrey A. Kerchner, Tobias Bittner, and Carsten Hofmann are full‐time employees of F. Hoffmann‐La Roche, Ltd. and own stock in F. Hoffmann‐La Roche, Ltd. David Holtzman, MD, the prior Department Head of Neurology where the research was conducted, is an inventor on patents for solanezumab, which was tested in the DIAN‐TU‐001 clinical trial. If solanezumab is approved as a treatment for AD or dominantly inherited AD, Washington University and Dr. Holtzman will receive part of the net sales of solanezumab from Eli Lilly and Company, which has licensed patents related to solanezumab from Washington University. All the other authors reported no conflicts of interest. Author disclosures are available in the supporting information.
Supporting information
SUPPORTING INFORMATION
SUPPORTING INFORMATION
ACKNOWLEDGMENTS
The authors gratefully acknowledge the outstanding commitment of the participants, family members, and caregivers whose participation was critical to the success of the DIAN‐TU trial. We thank the DIAN‐TU study team for their exceptional dedication and accomplishments, which ensured the success of the trial. We thank the DIAN‐EXR and DIAN‐OBS study teams for their support on recruitment and commitment to family members. We appreciate the robust intellectual collaboration between the DIAN‐TU investigators, participants and family members, F. Hoffmann‐La Roche, Ltd./Genentech, and Eli Lilly and Company, the DIAN‐TU Pharma Consortium (https://dian.wustl.edu/our‐research/the‐pharma‐consortium/), the NIH, and regulatory representatives who were critical in making this study possible. We thank the Alzheimer's Association, GHR Foundation, an anonymous organization, other industry partners (Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly and Company, Signant, and Cogstate), and regulatory representatives for their support. We also acknowledge Dr. Laurie Ryan from the National Institute on Aging for her key contributions in leadership and scientific guidance on this project. Research reported in this publication was supported by the National Institute on Aging of the National Institutes of Health under Award Numbers U01AG042791, U01AG042791‐S1 (FNIH and Accelerating Medicines Partnership), R01AG046179, R01AG053267‐S1. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This research was also supported by the Alzheimer's Association, Eli Lilly and Company, F. Hoffman‐LaRoche Ltd., Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly and Company, GHR Foundation, an anonymous organization. Cogstate, and Signant offered in‐kind support. The DIAN‐OBS was supported by the National Institute on Aging of the National Institutes of Health (DIAN, U19AG032438), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI).
Wang G, Li Y, Xiong C, et al. Evaluation of dose‐dependent treatment effects after mid‐trial dose escalation in biomarker, clinical, and cognitive outcomes for gantenerumab or solanezumab in dominantly inherited Alzheimer's disease. Alzheimer's Dement. 2022;14:e12367. 10.1002/dad2.12367
Guoqiao Wang and Yan Li contributed equally to this article.
REFERENCES
- 1. Bateman RJ, Benzinger TL, Berry S, et al. The DIAN‐TU Next Generation Alzheimer's prevention trial: adaptive design and disease progression model. Alzheimers Dement. 2017;13(1):8‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377(1):62‐70. [DOI] [PubMed] [Google Scholar]
- 3. Salloway S, Farlow M, McDade E, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer's disease. Nat Med. 2021;27(7)1187‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ostrowitzki S, Lasser RA, Dorflinger E, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer's disease. Alzheimers Res Ther. 2017;9(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med. 2018;378(4):321‐330. [DOI] [PubMed] [Google Scholar]
- 6. Berg L, Miller JP, Storandt M, et al. Mild senile dementia of the Alzheimer type: 2. Longitudinal assessment. Ann Neurol. 1988;23(5):477‐484. [DOI] [PubMed] [Google Scholar]
- 7. Breines E. The functional assessment scale as an instrument for measuring changes in levels of function of nursing home residents following occupational therapy. Can J Occup Ther. 1988;55(3):135‐140. [Google Scholar]
- 8. Folstein M. A practical method for grading the cognitive state of patients for the children. J Psychiatr Res. 1975;12:189‐198. [DOI] [PubMed] [Google Scholar]
- 9. Dumont R, Willis JO, Veizel K, Zibulsky J. Wechsler adult intelligence scale – fourth edition. Encyclopedia of Special Education: A Reference for the Education of Children, Adolescents, and Adults with Disabilities and Other Exceptional Individuals. Wechsler, D. WMS‐R: Wechsler Memory Scale. Revised: manual (Psychological Corporation, 1987) 2013.
- 10. Lim YY, Prang KH, Cysique L, Pietrzak RH, Snyder PJ, Maruff P. A method for cross‐cultural adaptation of a verbal memory assessment. Behav Res Methods. 2009;41(4):1190‐1200. [DOI] [PubMed] [Google Scholar]
- 11. Thompson TA, Wilson PH, Snyder PJ, et al. Sensitivity and test – retest reliability of the international shopping list test in assessing verbal learning and memory in mild Alzheimer's disease. Arch Clin Neuropsychol. 2011;26(5):412‐424. [DOI] [PubMed] [Google Scholar]
- 12. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benzinger TL, Blazey T, Jack CR, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci. 2013;110(47):E4502‐E4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Su Y, Blazey TM, Snyder AZ, et al. Partial volume correction in quantitative amyloid imaging. Neuroimage. 2015;107:55‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fitzmaurice GM, Laird NM, Ware JH. Applied Longitudinal Analysis. John Wiley & Sons; 2012. [Google Scholar]
- 16. Salloway S, Farlow M, McDade E, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer's disease. Nat Med. 2021;27(7):1187‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370(4):311‐321. [DOI] [PubMed] [Google Scholar]
- 18. Bateman RJ, Klunk WE. Measuring target effect of proposed disease‐modifying therapies in Alzheimer's disease. Neurotherapeutics. 2008;5(3):381‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knopman DS, Jones DT, Greicius MD, Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION
SUPPORTING INFORMATION