

Abstract
The worldwide pandemic of coronavirus disease 2019 (COVID-19) along with the various newly discovered major SARS-CoV-2 variants, including B.1.1.7, B.1.351, and B.1.1.28, constitute the Variant of Concerns (VOC). It's difficult to keep these variants from spreading over the planet. As a result of these VOCs, the fifth wave has already begun in several countries. The rapid spread of VOCs is posing a serious threat to human civilization. There is currently no specific medicine available for the treatment of COVID-19. Here, we present the findings of methods that used a combination of structure-assisted drug design, virtual screening, and high-throughput screening to swiftly generate lead compounds against Mpro protein of SARs-CoV-2. Therapeutics, in addition to vaccinations, are an essential element of the healthcare response to COVID-19's persistent threat. In the current study, we designed the efficient compounds that may combat all emerging variants of SARs-CoV-2 by targeting the common Mpro protein. The present study was aimed to discover new compounds that may be proposed as new therapeutic agents to treat COVID-19 infection without any adverse effects. For this purpose, a computational-based virtual screening of 352 in-house synthesized compounds library was performed through molecular docking and Molecular Dynamics (MD) simulation approach. As a result, four novel potent compounds were successfully shortlisted by implementing certain pharmacological, physiological, and ADMET criteria i.e., compounds 3, 4, 21, and 22. Furthermore, MD simulations were performed to evaluate the stability and dynamic behavior of these compounds with Mpro complex for about 30 ns. Eventually, compound 22 was found to be highly potent against Mpro protein and was further evaluated by applying 100 ns simulations. Our findings showed that these shortlisted compounds may have potency to treat the COVID-19 infection for which further experimental validation is proposed as part of a follow-up investigation.
Keywords: COVID-19, Mpro, Synthetic compounds, Virtual screening, Molecular docking and simulation
Graphical abstract

1. Introduction
The current ongoing pandemic of COVID-19 has appeared as one of the most life-threatening infections caused by SARS-CoV-2 [1]. It was initially diagnosed in Wuhan city, China (December 2019), associated with a bunch of respiratory diseases. The International Committee on Taxonomy of Viruses (ICTV) named this virus “SARS-CoV-2” and the disease “COVID-19”. Several countries have declared it as a pandemic in a very short span of time due to its rapid spread [2,3]. The World Health Organization (WHO) reported on July 16, 2021 that there are more than 180 million infections with 4 million deaths caused by Covid-19 around the globe [4].
COVID-19 is a positive sense ranging from 26 to 32 kb ssRNA containing virus (65–125 nm in diameter) in size [5]. Coronavirus genome is comprised of the 5′-untranslated region (UTR), replicase complex (ORF1a and ORF1ab), structural genes for spike (S), nucleocapsid (N) envelope (E), membrane (M) proteins, and several Open Reading Frames (ORFs) for accessory proteins introduced between four structural genes, ending with 3′-UTR with poly A tail. However, the two polypeptides generated from the first ORFs (pp1a and pp1ab) encode for several vital Nonstructural Proteins (NSPs), among which is NSP5 (a cysteine 3C-like protease) also known as main protease (Mpro) [6,7]. The Mpro protein mediates the production of important NSPs crucial for the viral infection cycle i.e. RNA-dependent RNA polymerase, methyltransferase, and helicase [8]. Hence, the main protease controls the major pathways and replication of the viral genome and it is an attractive drug target to halt the replication event in the virus life cycle [9,10]. The membrane association, fusion of membranes and entry of this pathogenic virus into the host cell is mediated by spike protein which is anchored by viral envelope. The spike proteins are large transmembrane glycoprotein which in some coronaviruses are cleaved by cellular proteases into two subunits [11]. There are S1 and S2 domains of spike protein which are described for HCoV-22E, HCoV-NL63 and SARS-CoV [12]. The S1 domain of spike protein mediates viral binding with susceptible host cell surface receptor/ACEII, while S2 domain comprises of hydrophobic peptide and coiled coil regions which facilitate membrane fusion followed by prior receptor association [13]. S-protein interacts with host angiotensin converting enzyme (II) receptor/ACE-II receptor. This S-protein/ACE-II interaction causes the fusing of host cell membrane with virus [14].
Currently, due to the mutations found in the SARS-CoV-2 genome, several different variants (alpha., beta, gamma, delta and recently Omicron) are recognized in different countries [15] having S-gene (+) with a higher rate of transmissibility and immune escape mechanism responsible for persevering a global threat [16]. Initially, the alpha variant that emerged from the Wuhan strain possesses 13 mutations that ultimately increases the transmissibility rate up to 50%. Another variant of the greatest concern is Beta or B.1.351, which was first emerged in South Africa, possessing nine mutations, decreases the neutralization aptitude of antibodies prompted by infection with earlier variants or vaccination [15]. Similarly, the third worldwide documented VOC, Gamma or P.1, is composed of 11 spike mutations [17]. Nevertheless, the emerging variant B.1.617 or delta derived from Alpha lineage B.1.617 is now internationally recognized as VOC, that is comprised of different mutational profiles [4]. These multiple mutations appear to give an advantage to variants [18] with increased transmission rate (e.g., within 10 s, 60% more transmissible than Alpha variant), rendering it as most dangerous and dominant variant globally [19,20]. The recently emerged Omicron variant also possessed 50 mutations (30 in spike proteins). Luckily, in all these Cov-II mutations, Mpro remained conserved without any prior mutations [21]. Therefore, Mpro protein remained a promising target to halt the potent COVID pathogenicity mechanism. Recent Bioinformatics studies have also uncovered the roles of miRNAs in the pathogenesis and in the infection mechanism of SARs-COV-2. The viral miRNAs are crucial in the infection as they facilitate the viral entry into host cells due to the low molecular weight and size. They are unperceived by the host immune system. They also target immune system, cytokine storm, and inflammatory response related host's genes. Thus, the interactions between the miRNA and other molecules may also be suitable intervention strategy for limiting the viral infection and in terms of prevention and cure [22].
Several vaccines are available since early 2022 however, the new viral variants with novel mutations and antigenic profiles are posing serious threats and diminishing the efficacy of the available vaccines (e.g., AstraZeneca, Sinovac, Sinopharm, and Pfizer's, etc) [23]. Collectively, these all vaccines are only ∼50% effective with lower optimal results against these variants [9,[24], [25], [26]]. Certain drug and vaccine designing approaches are used to develop efficacious drugs or vaccines against COVID-19 [[27], [28], [29]]. However, no such effective vaccine or drug candidate is yet designed that can completely treat CoV-II infections [15]. A new approach is imperative with insights into the initial strains of COVID-19 to investigate the mutation mechanism and look forward to developing new therapeutics. Therefore, we employed the computational based virtual screening, molecular docking, followed by the intensive ADMET profiling and MD simulation approach to predict the in-house synthesized multiple medicinally important class of chemical compounds as novel therapeutic candidates against SARS-CoV-2. In this study, an established drug target is used to discover new therapeutics against it while a machine learning approach can also be used to explore the chemical space if the drug target is known [[30], [31], [32]]. The prioritized compounds in this study belong to the tyramine, quinoline, thiourea and indole class of compounds. Quinolone are highly reported compounds for their diverse pharmacological role, used as anti-bacterial, antitumor, and antiparasitic [33]. Significantly, some of its derivatives are approved (FDA approved) against fungal infections, antiseptics, and amoebiasis etc (https://go.drugbank.com/categories/DBCAT002551). Furthermore, thiourea derivative are primarily used as second generation antibiotics to treat Graves, diabetes, and cancers [34]. Similarly, indole derivatives are widely tested against drug-resistant caner and multiple disease [35]. In comparison to the existing therapy and reported data for SARS-CoV-2, the derivatives of these compounds utilized in the current study are not previously studied, thus providing a new prospective for SARS-Cov-2 drug discovery. Overall, the current study provides an insight to combat COVID-19 using novel identified drug candidates as promising lead, optimized for drug discovery. We strongly believe that our findings may provide prolific information and better guidance for drug development against all the existing strains of the CoV-II virus including the newly emerged Omicron variant as well.
2. Material and methods
2.1. Dataset collection and preparation
The structures of synthetic compounds were drawn in ChemDraw3D. All 352 structures were subjected to energy minimization with MM9F forcefield available in Open Babel v.2.3.1. The minimized compounds were then saved in PDB format for further virtual screening analysis. The X-ray crystal structure of the main protease (PDB ID: 6Y2G) with its inhibitor was retrieved from RCSB Protein Data Bank [36]. The PDB 6Y2G was preferred because it is a complexed structure of SARS-CoV-2 Mpro protein with an inhibitor and having better resolution.
2.2. Ligand preparation
All 352 in-house synthetic compounds were subjected to Open Babel 2.3.1 [37] to convert in to PDBQT format. The PDBQT format is required for virtual screening as AutoDock Vina [38] read this format.
2.3. Protein/receptor preparation and grid generation
The X-ray crystal structure of the main protease (Mpro) was prepared with ADT tool of AutoDock Vina. The water molecules were removed, polar Hydrogens were added and non-polar Hydrogens were merged. Kollman charges were added to estimate the potential partial atomic charges followed by saving the receptor protein in the PDBQT format for further analysis. The prepared protein structure was used for grid generation using the AutoGrid program in AutoDock Vina. The receptor grid box of size x = 40, y = 40, and z = 40 dimensions were centered on the selected co-crystallized ligand of the protein within the active site. The grid was centered on x = −18.792, y = −6.95, and z = −26.325 with 0.375 nm spacing. The grid maps were prepared around amino acids Leu27, His41, Ser139, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, Met165, Glu166, Leu167, Val171, His172, and Ala173 by running AutoGrid 4. These amino acid residues were selected due to the presence of co-crystallized ligand and existence of catalytic dyad i.e. His41 and Cys145.
2.4. Virtual screening
The approach to discover the potent inhibitors against the SARS-CoV main protease (Mpro) employed the screening of a small library of synthetic compounds into the active site of the target protein. The AutoDock Vina was used to perform the screening of all 352 synthetic compounds. The compounds were docked with the receptor and 20 conformations were generated for each compound. The screening model was employed to select the compounds with potential of mediating the energetically favorable interactions with the active site of the target protein. The method was first validated by redocking the co-crystal ligand “ketoamide” to the Mpro structure (Fig. 1 and Supplementary Information heading 1 and Fig. 3). The crystal structure of Mpro was fetched from the protein data bank.
Fig. 1.

The 3D structure of Mpro, integrated with inhibitor (brown) embedded in enzyme's catalytic pocket.
Fig. 3.

Docking studies of compounds showing significant Hydrogen bonds and Hydrophobic interaction in 2D (3A) and 3D (3B) formats of compound 22. Showing potent interaction within the binding cavity of Mpro.
2.5. ADMET-tox properties estimation
The compound obtained after the virtual screening was narrowed down based on ADME-Tox (http://www.acdlabs.com/products/admet/tox/) properties. An online tool PreADMET (http://preadmet.bmdrc.org) was employed to perform ADME-Tox calculation. The properties were considered to screen the compounds that fulfill the requirement to define them as ADME-Tox competent and safe enough to administer them in-vivo or animal use. The estimated features to define molecules are safe based on ADME-Tox and include CMC_like_Rule, Lead_like_Rule_Violation_Fields, Rule_of_Five, Blood_Brain_Barrier (BBB), Buffer_solubility, Caco2, Plasma_Protein_Binding, Pure_Water_Solubility, Skin_Permeability, Carcino_Mouse, Carcino_Rat, and hERG_inhibitor. All screened compounds were evaluated for their drug-like nature under the rule of five, CMC-like-Rule, and Lead-like-Rule. ADME predictions such as oral drug absorption was based on Caco2-cell model, potential oral and transdermal drug delivery was based on Skin permeability. The drug distribution was predicted by Blood brain barrier(BBB) and Plasma protein binding model predicted efficacy and disposition. Toxicity predictions was based on carcino mouse and rat model that can readily classify the risk of mutagenicity.
2.6. Molecular docking experiments
The compounds that fulfilled the requirements of ADME-Tox properties were further subjected to molecular docking via AutoDock 4.2 tools. The specificity of the binding of the compounds with stable conformation within the binding site pocket was increased via the employment of same grid box which was used for virtual screening. The molecular docking was used to dock compounds with the following parameters: 250 number of GA runs, 250 population size, and the maximum number of evaluations = 10000000. The best-docked ligand conformation out of 250 conformations having the least binding energy was obtained complexed with Mpro in PDBQT format. The best-docked pose of the compounds was 2D plotted via LigPlus 1.4.5 [39] to get insights of 2D interactions, whereas the 3D interactions were generated and examined in UCSF Chimera v.1.14 [40].
2.7. MD simulations
The obtained docked conformations complexed with Mpro binding site were subjected to molecular dynamics simulation. The ligand and protein were prepared separately for MD simulation. The ligand was prepared via an online tool named Automated Topology Builder (ATB) V 3.0 [41]. The ATB server used GROMOS96 54A7 forcefield to minimize the ligand and the ligand topology files were obtained as an output. Additionally, the Mpro protein was converted in GROMACS file format with pdb2gmx using a forcefield of gromos96 54a7. Later, both ligand and protein in GROMACS formatted file were complexed. The ligand-Mpro complex was covered with a unit cell of dodecahedron box shape of size 1.0 and the spc216 water model of GROMACS was used to solvate the complex with water. System solvation was followed by the addition of four Na + ions in the system to make it neural. The molecular dynamics was performed in three steps on the prepared ligand-Mpro complex with GROMACS. First, the system's minimization was performed which consisted of 50,000 steps via the steepest descent minimization algorithm. The system minimization is intended to eradicate the major atomic clashes in the protein. Then the system was equilibrated to restrain protein and ligand whereas solvent and Na + ions were allowed to contact the protein. The total equilibration time was 100 ps along with the time step of 2 fs at 300 K. Finally, the MD production steps were performed without position restraining for a total of 30 ns with a timestep of 2 fs at 300 K. The trajectories were obtained after MD production step. They were used to analyze root mean square deviation (RMSD), root mean square fluctuations (RMSF), radius of gyration (Rg), and hydrogen bond analysis of the system. The binding free energy of compound 22 was evaluated via MMGBSA method by using gmx_MMPBSA tool v1.5.2 [42]. The following equation used to evaluate binding free energies:
| ΔGbind = Gcomplex – Gprotein – Gligand |
| = ΔEMM + ΔGGB + ΔGSA – TΔS | Eq (1) |
| = ΔEvdw + ΔEele + ΔGGB + ΔGSA – TΔS |
3. Results
3.1. Virtual screening of synthesized compounds
All 352 in-house synthesized compounds were virtually screened against the chain A of Mpro (since Mpro is a homodimeric enzyme). It resulted in shortlisting of the 22 compounds with the estimated lowest binding energy (≤−7.0 kcal/mol) in comparison with the binding energy of the co-crystallized ligand (−7.4 kcal/mol) (Fig. 2 ). All compounds obtained from the experiment of virtual screening were found in contact with the amino acids His41, Met49, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163-164, Asp187, and Arg188. Whereas the interactions of the compounds with the amino acid residues His41 and Cys145 were found to be more significant since these residues are also involved in the substrate catalysis [21]. The compounds along with their structure and binding energy are given in Table 1 .
Fig. 2.

Virtual screening of 352 in-house synthetic compounds against SARS-CoV-II Mpro. It shows that compounds mainly showed binding affinities from −5.8 to −6.11 kcal/mol, −6.38 to −6.92 kcal/mol. Additionally, >200 compounds showed binding energies > than −6.0 kcal/mol.
Table 1.
Top 22 molecules obtained on virtual screening via autodock vina, with the best binding affinity score.
| S. No. | Compound Structure | Binding Affinity (Kcal/mol) | Compound Class |
|---|---|---|---|
| 1 | 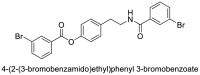 |
−7.5 | Tyramine |
| 2 |  |
−7.4 | Tyramine |
| 3 |  |
−7.3 | Quinoline |
| 4 |  |
−7.2 | Quinoline |
| 5 |  |
−7.3 | Quinoline |
| 6 | 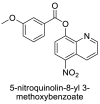 |
−7.2 | Quinoline |
| 7 |  |
−7.3 | Quinoline |
| 8 | 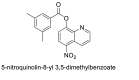 |
−7.3 | Quinoline |
| 9 |  |
−7.5 | Quinoline |
| 10 |  |
−7.6 | Quinoline |
| 11 | 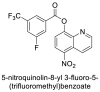 |
−7.4 | Quinoline |
| 12 | 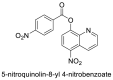 |
−7.7 | Quinoline |
| 13 |  |
−7.3 | Quinoline |
| 14 | 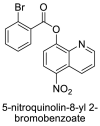 |
−8.1 | Quinoline |
| 15 |  |
−7.7 | Quinoline |
| 16 |  |
−7.4 | Quinoline |
| 17 | 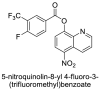 |
−7.3 | Quinoline |
| 18 |  |
−7.4 | Quinoline |
| 19 |  |
−7.2 | Quinoline |
| 20 |  |
−7.2 | Thiourea |
| 21 | 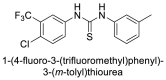 |
−7.2 | Thiourea |
| 22 | 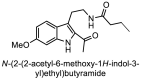 |
−7.5 | Indole |
3.2. Physicochemical properties and ADME-TOX prediction
The pharmacokinetics properties and toxicity profile of the compounds are necessarily required to certify the level of their efficacy along with their therapeutic and toxic effects. The examined properties of the initially obtained significant compounds through virtual screening are shown in Table 2 . The Blood-Brain Barrier (BBB) permeability defines the ability of the compound to penetrate the CNS [43]. The value of CNS > −2 was measured to infiltrate the Central Nervous System (CNS). Among the four lead compounds, compounds 3, 4, and 21 had CNS permeability of −1.4, −1.357, and −0.863 respectively, showing that they are accessible to CNS. Whereas compound 22 had the least −2.649 value i.e. impermeable to CNS and considered as non-toxic to CNS. All the four compounds were predicted as non-toxic and non-carcinogenic in AMES (assay to assess reverse mutation in Salmonella) and carcinogenic profile assessment. The shortlisted compounds followed the Lipinski rule of five where the molecular weight of compounds 2, 3, 21, and 22 were 352.604, 351.711, 344.789, and 304.39 g/mol respectively. That is in complete accord to the Lipinski's rule of five. All four lead-like compounds were found to be in an acceptable range according to the Lipinski rule of five, and their pharmacokinetic and toxicity profiles. Since compound 22 was impermeable to CNS, it was deemed more suitable for further analysis.
Table 2.
Pharmacological and toxic profiling of virtually significant compounds using Pre ADMET webserver.
| Parameter | Compound 3 | Compound 4 | Compound 21 | Compound 22 |
|---|---|---|---|---|
| CNS permeability | −1.4 | −1.357 | −0.863 | −2.649 |
| MW | 352.604 | 351.711 | 344.789 | 304.39 |
| Rule 0f five | Suitable | Suitable | Suitable | Suitable |
| CMC like rule | Qualified | Qualified | Not qualified | Qualified |
| Plasma protein binding | 98.426781 | 90.847276 | 91.408083 | 78.62002 |
| Caco2 | 48.126 | 48.548 | 55.282 | 33.0089 |
| Skin permeability | −2.72608 | −2.01596 | −1.63015 | −4.15853 |
| AMES toxicity | No | No | No | No |
| Carcino Mouse | No | Yes | No | No |
| Carcino Rat | No | No | No | Yes |
| CYP2C19 inhibition | Yes | Yes | ||
| CYP2C9 inhibition | Yes | Yes | No | No |
| CYP2D6 inhibition | No | No | No | No |
| CYP3A4 inhibition | Yes | Yes | No | No |
| CYP3A4 substrate | Yes | Yes | Yes | Yes |
| CYP2D6 substrate | No | No | No | No |
| P-glycoprotein inhibition | Yes | Yes | Yes | No |
3.3. Molecular docking studies
The ADME-Tox profile of compounds 3, 4, 21, and 22 was suitable for their possible consideration to be used as a safe drug candidate for in-vivo studies. The binding energies of compounds 3 and 4 were estimated as −7.17 and −6.6 kcal/mol respectively. Both compounds were interacted with the target binding site on CoV-II Mpro via mediating only significant hydrophobic interactions (Table 3 ) as seen in Supplementary Figs. 1A and 1B. Whereas compounds 21 and 22 were found as mediating the hydrogen bonds between the amino acid residue His163 and His164 while compound 21 formed an additional H-bond with Ser144 (Table 3). The compound 21, having trifluoromethyl group, formed one H-bond with amino acid Ser144 at a bond distance = 2.82 Å and second hydrogen bond with the His163 at a bond distance of 2.72 Å, whereas the N1 group of compound 21 formed an H-bond with oxygen (O) of His164 (bond distance of 2.78 Å) as shown in Supplementary Figure 1C. The O-3 group of compound 22 mediated an H-bond with NE2 of His163 with a bond distance of 2.84 Å, while the N2 group of compound 22 mediated an H-bond with (O) of His164 (bond distance 2.90 Å) as seen in Fig. 3 . The co-crystallized ligand “O6K” with the Main protease of SARS-CoV-II was re-docked with −7.97 kcal/mol as estimated binding energy. The O-26 group of O6K mediated an H-bond with SG of Cys145 (bond distance = 2.87 Å) whereas, O-40 of O6K mediated an H-bond with the O of the carbonyl group of Glu166 backbone (bond distance = 2.86 Å) as shown in Supplementary Fig. 1D.
Table 3.
Molecular docking of shortlisted compounds with SARS CoV-II Mpro (6Y2G) using AutoDock 4.2
| Compounds | Binding Energy (Kcal/mol) | Amino Acid (H-Bond) | Amino Acid (Hydrophobic) | Predicted Ki (μm) |
|---|---|---|---|---|
| 3 | −7.17 | – | His41, Phe140, Leu141, Ser144, Cys145, His163, His164, Met165, Glu166 | 12.37 |
| 4 | −6.6 | – | His41, Phe140, Leu141, Ser144, Cys145, His163, His164, Met165, Glu166, Asp187 | 14.52 |
| 21 | −6.67 | Ser144, His163, His164 | His41, Asn142, Phe140, Cys145, Met165, Glu166, Val186, Asp187, Arg188, Gln189, | 12.44 |
| 22 | −6.6 | His163, His164 | His41, Met49, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, Met165, Glu166, His172, Phe181, Asp187, | 11.53 |
| O6K | −7.97 | Cys145, Glu166 | His41, Met49, Phe140, Leu141, Asn142, Gly143, Ser144, His163, His164, Met165, Asp187, Arg188 | 14.56 |
In addition, the docking scores were used to make predictions for the inhibitory constant (K i) for these compounds. By measuring the K i, one may estimate that how likely a compound will inhibit an enzyme and lead to a clinically significant drug interaction with an enzyme. A compound inhibitory potential may often be predicted by calculating the K i in proportion to the inhibitor concentration in the body. In the current study, K i values for the lead compounds were predicted to be in the range of 11–14 μm (Table 3). This correlation study showed that the predicted K i and binding energy of the compounds were highly correlated (r 2 = 0.99), estimating the direct correlation of K i with predicted docking scores [44] (Fig. 4 ).
Fig. 4.

The correlation analysis of lead compounds docking scores and their predicted Ki, showing the strong correlation with R2 = 0.99.
3.4. Molecular dynamics simulation
The docked complex of Mpro and ligands (3, 4, 21, and 22) shortlisted by docking studies were further assessed for their stability analysis utilizing MD simulation for up to 30 and 100 ns. The simulation outcomes are highlighted in terms of RMSD, RMSF, hydrogen bond, and radius of gyration analysis.
The RMSD suggested that the lead compounds 3, 4, 21, and 22 were all within the permissible range of 0.25 nm, indicating that all detected lead compounds were tightly bound within Mpro active cavity. The protein-compound complex system proceeded to equilibrate between 12 and 30 ns within the RMSD range of 0.2–0.3 nm resulting in an average RMSD value of 0.25 nm, as per the RMSD graph (Fig. 4A). The Mpro in complex with compound 3 (black) showed stability after 13 ns of simulation with mild fluctuations. After 13 ns simulation, a modest increase in complex with 4 (red) and 21 (blue) was observed, resulting in overall stability within the range of 0.2–0.4 nm. The compound 22 complex (green) and reference compounds (yellow) showed the stable RMSD at 12 ns around 0.2 nm. These backbone variations observed in Mpro and ligand complexes indicate the conformational changes as shown by the RMSD.
The RMSF trajectories provide essential information regarding the stability of the complex. The large variance in the plots indicates that the interactions are more flexible and fragile. Low values or less fluctuation indicate well-structured complex regions with less distortion. However, the Mpro complex with compounds showed an identical pattern of interactions in the system as shown in Fig. 4B. The compounds complexed with Mpro showed stability of protein with slight high peaks observed as the constant pattern during the complete (30 ns) simulation period.
Furthermore, the radius of gyration was also analyzed to study the compactness of Mpro in the presence of compounds. The simulated Rg of these five (four selected and one reference) compounds varies from 2.17 to 2.3 nm as illustrated in Fig. 4C. The Rg value revealed the stability of the protein in the complex indicating the stable binding of these five molecules without any structural reforms.
Hydrogen bonding determines the strength of the interaction between ligands and proteins. Throughout the simulation, a continuous range of hydrogen bonds ∼5 was detected in these compounds when analyzed at 4 frames interval. Whereas compound 22 mediates ∼10 hydrogen bonds in 1790 frames with the Cys145 of catalytic site along with Gly143, Asn142, Glu166, Ser144, Gln189, Gly143, His41, Ser46 in 615, 484, 213, 202, 171, 15, 6, and 6 frames respectively (Fig. 6 ), indicating the most stable bonding with Mpro protein.
Fig. 6.

Hydrogen bonds mediated by ligand 22 with MPro of CoV-II. Significant atom types shown involve in mediation of H-bond with the amino acids of binding pocket of MPro for the 30 ns simulation, showing the potential interaction of compound 22 with enzyme.
During simulation, the Mpro protein in complex with four compounds was identified to be stable during 30 ns simulation. However, compound 22 outperformed the overall simulation studies and showed more stable interaction compared to other compounds and reference ligand and hence better possibilities for inhibiting Mpro as a prospective inhibitor. Therefore, 200-ns simulation of Apo protein (Mpro) and compound 22 were further performed to more accurately understand the stability and flexibility of compound 22 within the Mpro cavity. The RMSD results showed that compound 22 (red) remained stable after ∼30 ns of simulation with mild fluctuation within the range of 0.2–0.4 nm as given in Fig. 7 A, whereas Apo protein (black) fluctuated in the range of 0.2–0.3 nm. It shows the significant differences in the binding pattern of compound 22 with Mpro i.e., a fluctuation can be observed after the ligand is bound to Mpro (red).Importantly, during simulations analysis, the RMSD of compound 22 within the binding pocket was also evaluated to monitor the ligand stability and flexibility throughout the 200 ns simulation. It was observed that the compound 22 initially showed fluctuations between 10 and 15 ns stabilizing at 0.5 nm, while convergence was observed after 15 ns to 55ns. However, after 55 ns its showed stable interaction with in the pocket throughout the 200 ns simulations within the range of 0.65–0.8 nm. This value lowers with time, showing that compound 22 may change Mpro conformation in the binding area. Throughout the simulation, the RMSD of compound 22 fluctuated between 0.5 and 0.8 nm and stayed constant at 0.75 nm respectively as showed in Fig. 7B. The fluctuation and overall stability of compound 22 in Mpro binding cavity showed variable conformations changes suggesting the stability of its interaction. Additionally, the calculated Coulombic and Lennard jones dispersion/repulsion interaction energies for Mpro Protein and Compound 22 complex was calculated. It resulted in the estimated average energies as −115 and −112 kJ/mol respectively as Supplementary Fig. 4. The detail of these estimated energies are provided in Supplementary Table S1.
Fig. 7.

Molecular dynamics simulation of Compound 22 (A), Root mean square deviation (RMSD) of apo protein (black) and ligand bounded protein (red) within the binding pocket of Mpro. It shows the significant differences in the binding pattern of compound 22 with Mpro i.e., a fluctuation can be observed after the ligand bound to Mpro (Red) (B), Root Mean Square Fluctuation of proteins amino acid residues throughout 200 ns simulations showing stability of protein and compound with mild fluctuations (C), the RMSF analysis of Apo protein (Black) and Mpro-compound 22 complex (red) showing the clear fluctuations in the ligand bounded protein amino acids, as a significant peaks of active amino acids can be observed from 100 to 200 range of amino acids i.e., Asn142, Gly143, Ser144, Cys145, Glu166, and Gln189. (D) Radius of gyration (Rg) of proteins (Black) and ligand bounded protein (Red) indicating protein stability upon inhibitor interaction with Mpro. A significant compactness of protein can be observed after the compound 22 occupied the binding pocket of Mpro with a significant decrease in Rg can be observed in range of 2.15 nm compared to 2.25 nm of Apo protein (E), and persistent hydrogen bonds mediation of compound 22 with the significant amino acids of binding pocket of Mpro such as Asn142, Gly143, Ser144, Cys145 respectively. The results of hydrogen bonds are further supported by the RMSF fluctuation observed (in Fig. 7C) within the binding pocket of Mpro, required for the anchoring and catalysis of substrate.
The RMSF and Rg for compound 22 and Apo protein were also calculated and compared and found to be in an acceptable range as given in Fig. 7C (shown in black-compound 22 and shown in red-Apo protein) and 7D (shown in black-compound 22 and shown in red-Apo protein). Moreover, several persistent hydrogen bonds mediated by ligand with mainly Cys145 in 9698 frames, Asn119 in 5686 frames, δ2N of Asn142 with O1 and O2 of ligand 22 in 3988 and 155 frames respectively. Whereas, N of Gly143 mediated H-bond O1 and O2 of compound 22 in 4823 and 1811 frames respectively. Furthermore, N and γ-O of Ser144 mediated H-bond with O1 of ligand in 3125 and 73 frames respectively. Additionally, ε-N of Gln189 mediated H-bond with O3 of compound 22 in 184 frames, shown in Fig. 6E. These bonds were mainly observed between compound 22 and amino acids residues of Mpro binding pocket (Cys145, Gln189, Asn142, Ser144, Asn119, and Gly143) defining the possible significant in-silico inhibitory activity of compound 22. The binding free energy of compound 22 was estimated via last 100 frames of equilibrated simulation trajectories by gmx_MMPBSA [42]. In this study, the ΔGbind value of compound 22 was equaled to −22.13 kcal/mol as shown in Fig. 8 , ranging from −18 to −28.5 kcal/mol (Supplementary Table S2).
Fig. 8.

Binding free energy calculation for compound 22 via MMGBSA Eq (1), showing binding free energy as −28 kcal/mol within the binding cavity of Mpro for last 100 frames of 200 ns simulation.
4. Discussion
The novel coronavirus has resulted in an ongoing pandemic problem [45]. The need to discover the new drugs, that can potentially target the virus in patients suffering from the infection due to new types of emerging COVID-19 variants such as delta and most recently the omicron despite the availability of vaccines, still exists [46]. The viral genome of the SARS-CoV II encodes several structural and non-structural proteins [47]. The non-structural proteins also include main proteases (Mpro) [47]. The Mpro protein is one of the most suitable and attractive drug targets for the design and development of direct-acting anti-viral drugs (Fig. 1). The Mpro protein is a conserved key enzyme and plays a pivotal role in the process of viral replication and transcription [48]. Our study has taken Mpro as potential drug target to perform the structure-based identification of potential inhibitors from the in-house synthesized compound library (Table 1). The virtual screening, molecular docking and dynamic simulation approaches were adopted as they are time efficient, cost-effective and powerful tool in the development of drugs against disease.
4.1. Virtual screening, molecular docking analysis based on binding energy and ADME-tox profiling
The amino acid residues of the catalytic pockets of the Mpro are highly conserved and share more than 90% sequence similarity with 3CLpro of other coronaviruses. The two conserved amino acids His41 and Cys145 (catalytic dyad) are mainly responsible for the catalysis of the substrate in the binding site [49]. In the current study, structure-based virtual screening for Mpro determined 22 compounds as computationally suitable ligand from the in-house synthesized compound library. The compounds with the potential antiviral activity were ranked based on the estimated binding energy profiles by comparing the estimated binding energy of the co-crystallized ligand. The interaction profiles of these 22 compounds in stable conformation within the binding cavity of the Mpro revealed the Hydrogen bond interactions with the conserved catalytic dyad. It also revealed the H-bond, pi-pi, non-covalent, or hydrophobic interactions with other hot spot amino acids such as Met49, Phe140, Leu141, Asn142, Gly143, Ser144, His163-164, Asp187, and Arg188 as highlighted in Fig. 3 and Supplementary Fig. 1. Although all the 22 ligands have been docked with significant binding energies onto the Mpro target protein, but only compounds 3, 4, 21, and 22 were selected due to their appropriate ADMET-Tox profiles and physicochemical properties (Table 2). Therefore, the molecular interaction profiles of only four ligands (compounds 3, 4, 21, and 22) were analyzed via molecular docking studies (Table 3).
The existence of H-bond, hydrophobic (pi-pi stacking) and non-covalent (non-polar) interactions implied that all ligands were docked in their energetically favorable conformations. It also implied that the interatomic distances were within the range of 5 nm. Detailed analysis of the docking experiment revealed that compounds 3 and 4 were stabilized by pi-pi interactions and compounds 21 and 22 were stabilized with the H-bond interactions and pi-pi bond with the hot spot residues. In previous similar docking studies performed against the Mpro, the hotspot residues such as His164, Glu166, Asp187, His41, Gln189, and Cys145 were found in mediating polar and non-polar interactions i.e. H-bond interactions and pi-pi (hydrophobic) interactions [50]. This confirmed that the binding modes of all four compounds with the target Mpro are significant and their binding behavior is similar to potential inhibitors.
4.2. Molecular dynamics trajectory analysis
The MD simulation offered the detailed study of binding interactions of ligands to the targets on atomic levels. The RMSD, RMSF, Rg and number of hydrogen bonds analysis provided useful insights into the structural stabilities and binding modes of the ligands. The resulting graph generated by all these analyses are more often used to predict the compound's affinity for the active site of Mpro target protein. The root means square deviation (RMSD) value of Mpro and all four compounds remain stable after 12 ns depicting the convergence of simulations. All systems appeared to converge within 30 ns simulations, shown in Fig. 5A. The average value of RMSD varies between 0.14 and 0.38 nm. The maximum deviation occurred in the compound 4 simulation system and the minimum deviation was in compound 22. Overall, the observation suggested that docked complexes were stable. The RMSF plot given in Fig. 5B showed ligands 21 and 4 depicted the slightly significant fluctuation across different regions of residues compared to other ligands. Overall, all four ligand complexes and co-crystallized complexes exhibited the same fluctuation pattern (peaks) across the residues. The Rg calculated for all four complex systems and co-crystallized ligand complex varied between 2.125 and 2.325 nm. It suggested that the compactness of all systems are similar and binding patterns are stable (Fig. 5C). Moreover, the 200 ns simulation for compound 22 compared with 200 ns simulation for Apo protein resulted in stable interaction throughout the 200 ns simulation with mild fluctuation within the acceptable range of 0.5–0.75 nm. It showed the significant differences in the binding pattern of compound 22 with Mpro i.e., fluctuations can be observed after the ligand bound to Mpro (Red) in Fig. 7A. These results showed that the compound 22 is highly stable and can be used as a lead to discover novel therapeutics against COVID-19.
Fig. 5.

Molecular dynamics simulation study highlighting Root mean square deviation (RMSD) of protein backbone (A), Root Mean Square Fluctuation of proteins in complex with shortlisted four compounds after simulations (B), Radius of gyration of proteins (Rg) showing the protein stability during Mpro-inhibitor interaction (C). i.e., Compound 3 (black), 4 (red), 22 (green), 21 (blue), Co-crystalized ligand (yellow).
4.3. Hydrogen bond analysis
Hydrogen bonding determines the binding strength of the ligands to the proteins. The hydrogen bond formed between the protein backbone and the ligands were calculated throughout the simulation runtime. The number of hydrogen bonds formed contribute to the overall conformational stability of the docked complex. Overall, the compounds 3 (Supplementary Fig. 2A) and 4 (Supplementary Fig. 2B) showed less stable and insignificant intermolecular hydrogen bond interactions and compound 21 (Supplementary Fig. 2C) showed inconsistent hydrogen bonds in just two frames of MD simulation, with amino acid Ser144 and His172 of Mpro. The simulation analysis revealed many hydrogen bonds formed between amino acid residues of Mpro and ligand 22 shown in Fig. 5. The simulation experiments also revealed that compound 22 had the highest affinity for Mpro compared to the other ligands. The extended simulation from 30 ns to 100 ns of complex 22 indicated strong interactions due to the formation of many intermolecular hydrogen bonds. During 30 ns simulation, a constant and strong H-bond was observed between residue Cys145. The Ser144 was also found to be involved in mediating the H-bond within the 5 ns of simulation and afterward with minor breaks. The Gln189 mediated a weak bond between 0 and 3 ns and then 10–15 ns. Similarly, two strong H-bonds were observed after 20 ns between Asn142 and Gly143. The 200 ns simulation revealed that the H-bond between Cys145 is significant and stable throughout the simulation, only minor breaks observed, shown in Fig. 7E. The H-bond interactions exhibited by Asn142 was found more stable between 20 and 50 ns and then less stable after 55 ns whereas completely despaired after 160ns of simulation. The bond between Gly143 and compound 22 was more stable in 200 ns simulation as it appeared after 20 ns and remain consistent up to 55 ns, appeared and disappeared in many frames up to 160 ns while completely disappeared after 160 ns of simulation. The H-bond of Ser144 was unstable, formed in many frames throughout the simulation showing the facilitation of Ser144 in the ligand binding in active site of enzyme. A new H-bond between Asn119 and compound 22 appeared after 55 ns which became more stable after 175 ns till the end of 200 ns of simulation. The hydrogen bond analysis revealed that Asn119, Asn142, Gly143, Ser144, and Cys145 were mainly involved in the interaction and stabilization of compounds within the Mpro binding pocket. Root Mean Square Fluctuation (RMSF) of protein's amino acid residues throughout 200 ns simulations showed the stability of protein and compound with mild fluctuations ( Fig. 7C). Similarly the RMSF analysis of Apo protein (black) and Mpro-compound 22 complex (red) showed the distinct fluctuations in the ligand bound protein amino acids as significant peaks of active amino acids can be observed from 100 to 200 range of amino acids i.e., Asn142, Gly143, Ser144, Cys145, Glu166, and Gln189 (Fig. 7D). On the other hand, the Radius of Gyration (Rg) of proteins (black) and ligand bound protein (red) presented protein stability upon inhibitor interaction with Mpro. A significant compactness of protein can be observed after the compound 22 occupied the binding pocket of Mpro while a significant decrease in Rg was observed in the range of 2.15 nm compared to 2.25 nm of Apo protein (Fig. 7E), and persistent hydrogen bonds mediation of compound 22 with the significant amino acids of binding pocket of Mpro such as Asn142, Gly143, Ser144, Cys145. The results of hydrogen bonds are further supported by the RMSF fluctuation observed (in Fig. 7C) within the binding pocket of Mpro, required for the anchoring and catalysis of substrate. The H-bond analysis of co-crystallized ligand given in Supplementary Fig. 2D showed that the bond between His41, Asn119, and Glu166 with co-crystallized ligand is stable, whereas Cys145, Thr26, Gly143, and Ser144 are less stable. Hence, it is suggested that the number of hydrogen bonds formed during simulation is more in case of compound 22 than the co-crystallized ligand. The results of hydrogen bond analysis illustrated that the residue Cys145 is stable in compound 22-Mpro complex and His45 is stable for co-crystallized ligand-Mpro complex. Besides these observations, the residue Gly143, Ser144, and Asn119 are also stable in compound 22-Mpro complex (Fig. 7A, B, and 7C) and Asn119 is stable in co-crystallized-Mpro complex (Fig. 5A, B, and 5C). Furthermore, the result obtained from MMGBSA calculations revealed the stable binding of compound 22 with Mpro target site. The detailed information of the calculated contribution of energy term in Eq. (1) revealed that compound 22 binding to Mpro was primarily driven by van der Waals (ΔEvdw) and electrostatic interaction energies (ΔEele), but hampered by polor solvent energies (ΔGpol-sol), as summarized in Supplementary Table S3.
The analysis of results revealed that compound 22 showed a very strong molecular interaction profile with an amino acid residue of the binding site especially the catalytic dyad (Cys145 and His41) of the Mpro target enzyme. Hence the compound 22 may behave as a plausible candidate for designing direct antiviral inhibitors against SARS-CoV as it also showed the highest extent of safety profile. In future, these results will be followed up by performing in vitro experiments to demonstrate their effectiveness.
5. Conclusion
The main protease (Mpro) is a potential drug target for designing direct-acting anti-viral inhibitors. We adopted a virtual screening protocol to search novel inhibitors from an in-house synthesized library. The four compounds were identified as potential inhibitors based on ligand-protein binding pattern (lowest estimated binding energy) and ADMET-Tox prediction profiles. Furthermore, the structure stability and dynamic behavior of Mpro were explored through MD simulation upon binding with these four compounds. Overall, the whole study of simulation analysis manifested that the Mpro-compound 22 complex is more stable, and its simulation properties were comparable to that of co-crystallized Mpro complex. Compound 22 exhibited good intermolecular interaction and binding profiles with the conserved residues of the catalytic site particularly the Cys145 (that is the part of catalytic dyad). Therefore, our findings suggested that the compound 22 may have the ability to inhibit the replication process in SARs-CoV-II by directly targeting the Mpro enzyme. However, in vitro testing protocols are required to further support these results.
Declaration of competing interest
The authors declare that there is no conflict of interest.
Acknowledgements
The authors would like to acknowledge the Higher Education Commission of Pakistan for providing the financial support under National Research Program for Universities.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2022.106284.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R. New England journal of medicine; 2019. A Novel Coronavirus from Patients with Pneumonia in China. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saha I., Ghosh N., Maity D., Sharma N., Sarkar J.P., Mitra K. Genome-wide analysis of Indian SARS-CoV-2 genomes for the identification of genetic mutation and SNP. Infect. Genet. Evol. 2020;85 doi: 10.1016/j.meegid.2020.104457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi H., Han X., Jiang N., Cao Y., Alwalid O., Gu J., Fan Y., Zheng C. Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect. Dis. 2020;20(4):425–434. doi: 10.1016/S1473-3099(20)30086-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Organization W.H. vol. 45. June 2021. p. 22. (COVID-19 Weekly Epidemiological Update). 2021. [Google Scholar]
- 5.Shereen M.A., Khan S., Kazmi A., Bashir N., Siddique R. COVID-19 infection: origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020;24:91. doi: 10.1016/j.jare.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Organization W.H. World Health Organization; 2020. Laboratory Testing for Coronavirus Disease (COVID-19) in Suspected Human Cases: Interim Guidance, 19 March 2020. [Google Scholar]
- 7.Cui J., Li F., Shi Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17(3):181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zehra Z., Luthra M., Siddiqui S.M., Shamsi A., Gaur N.A., Islam A. Corona virus versus existence of human on the earth: a computational and biophysical approach. Int. J. Biol. Macromol. 2020;161:271–281. doi: 10.1016/j.ijbiomac.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020;94(7):e00127. doi: 10.1128/JVI.00127-20. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verheije M.H., Hagemeijer M.C., Ulasli M., Reggiori F., Rottier P.J., Masters P.S., Haan C.A.d. The coronavirus nucleocapsid protein is dynamically associated with the replication-transcription complexes. J. Virol. 2010;84(21):11575–11579. doi: 10.1128/JVI.00569-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson C.B., Farzan M., Chen B., Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022;23(1):3–20. doi: 10.1038/s41580-021-00418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riccio A., Santopolo S., Rossi A., Piacentini S., Rossignol J.-F., Santoro M.G. Impairment of SARS-CoV-2 spike glycoprotein maturation and fusion activity by nitazoxanide: an effect independent of spike variants emergence. Cell. Mol. Life Sci. 2022;79(5):1–21. doi: 10.1007/s00018-022-04246-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Essalmani R., Jain J., Susan-Resiga D., Andréo U., Evagelidis A., Derbali R.M., Huynh D.N., Dallaire F., Laporte M., Delpal A. Distinctive roles of furin and TMPRSS2 in SARS-CoV-2 infectivity. J. Virol. 2022:e00128. doi: 10.1128/jvi.00128-22. 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim H.X., Masomian M., Khalid K., Kumar A.U., MacAry P.A., Poh C.L. Identification of B-cell epitopes for eliciting neutralizing antibodies against the SARS-CoV-2 spike protein through bioinformatics and monoclonal antibody targeting. Int. J. Mol. Sci. 2022;23(8):4341. doi: 10.3390/ijms23084341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lazarevic I., Pravica V., Miljanovic D., Cupic M. Immune Evasion of SARS-CoV-2 emerging variants: what have we learnt so far? Viruses. 2021;13(7):1192. doi: 10.3390/v13071192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jalal K., Khan K., Basharat Z., Abbas M.N., Uddin R., Ali F., Khan S.A., Hassan S.S.u. Environmental Science and Pollution Research; 2022. Reverse Vaccinology Approach for Multi-Epitope Centered Vaccine Design against Delta Variant of the SARS-CoV-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Beltran W.F., Lam E.C., Denis K.S., Nitido A.D., Garcia Z.H., Hauser B.M., Feldman J., Pavlovic M.N., Gregory D.J., Poznansky M.C. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell. 2021;184(9):2372–2383. doi: 10.1016/j.cell.2021.03.013. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patone M., Thomas K., Hatch R., San Tan P., Coupland C., Liao W., Mouncey P., Harrison D., Rowan K., Horby P. The Lancet Infectious Diseases; 2021. Mortality and Critical Care Unit Admission Associated with the SARS-CoV-2 Lineage B. 1.1. 7 in England: an Observational Cohort Study. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis C., Logan N., Tyson G., Orton R., Harvey W., Haughney J., Perkins J., Peacock T., Barclay W.S., Cherepanov P. medRxiv; 2021. Reduced Neutralisation of the Delta (B. 1.617. 2) SARS-CoV-2 Variant of Concern Following Vaccination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies N.G., Abbott S., Barnard R.C., Jarvis C.I., Kucharski A.J., Munday J.D., Pearson C.A., Russell T.W., Tully D.C., Washburne A.D. Estimated transmissibility and impact of SARS-CoV-2 lineage B. 1.1. 7 in England. Science. 2021;372(6538) doi: 10.1126/science.abg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uddin R., Jalal K., Khan K., Ul-Haq Z. Re-purposing of hepatitis C virus FDA approved direct acting antivirals as potential SARS-CoV-2 protease inhibitors. J. Mol. Struct. 2022;1250 doi: 10.1016/j.molstruc.2021.131920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang S., Amahong K., Sun X., Lian X., Liu J., Sun H., Lou Y., Zhu F., Qiu Y. The miRNA: a small but powerful RNA for COVID-19. Briefings Bioinf. 2021;22(2):1137–1149. doi: 10.1093/bib/bbab062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell F., Archer B., Laurenson-Schafer H., Jinnai Y., Konings F., Batra N., Pavlin B., Vandemaele K., Van Kerkhove M.D., Jombart T. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Euro Surveill. 2021;26(24) doi: 10.2807/1560-7917.ES.2021.26.24.2100509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glover A., Heathcote J., Krueger D., Rios-Rull J.-V. Optimal age-based vaccination and economic mitigation policies for the second phase of the covid-19 pandemic. J. Econ. Dynam. Control. 2022;140:104306. doi: 10.1016/j.jedc.2022.104306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryckman T., Chin E.T., Prince L., Leidner D., Long E., Studdert D.M., Salomon J.A., Alarid-Escudero F., Andrews J.R., Goldhaber-Fiebert J.D. Outbreaks of COVID-19 variants in US prisons: a mathematical modelling analysis of vaccination and reopening policies. Lancet Public Health. 2021;6(10):e760–e770. doi: 10.1016/S2468-2667(21)00162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma K., Koirala A., Nicolopoulos K., Chiu C., Wood N., Britton P.N. Vaccines for COVID-19: where do we stand in 2021? Paediatr. Respir. Rev. 2021;39:22–31. doi: 10.1016/j.prrv.2021.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chukwudozie O.S., Duru V.C., Ndiribe C.C., Aborode A.T., Oyebanji V.O., Emikpe B.O. The relevance of bioinformatics applications in the discovery of vaccine candidates and potential drugs for COVID-19 treatment. Bioinf. Biol. Insights. 2021;15 doi: 10.1177/11779322211002168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ita K. Coronavirus disease (COVID-19): current status and prospects for drug and vaccine development. Arch. Med. Res. 2021;52(1):15–24. doi: 10.1016/j.arcmed.2020.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lv H., Shi L., Berkenpas J.W., Dao F.-Y., Zulfiqar H., Ding H., Zhang Y., Yang L., Cao R. Application of artificial intelligence and machine learning for COVID-19 drug discovery and vaccine design. Briefings Bioinf. 2021;22(6):bbab320. doi: 10.1093/bib/bbab320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hassam M., Shamsi J.A., Khan A., Al-Harrasi A., Uddin R. Prediction of inhibitory activities of small molecules against Pantothenate synthetase from Mycobacterium tuberculosis using Machine Learning models. Comput. Biol. Med. 2022;145 doi: 10.1016/j.compbiomed.2022.105453. [DOI] [PubMed] [Google Scholar]
- 31.Abbasi K., Razzaghi P., Poso A., Amanlou M., Ghasemi J.B., Masoudi-Nejad A. DeepCDA: deep cross-domain compound–protein affinity prediction through LSTM and convolutional neural networks. Bioinformatics. 2020;36(17):4633–4642. doi: 10.1093/bioinformatics/btaa544. [DOI] [PubMed] [Google Scholar]
- 32.Sajadi S.Z., Zare Chahooki M.A., Gharaghani S., Abbasi K. AutoDTI++: deep unsupervised learning for DTI prediction by autoencoders. BMC Bioinf. 2021;22(1):1–19. doi: 10.1186/s12859-021-04127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar S., Bawa S., Gupta H. Biological activities of quinoline derivatives. Mini Rev. Med. Chem. 2009;9(14):1648–1654. doi: 10.2174/138955709791012247. [DOI] [PubMed] [Google Scholar]
- 34.Shakeel A., Altaf A.A., Qureshi A.M., Badshah A. Thiourea derivatives in drug design and medicinal chemistry: a short review. Drug Des. Med. Chem. 2016;2(1):10. [Google Scholar]
- 35.Kumari A., Singh R.K. Medicinal chemistry of indole derivatives: current to future therapeutic prospectives. Bioorg. Chem. 2019;89 doi: 10.1016/j.bioorg.2019.103021. [DOI] [PubMed] [Google Scholar]
- 36.Zhang L., Lin D., Sun X., Rox K., Hilgenfeld R. 2020. X-Ray Structure of Main Protease of the Novel Coronavirus SARS-CoV-2 Enables Design of α-ketoamide Inhibitors. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel development team NextMove Software; Cambridge, UK: 2013. Open Babel. Access Interconvert Chemical Information. [Google Scholar]
- 38.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wallace A.C., Laskowski R.A., Thornton J.M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995;8(2):127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 40.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 41.Koziara K.B., Stroet M., Malde A.K., Mark A.E. Testing and validation of the Automated Topology Builder (ATB) version 2.0: prediction of hydration free enthalpies. J. Comput. Aided Mol. Des. 2014;28(3):221–233. doi: 10.1007/s10822-014-9713-7. [DOI] [PubMed] [Google Scholar]
- 42.Valdés-Tresanco M.S., Valdés-Tresanco M.E., Valiente P.A., Moreno E. gmx_MMPBSA: a new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theor. Comput. 2021;17(10):6281–6291. doi: 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
- 43.Jalal K., Khan K., Haleem D.J., Uddin R. Silico study to identify new monoamine oxidase type a (MAO-A) selective inhibitors from natural source by virtual screening and molecular dynamics simulation. J. Mol. Struct. 2022;1254 [Google Scholar]
- 44.Uddin R., Jalal K., Khan K. Re-purposing of hepatitis C virus FDA approved direct acting antivirals as potential SARS-CoV-2 protease inhibitors. J. Mol. Struct. 2022;1250 doi: 10.1016/j.molstruc.2021.131920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X., Jiang Z., Yuan X., Wang Y., Huang D., Hu R., Zhou J., Chen F. Nurses reports of actual work hours and preferred work hours per shift among frontline nurses during coronavirus disease 2019 (COVID-19) epidemic: a cross-sectional survey. International Journal of Nursing Studies Advances. 2021;3 doi: 10.1016/j.ijnsa.2021.100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karim S.S.A., Karim Q.A. The Lancet Infectious Diseases; 2021. Omicron SARS-CoV-2 Variant: a New Chapter in the COVID-19 Pandemic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang H., Rao Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021;19(11):685–700. doi: 10.1038/s41579-021-00630-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naqvi A.A.T., Fatima K., Mohammad T., Fatima U., Singh I.K., Singh A., Atif S.M., Hariprasad G., Hasan G.M., Hassan M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim. Biophys. Acta, Mol. Basis Dis. 2020;1866(10):165878. doi: 10.1016/j.bbadis.2020.165878. 165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferreira J.C., Fadl S., Villanueva A.J., Rabeh W.M. vol. 9. 2021. p. 491. (Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-chymotrypsin-like Protease). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh V.K., Chaurasia H., Kumari P., Som A., Mishra R., Srivastava R., Naaz F., Singh A., Singh R.K. Design, synthesis, and molecular dynamics simulation studies of quinoline derivatives as protease inhibitors against SARS-CoV-2. J. Biomol. Struct. Dynam. 2021:1–24. doi: 10.1080/07391102.2021.1946716. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.