

Abstract
RNF5 E3 ubiquitin ligase has multiple biological roles and has been linked to the development of severe diseases such as cystic fibrosis, acute myeloid leukemia, and certain viral infections, emphasizing the importance of discovering small-molecule RNF5 modulators for research and drug development. The present study describes the synthesis of a new benzo[b]thiophene derivative, FX12, that acts as a selective small-molecule inhibitor and degrader of RNF5. We initially identified the previously reported STAT3 inhibitor, Stattic, as an inhibitor of dislocation of misfolded proteins from the endoplasmic reticulum (ER) lumen to the cytosol in ER-associated degradation. A concise structure–activity relationship campaign (SAR) around the Stattic chemotype led to the synthesis of FX12, which has diminished activity in inhibition of STAT3 activation and retains dislocation inhibitory activity. FX12 binds to RNF5 and inhibits its E3 activity in vitro as well as promoting proteasomal degradation of RNF5 in cells. RNF5 as a molecular target for FX12 was supported by the facts that FX12 requires RNF5 to inhibit dislocation and negatively regulates RNF5 function. Thus, this study developed a small-molecule inhibitor and degrader of the RNF5 ubiquitin ligase, providing a chemical biology tool for RNF5 research and therapeutic development.
INTRODUCTION
RNF5 is a RING finger ubiquitin ligase involved in endoplasmic reticulum (ER)-associated degradation (ERAD) of misfolded proteins, including the premature degradation of cystic fibrosis transmembrane conductance regulator (CFTR) and its most common mutant, deletion of phenylalanine 508 (ΔF508 CFTR) (Younger et al., 2006). ΔF508 CFTR degradation leads to a deficiency of CFTR function at the cell surface, which is the primary cause of cystic fibrosis (CF) (Ward and Kopito, 1994; Ward et al., 1995; Younger et al., 2006; D’Antonio et al., 2019). Wild-type (wt) CFTR degradation has been linked to the development of chronic obstructive pulmonary disease (COPD) (Patel et al., 2020; Grand et al., 2021). The RNF5 gene is among the high-priority genes associated with COPD (Wain et al., 2017). Additionally, RNF5 is frequently hijacked by viruses to evade the immune response and facilitate viral replication. Paramyxoviruses, such as Newcastle disease virus, utilize their V protein as an adaptor to bring the mitochondrial antiviral-signaling protein (MAVS) to RNF5 for ubiquitination and degradation (Sun et al., 2019). Loss of MAVS diminishes the innate immune response against RNA viruses, as it is a signaling adaptor for the cytosolic RNA sensors RIG-1 and MDA5-mediated, microbial RNA-induced, and TLR-independent interferon response (Tan et al., 2018). RNF5 also regulates the stimulator of interferon genes (STING) during various viral infections. STING is a critical component of the cytosolic double-stranded DNA (dsDNA)-sensing cGAS-cGAMP-STING pathway. This pathway is activated in response to viral infection to induce the expression of type I interferons and thus mediates host defense against a range of DNA and RNA viruses (Ma and Damania, 2016; Li and Chen, 2018). Viral infection enhances RNF5 interaction with STING, leading to STING ubiquitination and proteasomal degradation, thus contributing to evasion of innate immune responses (Liu and Gao, 2018; Sun et al., 2019). During RNA virus infection, the JMJD6 protein recruits RNF5 to promote IRF3 ubiquitination and degradation, thereby attenuating the type I interferon response (Zhang et al., 2021). RNF5 has also been implicated in nonviral infections, including group A streptococcus infection, by regulating the level of the ATG4B protein, which is essential for LC3 maturation and autophagosome formation and thus controls the degree of infection (Kuang et al., 2012).
RNF5 has also been associated with cancer progression. In cancer, cGAS-cGAMP-STING signaling is activated by the accumulation of cytosolic dsDNA, a consequence of nuclear DNA damage, and induces tumor suppressor functions, including immune surveillance, cellular senescence, and cell death (Ma and Damania, 2016; Khoo and Chen, 2018; Li and Chen, 2018; Tan et al., 2018). However, some studies have reported that activation of the cGAS-cGAMP-STING pathway by chromosomal instability can conversely drive metastasis (Bakhoum and Cantley, 2018; Bakhoum et al., 2018). In breast cancer, increased expression of RNF5 is correlated with decreased survival. Knockdown of RNF5, which is highly expressed in several breast cancer cell lines such as MCF-7, produces increased sensitivity to DNA damage–inducing chemotherapeutics (Bromberg et al., 2007). In addition to inhibition of STING signaling, RNF5 has been linked to other cancer-related events such as the regulation of cell migration, an essential process in metastasis, through ubiquitination of the focal adhesion protein paxillin (Didier et al., 2003). An increased abundance of RNF5 contributes to acute myeloid leukemia development and survival (Khateb et al., 2021). RNF5 has also been implicated in pancreatic cancer, where it targets the tumor suppressor PTEN for degradation and accelerates tumor growth (Pitarresi et al., 2018). A recent study reported that altered unfolded protein response (UPR) signaling in RNF5 knockout (KO) mice coincided with altered gut microbiota composition and consequently limited tumor expansion (Li et al., 2019). RNF5 also controls the stability of the glutamine carrier proteins SLC1A5 and SLC38A2, which limit glutamine uptake, and renders tumor cells more sensitive to ER stress–inducing chemotherapy (Jeon et al., 2015).
The various roles exhibited by RNF5 in numerous disease pathologies highlight the importance of identifying small-molecule modulators of its E3 activity for research and assessment of RNF5 as a potential drug target. In this study, we report the synthesis and characterization of FX12, a selective small-molecule inhibitor and degrader of RNF5, which directly binds to RNF5, inhibits RNF5 E3 activity, and targets RNF5 for degradation via ERAD.
RESULTS
Identification of Stattic as a dislocation inhibitor
Transport of misfolded proteins from the ER lumen to the cytosol, known as retrotranslocation or dislocation, is crucial during ERAD. Previously, we established a dislocation-induced reconstituted green fluorescent protein (drGFP) reporter system to quantify the dislocation of misfolded protein substrates in living cells (Zhong and Fang, 2012). Using drGFP reporter screening of small-molecule libraries, we have previously identified two dislocation inhibitors that possess anti-flavivirus (Dengue and Zika viruses) activity (Rothan et al., 2019; Ruan et al., 2019). In this study, we identified Stattic, a previously characterized small-molecule inhibitor of STAT3 activation (Schust et al., 2006), as a dislocation inhibitor from the Library of Pharmacologically Active Compounds (LOPAC) (Figure 1A).
FIGURE 1:

Synthesis and characterization of FX12 as a dislocation inhibitor. (A) Chemistry optimization through SAR studies of Stattic. (B) drGFP assay. HeLa cells stably expressing NHK-drGFP reporter plasmids were treated with DMSO (background control), proteasome inhibitor BTZ (50 nM) alone (positive control), or BTZ in combination with series concentrations of the indicated compounds for 4 h before GFP intensities were measured in a microplate reader. GFP intensities were normalized to the positive control (set as 100%) after background subtraction. Error bars = SD for n = 3 biological replicates. (C) Cytotoxicity assay. HepG2 cells were treated with increasing concentrations of FX12 or its analogues for 24 h followed by measuring cell viability with WST-1. Cells treated with DMSO was used as control (set as 100%). Error bars = SD for n = 5 biological replicates. (D) IC50s derived from C.
Synthesis and characterization of FX12 as a novel dislocation inhibitor
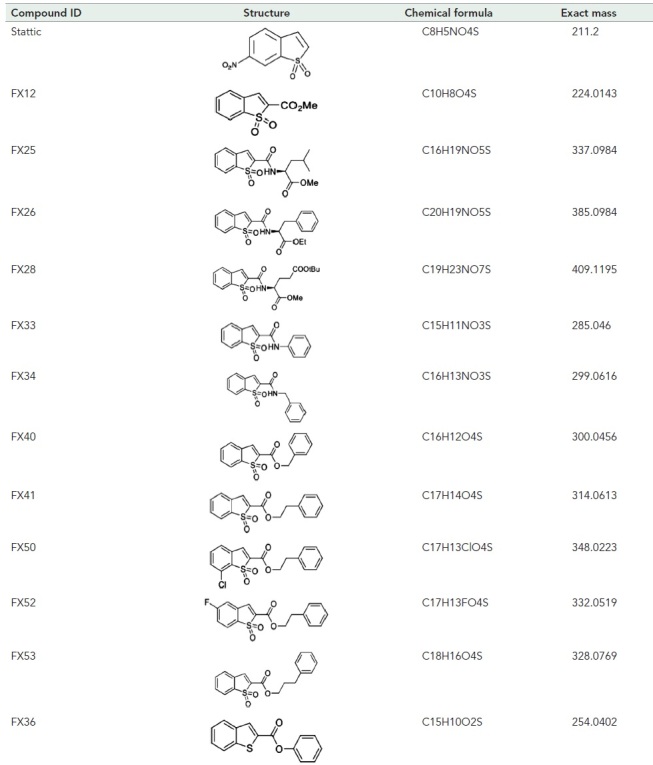
A concise SAR campaign around the Stattic chemotype was performed to diminish the STAT3 inhibitory activity while retaining activity in dislocation. The previous study showed that the nitro group in Stattic is important for STAT3 inhibitory activity (Schust et al., 2006). Therefore, the SAR effort was focused on benzo[b]thiophene 1,1-dioxide–based analogues (Figure 1, B–D, and Table 1). Eleven analogues exhibit excellent potencies in inhibiting the misfolded null Hong Kong variant of α-1-antitrypsin (NHK) dislocation in the drGFP assay, among which FX12 has the lowest cytotoxicity (Figure 1, B–D, and Table 1). FX12 inhibited NHK dislocation with an IC50 value of 2.7 μM, similar to that of Stattic (IC50 = 2.8 μM) (Figure 1, B and D). FX12 inhibited HepG2 cell growth with an IC50 value of 32.2 μM, which was higher than that of Stattic (10.8 μM) (Figure 1, C and D). Some other analogues showed better dislocation inhibition activity than FX12, such as FX40 and FX41, but all of those compounds had higher cytotoxicities (Figure 1D). On the basis of these results, we chose to focus on FX12 for further characterization.
TABLE 1:
Structures of Stattic analogues.
|
Time-lapse, live-cell imaging of drGFP confirmed that FX12 inhibited NHK dislocation (Figure 2A). ER–luminal substrates, such as NHK, require dislocation to the cytosolic surface of the ER to be ubiquitinated by an E3 ubiquitin ligase for subsequent proteasomal degradation (Ye et al., 2001; Hebert et al., 2010; Olzmann et al., 2013; Christianson and Ye, 2014; Sun and Brodsky, 2019). Therefore, inhibition of dislocation is expected to inhibit the ubiquitination and degradation of luminal ERAD substrates. We examined the effect of FX12 on NHK ubiquitination in cells. HeLa cells stably expressing HA-tagged NHK were treated with the proteasome inhibitor bortezomib (BTZ) alone or combined with FX12 to accumulate ubiquitinated NHK. Anti-HA reimmunoprecipitation revealed that FX12 efficiently inhibited NHK ubiquitination (Figure 2B). Consistently, cycloheximide (CHX) chase showed that NHK degradation was inhibited by FX12 (Figure 2C). In addition, FX12 did not affect the degradation of the cytosolic protein GFPu, whose degradation does not require dislocation (Bence et al., 2001) (Figure 2D). These results provide additional evidence that FX12 is a bona fide inhibitor of NHK dislocation.
FIGURE 2:

FX12 inhibits dislocation and has diminished activity against STAT3 activation. (A) Time-lapse imaging of NHK dislocation by drGFP. HeLa cells stably transfected with NHK-drGFP reporter plasmids were treated with BTZ (50 nM) alone or BTZ in combination with FX12 (10 μM) and imaged every 2 h. (B) FX12 inhibits NHK ubiquitination in cells. HeLa cells expressing NHK-HA were treated with BTZ (50 nM) or BTZ in combination with FX12 (10 μM). Reimmunoprecipitation of NHK-HA was performed to determine NHK ubiquitination. Nontransfected HeLa cells were used as negative control. (C) CHX chase. HEK-293T cells stably expressing HA-tagged NHK were treated with CHX in combination with DMSO or FX12 (10 μM) for the time indicated. The graph (right) shows relative band intensities of NHK measured by ImageJ and normalized to actin. The band intensities of NHK at time 0 were set as 100%. Error bars = SEM for n = 3 experiments. The p value was calculated from a one-tailed Student’s t test. *p < 0.05. (D) Effects of FX12 proteasomal degradation of cytosolic substrate GFPu. HEK-293T cells stably expressing GFPu treated with BTZ or FX12 for 6 h followed by processing for immunoblotting of GFPu and actin. (E) Immunoblotting for activated STAT3 (p-STAT3). HepG2 cells were treated with IL-6 in the presence of increasing amounts of FX12 or Stattic. DMSO was used as vehicle control. NT, nontreated.
FX12 is a derivative of benzo[b]thiophene 1,1-dioxide that does not have the nitro group required for Stattic activity (Schust et al., 2006). Therefore, we determined the effects of FX12 on IL6-stimulated STAT3 activation. Immunoblotting showed that FX12 at concentrations up to 20 μM did not inhibit IL6-induced phosphorylation of endogenous STAT3, compared with ∼75% inhibition by Stattic at 10 μM concentration (Figure 2E). Thus, FX12 is a dislocation inhibitor with diminished effects on STAT3 activation.
FX12 induces RNF5 degradation through ERAD
We determined the effects of FX12 on ERAD complex protein expression to seek the potential mechanism underlying its activity in dislocation. HeLa cells were treated with increasing amounts of FX12 for 16 h. A dose-dependent down-regulation of RNF5 protein was observed after the treatment (Figure 3A). Conversely, the levels of other ERAD components increased in a dose-dependent manner, such as Derlin1, Ube2j1, and BiP (Figure 3A), possibly resulting from the activation of adaptive UPR due to inhibition of dislocation. In support of UPR activation, PERK and Ire1α exhibited subtle changes in mobility and the activated ATF6 was increased (Figure 3A). Up-regulation of the C/EBP homologous protein (CHOP), an indicator of apoptotic UPR, was observed only in positive control cells treated with tunicamycin, but not in FX12-treated cells, suggesting that FX12 does not cause severe ER stress to activate apoptotic UPR even at concentrations that fully inhibit NHK dislocation (10–20 μM) (Figures 1C and 2A). A time-course study showed that the RNF5 protein was markedly down-regulated by FX12 9 h after the treatment (Figure 3B). Reverse transcription PCR (RT-PCR) showed that FX12 treatment caused little to no changes in RNF5 mRNA levels (Figure 3C), indicating that FX12-induced RNF5 protein down-regulation is not due to inhibition of its gene transcription.
FIGURE 3:

FX12 induces RNF5 degradation by ERAD in cells. (A, B) FX12 induces a time- and dose-dependent down-regulation of RNF5 protein in cells. (A) HeLa cells were treated with DMSO (control), increasing amounts of FX12, or tunicamycin (5 μg/ml) for 16 h. Asterisk in A indicates a nonspecific band. (B) HeLa cells were treated with FX12 (10 μM) for the indicated time. The indicated proteins were examined by immunoblotting. (C) FX12 does not affect RNF5 mRNA expression. HeLa cells were treated with DMSO or increasing amounts of FX12 for 16 h. RNF5 mRNA expression was examined by RT-PCR. Actin mRNA was examined as control. (D) FX12 induces proteasomal degradation of RNF5. HeLa cells were treated with FX12 (10 μM), BTZ (50 nM), and lysosome inhibitor CQ (50 μM) as indicated for 16 h. The indicated proteins were examined by immunoblotting. (E) FX12 increases RNF5 ubiquitination. HeLa cells transfected with FLAG-RNF5 were further treated with FX12 (10 µM) and p97/VCP inhibitor NMS-873 (NMS, 10 µM) as indicated for 4 h before reimmunoprecipitation with anti-FLAG antibody and blot for ubiquitin and the indicated proteins. The numbers under or within the blots in A, B, D, and E show the relative bind intensities. (F) NMS-873 inhibits FX12-induced RNF5 down-regulation. HeLa cells were treated as indicated, FX12 (10 µM) or in combination with NMS-873 (NMS, 10 µM) for 24 h followed by processing for immunoblotting.
Next, we determined whether RNF5 down-regulation is due to degradation. HeLa cells were treated with FX12 or inhibitor of one of the two major protein degradation pathways: proteasome or lysosome, or in combination. RNF5 protein was stabilized in cells treated by either the proteasome inhibitor BTZ or the lysosome inhibitor chloroquine (CQ) (Figure 3D, lane 1 vs. lanes 3 and 5), suggesting that both pathways are normally involved in RNF5 degradation. Interestingly, FX12-induced down-regulation of RNF5 was inhibited only by BTZ but not CQ, indicating that FX12 binding targets RNF5 to the proteasome for degradation (Figure 3D, lane 2 vs. lanes 4 and 6). Moreover, FX12 treatment directed all RNF5 protein to proteasomes for degradation (Figure 3D, lane 5 vs. lane 6). To determine whether the proteasome-dependent ERAD pathway degrades RNF5, we examined whether p97/VCP, a central regulator of ERAD, is involved in RNF5 degradation. The role of p97/VCP in ERAD is the extraction of ubiquitinated ERAD substrates from the ER for delivery to the proteasome for degradation. As such, HeLa cells were treated with FX12 in combination with the p97/VCP inhibitor NMS-873 (Magnaghi et al., 2013). As previously reported (Magnaghi et al., 2013; Walworth et al., 2016; Xia et al., 2016), inhibition of p97/VCP accumulated total ubiquitinated proteins (Figure 3E, lane 4) and FX12 treatment enhanced the accumulation (Figure 5E, lane 5), possibly by inducing ER stress due to inhibition of ERAD (Figure 2). Increased RNF5 ubiquitination was observed with NMS-873 treatment alone (Figure 3E, lane 9), consistent with a previous report that RNF5 autoubiquitinates and targets itself to ERAD (Huang et al., 2018). We did not see increases in RNF5 ubiquitination in FX12-treated cells (Figure 3E, lane 8), which is likely due to its rapid degradation. Importantly, RNF5 ubiquitination was increased by NMS-873 and further increased by simultaneous treatment with FX12 and NMS-873 (Figure 3E, lanes 9 and 10). In addition, NMS-873 also inhibited FX12-induced RNF5 down-regulation (Figure 3F). These results suggest that FX12 promoted RNF5 degradation by the p97/VCP-dependent ERAD pathway.
FIGURE 5:

The effects of FX12 on thermal stability of ERAD regulators as revealed by CETSA. (A–D) FX12 increases the thermal stability of Derlin1 and RNF5 but not Derlin2 in HEK-293T cells as revealed by CETSA. CETSA was performed using HeLa cells treated with DMSO or 20 μM FX12 at 37°C for 1 h. The band densities in immunoblots for each protein at the lowest temperature (27°C) were set as 100%. Error bars = SEM for n = 3 experiments. The p value for DMSO vs. FX12 was calculated from a one-tailed Student’s t test. p < 0.05 for B and C and p > 0.05 for D. (E–G) FX12 stabilizes RNF5 independent of Derlin1 in CETSA. CETSA was performed as in A–D except that HEK-293T cells with Derlin1 KO were used. Derlin1 KO is shown by immunoblot (F). Graph in G was generated as described in B and C. p < 0.05 for DMSO vs. FX12. (H) FX12 enhances RNF5 interaction with Derlin1. FLAG-tagged Derlin1 or Derlin2 was transiently expressed in HEK-293T cells. Cells were then treated with FX12 for 2 h and subjected to coimmunoprecipitation with anti-FLAG antibody.
FX12 binds to RNF5 and inhibits its E3 activity in vitro
FX12 is selectively targeting RNF5 for degradation, suggesting that RNF5 may directly bind to FX12. Surface plasmon resonance (SPR) was used to assess FX12 and RNF5 binding in vitro. RNF5 is predicted to have two transmembrane domains in its C-terminal region and an N-terminal cytosolic domain (amino acid [aa]1–117, RNF5N) harboring a RING finger (aa27–67) that confers its E3 activity (https://www.uniprot.org/uniprot/Q99942). If FX12 targets RNF5, one would expect that they directly bind in vitro, and thus we determined whether FX12 binds to the cytosolic domain of RNF5 (RNF5N). Two other benzo[b]thiophene 1,1-dioxide derivatives (FX41 and FX36) were analyzed as controls (Figure 4, A–D). The results showed that FX12 binds to RNF5N with a KD of 594 nM (Figure 4A). FX41 also showed a similar KD of 666 nM, whereas FX36 had no detectable binding (Figure 4, B and C), consistent with their activity in the drGFP assay (Figures 1, B and D, and 4D). These results indicate that FX12 and its active analogue FX41 directly bind to the N-terminal cytosolic domain of RNF5 in vitro.
FIGURE 4:

FX12 directly interacts with RNF5 and inhibits its E3 activity in vitro. (A–C) SPR analysis. Purified cytosolic tail of RNF5 (RNF5N) was immobilized on a sensor chip. The binding sensorgrams were obtained when FX12 (A), FX41 (B), or FX36 (C) at the indicated concentrations were flowed across the sensor chip. (D) Differential effects on NHK-drGFP by FX36 and FX41. (E) FX12 inhibits autoubiquitination of GST fusion of RNF5N but not GST-Hrd1C and -praja1 In vitro. (F) FX12 does not affect ubiquitin charging to E1 (UBA1) in vitro. (G) FX12 does not affect ubiquitin charging to E2 (UbcH5B) in vitro. (H, I) Transient overexpression of the RNF5 RING finger mutant (RINGm) inhibits NHK degradation (H). The graph (right) shows relative band intensities of NHK measured by ImageJ and normalized to actin. The band intensities of NHK at time 0 were set as 100%. Error bars = SEM for n = 3 experiments. The p value was calculated from a one-tailed Student’s t test. *p < 0.01.
We next assessed whether FX12 binding to RNF5 affected its E3 activity by in vitro ubiquitination assay reconstituted with glutathione S-transferase (GST)-RNF5N. GST-fusions of the cytosolic domain of another ERAD RING finger E3 Hrd1 (Hrd1C, aa235–617) and a non-ERAD RING finger E3 praja1 were tested as E3 controls. FX12 markedly inhibited the E3 activity of RNF5N but did not affect Hrd1C and Praja1 (Figure 4E). Importantly, FX12 did not affect ubiquitin charging to either ubiquitin-activating enzyme (E1) or ubiquitin-conjugating enzyme (E2) (Figure 4, F and G). These results suggest that FX12 binds to RNF5 and inhibits its E3 activity in vitro. FX12 inhibited NHK dislocation and degradation (Figure 2, A–C). If inhibition of RNF5 E3 activity by FX12 also occurs in cells, we would expect that the E3-inactive RNF5 inhibits NHK degradation. Therefore, we transiently expressed an E3-inactive RING finger mutant RNF5 (RNF5 RINGm) and determined its effects on NHK degradation. CHX chase showed that overexpression of RNF5 RINGm indeed inhibited NHK degradation (Figure 4, H and I). These results suggest that FX12 inhibits NHK dislocation and degradation by generating E3-inactive RNF5.
FX12 enhances the thermal stability of RNF5 in cells
The cellular thermal shift assay (CETSA) was used to determine the potential FX12-target engagement (Martinez Molina et al., 2013). CETSA is based on the biophysical principle of ligand-induced thermal stabilization of target proteins. A ligand-stabilized protein target can be detected in the soluble cellular fraction by immunoblotting or mass spectrometry (Martinez Molina et al., 2013; Jafari et al., 2014). Among major dislocation regulators examined, only RNF5 and Derlin1 showed increased thermal stabilities upon FX12 treatment (Figure 5, A–D). This may not be surprising because previous studies have shown that RNF5 and Derlin1 are interacting partners and stabilizing the target protein can lead to changes in the thermal stability of target-associated proteins (Younger et al., 2006; Morito et al., 2008). Next, a CETSA was performed in Derlin1 KO HEK-293T cells to determine whether FX12 could still stabilize RNF5 in cells lacking Derlin1. The results showed that FX12 stabilized RNF5 independent of Derlin1 (Figure 5, E–G). Interestingly, coimmunoprecipitation revealed that FX12 increased RNF5 interaction with Derlin1 but decreased its interaction with Derlin2 (Figure 5H). These results suggest that RNF5 is a molecular target for FX12 in cells and the increased thermal stability of Derlin1 is due to the FX12-enhanced RNF5-Derlin1 interaction.
FX12 improves the efficacy of VX809 and VX661 in the rescue of misfolded CFTR trafficking to the cell surface
RNF5 is a well-established E3 involved in the degradation of both misfolded wt-CFTR and ΔF508 CFTR (Younger et al., 2006). ΔF508 CFTR is a major cause of CF and exists in approximately 90% of CF patients (Younger et al., 2006). Nearly 99% of newly synthesized ΔF508 CFTR cannot fold correctly and is rapidly degraded by ERAD (Ward and Kopito, 1994). Moreover, only ∼25% of wt-CFTR reaches its correct localization at the cell surface, and the remaining protein is degraded, analogous to mutant CFTR, by ERAD (Ward and Kopito, 1994; Farinha and Amaral, 2005). We hypothesized that inhibition and degradation of RNF5 by FX12 should have a stabilizing effect on ΔF508 CFTR. BHK cells stably expressing HA-ΔF508 CFTR (Gentzsch et al., 2004) were used to test this hypothesis. As predicted, FX12 induced a dose-dependent degradation of RNF5 in BHK cells (Figure 6A), as seen in HeLa cells (Figure 3A). FX12 dose-dependent increases in both the ER core–glycosylated immature form (B form) and the complex-glycosylated mature form (C form) of ΔF508 CFTR were observed (Figure 6, A and C). The C form of CFTR represents the mature form trafficked through Golgi, suggesting that inhibition of RNF5 enhances the stability and improves cell surface trafficking of ΔF508 CFTR.
FIGURE 6:

FX12 enhances the effects of VX809 and VX661 on stabilization and trafficking of HA-wt-CFTR and -∆F508 CFTR. (A) FX12 down-regulates RNF5 accompanied by stabilization of HA-∆F508 CFTR in BHK cells. BHK cells stably expressing HA-∆F508 CFTR were treated with DMSO or increasing amounts of FX12 for 24 h followed by processing for immunoblotting. B form: ER core-glycosylated CFTR, C form: fully glycosylated mature form of CFTR. (B, C) FX12 enhances the effects of VX809 and VX661 in stabilization and trafficking of HA-wt-CFTR and ∆F508 CFTR. BHK cells stably expressing HA-wt-CFTR (B) or HA-∆F508 CFTR (C) were treated with FX12, VX809, VX661, or in combination as indicated for 24 h followed by processing for immunoblotting. BiP was blotted as loading control. (D) FX36 does not affect HA-∆F508 CFTR levels. BHK cells stably expressing HA-∆F508 CFTR were treated with FX36 or VX809 for 24 h followed by processing for immunoblotting. (E) Effects of FX12 and its analogues on ∆F508 CFTR stabilization and trafficking. HA-∆F508 CFTR BHK cells were treated with FX12 or its analogue (5 μM) alone in the presence of VX809 (5 μM) for 24 h followed by processing for immunoblotting. VCP was blotted as loading control. (F) ∆F508 CFTR ubiquitination. HEK-293T cells expressing GFP-tagged ∆F508 CFTR were treated with BTZ (50 nM) or BTZ in combination with FX12 (5 or 10 μM). Reimmunoprecipitation with anti-GFP antibody was performed to determine ∆F508 CFTR ubiquitination. Nontransfected HEK-293T cells were used as negative control. (G) FX12 inhibits HA-∆F508 CFTR degradation. BHK cells stably expressing HA-∆F508 CFTR were treated with DMSO or FX12 for 24 h, and the CHX chase was performed. The graph (right) shows relative band intensities of HA-∆F508 CFTR measured by ImageJ and normalized to VCP. The band intensities of HA-∆F508 CFTR at time 0 were set as 100%. Error bars = SEM for n = 3 experiments. The p value was calculated from a one-tailed Student’s t test. *p < 0.01.
The current U. S. Food and Drug Administration (FDA)-approved therapies for CF rescue the function of CFTR using correctors (e.g., VX809, VX661, and VX445) to improve ΔF508 CFTR folding and a potentiator (e.g., VX770) to increase CFTR channel activity (Dekkers et al., 2016; Davies et al., 2018; Keating et al., 2018). By slowing the degradation of misfolded CFTR through inhibition and degradation of RNF5, FX12 may cooperate with VX809 or VX661 to further improve cell surface trafficking of both wt-CFTR and ΔF508 CFTR. This possibility was tested by treating BHK cells stably expressing either HA-wt-CFTR or HA-ΔF508 CFTR (Gentzsch et al., 2004) with increasing amounts of FX12 with or without the folding correctors VX809 or VX661. Both VX809 and VX661 induced increases in the C form of wt-CFTR and ΔF508 CFTR, which is consistent with their activities in improving CFTR folding (Figure 6, B and C). Importantly, cotreatment with FX12 further increased the C form of both wt-CFTR and ΔF508 CFTR in a dose-dependent manner (Figure 6, B and C). As a negative control, FX36 that had no activity in drGFP also did not affect ΔF508 CFTR (Figures 4, C and D, and 6D). These results suggest that inhibition of RNF5 provides more wt-CFTR and ΔF508 CFTR for folding correctors to increase their trafficking to the surface of BHK cells.
To further investigate whether the effect of FX12 on CFTR is correlated with its dislocation inhibitory activity, we tested the effects of FX12 analogues on ΔF508 CFTR. Among 10 FX12 analogues showing inhibitory activities in NHK drGFP, seven increased the B form of ΔF508 CFTR, which was improved by cotreatment with VX809 (Figure 6E). The other three active analogues (FX33, FX34, and FX50) did not increase ΔF508 FTR, possibly due to their higher cytotoxicity (Figure 1, C and D). FX12 inhibits RNF5 E3 activity in vitro (Figure 4E). Consistently, we demonstrated that it inhibited ΔF508 CFTR ubiquitination and degradation in BHK cells (Figure 6, F and G).
Next, we tested the effect of FX12 on ΔF508 CFTR function in primary HBE cells obtained from a CF patient homozygous for ΔF508. Cells were grown at an air–liquid interface until differentiated. Then the well-differentiated ΔF508/ΔF508 HBE cultures were exposed for 24 h to basolaterally added corrector VX809 (5 µM) and/or FX12 (5 µM) in the presence and absence of potentiator VX770 (1 µM). CFTR function was then evaluated in Ussing chambers (Figure 7, A and B). Amiloride was added to inhibit the epithelial sodium channel. Treatment with VX809 significantly increased maximal CFTR (stimulated by subsequent addition of forskolin and acute addition of VX770) (Figure 7B). As previously observed, VX809+VX770–treated cells showed a smaller CFTR response than VX809-treated cells caused by destabilization of rescued ΔF508 CFTR in the presence of chronic treatments with VX770 (Cholon et al., 2014). Unlike VX809, FX12 did not enhance CFTR responses and did also not further enhance responses of VX809- or VX809+VX770–rescued cells. Biochemical rescue of ΔF508 was assessed by immunoprecipitation of CFTR followed by Western blotting (Figure 7C). Robust rescue of formation of mature ΔF508 (Figure 7C, band C) was detected when cells were treated with VX809, whereas a minor amount of band C was observed when cells were treated with FX12 (Figure 7C). Further studies are needed to optimize FX12 effects on CFTR function in ΔF508/ΔF508 HBE cells via adjusting drug dosage and treatment times and/or simultaneous targeting of alternative degradation pathways.
FIGURE 7:

Effects of VX809 and FX12 treatments on CFTR responses in well-differentiated ∆F508/∆F508 HBE cultures. Well-differentiated HBE cultures were treated for 24 h with VX809 and/or FX12 (5 µM each). (A) Representative short-circuit currents (Isc) recorded in Ussing chambers. In the chambers, the following compounds were added: amiloride (Amil, 100 μM) to inhibit ENaC, forskolin (Fsk, 10 μM) to activate CFTR, VX770 (1 μM) to potentiate CFTR, and CFTR-inhibitor (Inh-172, 10 μM) to inhibit CFTR. (B) CFTR responses (Fsk+VX770) are expressed as mean ± SEM (**p < 0.01; ***p < 0.001) (n = 4). Treatment with corrector compound VX809 (red) resulted in CFTR responses that were significantly different from those of vehicle control (black). Treatment with FX12 (dark blue) resulted in CFTR responses that were not significantly different from those of vehicle control. FX12 did not further increase CFTR responses of VX809-rescued CFTR (FX12+VX809 [turquoise] vs. VX809 [red]) or VX809+VX770–treated cultures (VX809+VX770+FX12 [orange] vs. VX809+VX770 [green]). (C) CFTR Western blot to analyze ∆F508 maturation. ∆F508/∆F508 HBE cultures were treated as above and CFTR was immunoprecipitated and subsequently analyzed by Western blotting. C = fully glycosylated mature CFTR, B = ER core–glycosylated immature CFTR.
RNF5 knockdown diminishes the inhibitory activity of FX12 in dislocation
If RNF5 is a target for FX12 to prevent dislocation, knocking it down should reduce FX12 activity. To test this possibility, the effects of RNF5 knockdown on dislocation were determined in HeLa cells that stably express the NHK-drGFP reporter (Zhong and Fang, 2012). RNF5 knockdown decreased FX12-mediated suppression of NHK dislocation (Figure 8, A–C), indicating that RNF5 is a molecular target for FX12 in cells. Next, the effects of FX12 on HA-ΔF508 CFTR stability and trafficking in RNF5 knockdown BHK cells were investigated for more proof (Figure 9). Derlin1, which forms a complex with RNF5 to recruit CFTR, was knocked down as a control (Figure 5H). Interestingly, knockdown of RNF5 stabilized ΔF508 CFTR more in the C form than in the B form, which was not seen in Derlin1 knockdown cells (Figure 9, C–F, lane 1). These results suggest that RNF5 not only is an E3 of misfolded CFTR but, in this system, may also play a role in its ER retention. Unlike in control and Derlin1 knockdown cells where FX12 treatment caused dose-dependent increases of both B and C forms of ΔF508 CFTR (Figures 9, C–F), FX12 treatment of RNF5 knockdown cells did not further increase ΔF508 CFTR (Figure 9, C–F). An FX12 dose-dependent shift from the C form to the B form of ΔF508 CFTR may be due to residual RNF5 protein in RNF5 knockdown cells (Figure 9, C and E). Next, we determined the effects of FX12 on paxillin, another substrate of RNF5. RNF5 ubiquitinates paxillin and inhibits paxillin localization to focal adhesion (Didier et al., 2003). Consistent with the previous report, FX12 treatment increases paxillin localization to focal adhesion (Figure 8D). These findings contribute to the growing body of evidence indicating that RNF5 is a molecular target for FX12 in cells.
FIGURE 8:

The effects of FX12 on NHK dislocation and ∆F508CFTR stabilization in RNF5 knockdown cells and paxillin localization to focal adhesions. (A–C) RNF5 knockdown diminishes the effects of FX12 on NHK dislocation. HeLa cells stably expressing the NHK-drGFP reporter were transfected with RNF5-targeting or control (NC) siRNA, and 24 h after siRNA transfection, the cells were treated with BTZ (50 nM) alone or BTZ in combination with FX12 (10, 20 μM) for 23 h followed by measuring drGFP fluorescence intensity on a TECAN F200 Pro multimode microplate reader. RNF5 knockdown is shown by immunoblotting in A. The graph is mean ± SD, n = 3 independent experiments (B). * p < 0.05 and p < 0.001 by Student’s t test. The representative drGFP images are shown in C. Bar = 200 µm. (D) HeLa cells cultured on coverslips were treated with DMSO (control) or FX12 (10 μM) for 24 h and then processed for anti-paxillin immunofluorescence staining. The green foci are focal adhesions. Bar = 10 μm.
FIGURE 9:

Knockdown of RNF5 but not Derlin1 diminishes the effects of FX12 on ∆F508CFTR stabilization. (A–C), RNF5 knockdown diminishes the effects of FX12 on HA-∆F508-CFTR stabilization. BHK cells stably expressing HA-∆F508-CFTR were transfected with siRNAs as indicated for 48 h. Cells were then treated with increasing amounts of FX12 or along with VX809 (5 µM) as indicated for 24 h. Knockdown efficiencies of RNF5 (A) and Derlin1 (B) were determined by immunoblotting. Control siRNA: siControl, RNF5 targeting siRNA: siRNF5, Derlin1 targeting siRNA: siDerlin1. (D–F), The band densities of B and C forms of HA-∆F508CFTR were normalized to the C band densities in the controls (lane 1, FX12 0 μM, set as 1) for each group and plotted.
DISCUSSION
To date, more than 20 ER membrane–spanning E3s have been identified, with some having a known role in ERAD (Neutzner et al., 2011; Fenech et al., 2020). The ERAD E3s, such as Hrd1, gp78, RNF5, and RNF185, act by organizing ER-membrane–anchored protein complexes (Fang et al., 2001; Amano et al., 2003; Sever et al., 2003; Kikkert et al., 2004; Younger et al., 2006; Morito et al., 2008; Bernardi et al., 2010; Fenech et al., 2020; van de Weijer et al., 2020) and have the potential to form different subcomplexes (Hwang et al., 2017). Cell type and disease–specific expression of ERAD complex proteins has also been observed (Amano et al., 2003; Bhattacharya and Qi, 2019). The variety of ERAD complexes may have evolved to dispose of a structurally diverse set of misfolded proteins or control protein levels associated with maintaining physiologic functions. The existence of cell type and disease–specific ERAD complexes suggests that small-molecule ERAD E3 inhibitors, if developed, could modulate specific cellular functions or halt disease progression (Bhattacharya and Qi, 2019). However, this promise has yet to be realized, as there are few reports of such inhibitors in the literature. The novel RNF5 inhibitor and degrader FX12 described in this paper could be a valuable tool for studying the roles of RNF5 in physiology and disease and evaluating RNF5 as a therapeutic target.
Previous studies supported the importance of ERAD E3 inhibitors. Hrd1 was the first ERAD E3 with a reported small-molecule modulator, LS-102, identified as a selective inhibitor of its autoubiquitination activity with an in vitro IC50 of 35 μM (Yagishita et al., 2012). LS-102 treatment improves rheumatoid arthritis and alleviates obesity in mouse models (Amano et al., 2003; Yagishita et al., 2012; Fujita et al., 2015), which agrees with the known roles of Hrd1 in pathogenesis (Amano et al., 2003; Bournat and Brown, 2010). Recently, a small-molecule inhibitor of RNF5, inh-2, was identified by ligand docking and virtual screening of the RING finger of the RNF5 structure obtained by homology modeling (Sondo et al., 2018). Inh-2 modulates the known downstream pathways of RNF5, including stabilization of ΔF508 CFTR and improves ΔF508 CFTR activity. However, it is not known whether inh-2 exerts its activities by directly binding to RNF5 (Sondo et al., 2018).
We have provided evidence that FX12, as a newly identified RNF5 E3 inhibitor and degrader, binds directly to RNF5 and inhibits RNF5 E3 activity in vitro (Figure 4, A and E). CETSA experiments support a direct FX12-RNF5 engagement in a cellular environment, as indicated by thermal stabilization of RNF5 in HEK-293T cells (Figure 5, A–G). Moreover, RNF5 knockdown tests show that FX12 targets RNF5 to produce its biological effects (Figures 8 and 9). CETSA and in vitro ubiquitination assays have also demonstrated a certain degree of target selectivity of FX12 (Figures 4E and 5, A–G). Consistent with the role of RNF5 as an E3 for misfolded CFTR, FX12 stabilizes ΔF508 CFTR and decreases its ubiquitination as well as increasing paxillin localization to focal adhesions (Figures 6, 7C, 8D, and 9). Moreover, FX12 enhances the FDA-approved CF therapeutics VX809 and VX661 in rescuing cell surface expression of ΔF508 CFTR in BHK cells (Figures 6 and 8F). However, unlike with inh-2, to date we could not demonstrate the improvement in ΔF508 CFTR channel activity by FX12 in patient-derived HBE cells (Figure 7). This discrepancy warrants further investigation. Inh-2 is a RNF5 inhibitor, but FX12is not only an inhibitor but also a degrader of RNF5. This differential activity may affect the channel activity of stabilized ΔF508 CFTR. In addition, the potential differential expression of RNF5 and/or its complexes could also contribute to the different responses between BHK and HBE cells. As multiple pathways have been shown to be involved in ΔF508 CFTR degradation in primary cells, additional maneuvers may be required to rescue the misfolded protein in these cultures. Because wt-CFTR has a longer half-life than ΔF508 CFTR, it might be a more suitable target in diseases such as COPD than ΔF508 CFTR in CF for which highly effective modulator combinations have become available recently. In cancer, inhibition of RNF5 has either beneficial or detrimental effects depending on cancer types (Bromberg et al., 2007; Jeon et al., 2015; Pitarresi et al., 2018; Gao et al., 2019; Li et al., 2019; Khateb et al., 2021). FX12 could be a useful chemical biology tool to study the role of RNF5 in different types of cancers and evaluate RNF5 as a therapeutic target for certain types of cancer.
Studies using the drGFP reporter demonstrated dislocation of NHK, a well-characterized substrate for Hrd1. Surprisingly, RNF5 was identified as an FX12 target because it has not been shown to regulate NHK degradation. Our data suggest that FX12 may inhibit NHK dislocation by an indirect mechanism. drGFP assays were performed in cells treated with FX12 for up to 6 h (Figures 1B and 2A). During the 6 h treatment, RNF5 protein was not down-regulated (Figure 3B), but its E3 activity is likely inhibited by FX12 based on an in vitro assay (Figure 4E). We speculated that the E3-inactive RNF5 might inhibit Hrd1-mediated ERAD by its abnormal interactions with Hrd1 complex proteins. In support of this possibility, we did see that FX12 enhanced RNF5-Derlin1 interaction (Figure 5H). Moreover, overexpression of the RNF5 E3–inactive mutant markedly inhibited NHK degradation (Figure 4, H and I). FX12 significantly down-regulated RNF5 protein 9 h after FX12 treatment (Figure 3B), but the underlying mechanism is not fully understood. FX12 binding might alter the normal structure of RNF5. Thus, FX12-bound RNF5 may be recognized as a misfolded ER protein and degraded by ERAD mediated by an E3 ubiquitin ligase that remains to be identified.
RNF5, like other ERAD E3s, forms protein complexes with membrane-spanning proteins. Although we demonstrated a direct interaction between FX12 and RNF5 in vitro, we cannot rule out the possibility that the FX12 binding site may be formed by RNF5 and its interacting proteins in cells. This binding model may explain why FX12 retains residual effects on ΔF508 CFTR stabilization in RNF5 knockdown cells (Figure 9, C and E), although the residual activity may also be explained by incomplete knockdown of RNF5. FX12 hijacks ERAD to initiate degradation of RNF5, rather than only inhibiting RNF5 E3 activity, which has important implications. In drug discovery and development, small-molecule–induced degradation of pathogenic proteins is actively being pursued, exemplified by proteolysis-targeting chimeric molecules (PROTACs) and molecular glues (Gadd et al., 2017; Gu et al., 2018; Smith et al., 2019; Isobe et al., 2020; Lv et al., 2020; Slabicki et al., 2020; Zeng and Han, 2020). A PROTAC is a designed, bifunctional small molecule that links its target protein to a known E3, hijacking the E3 to degrade the target protein. This study indicates that FX12 hijacks the protein quality control mechanism to degrade RNF5. Theoretically, this approach may be used to target other critical pathogenic proteins that access the secretory pathway by directly targeting them to ERAD for degradation without needing to design a bifunctional PROTAC molecule. In support of this possibility, a previous study reported that metformin hijacks ERAD to degrade PD-L1 (Cha et al., 2018). Because protein quality control degradation also occurs in the cytoplasm and the nucleus, the same approach may also be used to remove disease-causing proteins in these subcellular compartments. The advantage of hijacking protein quality control degradation to remove disease proteins is that there is no need to design bimodal PROTACs. Instead, small-molecule monovalent degraders can be identified by screening compound libraries using cell lines that stably express the disease protein reporters.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Cell culture
Cells from human cell lines including HeLa, HepG2, and HEK-293T were purchased from the American Type Culture Collection (ATCC; https//www.atcc.org). Previously published cell lines used in this study include a HeLa cell line stably expressing a drGFP reporter for NHK dislocation (NHK-drGFP) (Zhong and Fang, 2012) and BHK cells stably expressing extope HA-tagged wt-CFTR or ΔF508 CFTR (Gentzsch et al., 2004). Primary HBE cells were purchased from Scott H. Randell (Marsico Lung Institute, The University of North Carolina at Chapel Hill). The obtained HBE cells were prepared from explant lungs from patients homozygous for the ΔF508 mutation under protocols approved by the University of North Carolina at Chapel Hill Biomedical Institutional Review Board (Fulcher and Randell, 2013). These cells were expanded in BEGM (Lonza) and then cultured at air–liquid interface on 12 mm Millicell inserts (Millipore) in modified BEBM (Lonza) for 24 d until differentiated.
Antibodies and reagents
Antibodies.
The following antibodies were purchased from MilliporeSigma: mouse monoclonal antibodies, anti-HA (H3663, Clone HA7), mouse monoclonal anti–β-actin−peroxidase (A3854, clone AC-15), rabbit polyclonal anti-Sel1L (S3699), rabbit polyclonal anti-Derlin1 (D4443), rabbit polyclonal anti-PDI (P7372), mouse monoclonal anti-FLAG (F3165, clone M2), and ANTI-FLAG M2 Affinity Gel (A2220). Antibodies from Santa Cruz Biotechnology include mouse monoclonal anti–ubiquitin-HRP (sc-8017-HRP, clone P4D1), mouse monoclonal anti-GRP94 (sc-393402, clone H-10), mouse monoclonal anti-paxillin (sc-365379, clone B-2), mouse monoclonal anti-RNF5 (sc-81716, clone 22B3), mouse monoclonal anti-ATF6α (sc-166659, clone F-7), and mouse monoclonal anti-Ube2j1 (sc-377002, clone B-6). Other antibodies were from different sources, including mouse monoclonal anti-HA.11 (clone 16B12) (ENZ-ABS120; Enzo Lifesciences), rabbit polyclonal anti-RNF5 (PA5-71703; ThermoFisher), rabbit monoclonal anti-Hrd1 (clone D3O2A) (14773; Cell Signaling Technology), mouse monoclonal anti-BiP/GRP78 (clone 40/BiP) (610979; BD Biosciences), rabbit monoclonal anti-OS9 (clone EPR4272(2)) (ab109510; Abcam), and anti-HA Affinity Matrix (11815016001; Roche). Mouse monoclonal anti-gp78 (clone 2G5) was previously published (Ballar et al., 2006). CFTR antibodies 596 and 217 were obtained from Tim Jensen through the University of North Carolina CFTR antibody program.
Chemicals.
MG132 (474787), bortezomib (5043140001), Stattic (S7947), CHX (C7698), tunicamycin (T7765), hygromycin B (H3274), and isopropyl β-d-1-thiogalactopyranoside (I6758) were purchased from MilliporeSigma; CFTR modulators VX809 (HY-13262; MedChemExpress; S1565; Selleck), VX661 (HY-15448; MedChemExpress), and VX770 (9582; BioVision; S1144; Selleck).
Recombinant proteins.
Interleukin-6 (human) (rec) (RDX-RCP9298; Axxora), UbcH5b (E2-622-100; R&D Systems), UBA1 (E-305-025; R&D Systems).
Other reagents.
SuperSignal West Pico PLUS Chemiluminescent Substrate (34578; ThermoFisher) and Lipofectamine RNAiMAX Reagent (13778150; ThermoFisher); and small interfering RNA (siRNA) targeting Derlin1 and RNF5 synthesized based on previous reports (Lilley and Ploegh, 2005; Delaunay et al., 2008).
Constructs
pFLAG-Derlin-1 and pFLAG-Derlin-2 were constructed by inserting the cDNA fragments encoding the open reading frames (ORF) of human Derlin-1 and Derlin-2, respectively, amplified by PCR and cut with HindIII/XhoI, to the HindIII/SalI sites of pFLAG-CMV-6a. pFLAG-RNF5 was constructed by inserting the cDNA fragments encoding the ORF of human RNF5 to the BglII/SalI sites of pFLAG-CMV-6C. In the pGEX-RNF5N plasmid, the cDNA fragment encoding the N-terminal 117 aa of human RNF5 was amplified by PCR, cut with BglII/XhoI, and then inserted into the BamHI/SalI sites of pGEX-4T-3. In the pGEX-Hrd1C plasmid, the BclI/NotI fragment of human Hrd1 cDNA, encoding the C-terminal portion (aa235–617) of human Hrd1 was inserted into the BamHI/NotI sites of pGEX-5X-1. pGEX-praja1 was previously published (Lorick et al., 1999).
Purification of recombinant proteins
Each of pGEX-RNF5N, pGEX-Hrd1C, and pGEX-praja1 was transformed into BL21(DE3). Single clones of BL21(DE3) transformed were cultured overnight in Lysogeny Broth (LB) medium. Then the overnight culture was inoculated (1:100) to fresh LB medium and cultured at 37°C until the OD600 reached 0.4–0.6. The expression of GST-tagged proteins was induced with 0.2 mM isopropyl β-d-thiogalactoside (IPTG) at 37°C for 1 h. The bacteria were lysed in lysis buffer (50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% Triton X-100, 5 mM dithiothreitol) containing 200 µg/ml lysozyme with sonication. The lysates were cleared by centrifugation at 20,000 × g for 20 min. To purify the GST-tagged proteins, the cleared lysates were bound to Glutathione Sepharose 4B (GE Healthcare) for 2 h at 4°C with rotation. The beads were washed with 20 column volumes (CV) of lysis buffer and then with 5 CV of 50 mM Tris-HCl, pH 8.0. The GST-tagged proteins were eluted from the beads with 10 mM reduced glutathione in 50 mM Tris-HCl, pH 8.0. The elution fractions containing proteins were dialyzed against 1× phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4). To prepare GST-cleaved RNF5N, GST-RNF5N was bound to Glutathione Sepharose 4B and the beads were washed with 20 CV of lysis buffer and then with 10 CV 1× PBS. Then the beads were incubated with 10 U thrombin per milligram of GST-RNF5N at 4°C overnight with gentle rotation. Thrombin was removed from the supernatant by incubating the latter with p-aminobenzamidine–agarose for 30 min.
Establishment of stable expression and KO cell lines
HeLa cells stably expressing HA-tagged NHK were established as previously reported (Zhong and Fang, 2012). HeLa cells were seeded at 5 × 105 in 100 mm dishes in complete DMEM supplemented with 10% fetal bovine serum (FBS) and cultured in 37°C, 5% CO2. The next day, 10 µg of pCIneo-NHK-HA together with 0.5 µg of pBABE-puro was transfected into the cells with Lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection, cells were selected with puromycin (1 μg/ml) for 14 d. Single stable clones surviving in the selection were trypsinized and transferred to individual wells in 24-well plates containing the culture medium for further culture. The single clones were screened for expression of HA-tagged NHK by immunoblotting with an anti-HA tag antibody.
Derlin1 KO cell lines were generated in HEK-293T cells using CRISPR-Cas9. The CRISPR single-guide RNA expression plasmids targeting Derlin1 (NM_001134671.2) were obtained commercially (HCP255023-SG01-3; GeneCopia.com). Derlin1-targeting plasmids were transfected into HEK-293T cells cultured in DMEM supplemented with 10% FBS in a 100 mm dish. Twenty-four hours after transfection, cells were selected with hygromycin B (250 µg/ml) for 7 d. Derlin1 KO was determined by screening the single clones for Derlin1 expression with an anti-Derlin1 antibody in immunoblotting.
Identification of small-molecule inhibitors of ER protein dislocation
We screened LOPAC small-molecule library (SigmaAldrich.com) for dislocation inhibitors using the HeLa cells stably expressing the NHK-drGFP reporter (Zhong and Fang, 2012). NHK-drGFP reporter cells were seeded at 2 × 104/well in a black wall, clear-bottom 96-well plate (3603; Costar.com) and cultured overnight. The next day, culture medium containing the proteasome inhibitor BTZ (1 µM) alone or together with each compound (10 μM) was added to the cells for an additional 4 h. Wells containing medium alone served as background control. After replaced the medium with 1× PBS, drGFP fluorescence intensity was measured on a TECAN F200 Pro multimode microplate reader using excitation = 488 nm and emission = 525 nm. Compounds that exhibited >70% inhibition compared with BTZ-alone samples after the background extraction were subject to measurement of auto green fluorescence, and fluorescent compounds were eliminated. The remaining compounds were retested in an NHK-drGFP assay and were denoted as dislocation inhibitors when confirmed. All dislocation inhibitors were further verified independently in the InCell Analyzer 2200 wide-field imaging system at the National Center for Advancing Translational Sciences (NCATS) using a different batch of compounds at 10 μM concentration.
Live-cell imaging and quantification of drGFP
NHK-drGFP reporter cells were seeded at 2 × 104/well in 96-well plates. After overnight culture, the cells were washed once with PBS and then treated with BTZ (1 μM) alone or BTZ (1 μM) together with increasing concentrations of Stattic or its analogue. Live-cell images were acquired immediately after the addition of the inhibitors and then every 30 min on the IncuCyte Live-Cell Analysis System under a 20× objective lens (Sartorius.com), and the fluorescence intensities were quantified in IncuCyte software or on a TECAN F200 Pro Multimode microplate reader.
Synthesis of Stattic analogues
FX12 was synthesized using methyl benzo[b]thiophene-2-carboxylate as the starting material. Briefly, a solution of methyl benzo[b]thiophene-2-carboxylate (192 mg, 1 mmol) in dichloromethane (10 ml) was added 3-chloroperbenzoic acid (m-CPBA, 2 mmol) in portions over 20 min. The reaction was stirred at 50°C for 5 h and then cooled to room temperature. To the reaction mixture was added 0.5 M sodium hydroxide (20 ml). The solution was stirred for another 15 min. The aqueous layer was removed. The organic phase was washed using brine, dried over Na2SO4, and concentrated. The crude material was purified by flash chromatography to give compound FX12: 1H NMR (Varian INOVA 400 MHz, CDCl3): δ 7.99 (d, J = 0.8 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.71-7.63 (m, 2H), 7.53 (dd, J = 1.6 Hz, J = 7.2 Hz, 1H) and 13C NMR (Varian INOVA 100 MHz, CDCl3): δ 154.4, 140.3, 134.9, 134.1, 133.4, 127.4, 127.2, 122.2, 53.4; and HRMS (ESI, JEOL AccuTOF with ESI/APCI ion sources coupled to an Agilent 1100 HPLC system): The exact mass calculated for C10H9O4S [M+H]+ 225.0222, found 225.0224.
FX36: Phenyl benzo[b]thiophene-2-carboxylate.
To a solution of the benzo[b]thiophene-2-carboxylic acid (2 mmol), phenol (2 mmol), and 4-dimethylaminopyridine (1.0 equiv.) in dichlormethane (0.5 mmol/ml) was added N, N-dicyclohexylcarbodiimide (1 equiv.) at 0°C. After the reaction mixture had been stirred at room temperature overnight, it was filtered through a pad of silica gel and purified by flash chromatography to afford FX36. 1H NMR (400 MHz, CDCl3): δ 8.31 (s, 1H), 7.98 (t, J = 7.6 Hz, 2H), 7.56-7.49 (m, 4H), 7.37-7.31 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 161.3, 150.6, 142.6, 138.6, 132.7, 131.9, 129.5, 127.3, 126.1, 125.8, 125.1, 122.8, 121.6.
FX41: Phenethyl cinnamate 1, 1-dioxide.
Step 1: To a solution of the benzo[b]thiophene-2-carboxylic acid (2 mmol), 2-phenylethan-1-ol (2 mmol), and 4-dimethylaminopyridine (1.0 equiv.) in dichlormethane (0.5 mmol/ml) was added N, N-dicyclohexylcarbodiimide (1 equiv.) at 0°C. After the reaction mixture had been stirred at room temperature overnight, it was filtered through a pad of silica gel and purified by flash chromatography to afford precursor phenethyl benzo[b]thiophene-2-carboxylate. 1H NMR (400 MHz, CDCl3): δ 8.06 (s, 1H), 7.897.87 (m, 2H), 7.49-7.27 (m, 7H), 4.57 (t, J = 6.8 Hz, 2H), 3.11 (t, J = 7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 162.7, 142.2, 138.7, 137.7, 133.6, 130.6, 129.0, 128.6, 126.9, 126.7, 125.6, 124.9, 122.8, 66.0, 35.2. Step 2: This step was carried out entirely in accordance with general method A. 1H NMR (400 MHz, CDCl3): δ 7.92 (s, 1H), 7.75 (d, J = 6.8 Hz, 1H), 7.68-7.60 (m, 2H), 7.51 (d, J = 7.2 Hz, 1H), 7.35-7.25 (m, 5H), 4.56 (t, J = 7.2 Hz, 2H), 3.10 (t, J = 6.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 158.5, 139.9, 138.1, 137.1, 134.7, 133.9, 133.2, 129.2, 128.6, 128.1, 127.4, 126.8, 121.9, 66.8, 34.9. HRMS (ESI): Exact mass calculated for C16H16NO4S [M+NH4]+ 332.0957, found 332.0964.
CHX chase
The CHX chase was performed as previously reported (Fang et al., 2001). HeLa cells stably expressing HA-tagged NHK or -CD3δ were seeded (2 × 105 cells/well) in a 12-well plate and cultured overnight. Cells were then incubated with CHX (50 μg/ml) alone or along with FX12 for the indicated time (Figure 2C) and then were harvested and processed for immunoblotting with anti-HA and anti–β-actin antibodies.
Immunoprecipitation
HEK-293T cells were transfected with pFLAG-Derlin-1 or pFLAG-Derlin-2 or vector as control. The cells were treated with FX12 for 2 h as indicated the next day. Cells were lysed in 0.2% NP-40 in cell lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA [ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid)]. Proteins (total 700 µg) were incubated with 10 µl of ANTI-FLAG M2 Affinity Gel for 2 h at 4°C with rotation. The beads were washed three times with cell lysis buffer containing 0.2% NP-40 before being processed for immunoblotting. Immunoprecipitation and immunoblotting of CFTR from HBE cells were performed as described previously (Cholon et al., 2014; Gentzsch et al., 2016, 2017; McCravy et al., 2020; He et al., 2021).
Reimmunoprecipitation
NHK ubiquitination was analyzed as previously reported (Zhong and Fang, 2012). Briefly, HeLa cells expressing NHK-HA were treated with BTZ (1 μM) or BTZ + FX12 (10 μM) to prevent degradation of ubiquitinated NHK. After the treatment, cells were lysed in cell lysis buffer containing 0.2% NP-40 followed by immunoprecipitation with the Anti-HA Affinity Matrix. To remove any protein that may associate with NHK-HA, the immunoprecipitates were denatured with 2% SDS, and the beads were removed by centrifugation. The supernatants were then diluted 20 times in native lysis buffer from which NHK-HA was reimmunoprecipitated, followed by immunoblotting for HA and ubiquitin.
To analyze RNF5 ubiquitination, HeLa cells were transfected with pFLAG-RNF5. The next day, transfected cells were treated with FX12 and NMS873 as indicated. After the treatment, cells were lysed in cell lysis buffer containing 0.2% NP-40 followed by immunoprecipitation with µl of ANTI-FLAG M2 Affinity Gel for 2 h at 4°C with rotation. The immunoprecipitates were denatured with 2% SDS, and the beads were removed by centrifugation. The supernatants were then diluted 20 times in native lysis buffer from which FLAG-RNF5 was reimmunoprecipitated, followed by immunoblotting for HA and ubiquitin.
CETSA
The CETSA was performed following a previously reported protocol (Martinez Molina et al., 2013; Reinhard et al., 2015). HeLa cells were treated with 10 μM FX12 or dimethyl sulfoxide (DMSO) control at 37°C for 2 h. After the treatment, cells were trypsinized and then neutralized with culture medium. After brief centrifugation at 1000 × g for 5 min, the cells were washed once and then resuspended with 1× PBS. Multiple aliquots of cell suspension were heated at increasing temperatures for 3 min before cooling to room temperature (25°C) for 3 min. An equal volume of 1× PBS containing 0.8% NP-40 was then added to the cells to lyse them. The samples were centrifuged at 20,000 × g for 20 min to separate soluble fractions from precipitated proteins. The soluble fractions were incubated with a 4× SDS sample buffer for 30 min at 37°C before being processed for immunoblotting. The following ERAD complex proteins were examined, including the major candidate target proteins in ERAD complexes, including RNF5, gp78, Hrd1, Sel1L, Derlin1, Derlin2, VIMP, Ube2j1, BiP, grp94, PDI, and OS9.
SPR
The binding of FX12 and another Stattic analogue, FX41, with the purified recombinant cytosolic domain of RNF5N (aa1–117) was measured using Biacore. Sensorgrams were obtained for 62.5, 125, 250, or 500 nM compounds against RNF5N immobilized on the CM5 sensor chip. Purified RNF5(aa1–117) was coupled to the surface of Biacore CM5 sensor chips by direct immobilization. Ligands were performed at a flow rate of 30 µl/min in HBS running buffer. The association was recorded by SPR with a Biacore T200 (Biacore, USA).
In vitro ubiquitination and ubiquitin charging assays
In vitro ubiquitination was performed following our previously reported protocol (Fang et al., 2001). Briefly, 500 ng of GST-RNF5, GST-Hrd1c, or GST-Praja1 was immobilized on glutathione beads. Ubiquitination assays were carried out by adding 20 ng each of human E1 and E2 (Ube2j1) and ubiquitin (2 μg) in ubiquitination buffer containing 50 mm Tris-HCl, pH 7.4, 2 mM ATP, and 5 mM MgCl2 with an increasing amount of FX12 (0, 5, 10 μM). Reactions were carried out in 20 μl for 30 min at 30°C followed by processing for immunoblotting. FX12 does not affect ubiquitin charging to E1 and E2. E1 assay: 0.2 μg of recombinant UBA1 (E1) was mixed with ubiquitin, reaction buffer with or without ATP, and DMSO or FX12 (10 μM final) on ice as indicated. The reactions were incubated at 37°C for 30 min. E2 assay: 0.5 μg of recombinant UbcH5b (E2) was mixed with ubiquitin, reaction buffer, and DMSO or FX12 (10 μM final) on ice as indicated. Recombinant UBA1 (E1) (0.2 μg) was added to the reactions as indicated. The reactions were incubated at 37°C for 30 min. UbcH5b was detected by Ponceau S staining.
Measurement of CFTR activity in Ussing chambers
Ion transport was measured as short-circuit current (Isc) in a modified Ussing chamber system using Acquire and Analyze software (Physiological Instruments) as previously described (Cholon et al., 2014; Gentzsch et al., 2016, 2017; He et al., 2021) in a bilateral Krebs bicarbonate–Ringers (KBR) solution. Bridges were equilibrated in KBR bubbled with CO2 and maintained at 37°C, and each chamber was zeroed against a blank insert. Changes in Isc were measured. Amiloride (100 μM; Sigma-Aldrich) was added to the apical bath to inhibit the epithelial sodium channel ENaC. Bilateral addition of forskolin (10 μM; Sigma-Aldrich) was followed by apical addition of potentiator compound VX770 (1 μM; Selleck Chemicals) to stimulate CFTR channel activity. CFTR inhibitor-172 (10 μM; Sigma-Aldrich) was then apically introduced to inhibit CFTR. Transepithelial resistance (in Ω·cm2) was monitored to assess monolayer integrity. Traces were plotted using Origin Graphic software. Changes in Isc were calculated using Microsoft Excel. Bar graphs were plotted in Prism.
Supplementary Material
Acknowledgments
We thank Douglas Cyr for his advice during this study. S.F. was funded by the National Institute of General Medical Sciences (UO1GM117175), M.G. was supported by Preclinical Core funding (P30DK065988) from the National Institute of Diabetes and Digestive and Kidney Diseases. D.C.T., G.R., D. T., C.A.L., A.S., and M.J.H. were supported by the intramural research program, National Center for Advancing Translational Sciences, National Institutes of Health.
Abbreviations used:
- BTZ
bortezomib
- CQ
chloroquine
- drGFP
dislocation-induced reconstituted green fluorescent protein
- HBE
primary human bronchial epithelial
- Hrd1C
aa235–617 of Hrd1
- NHK
null Hong Kong variant of α-1-antitrypsin
- RNF5N
N-terminal cytosolic domain aa1–117 of RNF5
- RNF5 RINGm
E3-inactive RING finger mutant RNF5.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E22-06-0233) on September 8, 2022.
REFERENCES
- Amano T, Yamasaki S, Yagishita N, Tsuchimochi K, Shin H, Kawahara K, Aratani S, Fujita H, Zhang L, Ikeda R, et al. (2003). Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev 17, 2436–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Cantley LC (2018). The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 174, 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballar P, Shen Y, Yang H, Fang S (2006). The role of a novel p97/valosin-containing protein-interacting motif of gp78 in endoplasmic reticulum-associated degradation. J Biol Chem 281, 35359–35368. [DOI] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. [DOI] [PubMed] [Google Scholar]
- Bernardi KM, Williams JM, Kikkert M, van Voorden S, Wiertz EJ, Ye Y, Tsai B (2010). The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol Biol Cell 21, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Qi L (2019). ER-associated degradation in health and disease—from substrate to organism. J Cell Sci 132, jcs.232850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournat JC, Brown CW (2010). Mitochondrial dysfunction in obesity. Curr Opin Endocrinol Diabetes Obes 17, 446–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg KD, Kluger HM, Delaunay A, Abbas S, DiVito KA, Krajewski S, Ronai Z (2007). Increased expression of the E3 ubiquitin ligase RNF5 is associated with decreased survival in breast cancer. Cancer Res 67, 8172–8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, Li CW, Kim T, Chang SS, Lee HH, et al. (2018). Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell 71, 606–620. e607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholon DM, Quinney NL, Fulcher ML, Esther CR Jr, Das J, Dokholyan NV, Randell SH, Boucher RC, Gentzsch M (2014). Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci Transl Med 6, 246ra296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Ye Y (2014). Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol 21, 325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antonio M, Reyna J, Jakubosky D, Donovan MK, Bonder MJ, Matsui H, Stegle O, Nariai N, D’Antonio-Chronowska A, Frazer KA (2019). Systematic genetic analysis of the MHC region reveals mechanistic underpinnings of HLA type associations with disease. eLife 8, e48476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JC, Moskowitz SM, Brown C, Horsley A, Mall MA, McKone EF, Plant BJ, Prais D, Ramsey BW, Taylor-Cousar JL, et al. (2018). VX-659-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med 379, 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers JF, Berkers G, Kruisselbrink E, Vonk A, de Jonge HR, Janssens HM, Bronsveld I, van de Graaf EA, Nieuwenhuis EE, Houwen RH, et al. (2016). Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci Transl Med 8, 344ra384. [DOI] [PubMed] [Google Scholar]
- Delaunay A, Bromberg KD, Hayashi Y, Mirabella M, Burch D, Kirkwood B, Serra C, Malicdan MC, Mizisin AP, Morosetti R, et al. (2008). The ER-bound RING finger protein 5 (RNF5/RMA1) causes degenerative myopathy in transgenic mice and is deregulated in inclusion body myositis. PLoS One 3, e1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didier C, Broday L, Bhoumik A, Israeli S, Takahashi S, Nakayama K, Thomas SM, Turner CE, Henderson S, Sabe H, Ronai Z (2003). RNF5, a RING finger protein that regulates cell motility by targeting paxillin ubiquitination and altered localization. Mol Cell Biol 23, 5331–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM (2001). The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci USA 98, 14422–14427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha CM, Amaral MD (2005). Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol Cell Biol 25, 5242–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech EJ, Lari F, Charles PD, Fischer R, Laetitia-Thezenas M, Bagola K, Paton AW, Paton JC, Gyrd-Hansen M, Kessler BM, Christianson JC (2020). Interaction mapping of endoplasmic reticulum ubiquitin ligases identifies modulators of innate immune signalling. eLife 9, e57306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Yagishita N, Aratani S, Saito-Fujita T, Morota S, Yamano Y, Hansson MJ, Inazu M, Kokuba H, Sudo K, et al. (2015). The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC-1beta. EMBO J 34, 1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcher ML, Randell SH (2013). Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol Biol 945, 109–121. [DOI] [PubMed] [Google Scholar]
- Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M, Ciulli A (2017). Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Xuan C, Jin M, An Q, Zhuo B, Chen X, Wang L, Wang Y, Sun Q, Shi Y (2019). Ubiquitin ligase RNF5 serves an important role in the development of human glioma. Oncol Lett 18, 4659–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch M, Chang XB, Cui L, Wu Y, Ozols VV, Choudhury A, Pagano RE, Riordan JR (2004). Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol Biol Cell 15, 2684–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch M, Martino M, Cholon DM, Quinney NL, Boyles SE, Ribeiro CM (2017). Impact of the inflammatory cystic fibrosis airway milieu on pharmacological function and rescue of CFTR. Pediatr Pulmonol 52, 228. [Google Scholar]
- Gentzsch M, Ren HY, Houck SA, Quinney NL, Cholon DM, Sopha P, Chaudhry IG, Das J, Dokholyan NV, Randell SH, Cyr DM (2016). Restoration of R117H CFTR folding and function in human airway cells through combination treatment with VX-809 and VX-770. Am J Physiol Lung Cell Mol Physiol 311, L550–L559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grand DL, Gosling M, Baettig U, Bahra P, Bala K, Brocklehurst C, Budd E, Butler R, Cheung AK, Choudhury H, et al. (2021). Discovery of Icenticaftor (QBW251), a cystic fibrosis transmembrane conductance regulator potentiator with clinical efficacy in cystic fibrosis and chronic obstructive pulmonary disease. J Med Chem 64, 7241–7260. [DOI] [PubMed] [Google Scholar]
- Gu S, Cui D, Chen X, Xiong X, Zhao Y (2018). PROTACs: an emerging targeting technique for protein degradation in drug discovery. Bioessays 40, e1700247. [DOI] [PubMed] [Google Scholar]
- He L, Kennedy AS, Houck S, Aleksandrov A, Quinney NL, Cyr-Scully A, Cholon DM, Gentzsch M, Randell SH, Ren HY, Cyr DM (2021). DNAJB12 and Hsp70 triage arrested intermediates of N1303K-CFTR for endoplasmic reticulum-associated autophagy. Mol Biol Cell 32, 538–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Bernasconi R, Molinari M (2010). ERAD substrates: which way out? Semin Cell Dev Biol 21, 526–532. [DOI] [PubMed] [Google Scholar]
- Huang EY, To M, Tran E, Dionisio LTA, Cho HJ, Baney KLM, Pataki CI, Olzmann JA (2018). A VCP inhibitor substrate trapping approach (VISTA) enables proteomic profiling of endogenous ERAD substrates. Mol Biol Cell 29, 1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Walczak CP, Shaler TA, Olzmann JA, Zhang L, Elias JE, Kopito RR (2017). Characterization of protein complexes of the endoplasmic reticulum-associated degradation E3 ubiquitin ligase Hrd1. J Biol Chem 292, 9104–9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe Y, Okumura M, McGregor LM, Brittain SM, Jones MD, Liang X, White R, Forrester W, McKenna JM, Tallarico JA, et al. (2020). Manumycin polyketides act as molecular glues between UBR7 and P53. Nat Chem Biol 16, 1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P, Martinez Molina D (2014). The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 9, 2100–2122. [DOI] [PubMed] [Google Scholar]
- Jeon YJ, Khelifa S, Ratnikov B, Scott DA, Feng Y, Parisi F, Ruller C, Lau E, Kim H, Brill LM, et al. (2015). Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell 27, 354–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, Ramsey BW, Rowe SM, Sass LA, Tullis E, et al. (2018). VX-445-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med 379, 1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khateb A, Deshpande A, Feng Y, Finlay D, Lee JS, Lazar I, Fabre B, Li Y, Fujita Y, Zhang T, et al. (2021). The ubiquitin ligase RNF5 determines acute myeloid leukemia growth and susceptibility to histone deacetylase inhibitors. Nat Commun 12, 5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo LT, Chen LY (2018). Role of the cGAS-STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep 19, e46935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van Voorden S, Thanedar S, Roitelman J, Chau V, Wiertz E (2004). Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem 279, 3525–3534. [DOI] [PubMed] [Google Scholar]
- Kuang E, Okumura CY, Sheffy-Levin S, Varsano T, Shu VC, Qi J, Niesman IR, Yang HJ, Lopez-Otin C, Yang WY, et al. (2012). Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet 8, e1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tinoco R, Elmen L, Segota I, Xian Y, Fujita Y, Sahu A, Zarecki R, Marie K, Feng Y, et al. (2019). Gut microbiota dependent anti-tumor immunity restricts melanoma growth in Rnf5(-/-) mice. Nat Commun 10, 1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL (2005). Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc Natl Acad Sci USA 102, 14296–14301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Gao C (2018). Regulation of MAVS activation through post-translational modifications. Curr Opin Immunol 50, 75–81. [DOI] [PubMed] [Google Scholar]
- Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM (1999). RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci USA 96, 11364–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv L, Chen P, Cao L, Li Y, Zeng Z, Cui Y, Wu Q, Li J, Wang JH, Dong MQ, et al. (2020). Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. eLife 9, e59994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Damania B (2016). The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe 19, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnaghi P, D’Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, et al. (2013). Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol 9, 548–556. [DOI] [PubMed] [Google Scholar]
- Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P (2013). Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87. [DOI] [PubMed] [Google Scholar]
- McCravy MS, Quinney NL, Cholon DM, Boyles SE, Jensen TJ, Aleksandrov AA, Donaldson SH, Noone PG, Gentzsch M (2020). Personalised medicine for non-classic cystic fibrosis resulting from rare CFTR mutations. Eur Respir J 56, 2000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morito D, Hirao K, Oda Y, Hosokawa N, Tokunaga F, Cyr DM, Tanaka K, Iwai K, Nagata K (2008). Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol Biol Cell 19, 1328–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutzner A, Neutzner M, Benischke AS, Ryu SW, Frank S, Youle RJ, Karbowski M (2011). A systematic search for endoplasmic reticulum (ER) membrane-associated RING finger proteins identifies Nixin/ZNRF4 as a regulator of calnexin stability and ER homeostasis. J Biol Chem 286, 8633–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Kopito RR, Christianson JC (2013). The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol 5, a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SD, Bono TR, Rowe SM, Solomon GM (2020). CFTR targeted therapies: recent advances in cystic fibrosis and possibilities in other diseases of the airways. Eur Respir Rev 29, 190068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitarresi JR, Liu X, Avendano A, Thies KA, Sizemore GM, Hammer AM, Hildreth BE 3rd, Wang DJ, Steck SA, Donohue S, et al. (2018). Disruption of stromal hedgehog signaling initiates RNF5-mediated proteasomal degradation of PTEN and accelerates pancreatic tumor growth. Life Sci Alliance 1, e201800190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard FB, Eberhard D, Werner T, Franken H, Childs D, Doce C, Savitski MF, Huber W, Bantscheff M, Savitski MM, Drewes G (2015). Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat Methods 12, 1129–1131. [DOI] [PubMed] [Google Scholar]
- Rothan HA, Zhong Y, Sanborn MA, Teoh TC, Ruan J, Yusof R, Hang J, Henderson MJ, Fang S (2019). Small molecule grp94 inhibitors block dengue and Zika virus replication. Antiviral Res 171, 104590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J, Rothan HA, Zhong Y, Yan W, Henderson MJ, Chen F, Fang S (2019). A small molecule inhibitor of ER-to-cytosol protein dislocation exhibits anti-dengue and anti-Zika virus activity. Sci Rep 9, 10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schust J, Sperl B, Hollis A, Mayer TU, Berg T (2006). Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 13, 1235–1242. [DOI] [PubMed] [Google Scholar]
- Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA (2003). Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell 11, 25–33. [DOI] [PubMed] [Google Scholar]
- Slabicki M, Kozicka Z, Petzold G, Li YD, Manojkumar M, Bunker RD, Donovan KA, Sievers QL, Koeppel J, Suchyta D, et al. (2020). The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 585, 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD, Crews CM (2019). Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondo E, Falchi F, Caci E, Ferrera L, Giacomini E, Pesce E, Tomati V, Mandrup Bertozzi S, Goldoni L, Armirotti A, et al. (2018). Pharmacological inhibition of the ubiquitin ligase RNF5 rescues F508del-CFTR in cystic fibrosis airway epithelia. Cell Chem Biol 25, 891–905.e898. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zheng H, Yu S, Ding Y, Wu W, Mao X, Liao Y, Meng C, Ur Rehman Z, Tan L, et al. (2019). Newcastle disease virus V protein degrades mitochondrial antiviral signaling protein to inhibit host type I interferon production via E3 ubiquitin ligase RNF5. J Virol 93, e00322-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Brodsky JL (2019). Protein quality control in the secretory pathway. J Cell Biol 218, 3171–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Sun L, Chen J, Chen ZJ (2018). Detection of microbial infections through innate immune sensing of nucleic acids. Annu Rev Microbiol 72, 447–478. [DOI] [PubMed] [Google Scholar]
- van de Weijer ML, Krshnan L, Liberatori S, Guerrero EN, Robson-Tull J, Hahn L, Lebbink RJ, Wiertz E, Fischer R, Ebner D, Carvalho P (2020). Quality control of ER membrane proteins by the RNF185/Membralin ubiquitin ligase complex. Mol Cell 80, 374–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wain LV, Shrine N, Artigas MS, Erzurumluoglu AM, Noyvert B, Bossini-Castillo L, Obeidat M, Henry AP, Portelli MA, Hall RJ, et al. (2017). Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet 49, 416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth K, Bodas M, Campbell RJ, Swanson D, Sharma A, Vij N (2016). Dendrimer-based selective proteostasis-inhibition strategy to control NSCLC growth and progression. PLoS One 11, e0158507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CL, Kopito RR (1994). Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem 269, 25710–25718. [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR (1995). Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127. [DOI] [PubMed] [Google Scholar]
- Xia D, Tang WK, Ye Y (2016). Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 583, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagishita N, Aratani S, Leach C, Amano T, Yamano Y, Nakatani K, Nishioka K, Nakajima T (2012). RING-finger type E3 ubiquitin ligase inhibitors as novel candidates for the treatment of rheumatoid arthritis. Int J Mol Med 30, 1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2001). The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656. [DOI] [PubMed] [Google Scholar]
- Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM (2006). Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 126, 571–582. [DOI] [PubMed] [Google Scholar]
- Zeng Z, Han T (2020). Discovering nature’s super glue. Nat Chem Biol 16, 1155–1156. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang Q, Yang F, Zhu Z, Duan Y, Yang Y, Cao W, Zhang K, Ma J, Liu X, Zheng H (2021). JMJD6 negatively regulates cytosolic RNA induced antiviral signaling by recruiting RNF5 to promote activated IRF3 K48 ubiquitination. PLoS Pathog 17, e1009366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Fang S (2012). Live cell imaging of protein dislocation from the endoplasmic reticulum. J Biol Chem 287, 28057–28066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.